

Abstract
Repair pathways of covalent DNA damage are understood in considerable detail due to decades of brilliant biochemical studies by many investigators. An important feature of these experiments is the defined adduct location on oligonucleotide or plasmid substrates that are incubated with purified proteins or cell free extracts. With some exceptions, this certainty is lost when the inquiry shifts to the response of living mammalian cells to the same adducts in genomic DNA. This reflects the limitation of assays, such as those based on immunofluorescence, that are widely used to follow responding proteins in cells exposed to a DNA reactive compound. The lack of effective reagents for adduct detection means that the proximity between responding proteins and an adduct must be assumed. Since these assumptions can be incorrect, models based on in vitro systems may fail to account for observations made in vivo. Here we discuss the use of a detection tag to address the problem of lesion location, as illustrated by our recent work on replication dependent and independent responses to interstrand crosslinks.
Keywords: DNA Damage Response, interstrand crosslink, antigen tag, replication stress, replication independent repair
1. Introduction
It is an oft cited truism that cells are continually exposed to DNA reactive agents from both endogenous and exogenous sources [1]. Throughout the cell cycle helix distorting lesions can activate the DNA Damage Response (DDR), a vast assembly of proteins and other factors [2;3]. The DDR has consequences that are local to the lesion (e.g. repair of the damage, modulation of chromatin structure) and more global (stimulation of stress response pathways, signaling to neighboring cells, etc.). The DDR can also be induced by the encounter of replication forks with blocking lesions, or by inhibitors of DNA synthesis. Again, the response includes functions with activity in the vicinity of the inducing lesion, as well as the stimulation of stress pathways with influence throughout the cell. One of the challenges for the investigator studying cellular responses to DNA damage in the mammalian genome is to distinguish local from global effects, and replication fork dependent from independent events.
The best understood component of the DDR is repair, a process immediate to the lesion. For covalent adducts, such as those introduced by ultraviolet light, or bulky aromatic hydrocarbons, or base oxidation products, the molecular steps involved in replication independent repair are understood in great detail. This reflects the spectacular advances in the biochemistry and genetics of repair during the long history of the field, driven, in part, by experimental approaches that follow the processing of model substrates carrying defined adducts, incubated with purified proteins, or cell extracts. These assays report endpoints at single nucleotide precision. Consequently, the spatial relationship between a lesion and molecular operations fundamental to repair pathways (incision, excision, resection, etc.) can be accurately defined. There are also methods for detection, again at single nucleotide resolution, of a few kinds of DNA damage in the mammalian genome [4;5]. Recently, next-generation sequencing of Nucleotide Excision Repair (NER) intermediates produced genomic location and repair maps of adducts introduced by UV or benzo[a]pyrene in human cells [6;7]. The success of this approach was based on two essentials: clearly defined intermediates in the repair pathway, and effective reagents for their capture. However, these requirements are not met for most lesions.
Another experimental strategy for following cellular responses to DNA damage is based on immunofluorescence (IF). With a few exceptions, the direct relationship between brightly colored foci and the physical location of a lesion is inferred rather than demonstrated. This follows from the lack of effective detection and display strategies for the adducts in question. Consequently, surrogate markers are commonly taken as denoting sites of damage in DNA. The best known, the phosphorylated form of the histone variant H2AX (γ-H2AX), is routinely taken as a prima facie evidence of a double strand break (DSB). While this is often a reasonable conclusion, in a formal sense, the appearance of γ-H2AX simply reflects the activity of one or more of the kinases (ATM, ATR, DNA-PK) for which H2AX is a substrate. These kinases can be activated by DNA structures other than DSBs [8–12]. Thus, interpretation of foci of γ-H2AX and similar markers is compromised by ambiguity in the identity of the initiating structure. Furthermore, DDR induced alterations in chromatin spread over great distances [2;13], resulting in uncertainty as to the location of the provocative lesion. For research on the local consequences of DNA damage it would be desirable to have reagents that could directly display the location of lesions within affected cells.
Remarkably, despite the long history of the DNA repair field, and the power and popularity of immunofluorescence technologies, effective methods for directly marking lesions in situ are limited to relatively few targets. For example, DSBs induced by ionizing radiation can be displayed in fixed cells by an end labeling technique described only recently [14]. This approach has been used in conjunction with the antibody-based Proximity Ligation Assay (PLA) [15] to identify DDR factors proximal to DSBs. Antibodies against ultraviolet photoproducts were developed many years ago [16]. They played an important role in a well-known study of the dynamics of nucleotide excision repair factors responding to UV lesions [17], and in subsequent reports from other laboratories [18;19]. They, and antibodies against benzo[a]pyrene adducts, were essential reagents in the production of genomic repair maps mentioned above [7]. In a study of Base Excision Repair (BER) antibodies against 8-oxoG were combined with laser localization to characterize protein recruitment to oxidized base damage [20].
The universe of DNA adducts is enormously greater than these few examples, including many formed by compounds of considerable relevance to human health. Among them are the interstrand crosslinks (ICLs), highly toxic lesions that are the focus of this article. Drugs that induce them, such as cisplatin, mitomycin C, and the nitrogen mustards, are clinically significant in cancer chemotherapy. Another, psoralen, has been used for thousands of years to treat skin disorders [21;22]. Despite many years of use (nitrogen mustard was the first cancer chemotherapy drug), our knowledge of ICL repair, although improving in recent years, is far from complete. In view of the importance of these drugs, an understanding of their repair pathways would support improved treatment protocols and might reduce their considerable side effects.
Although antibodies against DNA modifications introduced by these compounds have been described, they have not been widely used for detailing cellular responses to ICLs. For example, measurements of different cisplatin adducts are understandably dominated by the major adducts, which are single strand reaction products [23;24] [25]. Antibodies against psoralen adducts were generated decades ago [26], but suffered from specificity problems and have not been utilized in recent years.
In the absence of effective immunologic reagents, there are other established methods for studying repair of these important structures. The alkaline comet assay, based on the resistance of crosslinked DNA to denaturation, has been used to follow the key step in ICL repair- the “unhooking” of the two strands [27–29]. However, we have found that the concentrations of crosslinking compound needed to compact nuclei in alkali (the starting point of the unhooking assay) are extremely toxic. As a result, the analysis of unhooking activity is, in effect, conducted in biochemically competent cells that are dead or dying. Indeed, it has been suggested that apoptotic cells might confound interpretation of comet assays [30]. More recently, an alkaline unwinding assay has been used to analyze ICL repair dynamics [31]. In accord with our experience, these authors reported that the cells were able to unhook many more ICLs than the level that reduced cell survival by 50%. Furthermore, while the alkaline assays might be instructive, they cannot be used for in situ assays.
The most productive approaches for studying the repair of ICLs have been based on plasmids carrying defined ICLs introduced into live cells or cell free extracts [32–37]. This strategy has been very powerful, and has received considerable attention, but cannot be informative as to events at the level of genomic ICLs in live cells.
2. Visualizing the location of genomic ICLs
To address questions about replication dependent and independent responses to ICLs, we developed an approach for marking their location following formation in living cells. Our experimental schemes exploit the properties of psoralen. Psoralens are intercalators that require photoactivation, by long wave UV light (UVA), in order to react with DNA [38]. In contrast to the other common crosslinking compounds, psoralens, such as the trimethyl derivative (TMP), form a very high proportion of ICLs relative to monoadducts, in excess of 90% [39;40]. Additionally, they can be chemically modified without loss of crosslinking activity, and so can be joined to moieties that afford easy detection. For example, psoralens linked to biotin have been employed as labeling reagents and commercial versions are available. The biotin conjugates would appear to offer an obvious tool for display of psoralen adducts in cellular DNA. However, biotin is a cofactor for carboxylases, and we found that most of the covalently bound biotin-psoralen after UVA was cytoplasmic/mitochondrial [41], indicating that the localization of the compound was driven by biotin. Ideally, the tag should be a passive element and not interfere with the interaction of psoralen and DNA. We also prepared the digoxigenin conjugate of TMP (Dig-TMP) (Fig 1). Digoxigenin is a plant sterol and is not found in mammalian cells. It has been used for many years as an immunotag and there are commercial antibodies against it. This conjugate had the same high ratio of genomic ICLs: monoadducts as the parent compound and gave a largely nuclear signal in cells exposed to a UVA lamp [41;42]. The Dig marker was removed over time in repair proficient cells, but not in repair deficient cells. This indicated that the tag was inherently stable in the cellular environment, and that the persistence of the ICLs was a function of the repair competence of the cell. We have made extensive use of this derivative to address two issues: the events that follow collisions of replication forks with the powerful block presented by an ICL, and; replication independent responses to ICLs.
Fig.1.
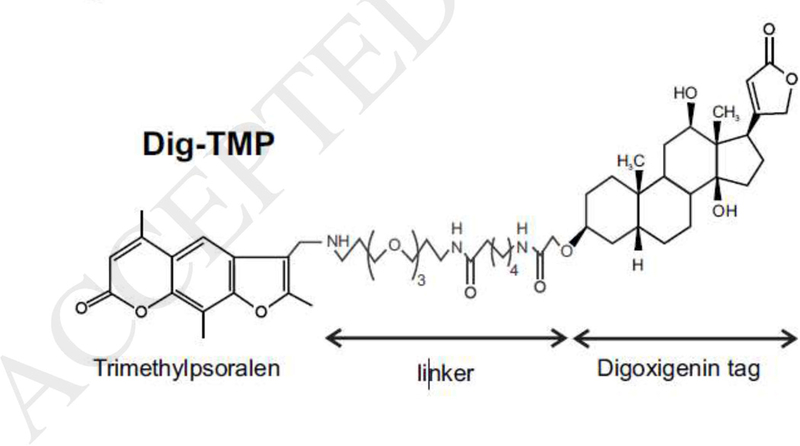
The structure of Digoxigenin tagged psoralen
3. The encounter of replication forks with blocks to the replisome
It is well established that passage of the replisome through the eukaryotic genome is not as straightforward as depicted in the textbooks. Indeed, it has become a common theme of publications on the topic to begin by noting the numerous impediments faced by the replication machinery [43]. These include non-covalent blocks [44] such as alternate DNA structures [45], DNA protein complexes [46], nucleotide depletion [47], encounters with RNA polymerase [48], among others. Of course, there are the well-known perturbations to the double helix: nicks and breaks in DNA [49], and the many covalent modifications introduced by endogenous and exogenous DNA reactive agents [1]. Failure to successfully negotiate these challenges has multiple consequences- genome rearrangement, mutagenesis, quiescence, senescence, apoptosis, induction of inflammatory pathways, neoplasia, etc, [50] [51;52]. ICLs, which block unwinding of DNA, are particularly challenging obstacles to the replisome, with severe consequences for cells that fail to resolve them.
3.1. The encounter of replication forks with ICLs
The interpretation of many studies of cellular responses to genomic ICLs assumes a replication fork encounter as the initiating event. This is the standard explanation for the appearance of phase foci of DDR proteins in cells treated with a crosslinking compound (but see below). Another popular experimental approach involves replication tract analysis in DNA fibers from cells exposed to a crosslinking agent. Typically, tract length declines, due, it is argued, to replisome collisions with blocks [53]. Although widely used, tract length measurements cannot distinguish the consequences of a “global” stress (that affects all replication forks, regardless of an encounter with a DNA lesion) from a “local” response (that reflects the collision of replisomes with an obstruction in the DNA). As a result, the interpretation of many of these studies is necessarily conjectural as to an actual physical interaction of the replisome with a lesion. Furthermore, while there has been considerable attention to the structure and functional biochemistry of the replisome (see below) there have been far fewer studies on the composition of the complex following encounters with blocks in live cells.
To distinguish global from local effects of replication fork encounters with DNA adducts, we treated cells with Dig -TMP/UVA. Then they were pulsed with nucleoside analogues, which were incorporated into DNA during S phase, and displayed on fibers by immunofluorescence. he Dig tag was detected with an immunequantum dot. These have received extensive use for single molecule analyses [54]. In our experiments less than 10% of tracts had an encounter with ICLs [42]. However, comparison of the lengths of tracts from cells treated with only UVA with those exposed to Dig-TMP/UVA revealed that tracts without an ICL encounter (from cells treated with Dig-TMP/UVA) were shorter on average than tracts from cells exposed to only UVA. Those with an encounter were shorter still. This argued that a global stress response reduced progression of all the forks, not just, as anticipated, those that made physical contact with the adduct. How global stress slows fork progression has yet to be determined.
The replication tract analysis also revealed an unexpected result. Based on traditional models and more recent work from the Walter laboratory we expected to find evidence for single and double fork (two forks from either side colliding at the ICL) events [55;56]. These forks would be either from replisomes moving at the time of treatment, or from dormant origins activated by the replication stress. While both single and double fork collisions were observed, there was another pattern, one that we termed fork traverse. It was consistent with the restart of replication on the side of the ICL distal to the initial encounter (Fig. 2A, B) [42], and was the most frequent result. This was surprising, since ever since their discovery ICLs have been regarded as insurmountable blocks to replication [57]. Restart required only a few minutes and was promoted by the translocase activity of FANCM protein, a member of the Fanconi Anemia family of proteins.
Fig. 2. Interpretation of patterns of tracts with encounters with an ICL.
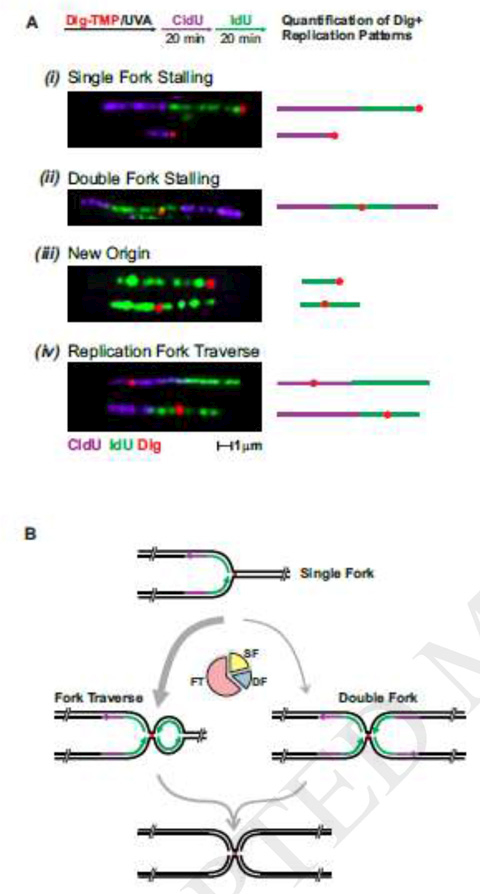
A. Experimental scheme and representative images of the patterns observed. The replication restart pattern is termed fork traverse.
B. Scheme summarizing the possible outcomes observed when the replication fork encounters a Dig-TMP/UVA induced ICL. The pie chart shows the relative frequency of the major patterns in wt cells. FT, replication fork traverse, SF, single fork, DF, double fork.
3.2. The challenge posed by replisome structure
The concept of a replication traverse of an ICL would appear to be challenged by the structure of the replisome. The core of the apparatus is a heterohexameric offset open ring formed by the MCM (M) proteins [58;59]. The MCM complex is loaded on duplex DNA only in G1 phase cells and activated to a functional helicase in S phase by the binding of additional proteins, including CDC45 (C) and the GINS (G) complex [60–62]. The assembly of the active MG complex is accompanied by melting of DNA and the encirclement of the template for leading strand synthesis by the MCM ring, which is locked by GINS, CDC45, and associated proteins. he collision of the locked ring with an ICL would seem to validate the longstanding view of ICLs as impassable by the replisome [63]. So, how might replication restart past this block?
There are several candidate explanations. We found that adducts introduced by Dig-angelicin/UVA (which can only form monoadducts) did not impede replication. Thus, conversion of an ICL to a monoadduct, via unhooking by repair activities, would relieve the block. However, the restart patterns were observed in repair deficient cells. Additionally, in a contemporary version of the Meselson Stahl experiment, we demonstrated that both parental template strands were associated with replication tracts that encountered an ICL. Replication past an unhooked ICL would produce a replication tract associated with only one of the template strands, as shown in the experiment with Dig-angelicin [42].
Another possibility- that a CMG might assemble de novo on the distal side of the ICL- seems unlikely since origin licensing is confined to G1 phase. A previously loaded replisome at a “dormant origin” might be activated downstream of the ICL. However, the traverse pattern frequency was unaffected by incubation of cells with inhibitors of dormant origin activation. Additionally, knockdown of MCM proteins to levels that precluded dormant origin activation also failed to influence traverse frequency [42]. RNA in R loops can serve as a primer and can rescue stalled forks [64]. However, we find that incubation with different RNA polymerase inhibitors, which suppress R loop formation, has no effect on the encounter patterns (Huang, et al., unpublished). Furthermore, R loops rise in FANCM deficient cells [65], but traverse frequencies declined in these cells. The restart patterns were unaffected by repair competence of the cells or by the absence of homologous recombination functions. In the absence of compelling alternate proposals, we have turned our attention to the possibility that the explanation for the restart lies in the CMG complex.
Some insight into this problem comes from the Costanzo laboratory. They followed replication on chromatin in a cell free extract from Xenopus eggs. They added S1 nuclease to the mixture on the anticipation that single strand DNA at replication forks would be cleaved, generating a double strand break and collapsing the fork. They found that this was accompanied by a loss of the GINS complex from the CMG, which would inactivate the replisome [66]. Reloading of the GINS proteins, reconstruction of the CMG, and restart of the forks was dependent on RAD51 and the nuclease activity of MRE11. These results indicate that collapse of the fork triggers a modulation of the protein composition of the CMG, marked by release of the GINS proteins (and perhaps others). One of the most important findings was that the process was reversible-following reconstruction of the fork the GINS proteins could re-associate, reactivating the replisome.
In recent work we examined the composition of CMG complexes in cells exposed to psoralen/UVA. The bulk of the replisomes showed no change in GINS association. However, as noted above, less than 10% of replisomes have a collision with an ICL under the conditions of the experiment. When the analysis was extended to those replisomes that had an ICL encounter we found that the GINS proteins were lost from the CMG complexes (Huang et al, unpublished). These results were in accord with those from the Costanzo group. Because the challenge to the replication machinery was quite different in the two systems, it seems likely that multiple forms of replication stress can modulate the composition of the replisome. Furthermore, since the GINS complex is involved in closing the MCM ring, the loss of these proteins would be consistent with opening of the ring. This would be an essential intermediate in a scheme in which the open ring (“CM”) structure could move past the ICL , prior to reconstruction, ring closure, and the restart of replication.
Although the notion of replication restart past an intact ICL was unexpected, the observation supports what might be termed the “replication imperative”. Since the ICLs were intact at the time of the restart, the results imply an evolutionary advantage to completing S phase, leaving adduct removal for a later time. Restart of replication downstream of single strand blocks, such as UV photoproducts, is a well- established mechanism through which cells finish DNA synthesis, reserving repair of the block to a post replication period [67–69]. Biology appears to have extended this principle to ICLs as well.
4. Replication independent responses to ICLs
Although replication dependent responses to ICLs have dominated research in recent years, it should be noted that most cells in the body are not in active cycle, and a substantial fraction of cells in many tumors are dormant [70]. Given the prominence of ICL forming drugs in cancer chemotherapy, it is important to understand how cells respond to ICLs outside the context of a replication fork. To address these questions, we took advantage of the UVA dependent reactivity of psoralen. We employed a confocal microscope equipped with a 365 nm laser to direct ICL formation to defined regions of interest (ROI) in nuclei in living cells [71]. This method raised the question of how the frequency and distribution on genomic DNA of laser localized ICLs compared to those from psoralen activated by a conventional UVA lamp (used in our replication tract experiments). To make this comparison we took advantage of the DNA fiber technique described above. We measured inter-adduct distances on fibers prepared from cells exposed to Dig-TMP and a UVA lamp. This measurement revealed inter-ICL distances ranging from less than 5 kb to more than 180 kb. In order to make the same determination in DNA from cells exposed to the Dig-TMP/laser combination, we introduced the localized ICLs into a cluster of cells in the immediate vicinity of a mark on the center of the culture plate. Those cells were harvested, fibers prepared, and the Dig tag displayed with an immunoquantum dot, as before. This analysis revealed a striking similarity of ICL spacing and distribution on the fibers from cells exposed to the two light sources [72]. This result was important because it indicated that the laser localized ICLs were well separated, rather than clustered close to one another. Consequently, results of experiments that monitored protein recruitment and L repair could be interpreted as reflecting events at individual ICLs.
The laser localization approach allows for control of the time of crosslink formation, such that the kinetics of protein recruitment to the ICL stripe can be determined with a defined start time. The decline of the intensity of the Dig tag as a function of time after ICL formation serves as a measure of the efficiency of ICL repair, which is influenced by the activity (or lack thereof) of relevant proteins (Fig 3A). Our results demonstrate ICL repair throughout the cell cycle, including in G1 phase cells [71]. Furthermore, the psoralen ICLs induce a conventional DDR, in which the Fanconi Anemia (FA) proteins, and associated factors participate [73–75].
Fig. 3. Strategies designed to study replication independent ICL repair employing laser localized tagged ICLs.
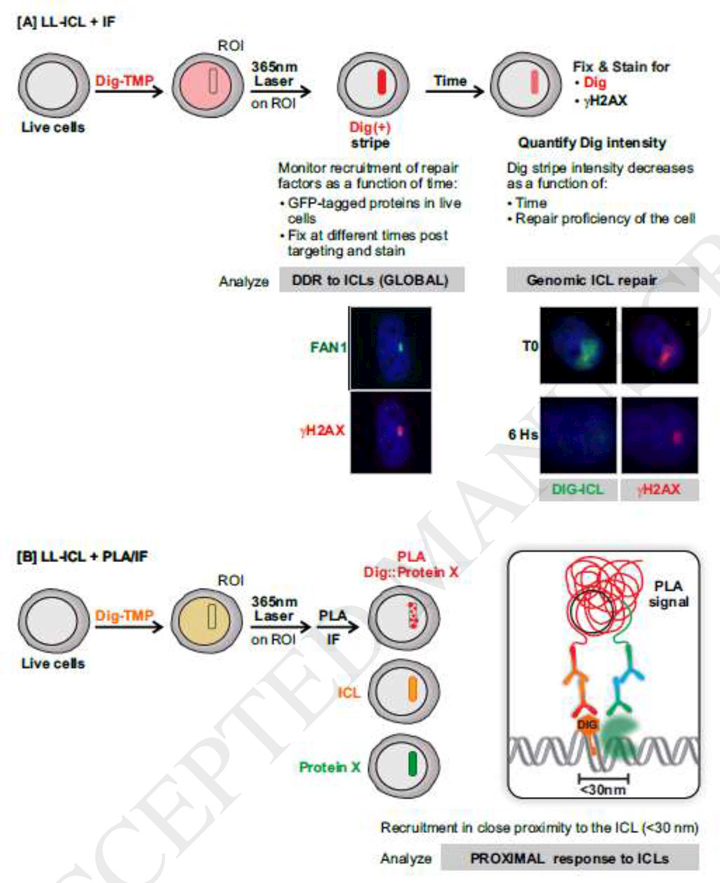
A. Laser-localized ICLs combined with immunofluorescence to study DDR activation by, and repair of, laser localized ICLs. γ-H2AX and the FAN1 nuclease appear in ICL stripes. Monitoring the Dig tag reveals repair of the ICL and the greater persistence of γ-H2AX.
B. Laser-localized ICL induction and proximity ligation assay (PLA) to distinguish DDR proteins that are proximal or distal to the tagged ICL.
We find that the induction of the DDR by the psoralen ICLs is pan cell cycle (Paramasivam et al. unpublished). It is dependent on ATR, not ATM or DNAPK, arguing against DSBs as the inducing lesion. ATR activation is generally associated with stalled replication forks and stretches of single strand DNA coated by RPA [76]. ssDNA appears in cells at sites of DNA repair [77], after the restart of DNA synthesis downstream of polymerase blocking adducts [68;78], and in cells in which nucleotide pools are depleted [79]. The prevailing experimental model of replication stress involves treatment of cells with hydroxyurea, which inhibits ribonucleotide reductase, thus depriving the replisome of dNTPs. Unwinding by the CMG helicase is not immediately inhibited, with the result that stretches of ssDNA, coated by RPA, appear. This experimental model has been very influential such that ATR activation is often associated with replication stress due to inhibitors of DNA synthesis. Furthermore, the activation of ATR by RPA coated single strand DNA is so well established that it is often assumed that the inverse is true-that ATR activation implies the presence of ssDNA. However, ATR can be activated by DNA structures that are independent of replication and RPA. These include bulky DNA adducts, mediated in part by Topoisomerase II-Binding Protein (TOPBP1) [80–82]. Recent work also implicates mechanical stress [83]. These inducers are effective throughout the cell cycle, and so ATR can be activated without the involvement of the replication fork [84].
Our results indicate that psoralen ICLs are sufficiently distorting to trigger an ATR driven DDR. that begins within seconds of formation. However, the kinetics of replication fork encounters are different. It is unlikely that an ICL will be introduced in the immediate path of the replisome. We find that replication fork collisions accumulate over a much longer period, reaching a plateau after about an hour [42]. Consequently, the replisome will encounter an ICL in the context of an already active DDR cascade. The models invoking activation of the DDR by fork stalling are so dominant that little attention has been given to the influence on replisomes of entry into a chromatin region already marked by the DDR. On the other hand, some lesions, such as ICLs formed by mitomycin C [85], are relatively non-distorting, and unlikely to trigger a DDR, except as they block replication or transcription. In these circumstances the conventional models would apply. Most studies of ICL inducing compounds ignore these distinctions.
5. The interaction of tagged ICLs and DDR proteins
The limit of resolution of confocal fluorescence microscopy is 200 nm. As noted above, this introduces substantial ambiguity in the interpretation of immunofluorescent foci in terms of the spatial relationship between an inducing lesion and repair and DDR proteins. he PLA between a tagged lesion (a DSB in the example from Galbiati et al [14]) and a protein of interest would appear to offer an improvement. We have employed the PLA between the tag on the ICL and DDR proteins as a means of distinguishing those that are in close proximity to the lesion from those that are not (see Fig. 3B). We found that while some DDR proteins were proximal to the ICLs, others were not. The assay is quantitative and can be used to determine the genetic requirements for lesion proximal recruitment of proteins of interest. Although the limit of resolution of the PLA is 30–40 nm, this is imposed by the size of the primary and secondary antibodies that are the basis of the assay. This limit will be reduced as smaller detection reagents, such as camelid nanobodies or aptamers become available [86–88].
In this article we have discussed the utility of a tagged DNA reactive compound for addressing a questions about the response to genomic adducts in living mammalian cells. Although our experience is limited to the psoralens, this is a generalizable approach that should be applicable to a variety of different lesions, particularly in the realm of genotoxic chemotherapy drugs, and their numerous derivatives. The use of a tag, for which commercial antibodies are available, eliminates the need to generate and characterize antibodies de novo for each adduct of interest. Furthermore, the progress in super resolution microscopy offers the promise of continued refinements in our analysis of cellular responses to genomic lesions.
Acknowledgements
This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging (Z01 AG000746–08), and by the Fundamental Research Funds for the Central Universities from Hunan University (531109010055) and the National Natural Science Foundation of China (21708007.
Abbreviation
- DDR
DNA Damage Response
- Dig
digoxigenin
- ICL
interstrand crosslink
- NER
Nucleotide Excision Repair
- IF
Immunofluorescence
- DSB
Double Strand Break
- PLA
Proximity Ligation Assay
- BER
Base xcision Repair
- 8-oxoG
8-Oxo-2’ deoxyguanosine
- MCM
Minichromosome Maintenance
- GINS
Go-Ichi-Ni-San
- FA
Fanconi Anemia
- DNA-PK
DNA dependent protein kinase
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia-mutated and rad3-related
- RPA
replication protein A
- ROI
regions of interest
- UVA
long wave ultraviolet light (315–400 nm)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
There are no conflicts of interest
References
- [1].Lindahl T. The Intrinsic Fragility of DNA (Nobel Lecture), Angew.Chem.Int.Ed Engl, 55, (2016) 8528–8534. [DOI] [PubMed] [Google Scholar]
- [2].Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives, Mol.Cell, 40, (2010) 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lu WT, Hawley BR, Skalka GL, Baldock RA, Smith EM, Bader AS, Malewicz M, Watts FZ, Wilczynska A, Bushell M. Drosha drives the formation of DNA:RNA hybrids around DNA break sites to facilitate DNA repair, Nat.Commun, 9, (2018) 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Besaratinia A, Pfeifer GP. DNA-lesion mapping in mammalian cells, Methods, 48, (2009) 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hu J, Adar S, Selby CP, Lieb JD, Sancar A. Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution, Genes Dev, 29, (2015) 948–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hu J, Adebali O, Adar S, Sancar A. Dynamic maps of UV damage formation and repair for the human genome, Proc.Natl.Acad.Sci.U.S.A, 114, (2017) 6758–6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li W, Hu J, Adebali O, Adar S, Yang Y, Chiou YY, Sancar A. Human genome-wide repair map of DNA damage caused by the cigarette smoke carcinogen benzo[a]pyrene, Proc.Natl.Acad.Sci.U.S.A, 114, (2017) 6752–6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation, Nature, 421, (2003) 499–506. [DOI] [PubMed] [Google Scholar]
- [9].Rybak P, Hoang A, Bujnowicz L, Bernas T, Berniak K, Zarebski M, Darzynkiewicz Z, Dobrucki J. Low level phosphorylation of histone H2AX on serine 139 (gammaH2AX) is not associated with DNA double-strand breaks, Oncotarget, 7, (2016) 49574–49587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Soutoglou E, Misteli T. Activation of the cellular DNA damage response in the absence of DNA lesions, Science, 320, (2008) 1507–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks, Proc.Natl.Acad.Sci.U.S.A, 103, (2006) 9891–9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Baure J, Izadi A, Suarez V, Giedzinski E, Cleaver JE, Fike JR, Limoli CL. Histone H2AX phosphorylation in response to changes in chromatin structure induced by altered osmolarity, Mutagenesis, 24, (2009) 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tang JB, Greenberg RA. Connecting the Dots: Interplay between Ubiquitylation and SUMOylation at DNA Double-Strand Breaks, Genes Cancer, 1, (2010) 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Galbiati A, Beausejour C, d’Adda di FF. A novel single-cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues, Aging Cell, 16, (2017) 422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Soderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, Larsson LG, Landegren U. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay, Methods, 45, (2008) 227–232. [DOI] [PubMed] [Google Scholar]
- [16].Clarkson JM, Mitchell DL, Adair GM. The use of an immunological probe to measure the kinetics of DNA repair in normal and UV-sensitive mammalian cell lines, Mutat.Res, 112, (1983) 287–299. [DOI] [PubMed] [Google Scholar]
- [17].Volker M, Mone MJ, Karmakar P, van HA, Schul W, Vermeulen W, Hoeijmakers JH, VAN DR, van Zeeland AA, Mullenders LH. Sequential assembly of the nucleotide excision repair factors in vivo, Mol.Cell, 8, (2001) 213–224. [DOI] [PubMed] [Google Scholar]
- [18].Oh KS, Imoto K, Emmert S, Tamura D, Digiovanna JJ, Kraemer KH. Nucleotide excision repair proteins rapidly accumulate but fail to persist in human XP-E (DDB2 mutant) cells, Photochem.Photobiol, 87, (2011) 729–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sertic S, Roma S, Plevani P, Lazzaro F, Muzi-Falconi M. Study of UV-induced DNA Repair Factor Recruitment: Kinetics and Dynamics, Methods Mol.Biol, 1672, (2018) 101–105. [DOI] [PubMed] [Google Scholar]
- [20].Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, Wilson SH, Yasui A. In situ analysis of repair processes for oxidative DNA damage in mammalian cells, Proc.Natl.Acad.Sci.U.S.A, 101, (2004) 13738–13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Benedetto AV. The psoralens. An historical perspective, Cutis, 20, (1977) 469–471. [PubMed] [Google Scholar]
- [22].Fitzpatrick TB, Pathak MA. Research and development of oral psoralen and longwave radiation photochemotherapy: 2000 B.C.−1982 A.D, Natl.Cancer Inst.Monogr, 66, (1984) 3–11. [PubMed] [Google Scholar]
- [23].Liedert B, Pluim D, Schellens J, Thomale J. Adduct-specific monoclonal antibodies for the measurement of cisplatin-induced DNA lesions in individual cell nuclei, Nucleic Acids Res, 34, (2006) e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Meczes EL, Azim-Araghi A, Ottley CJ, Pearson DG, Tilby MJ. Specific adducts recognised by a monoclonal antibody against cisplatin-modified DNA, Biochem.Pharmacol, 70, (2005) 1717–1725. [DOI] [PubMed] [Google Scholar]
- [25].Fichtinger-Schepman AM, van Oosterom AT, Lohman PH, Berends F. cis-Diamminedichloroplatinum(II)-induced DNA adducts in peripheral leukocytes from seven cancer patients: quantitative immunochemical detection of the adduct induction and removal after a single dose of cis-diamminedichloroplatinum(II), Cancer Res, 47, (1987) 3000–3004. [PubMed] [Google Scholar]
- [26].Santella RM, Gasparro FP, Edelson RL. Quantification of methoxsalen-DNA adducts with specific antibodies, IARC Sci.Publ, (1986) 127–139. [PubMed] [Google Scholar]
- [27].Almeida GM, Duarte TL, Steward WP, Jones GD. Detection of oxaliplatin-induced DNA crosslinks in vitro and in cancer patients using the alkaline comet assay, DNA Repair (Amst), 5, (2006) 219–225. [DOI] [PubMed] [Google Scholar]
- [28].Rothfuss A, Grompe M. Repair Kinetics of Genomic Interstrand DNA Cross-Links: Evidence for DNA Double-Strand Break-Dependent Activation of the Fanconi Anemia/BRCA Pathway, Mol.Cell Biol, 24, (2004) 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Richards S, Liu ST, Majumdar A, Liu JL, Nairn RS, Bernier M, Maher V, Seidman MM. Triplex targeted genomic crosslinks enter separable deletion and base substitution pathways Nucleic Acids Res, 33, (2005) 5382–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kumaresan KR, Sridharan DM, McMahon LW, Lambert MW. Deficiency in incisions produced by XPF at the site of a DNA interstrand cross-link in Fanconi anemia cells, Biochemistry, 46, (2007) 14359–14368. [DOI] [PubMed] [Google Scholar]
- [31].Vare D, Johansson F, Persson JO, Erixon K, Jenssen D. Quantification and repair of psoralen-induced interstrand crosslinks in human cells, Toxicol.Lett, 226, (2014) 343–350. [DOI] [PubMed] [Google Scholar]
- [32].Shen X, Do H, Li Y, Chung WH, Tomasz M, de Winter JP, Xia B, Elledge SJ, Wang W, Li L. Recruitment of fanconi anemia and breast cancer proteins to DNA damage sites is differentially governed by replication, Mol.Cell, 35, (2009) 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Raschle M, Knipsheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, Walter JC. Mechanism of replication-coupled DNA interstrand crosslink repair, Cell, 134, (2008) 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Semlow DR, Zhang J, Budzowska M, Drohat AC, Walter JC. Replication-Dependent Unhooking of DNA Interstrand Cross-Links by the NEIL3 Glycosylase, Cell, 167, (2016) 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Williams HL, Gottesman ME, Gautier J. Replication-Independent Repair of DNA Interstrand Crosslinks, Mol.Cell, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Enoiu M, Ho TV, Long DT, Walter JC, Scharer OD. Construction of plasmids containing site-specific DNA interstrand cross-links for biochemical and cell biological studies, Methods Mol.Biol, 920, (2012) 203–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kato N, Kawasoe Y, Williams H, Coates E, Roy U, Shi Y, Beese LS, Scharer OD, Yan H, Gottesman ME, Takahashi TS, Gautier J. Sensing and Processing of DNA Interstrand Crosslinks by the Mismatch Repair Pathway, Cell Rep, 21, (2017) 1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hearst JE. Psoralen photochemistry, Annu.Rev.Biophys.Bioeng, 10, (1981) 69–86. [DOI] [PubMed] [Google Scholar]
- [39].Lai C, Cao H, Hearst JE, Corash L, Luo H, Wang Y. Quantitative analysis of DNA interstrand cross-links and monoadducts formed in human cells induced by psoralens and UVA irradiation, Anal.Chem, 80, (2008) 8790–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cao H, Hearst JE, Corash L, Wang Y. LC-MS/MS for the detection of DNA interstrand cross-links formed by 8-methoxypsoralen and UVA irradiation in human cells, Anal.Chem, 80, (2008) 2932–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Thazhathveetil AK, Liu ST, Indig FE, Seidman MM. Psoralen conjugates for visualization of genomic interstrand cross-links localized by laser photoactivation, Bioconjug.Chem, 18, (2007) 431–437. [DOI] [PubMed] [Google Scholar]
- [42].Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, Seidman MM. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks, Mol.Cell, 52, (2013) 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zeman MK, Cimprich KA. Causes and consequences of replication stress, Nat.Cell Biol, 16, (2014) 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gadaleta MC, Noguchi E. Regulation of DNA Replication through Natural Impediments in the Eukaryotic Genome, Genes (Basel), 8, (2017). [Google Scholar]
- [45].Rizzo A, Salvati E, Porru M, D’Angelo C, Stevens MF, D’Incalci M, Leonetti C, Gilson E, Zupi G, Biroccio A. Stabilization of quadruplex DNA perturbs telomere replication leading to the activation of an ATR-dependent ATM signaling pathway, Nucleic Acids Res, 37, (2009) 5353–5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Beuzer P, Quivy JP, Almouzni G. Establishment of a replication fork barrier following induction of DNA binding in mammalian cells, Cell Cycle, 13, (2014) 1607–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development, Cell, 145, (2011) 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Garcia-Muse T, Aguilera A. Transcription-replication conflicts: how they occur and how they are resolved, Nat.Rev.Mol.Cell Biol, 17, (2016) 553–563. [DOI] [PubMed] [Google Scholar]
- [49].Kitao H, Iimori M, Kataoka Y, Wakasa T, Tokunaga E, Saeki H, Oki E, Maehara Y. DNA replication stress and cancer chemotherapy, Cancer Sci, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Venkatachalam G, Surana U, Clement MV. Replication stress-induced endogenous DNA damage drives cellular senescence induced by a sub-lethal oxidative stress, Nucleic Acids Res, 45, (2017) 10564–10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wunderlich R, Ruehle PF, Deloch L, Unger K, Hess J, Zitzelsberger H, Lauber K, Frey B, Gaipl US. Interconnection between DNA damage, senescence, inflammation, and cancer, Front Biosci.(Landmark.Ed), 22, (2017) 348–369. [DOI] [PubMed] [Google Scholar]
- [52].Barr AR, Cooper S, Heldt FS, Butera F, Stoy H, Mansfeld J, Novak B, Bakal C. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression, Nat.Commun, 8, (2017) 14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Vare D, Groth P, Carlsson R, Johansson F, Erixon K, Jenssen D. DNA interstrand crosslinks induce a potent replication block followed by formation and repair of double strand breaks in intact mammalian cells, DNA Repair (Amst), 11, (2012) 976–985. [DOI] [PubMed] [Google Scholar]
- [54].Kong M, Beckwitt EC, Springall L, Kad NM, Van HB. Single-Molecule Methods for Nucleotide Excision Repair: Building a System to Watch Repair in Real Time, Methods Enzymol, 592, (2017) 213–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Knipscheer P, Raschle M, Scharer OD, Walter JC. Replication-coupled DNA interstrand cross-link repair in Xenopus egg extracts, Methods Mol.Biol, 920, (2012) 221–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang J, Walter JC. Mechanism and regulation of incisions during DNA interstrand cross-link repair, DNA Repair (Amst), 19, (2014) 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Marmur J, Grossman L. Ultraviolet light induced linking of deoxyribonucleic acid strands and its reversal by photoreactivating enzyme, Proc.Natl.Acad.Sci.U.S.A, 47, (1961) 778–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhai Y, Tye BK. Structure of the MCM2–7 Double Hexamer and Its Implications for the Mechanistic Functions of the Mcm2–7 Complex, Adv.Exp.Med.Biol, 1042, (2017) 189–205. [DOI] [PubMed] [Google Scholar]
- [59].Abid AF, Costa A. The MCM Helicase Motor of the Eukaryotic Replisome, J.Mol.Biol, 428, (2016) 1822–1832. [DOI] [PubMed] [Google Scholar]
- [60].Yeeles JT, Deegan TD, Janska A, Early A, Diffley JF. Regulated eukaryotic DNA replication origin firing with purified proteins, Nature, 519, (2015) 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Georgescu R, Yuan Z, Bai L, de Luna Almeida SR, Sun J, Zhang D, Yurieva O, Li H, O’Donnell ME. Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation, Proc.Natl.Acad.Sci.U.S.A, 114, (2017) E697–E706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Costa A, Ilves I, Tamberg N, Petojevic T, Nogales E, Botchan MR, Berger JM. The structural basis for MCM2–7 helicase activation by GINS and Cdc45, Nat.Struct.Mol.Biol, 18, (2011) 471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Trakselis MA, Seidman MM, Brosh RM Jr. Mechanistic insights into how CMG helicase facilitates replication past DNA roadblocks, DNA Repair (Amst), 55, (2017) 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Camps M, Loeb LA. Critical role of R-loops in processing replication blocks, Front Biosci, 10, (2005) 689–698. [DOI] [PubMed] [Google Scholar]
- [65].Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez MD, Liang CC, Cohn MA, Gibbons RJ, Deans AJ, Niedzwiedz W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription, Mol.Cell, 60, (2015) 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hashimoto Y, Puddu F, Costanzo V. RAD51 and MRE11 dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks, Nat.Struct.Mol.Biol, 19, (2011) 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Rupp WD, Howard-Flanders P. Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation, J.Mol.Biol, 31, (1968) 291–304. [DOI] [PubMed] [Google Scholar]
- [68].Lehmann AR, Fuchs RP. Gaps and forks in DNA replication: Rediscovering old models, DNA Repair (Amst), 5, (2006) 1495–1498. [DOI] [PubMed] [Google Scholar]
- [69].Yeeles JT, Poli J, Marians KJ, Pasero P. Rescuing stalled or damaged replication forks, Cold Spring Harb.Perspect.Biol, 5, (2013) a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Evans EB, Lin SY. New insights into tumor dormancy: Targeting DNA repair pathways, World J.Clin.Oncol, 6, (2015) 80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Muniandy PA, Thapa D, Thazhathveetil AK, Liu ST, Seidman MM. Repair of laserlocalized DNA interstrand cross-links in G1 phase mammalian cells, J Biol.Chem, 284, (2009) 27908–27917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Huang J, Gali H, Paramasivam M, Muniandy P, Gichimu J, Bellani MA, Seidman MM. Single Molecule Analysis of Laser Localized Interstrand Crosslinks, Front Genet, 7, (2016) 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Yan Z, Guo R, Paramasivam M, Shen W, Ling C, Fox D III, Wang Y, Oostra AB, Kuehl J, Lee DY, Takata M, Hoatlin ME, Schindler D, Joenje H, de Winter JP, Li L, Seidman MM, Wang W. A Ubiquitin-Binding Protein, FAAP20, Links RNF8- Mediated Ubiquitination to the Fanconi Anemia DNA Repair Network, Mol.Cell, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Thongthip S, Bellani M, Gregg SQ, Sridhar S, Conti BA, Chen Y, Seidman MM, Smogorzewska A. Fan1 deficiency results in DNA interstrand cross-link repair defects, enhanced tissue karyomegaly, and organ dysfunction, Genes Dev, 30, (2016) 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Jiang Q, Paramasivam M, Aressy B, Wu J, Bellani M, Tong W, Seidman MM, Greenberg RA. MERIT40 cooperates with BRCA2 to resolve DNA interstrand crosslinks, Genes Dev, 29, (2015) 1955–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Nam EA, Cortez D. ATR signalling: more than meeting at the fork, Biochem.J, 436, (2011) 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Marteijn JA, Bekker-Jensen S, Mailand N, Lans H, Schwertman P, Gourdin AM, Dantuma NP, Lukas J, Vermeulen W. Nucleotide excision repair-induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response, J Cell Biol, 186, (2009) 835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Heller RC, Marians KJ. Replication fork reactivation downstream of a blocked nascent leading strand, Nature, 439, (2006) 557–562. [DOI] [PubMed] [Google Scholar]
- [79].Kottemann MC, Conti BA, Lach FP, Smogorzewska A. Removal of RTF2 from Stalled Replisomes Promotes Maintenance of Genome Integrity, Mol.Cell, 69, (2018) 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Choi JH, Lindsey-Boltz LA, Sancar A. Reconstitution of a human ATR-mediated checkpoint response to damaged DNA, Proc.Natl.Acad.Sci.U.S.A, 104, (2007) 13301–13306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Unsal-Kacmaz K, Makhov AM, Griffith JD, Sancar A. Preferential binding of ATR protein to UV-damaged DNA, Proc.Natl.Acad.Sci.U.S.A, 99, (2002) 6673–6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts, Mol.Cell, 22, (2006) 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kumar A, Mazzanti M, Mistrik M, Kosar M, Beznoussenko GV, Mironov AA, Garre M, Parazzoli D, Shivashankar GV, Scita G, Bartek J, Foiani M. ATR mediates a checkpoint at the nuclear envelope in response to mechanical stress, Cell, 158, (2014) 633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kemp MG, Sancar A. ATR Kinase Inhibition Protects Non-cycling Cells from the Lethal Effects of DNA Damage and Transcription Stress, J.Biol.Chem, 291, (2016) 9330–9342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Muniandy PA, Liu J, Majumdar A, Liu ST, Seidman MM. DNA interstrand crosslink repair in mammalian cells: step by step, Crit Rev.Biochem.Mol.Biol, 45, (2010) 23–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Dmitriev OY, Lutsenko S, Muyldermans S. Nanobodies as Probes for Protein Dynamics in Vitro and in Cells, J.Biol.Chem, 291, (2016) 3767–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Pleiner T, Bates M, Gorlich D. A toolbox of anti-mouse and anti-rabbit IgG secondary nanobodies, J.Cell Biol, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].McKeague M. Aptamers for DNA Damage and Repair, Int.J.Mol.Sci, 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]