

Abstract
Aims
Macrophage phagocytosis of dead cells is a prerequisite for inflammation resolution. Because CXCL4 induces macrophage phagocytosis in vitro, we examined the impact of exogenous CXCL4 infusion on cardiac wound healing and macrophage phagocytosis following myocardial infarction (MI).
Methods and results
CXCL4 expression significantly increased in the infarct region beginning at Day 3 post-MI, and macrophages were the predominant source. Adult male C57BL/6J mice were subjected to coronary artery occlusion, and MI mice were randomly infused with recombinant mouse CXCL4 or saline beginning at 24 h post-MI by mini-pump infusion. Compared with saline controls, CXCL4 infusion dramatically reduced 7 day post-MI survival [10% (3/30) for CXCL4 vs. 47% (7/15) for saline, P < 0.05] as a result of acute congestive heart failure. By echocardiography, CXCL4 significantly increased left ventricular (LV) volumes and dimensions at Day 5 post-MI (all P < 0.05), despite similar infarct areas compared with saline controls. While macrophage numbers were similar at Day 5 post-MI, CXCL4 infusion increased Ccr4 and Itgb4 and decreased Adamts8 gene levels in the infarct region, all of which linked to CXCL4-mediated cardiac dilation. Isolated Day 5 post-MI macrophages exhibited comparable levels of M1 and M4 markers between saline and CXCL4 groups. Interestingly, by both ex vivo and in vitro phagocytosis assays, CXCL4 reduced macrophage phagocytic capacity, which was connected to decreased levels of the phagocytosis receptor CD36. In vitro, a CD36 neutralizing antibody (CD36Ab) significantly inhibited macrophage phagocytic capacity. The combination of CXCL4 and CD36Ab did not have an additive effect, indicating that CXCL4 regulated phagocytosis through CD36 signalling. CXCL4 infusion significantly elevated infarct matrix metalloproteinase (MMP)-9 levels at Day 5 post-MI, and MMP-9 can cleave CD36 as a down-regulation mechanism.
Conclusion
CXCL4 infusion impaired macrophage phagocytic capacity by reducing CD36 levels through MMP-9 dependent and independent signalling, leading to higher mortality and LV dilation.
Keywords: Myocardial infarction , CXCL4 , Macrophage , Phagocytosis , Matrix metalloproteinases
1. Introduction
Following myocardial infarction (MI), the left ventricle undergoes a cardiac wound healing process, consisting of overlapping inflammatory, proliferative, and maturation phases.1 Macrophages play an indispensable role during each stage of cardiac repair. During the early inflammatory phase, macrophages display a pro-inflammatory phenotype and produce a variety of pro-inflammatory cytokines, chemokines, and matrix metalloproteinases (MMPs).2,3 In the proliferative and maturation phase, macrophages exhibit an anti-inflammatory phenotype and generate multiple anti-inflammatory, pro-angiogenic, and pro-reparative factors.3–5 In addition, macrophages can engulf and clear dead cell and tissue debris, which is important for inflammation resolution.4,6 Macrophage depletion impairs cardiac repair in adult mouse hearts and myocardial regeneration in mouse neonates.7–9 Macrophages have recently been shown to promote myocardial electrical conduction in the steady state, indicating a wide array of macrophage roles.10 Therefore, modulating macrophage cell physiology may represent a novel way to improve post-MI cardiac wound healing.2
Platelet factor 4 (PF4) is a platelet protein first discovered by Deutsch et al. in 1955.11 Subsequently, PF4 was classified as a member of the CXC chemokine family, known as CXCL4. CXCL4 has a relatively weak chemoattractant role and exhibits strong pro-inflammatory features.12 CXCL4 deletion attenuates atherosclerosis and mesenteric ischaemia/reperfusion injury in mouse models by inhibiting inflammation.13,14 CXCL4 and CCL5 have functional synergism, and disrupting the CCL5-CXCL4 interaction inhibits monocyte recruitment and attenuates atherosclerosis in hyperlipidaemic mice.15,16In vitro CXCL4 elicits macrophage phagocytosis.17
As enhanced inflammation and impaired phagocytosis are detrimental for post-MI cardiac repair,18–20 our initial hypothesis was that CXCL4 infusion in vivo would stimulate macrophage phagocytosis to subsequently reduce inflammation and orchestrate post-MI cardiac repair. Interestingly, CXCL4 infusion led to high mortality and left ventricular (LV) dilation post-MI, indicating detrimental mechanisms of CXCL4, which were explored in the current study.
2. Methods
Detailed descriptions of the materials and methods, and supplementary tables and figures are available in the Supplementary material online.
2.1 Querying the mouse heart attack research tool (mHART) 1.0 database and tissue bank
We queried our mouse heart attack research tool (mHART) 1.0 database (see Supplementary material online for details) to determine post-MI CXCL4 gene expression patterns and used Day 5 post-MI tissue sections of C57BL/6J mice from our tissue bank for immunohistochemistry analysis.21,22 To examine CXCL4 protein changes after MI, immunohistochemistry was performed according to the guidelines and as described previously using a rat anti-mouse CXCL4 (1:50, MAB595, R&D).21,23 Staining quantification was calculated as the percentage of positively stained area to total area. Platelets were stained using a rat anti-mouse CD41 antibody (1:100, Ab33661, Abcam). To evaluate CXCL4 cell localization, multiplexed immunofluorescence was performed using the Opal Multiplex Immunohistochemistry Kit (Perkin Elmer).24 After antigen retrieval, sections were blocked with serum and incubated with a macrophage antibody Mac3 (1:100, CL8493AP, Cedarlane), followed by a horseradish peroxidase conjugated anti-rat IgG, and fluorophore Opal620 (1:100, FP1495A, PerkinElmer). The above steps were repeated to label neutrophils (1:100, CL8993AP, Cedarlane) plus fluorophore Opal 520 (1:100, FP1487A, PerkinElmer) and CXCL4 plus fluorophore Opal690 (1:100, FP1497A, PerkinElmer). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, 1:10, FP1490, PerkinElmer). Images were acquired using the Mantra™ Quantitative Pathology Imaging microscope.24 The quantitative analysis was performed using the inForm software.
2.2 CXCL4 mRNA expression in peritoneal macrophages and infarct macrophages
Peritoneal macrophages were primed to the pro-inflammatory M1 phenotype with lipopolysaccharide (1 µg/mL, L2880, Sigma) plus interferon-γ (20 ng/mL, 485-MI, R&D) or to the anti-inflammatory M2 subtype with interleukin-4 (20 ng/mL, 404-ML, R&D).21 Unstimulated cells served as M0. The whole transcriptome analysis was performed using RNA-seq, as described previously.24,25CXCL4 data were normalized to values of M0 and reported as fold change. We queried our published macrophage RNA-seq dataset and exported the data of CXCL4 mRNA expression in macrophages from days 0, 1, and 3 post-MI.26
2.3 Mice, MI surgery, and treatment
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Mississippi Medical Center and were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (Eighth edition; revised 2011). To examine the effect of CXCL4 infusion, male C57BL/6J mice (3-6 months of age) were used. MI surgery was performed by permanent ligation of the left coronary artery, according to the guidelines and as previously described.21,27,28 Buprenorphine (0.1 mg/kg) was intraperitoneally administered at the time of MI. At Day 1 (24 h) post-MI, mice underwent echocardiography assessment to confirm MI, and CXCL4 (595-P4-025, R&D) or saline (negative control) was randomly infused subcutaneously via osmotic mini-pump (Alzet). Cohort 1 were post-MI mice administered 2.5, 5, 25, or 50 μg/kg/day CXCL4 and sacrificed at Day 7 post-MI. Cohort 2 were post-MI mice administered 25 μg/kg/day CXCL4 and sacrificed at Day 5 post-MI. CXCL4 infusion at 2.5 µg/kg/day had no significant effect on post-MI 7 day survival, indicating this dose is below pathological levels. All three other doses (5, 25, or 50 µg/kg/day) showed a 10% survival at 7 day post-MI. We selected the middle dose (25 µg/kg/day) to elucidate the underlying mechanisms. As controls, CXCL4 (25 μg/kg/day) was administered to non-MI mice for 4 days. Mice were euthanized with 5% inhalational isoflurane, followed by exsanguination and removal of the heart.
2.4 Electrocardiogram, echocardiography, survival, and necropsy examination
Mice were anaesthetized with 1.5% inhalational isoflurane. After 5 min for stabilization, electrocardiogram (EKG) was recorded in lead I, II, and III configuration for 30 min. Isoproterenol (2 mg/kg, 1351005 USP) was administered by intraperitoneal injection, and EKG was recorded for another 30 min.29,30 LV physiology was measured using the Vevo 2100 system (VisualSonics) according to the guidelines for measuring cardiac physiology in mice.24,27,31 All images were captured at heart rates >400 bpm and at body temperature 35–37°C to achieve physiologically relevant measurements. Three cardiac cycles were analysed and averaged. The echocardiography assessor was blinded to experimental groups. The mice were checked daily for survival analysis. Autopsy was performed on non-surviving mice, and cardiac rupture was confirmed if there was a blood clot in the thoracic cavity or the LV rupture site was observed.21
At sacrifice, the mice were anaesthetized with 2% inhalational isoflurane, and the heart was arrested in diastole using cardioplegic solution.21 The left ventricle was sliced into apex, middle, and base pieces, and stained with 1% 2, 3, 5-triphenyltetrazolium chloride for infarct area evaluation. Infarct area was calculated as the percentage of infarct area to total LV area. The infarct regions from the apex and base were isolated for biochemical analysis. The middle section was fixed in 10% zinc formalin for histological examination.
2.5 Human subjects
All participants gave written consent before participation in the study. The investigation conformed to the principles outlined in the Declaration of Helsinki. The human subject protocol was approved by the Institutional Review Board at the University of Mississippi Medical Center (IRB# 2013-0164). Plasma was collected within 24 h after admission in MI patients (n = 50). Echocardiography was acquired during admission. Plasma CXCL4 levels were quantified using dot immunoblotting. Patient characteristics are listed in Supplementary material online, Table S3.
2.6 Quantitative real time PCR
Gene array for Inflammatory Cytokines and Receptors (Qiagen) and for Extracellular Matrix and Adhesion Molecules (Qiagen) were performed to quantify gene expression levels.21,32 For individual markers, gene expression was measured using Taqman Gene Expression Master Mix plus individual primers. The gene levels were normalized to the reference gene hprt1.21 MIQE guidelines were followed for all the PCR experiments and analysis.33
2.7 Immunohistochemistry
Immunohistochemical staining procedures were performed as described previously and followed the guidelines for authors and reviewers on antibody use in physiology studies.21,23,32 The following antibodies were used: rat anti-mouse neutrophil antibody (1:100, CL8993AP, Cedarlane) and macrophage antibody Mac3 (1:100, CL8493AP, Cedarlane). Staining quantification was calculated as the percentage of positively stained area to total area.
2.8 Protein extraction, enzyme-linked immunosorbent assay (ELISA), and immunoblotting
The guidelines for authors and reviewers on antibody use in physiology studies were followed.23 Total protein was extracted using reagent type 4 plus 1× protease inhibitor cocktail. Protein concentrations were determined by the Quick Start™ Bradford Protein Assay (Bio-Rad). CXCL4 levels in the infarct were measured using Mouse CXCL4 Quantikine ELISA Kit (MCX400, R&D) according to manufacturer instructions. Immunoblotting procedures were performed as described previously.21,32 The densitometry of the total protein was used as the internal loading control for each lane, and results quantified as the densitometry of the protein of interest divided by the densitometry of the total protein. Protein expression levels were quantified using the following antibodies: CD36 (1:2000, NB400-144, Novus), CD163 (1:1000, ab182422, Abcam), low-density lipoprotein receptor-related protein 1 (LRP1, 1:1000, ab92544, Abcam), myeloid-epithelial-reproductive tyrosine kinase (MERTK, 1:1000, ab95925, Abcam), mannose receptor C-type 1 (MRC1/CD206, 1:1000, ab64693, Abcam), macrophage scavenger receptor 1 (MSR1, 1:1000, ab151707, Abcam), MMP-8 (1:1000, ab81286, Abcam), and MMP-9 (1:1000, AF909, R&D).
2.9 Macrophage isolation, ex vivo phagocytosis, and immunoblotting
Macrophages at Day 0 and Day 5 post-MI were isolated using magnetic microbeads as previously described.24,27 CD11b+Ly6G− macrophages were collected for further analysis. Isolated post-MI macrophages were used for M1 and M4 phenotype evaluation or stained with a macrophage specific marker Mac-3 to evaluate purity or used to assess phagocytic capacity. After fixation, permeabilization, and blocking, macrophages were stained with a rabbit anti-α-actinin antibody (myocyte marker, 1:50, D6F6, CST) and a rat anti-Ly-6B.2 (neutrophil marker, 1:100, CL8993AP, Cedarlane), followed by incubation with corresponding fluorescent secondary antibodies. Nuclei were stained with DAPI. The data were presented as percentage of α-actinin+ or Ly-6B.2+ macrophages to total macrophages to indicate macrophage phagocytosis of myocytes or neutrophils. Immunoblotting for macrophage CXCL4 (1:250, MAB595, R&D) and CD36 (1:2000, NB400-144, Novus) was performed.
2.10 Peritoneal macrophage isolation and stimulation
Unelicited peritoneal macrophages were isolated as described previously.21 The cells were stimulated with or without 5 μg/mL recombinant mouse CXCL4 (595-P4-025, R&D) protein for 4 h. The cells were washed with 1× phosphate-buffered saline and used for M1, M2, and M4 phenotype evaluation. The data were represented as fold changes to unstimulated control group.
2.11 In vitro phagocytosis assay
Phagocytosis was performed according to manufacturer instructions (V-6694, Molecular Probes). Isolated peritoneal macrophages were stimulated with CXCL4 (5 μg/mL, 595-P4-025, R&D), a CD36 neutralizing antibody (1 μg/mL, MABF956, Millipore), or CXCL4 (5 μg/mL) + CD36 antibody (1 μg/mL) for 20 h.34 Unstimulated cells served as controls. The cells were then incubated with the fluorescent Escherichia coli BioParticle suspension for 2 h. Trypan blue suspension was added to stop the phagocytosis process. The cells were fixed with 100% ethanol. Nuclei were stained with DAPI. The intensity of fluorescence per cell was calculated, and the data were presented as fold changes to controls.
2.12 Picrosirius red staining
Picrosirius red (PSR) staining was carried out, as previously described.35 Staining quantification was calculated as the percentage of positively stained area to total area. Six to ten random scans in the infarct region per section were analysed and averaged.
2.13 Statistical analyses
Statistics were reported according to statistical considerations in reporting cardiovascular research.36 All experiments were performed and analysed in a blinded design. Data are presented as mean ± SEM. Two group comparisons were analysed by unpaired or paired t-test. The in vitro studies used paired t-test, because the same set of cells were stimulated under different conditions. Other experiments used unpaired t-test, because the samples were collected from different biological sets. Multiple group comparisons were performed using one-way or two-way analysis of variance (ANOVA) with corresponding post-test. The survival rate was analysed by Kaplan–Meier survival analysis and compared by the log-rank test. The χ2 test was used for rupture rate analysis. Volcano plot analysis and Pearson correlation analysis were performed using GraphPad Prism 7.02. A value of P < 0.05 was considered statistically significant. For RNA-seq data, the false discovery rate (FDR) adjusted P-value was used.
3. Results
3.1 Infarct CXCL4 levels increased post-MI due to macrophage influx
To investigate the cardiac CXCL4 response to MI, we queried the mHART 1.0 database and used banked LV tissue sections to evaluate CXCL4 mRNA and protein levels.22 As shown in Figure 1A , infarct CXCL4 mRNA levels significantly increased at Day 3, peaked at days 5 and 7 (all P < 0.05 vs. Day 0), returning to baseline levels by Day 28. Consistently, immunohistochemical staining of Day 5 post-MI LV tissue revealed that infarct CXCL4 protein increased by approximately 12-fold at Day 5 post-MI, compared with Day 0 controls (Figure 1B, P < 0.05 vs. Day 0). We stained for platelets in the Day 5 infarcted heart, using spleen as a positive control. There was no evidence of positive staining for CD41, a specific platelet marker (see Supplementary material online, Figure S1), indicating that CXCL4 is not likely deriving from platelets at Day 5 post-MI. The quantitative analysis of multiplex staining of CXCL4, macrophages, and neutrophils revealed that 72 ± 5% of infarct CXCL4 co-localized to macrophages, 4 ± 1% to neutrophils, and 24 ± 4% to other cell types (Figure 1C). Within infarct macrophages, 55 ± 6% of macrophages were CXCL4+, while 45 ± 6% were CXCL4− (Figure 1D). The RNA-seq data demonstrated that Day 3 infarct macrophages had higher levels of CXCL4 mRNA than Day 0 cells (see Supplementary material online, Figure S2). Compared with Day 0 macrophages, Day 5 infarct macrophages produced 70-fold more CXCL4 protein (Figure 1E). To study CXCL4 expression in distinct macrophage phenotypes, peritoneal macrophages were primed to M1 or M2. Compared with M0 (unstimulated controls), M1 showed significantly lower expression of CXCL4 (Figure 1F).
Figure 1.

Macrophages were the major source of CXCL4 in Day 5 LV infarct, and exogenous CXCL4 infusion decreased 7 day post-MI survival. (A) Infarct CXCL4 mRNA expression significantly increased at Day 3 (n = 19), peaked at Days 5 (n = 17) and 7 (n = 24), returning to baseline levels by Day 28 post-MI (n = 7), compared with Day 0 (n = 32). One-way ANOVA was used. (B) Infarct CXCL4 protein increased by approximately 12-fold at Day 5 post-MI. Arrows indicate positive staining (brown dots). Scale bar, 50 µm. n = 6–9/group. *P < 0.05 vs. Day 0. Unpaired t-test was used. (C) Representative images of multiplex staining of CXCL4 (green), macrophages (orange), and neutrophils (red). Nuclei were stained with DAPI (blue). Scale bar, 50 µm. The quantitative analysis of multiplex staining of CXCL4, macrophages, and neutrophils revealed that 72 ± 5% of infarct CXCL4 co-localized to macrophages, 4 ± 1% to neutrophils, and 24 ± 4% to other cell types. n = 6/group. (D) Within infarct macrophages, 55 ± 6% of macrophages were CXCL4+, while 45 ± 6% were CXCL4−. n = 6/group. (E) Compared with Day 0 macrophages, Day 5 infarct macrophages produced 70-fold more CXCL4 protein. n = 5/group. *P < 0.05 vs. Day 0. Unpaired t-test was used. (F) M1 peritoneal macrophages demonstrated lower CXCL4 mRNA levels than M0, evaluated by RNA-seq. n = 4/group. *FDR adjusted P = 0.001 vs. M0. One-way ANOVA was used. (G) CXCL4 infusion decreased 7 day post-MI survival to 10%, compared with 47% for the saline group. n = 15–30/group. *P < 0.05 vs. saline. The survival rate was analysed by Kaplan–Meier survival analysis and compared by the log-rank test. (H) Lung oedema index, the ratio of wet lung weight to tibia length, was significantly higher in CXCL4 treated non-surviving mice than saline controls. n = 8–27/group. *P < 0.05 vs. saline. Unpaired t-test was used.
3.2 Exogenous CXCL4 infusion decreased 7 day post-MI survival
In a dosing experiment, we infused recombinant mouse CXCL4 protein (2.5, 5, 25, or 50 µg/kg/day) or saline (negative control) to MI mice at 24 h post-MI by osmotic mini-pump. CXCL4 infusion at 2.5 µg/kg/day had no significant effect on post-MI 7 day survival (see Supplementary material online, Figure S3), indicating this dose is below pathological levels. All three other doses (5, 25, or 50 µg/kg/day) resulted in reduced 7 day post-MI survival to 10% (1/10 for each of the three doses, combined 3/30), compared with 47% for the saline controls (7/15, P < 0.05, Figure 1G). Cardiac rupture, arrhythmias, and acute congestive heart failure are the major causes of post-MI mortality. The decreased survival rate was not due to an increase in rupture rates (see Supplementary material online, Figure S4A). Saline and CXCL4 treated mice both showed mild atrioventricular block at Day 5 post-MI with or without isoproterenol induction (see Supplementary material online, Figure S4B), with no evidence of ventricular arrhythmias, indicating that the mortality in the CXCL4 treated mice was not due to increased incidence of arrhythmias. Non-surviving mice in the CXCL4 group had a higher lung oedema index (wet lung weight: tibia length, Figure 1H), indicative of more severe congestive heart failure. Therefore, CXCL4 induced mortality was due to acute congestive heart failure.
3.3 Exogenous CXCL4 infusion aggravated post-MI LV dilation
Since CXCL4 infusion induced a 90% mortality at Day 7 post-MI, and most mice died between days 5 and 7, we evaluated the underlying mechanisms at Day 5 post-MI as the time of mechanism origin. To confirm the recombinant CXCL4 protein homed to the infarct zone, we measured infarct CXCL4 protein. By ELISA, CXCL4 infusion (25 µg/kg/day) increased infarct CXCL4 concentrations by 64% at Day 5 post-MI (Figure 2A, P < 0.05 vs. saline). CXCL4 infusion did not increase Cxcl4 gene levels, as expected (see Supplementary material online, Table S1). Infarct areas were similar between saline and CXCL4 treated Day 5 post-MI mice (53 ± 2% for saline vs. 53 ± 4% for CXCL4, P = 0.95, Figure 2B), consistent with CXCL4 infusion being initiated at 24 h post-MI, a time after myocardial salvage would be probable.
Figure 2.

Exogenous CXCL4 infusion exacerbated LV dilation at Day 5 post-MI. (A) CXCL4 infusion increased CXCL4 levels by 64% in the Day 5 infarct. Unpaired t-test was used. (B) Infarct areas were comparable between saline and CXCL4 treated mice. Unpaired t-test was used. (C) LV volumes and dimensions in CXCL4 treated mice were higher than saline controls at Day 5 post-MI. Ejection fraction, fractional shortening, and LV anterior wall thickness in systole were similar between groups. n = 8–9/group. *P < 0.05 vs. saline and #P < 0.05 vs. respective Day 0. Two-way ANOVA was used. (D and E) Plasma CXCL4 levels in patients with MI positively correlated with end diastolic dimension (EDD) and negatively correlated with interventricular septal thickness in diastole (IVSd). n = 50. Pearson correlation analysis was used.
Saline and CXCL4 treated mice displayed similar LV dilation and pathophysiology responses at 24 h post-MI (the time of intervention), confirming the grouping was unbiased. CXCL4 treated mice at Day 5 post-MI showed significantly higher end-systolic and end-diastolic volumes and dimensions (Figure 2C, all P < 0.05) than saline controls, indicating that CXCL4 infusion exacerbated post-MI LV dilation. Fractional shortening, ejection fraction, and wall thickness at Day 5 post-MI were comparable between saline and CXCL4 treated animals (Figure 2C). To address potential effects of CXCL4 on cardiomyocytes, we infused exogenous CXCL4 into non-MI mice for 4 days. There was no effect on cardiomyocytes, indicated by no change in wall thickness or other cardiac physiology variables (see Supplementary material online, Table S2). This indicates that the increase in LV dilation at Day 5 post-MI in CXCL4 treated mice was not due to direct actions of CXCL4 on the myocyte.
Plasma CXCL4 levels are elevated in patients with acute coronary syndrome and are associated with platelet counts and C-reactive protein concentrations.37 We measured plasma CXCL4 levels in MI patients within 24 h of admission (see Supplementary material online, Table S3), and performed correlation analysis to patient characteristics and cardiac physiology parameters. Plasma CXCL4 levels positively correlated with end diastolic dimension (Figure 2D) and negatively correlated with interventricular septal thickness in diastole (Figure 2E). This is consistent with the mouse data showing that CXCL4 infusion enhances post-MI LV dilation.
3.4 The impact of exogenous CXCL4 infusion on post-MI inflammation
We explored the impact of CXCL4 infusion on inflammation by examining 84 inflammatory cytokine and receptor genes in the infarct region of Day 5 post-MI mice. CXCL4 decreased expression of Ccl3 and Ltb and increased Ccr4 (Figure 3A, all P < 0.05 compared with saline). All three genes are pro-inflammatory factors, and Ccr4 significantly and positively correlated with LV dilation irrespective of CXCL4 infusion (Figure 3B). Saline and CXCL4 treated mice showed similar numbers of neutrophils and macrophages in the Day 5 infarct (Figure 3C and D), indicating that the impact of CXCL4 on these 3 pro-inflammatory cytokines was not due to alterations in leukocyte cell numbers.
Figure 3.

The impact of exogenous CXCL4 infusion on the inflammatory response. (A) The volcano plot showed that out of 84 inflammatory mediators examined by gene array, CXCL4 treated mice displayed lower Ccl3 and Ltb and higher Ccr4 in Day 5 LV infarct. n = 9/group. Unpaired t-test were used. FC, fold change. (B) Ccr4 gene levels positively correlated with end systolic dimension (ESD). n = 17. Pearson correlation analysis was performed. (C and D) Infarct neutrophil and macrophage numbers were similar between saline and CXCL4 groups. Arrows indicate positive staining. n = 9/group. Unpaired t-test was used. (E) In vitro, CXCL4 treatment up-regulated Ccl3 expression in peritoneal macrophages. n = 6–9/group. *P < 0.05 vs. negative control. Paired t-test was used.
To investigate whether CXCL4 can directly regulate macrophage expression of the three genes differently expressed in saline and CXCL4 treated mice, we stimulated unelicited peritoneal macrophages with recombinant CXCL4 (5 µg/mL) for 4 h and measured gene expression using quantitative real time PCR. Interestingly, in vitro CXCL4 enhanced macrophages to express Ccl3 (Figure 3E, P < 0.05), while in vivo CXCL4 treated mice showed reduced Ccl3 in the infarct region. CXCL4 stimulation in vitro demonstrated no direct impact on Ltb or Ccr4 expression in macrophages. These results indicate that for these three factors, there is a divergence between in vitro CXCL4 stimulation by a single factor and in vivo multi-cell stimulation by CXCL4 within a complex microenvironment containing other factors.
3.5 In vitro CXCL4 elicited macrophages to express pro-inflammatory cytokines
Gleissner et al.38 previously reported that CXCL4 induces monocyte differentiation into a unique macrophage M4 phenotype, which overexpresses M1 and M2 markers. Using the same macrophages stimulated above, we measured a panel of M1, M2, and M4 markers. In vitro, CXCL4 significantly up-regulated the expression of all M1 markers examined (Figure 4A, all P < 0.05), which include Ccl3, Ccl5, Il1β, and Il6; in contrast, M2 markers, including Arg1, Fizz1, Tgfβ1, and Ym1, were not affected by CXCL4 stimulation (see Supplementary material online, Figure S5). Consistent with literature,38 CXCL4 up-regulated Tnfα and down-regulated Mmp8 (Figure 4A, both P < 0.05); CXCL4 did not affect Il10 and Mmp12 expression. Post-MI macrophages isolated from Day 5 saline and CXCL4 infarcts demonstrated no differences in M1 and M4 marker expression (Figure 4B). This indicates that the in vivo complexity of inflammation is not recapitulated by the single CXCL4 in vitro stimulus.
Figure 4.

In vitro CXCL4 treatment induced macrophage production of pro-inflammatory M1 markers. (A) CXCL4 treatment (5 µg/mL for 4 h) significantly up-regulated all pro-inflammatory M1 markers examined and M4 marker Tnfα, and down-regulated M4 marker Mmp8. n = 6/group. *P < 0.05 vs. negative control. Paired t-test was used. (B) Day 5 post-MI macrophages in saline and CXCL4 groups exhibited comparable expression of M1 and M4 markers. n = 6/group. Unpaired t-test was used.
3.6 Exogenous CXCL4 infusion decreased ex vivo macrophage phagocytic capacity
Macrophages engulf dead cell and tissue debris, a process termed phagocytosis or efferocytosis (clearance of apoptotic cells).19 To evaluate the effect of CXCL4 on macrophage phagocytic capacity, cardiac macrophages (CD11b+Ly6G−) were isolated from Day 5 infarcts and stained with the macrophage specific marker Mac-3 to assess cell purity.27,32 These CD11b+Ly6G− macrophages were Mac-3+ (Figure 5A), indicating >95% cell purity. To evaluate the presence of myocytes or neutrophils within the macrophage (indicating active in vivo phagocytosis), immunocytochemistry was performed on the macrophages using the myocyte marker (α-actinin, Figure 5B) and the neutrophil marker (Ly-6B.2, Figure 5C) antibody. As shown in Figure 5B and D, 35 ± 4% of macrophages were phagocytic for myocytes in the saline MI group, which was significantly reduced by CXCL4 treatment (Figure 5B and D, P < 0.05). Likewise, 18 ± 1% of macrophages were phagocytic for neutrophils in the saline MI group, which was significantly reduced by CXCL4 treatment (Figure 5C and D, P < 0.05). These data indicate that CXCL4 infusion impaired macrophage phagocytic capacity. This result contradicts previous published in vitro studies demonstrating that CXCL4 induces phagocytosis, indicating the complex in vivo environment alters the direct in vitro effect of CXCL4.17
Figure 5.

Exogenous CXCL4 infusion decreased macrophage phagocytosis by inhibiting CD36 expression. (A) Isolated macrophages (CD11b+Ly6G−) from Day 5 infarct were all Mac-3+, a specific marker for macrophages, indicating high purity. (B–D) Macrophage (DAPI+) phagocytosis of myocytes (green) and neutrophils (red) were decreased after CXCL4 treatment. Yellow arrows indicate phagocytic macrophages. n = 7–9/group. Scale bar, 50 µm. (E) Out of six phagocytosis markers measured, Cd36 expression was lower in macrophages isolated from the Day 5 infarct of the CXCL4 group. n = 6/group. (F) CXCL4 infusion significantly reduced CD36 protein levels in Day 5 LV infarct. n = 9/group. (G) CXCL4 infusion significantly reduced CD36 protein levels in Day 5 infarct macrophages. n = 5–6/group. *P < 0.05 vs. saline. Unpaired t-test was used.
3.7 CD36 mediated CXCL4 inhibition of macrophage phagocytosis
To explore the mechanisms by which CXCL4 infusion decreased macrophage phagocytic capacity, we measured six well-known phagocytosis related markers (Cd36, Cd163, Lrp1, Mertk, Mrc1, and Msr1) in Day 5 infarct macrophages. Out of the six markers examined, only Cd36 gene expression was statistically decreased in response to CXCL4 treatment (Figure 5E, P < 0.05). The protein levels of these markers in Day 5 infarct left ventricle were quantified by immunoblotting. In agreement with the mRNA results, only CD36 protein levels out of the six proteins were decreased after CXCL4 infusion (Figure 5F and Supplementary material online, Figure S6). In addition, Day 5 infarct macrophages from CXCL4 treated mice exhibited lower CD36 protein levels than saline controls (Figure 5G). This indicates that CD36 mediates CXCL4 effects on phagocytosis.
3.8 CXCL4 inhibited in vitro macrophage phagocytosis capacity through CD36 signalling
To evaluate whether CXCL4 directly decreased macrophage phagocytosis, unelicited peritoneal macrophages were isolated and stimulated with CXCL4 for 20 h. Following washing to remove CXCL4, the cells were incubated with fluorescent E. coli bioparticles to measure phagocytosis. The untreated macrophages efficiently phagocytized bioparticles, an effect significantly inhibited by CXCL4 stimulation (Figure 6A, P < 0.05 vs. control) and in line with the ex vivo findings. The phagocytic capacity was quantified as fluorescence intensity per cell; and cell numbers were not different between groups (P = 0.61). Thus, the difference in phagocytosis was not due to a difference in cell number. Out of six markers examined, both Cd36 and Mertk were down-regulated in CXCL4 pre-treated in vitro macrophages (Figure 6B, P < 0.05), which further confirmed that CD36 signalling may mediate CXCL4 effects on phagocytosis. The fact that Mertk was down-regulated in vitro and not ex vivo highlights that while the direct effect of CXCL4 on phagocytosis involves both Cd36 and Mertk, in vivo conditions mask the Mertk effect.
Figure 6.

In vitro CXCL4 pretreatment inhibited phagocytic capability of macrophages through CD36 signalling. (A) Macrophage phagocytosis of fluorescent Escherichia coli BioParticles was significantly lower in cells pre-stimulated with 5 µg/mL recombinant CXCL4 protein for 20 h. (B) CXCL4 pretreatment significantly inhibited macrophage expression of Cd36 and Mertk. n = 6–10/group. *P < 0.05 vs. control. Paired t-test was used. (C) Either CXCL4 or CD36Ab significantly inhibited macrophage phagocytic capacity. The combination of CXCL4 and CD36Ab did not have an additive effect. CD36Ab, CD36 neutralizing antibody. Scale bar, 50 µm. n = 6–13/group. P < 0.05 vs. control. One-way ANOVA was used.
To test if the phagocytosis decrease induced by CXCL4 occurred through CD36 signalling, macrophages were incubated with CXCL4, a CD36 neutralizing antibody (CD36Ab), or both. Compared with untreated controls, either CXCL4 or CD36Ab significantly inhibited macrophage phagocytic capacity (Figure 6C). The combination of CXCL4 and CD36Ab was not additive (Figure 6C), indicating that CXCL4 regulated phagocytosis through CD36 signalling.
3.9 Exogenous CXCL4 infusion increased MMP-8 and MMP-9 levels in the Day 5 infarct
Extracellular matrix (ECM) turnover is a necessary component of post-MI scar formation. We performed gene array analysis for 84 ECM and adhesion molecules in the infarct of Day 5 post-MI mice. CXCL4 infusion down-regulated the expression of Adamts8, Lama1, and Timp3, and up-regulated Itgb4 (Figure 7A, all P < 0.05). By correlation analysis, Adamts8 negatively and Itgb4 positively correlated with LV dilation irrespective of CXCL4 infusion (Figure 7B and C). Of note, CXCL4 infusion did not significantly affect col1 or col3 gene expression (see Supplementary material online, Table S4), two major components of the infarct scar. Collagen protein levels in the infarct, evaluated by PSR staining, were similar between saline and CXCL4 treated mice (Figure 7D). Collagen quantity, therefore, was not influenced by CXCL4 infusion. Tgfb1, the major pro-fibrotic cytokine, was not significantly affected by CXCL4 infusion (see Supplementary material online, Table S1).
Figure 7.

The impact of exogenous CXCL4 infusion on ECM. (A) Volcano plot demonstrated that out of 84 ECM molecules evaluated by gene array, CXCL4 treated mice displayed lower Adamts8, Lama1, and Timp3 and higher Itgb4 in Day 5 LV infarct. n = 9/group. Unpaired t-test was used. (B) Adamts8 negatively correlated with end diastolic dimension (EDD). (C) Itgb4 positively correlated with end diastolic volume (EDV). n = 17. Pearson correlation analysis was performed for B and C. (D) PSR staining demonstrated no statistical difference in total collagen content between saline and CXCL4 LV infarct. Arrows indicate positive staining. (E) MMP-8 and MMP-9 levels in Day 5 infarct were significantly higher in CXCL4 treated mice than saline controls. n = 9/group. *P < 0.05 vs. saline. Unpaired t-test was used. (F) MMP-8, but not MMP-9, negatively correlated with LV infarct wall thickness (posterior wall in diastole). n = 17. Pearson correlation analysis was used.
MMPs regulate many aspects of cardiac remodelling, and MMP-9 in particular has been shown to cleave CD36 as a mechanism to co-ordinate neutrophil apoptosis and removal.34 For the reason, we measured a panel of MMPs and their tissue inhibitors of metalloproteinases.39–41 None of the MMP or TIMP genes were different between saline and CXCL4 treated infarcts (see Supplementary material online, Table S4). CXCL4 infusion increased active MMP-8 and MMP-9 levels in the infarct (Figure 7E, both P < 0.05), and both MMPs can degrade ECM to promote LV dilation. MMP-8, but not MMP-9, negatively correlated with LV infarct wall thickness (Figure 7F).
4. Discussion
This study explored the impact of exogenous CXCL4 infusion on LV remodelling and physiology following MI, focusing on the reparative wound healing responses. The salient findings of this study were: (i) CXCL4 levels increased in the infarct due to macrophage infiltration; (ii) CXCL4 infusion decreased post-MI survival and aggravated LV dilation, in part by increasing MMP-8 and MMP-9; Ccr4 and Itgb4 positively correlated with LV dilation, while Adamts8 negatively correlated with LV dilation; and (iii) CXCL4 infusion decreased in vivo macrophage phagocytosis of myocytes and neutrophils, an effect mediated by CD36. This is the first study to monitor in vivo phagocytosis of post-MI myocytes and neutrophils in mice and assign a role for CXCL4 in the process. After MI, inflammation induces hyper-permeability of the microcirculation in the ischaemic area, which may induce plasma CXCL4 leakage to the infarct area. Other regions, including skeletal muscle, liver, and kidney may have increased CXCL4 levels as well. The in vitro results indicate that CXCL4, regardless of the source, directly affects cardiac macrophage physiology.
CXCL4 expression is up-regulated in atherosclerosis, inflammatory bowel disease, and mesenteric ischaemia/reperfusion injury.14,42 The current study adds to the literature by showing that CXCL4 levels are dramatically increased after MI, peaking at Day 5, and macrophages are the major source. Moreover, Day 5 MI macrophages produce more CXCL4 proteins than Day 0 resident cardiac macrophages. The expression of CXCL4 in cells other than platelets is not well established.16,43,44 Whether CXCL4 is derived from other major cell types in the MI heart (fibroblasts and endothelial cells) needs to be investigated.
An appropriate degree of inflammation is needed to promote optimal cardiac repair. Excessive inflammation can further damage normal tissue, while insufficient inflammation compromises the wound healing response.19,20,45,46 CXCL4 is a strong pro-inflammatory chemokine that induces inflammatory cytokine production in vascular smooth muscle cells by krüppel-like factor 4-dependent mechanisms.47 Platelet-derived CXCL4 decreases neutrophil apoptosis after arterial occlusion.48 CXCL4 deletion decreases neutrophil and monocyte infiltration in a mouse model of mesenteric ischaemia/reperfusion injury.13 CXCL4−/− mice are protected from acute lung injury induced by acid aspiration by improving barrier function without affecting neutrophil migration.49 Therefore, some of the lung effects observed in the CXCL4 non-surviving group could also be due to direct effects on the lung. Our study showed no effect on macrophage and neutrophil numbers; the major effect was on macrophage phenotype. There was a divergence between in vitro and in vivo CXCL4 induced M1 and M4 marker expression in macrophages, with in vitro stimulated macrophages showing robust up-regulation of M1 markers. Post-MI macrophages produced similar levels of M1 markers in saline and CXCL4 treated mice. One explanation for the discrepancy between ex vivo and in vitro data is that CXCL4 can induce pro-inflammatory gene expression in unactivated macrophages (in vitro unstimulated cells) but not in already activated macrophages (post-MI cells). Another explanation is that the levels of endogenous CXCL4 were sufficient to induce M1 expression, and the further elevation of CXCL4 did not have an additive effect. While CXCL4 is a strong in vitro stimulus, the in vivo MI complexity does not recapitulate the single stimulus effect. This indicates that CXCL4 effects on LV dilation and mortality in vivo were not entirely due to effects on polarization.
Phagocytosis and clearance of dead cell and tissue debris by macrophages is a prerequisite for post-MI cardiac repair.6 Defects in phagocytosis exacerbate adverse cardiac remodelling, leading to worse LV physiology. Dr. Thorp’s group has reported that MERTK deletion substantially impairs macrophage phagocytosis of apoptotic cardiomyocytes, resulting in worse wound healing.18 CXCL4 infusion suppressed macrophage phagocytosis of both myocytes and neutrophils, which partially explained the impaired survival and enhanced LV dilation in mice treated with CXCL4. While CXCL4-induced macrophages have been shown to be almost completely devoid of phagocytic capacity,38 in another study CXCL4 was reported to induce phagocytosis17 A difference between the two studies is when CXCL4 was given (simultaneous vs. pre-administration), indicating that CXCL4 may rapidly and directly induce phagocytosis initially and inhibit later. Supplementary material online, Table S5 compares in vivo, ex vivo, and in vitro results for phagocytosis and M1 markers induced by CXCL4, to highlight similarities and differences in response.
Apoptotic cells express multiple find me and eat me signals (e.g. lipid mediators and nucleotides) that can attract phagocytic cells and bind to phagocytosis-associated receptors on the cells, including CD36, CD163, LRP1, MERTK, MRC1, and MSR1. Binding to these receptors initiates the phagocytosis process.18,50,51 Our in vivo, ex vivo, and in vitro data demonstrated that CXCL4 down-regulated CD36 expression at both the cell and tissue level. Consistently, Gleissner et al.38 reported that in vitro CXCL4 significantly decreased Cd36 expression in monocyte-derived macrophages. CD36, also known as fatty acid translocase, is involved in the uptake of fatty acid across the plasma membrane in heart and skeletal muscle.52 CD36 is a scavenger receptor implicated in atherosclerosis.50 CD36 is important for macrophage phagocytosis of apoptotic neutrophils in the MI heart, and in vitro blocking CD36 reduces macrophage phagocytosis.34 Our findings that the combination of CXCL4 and CD36Ab did not have an additive effect indicate that CXCL4 regulated phagocytosis through CD36 signalling. Our in vitro studies used E. coli BioParticle as the substrate for macrophage phagocytosis. Whether the effects of CXCL4 on phagocytosis are also mediated through CD36 interactions with other ligands (e.g. oxidized low density lipoprotein) remain to be explored. CXCL4 treatment down-regulates CD163 in human macrophages in vitro.51 Our study demonstrated comparable CD163 expression in macrophages in saline and CXCL4 treated mice. This indicates that the impact of CXCL4 on CD163 may be compensated by other unknown mechanisms, or it is context dependent. These results also highlight that different phagocytosis mechanisms may be in play under different pathological conditions. Therefore, the primary effect of CXCL4 infusion was to inhibit macrophage phagocytosis by limiting CD36 signalling, which enhanced post-MI LV dilation and mortality.
New collagen starts to be synthesized to form the reparative scar at around Day 3 post-MI. CXCL4 infusion did not affect collagen expression and deposition at Day 5 post-MI. Interestingly, MMP-8 and MMP-9 were higher in mice treated with CXCL4; at this time, both MMPs are returning towards baseline values. We have previously shown that CD36 is an in vivo substrate of MMP-9 in the post-MI heart.34 MMP-9 can cleave CD36, leading to reduced CD36 in CXCL4 treated animals. This may represent another mechanism that CXCL4 inhibits CD36 levels and subsequently impairs macrophage phagocytosis.
By the law of LaPlace, increased dilation raises LV wall stress and tension. We have previously reported that left ventricles with higher dilation values at Day 3 post-MI have elevated rupture rates and adverse remodelling.21 In the current study, Ccr4 and Itgb4 positively and Adamts8 negatively correlated with LV dilation, and these genes were affected by CXCL4 treatment. Ccr4 (CD194) is a receptor for the pro-inflammatory chemokines Ccl2, Ccl4, Ccl5, Ccl17, and Ccl22.53 The strong correlation between Ccr4 and end diastolic dimensions indicates that CXCL4-associated inflammation regulates post-MI LV dilation. Itgb4/CD104 is an integrin that mediates cell-matrix or cell–cell adhesion. Adamts8 is a protease involved in ECM proteolysis. Their roles in the post-MI heart, however, have not been studied. Studies are needed to validate whether and how these factors directly contribute and mediate CXCL4-associated post-MI LV dilation. One limitation is that CXCL4 was given systemically and is likely to bind to proteoglycans.51 Whether CXCL4 effects on phagocytosis is mediated via binding to proteoglycans needs to be investigated.
In conclusion, this study provides the first evidence that early exogenous CXCL4 infusion inhibits macrophage phagocytosis of myocytes and neutrophils by suppressing CD36 expression through MMP-9 dependent and independent mechanisms and regulate Ccr4, Itgb4, and Adamts8 expression to increase mortality and LV dilation (Figure 8). Studies are warranted to investigate whether blocking endogenous CXCL4 is beneficial for post-MI cardiac wound healing.
Figure 8.
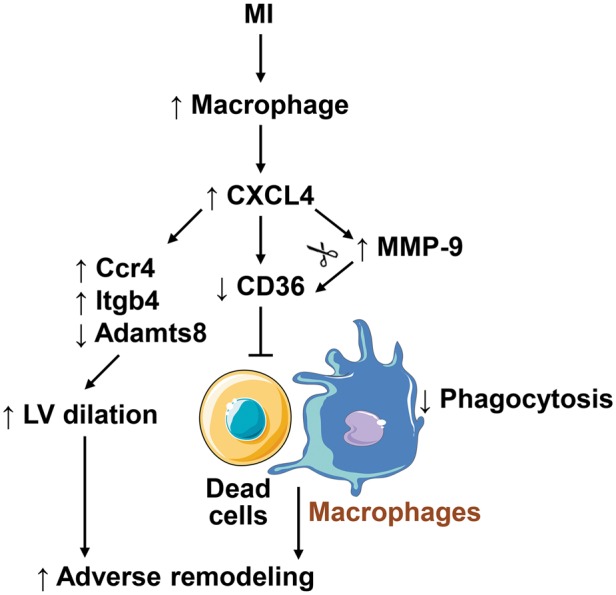
A diagram illustrating the mechanisms of action of CXCL4 in regulating LV remodelling following MI. MI triggers macrophage infiltration into the infarct area, which releases abundant CXCL4. CXCL4 decreases CD36 expression in MMP-9 independent (direct) and dependent (cleavage of CD36) modes to inhibit macrophage phagocytosis of dead myocytes and neutrophils, which results in impaired removal of debris and adverse remodelling post-MI. In addition, CXCL4 up-regulates Ccr4 and Itgb4 and down-regulates Adamts8, which may result in LV dilation. The cell images are from Servier Medical Art (www.servier.com).
Conflict of interest: none declared.
Funding
This work was supported by the National Institutes of Health [HL075360, HL129823, and GM114833 to M.L.L., HL111600 to C.M.R., HL051971, GM104357, and GM115428]; by the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development [5I01BX000505 to M.L.L. and IK2BX003922 to K.Y.D.]; and by the American Heart Association [15SDG22930009 to Y.M.]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the Veterans Administration, or the American Heart Association.
Supplementary Material
Footnotes
Time for primary review: 23 days
References
- 1. Ma Y, Iyer RP, Jung M, Czubryt MP, Lindsey ML.. Cardiac fibroblast activation post-myocardial infarction: current knowledge gaps. Trends Pharmacol Sci 2017;38:448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dassanayaka S, Jones SP.. Recent developments in heart failure. Circ Res 2015;117:e58–e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ma Y, Mouton AJ, Lindsey ML.. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl Res 2018;191:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lindsey ML, Saucerman JJ, DeLeon-Pennell KY.. Knowledge gaps to understanding cardiac macrophage polarization following myocardial infarction. Biochim Biophys Acta 2016;1862:2288–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tourki B, Halade G.. Leukocyte diversity in resolving and nonresolving mechanisms of cardiac remodeling. FASEB J 2017;31:4226–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kain V, Halade GV.. Big eater macrophages dominate inflammation resolution following myocardial infarction. J Mol Cell Cardiol 2015;87:225–227. [DOI] [PubMed] [Google Scholar]
- 7. Leblond AL, Klinkert K, Martin K, Turner EC, Kumar AH, Browne T, Caplice NM.. Systemic and cardiac depletion of M2 macrophage through CSF-1R signaling inhibition alters cardiac function post myocardial infarction. PLoS One 2015;10:e0137515.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Couto G, Liu W, Tseliou E, Sun B, Makkar N, Kanazawa H, Arditi M, Marbán E.. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J Clin Invest 2015;125:3147–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, Sadek HA, Olson EN.. Macrophages are required for neonatal heart regeneration. J Clin Invest 2014;124:1382–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wulfers EM, Seemann G, Courties G, Iwamoto Y, Sun Y, Savol AJ, Sager HB, Lavine KJ, Fishbein GA, Capen DE, Da Silva N, Miquerol L, Wakimoto H, Seidman CE, Seidman JG, Sadreyev RI, Naxerova K, Mitchell RN, Brown D, Libby P, Weissleder R, Swirski FK, Kohl P, Vinegoni C, Milan DJ, Ellinor PT, Nahrendorf M.. Macrophages facilitate electrical conduction in the heart. Cell 2017;169:510–522.e520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deutsch E, Johnson SA, Seegers WH.. Differentiation of certain platelet factors related to blood coagulation. Circ Res 1955;3:110–115. [DOI] [PubMed] [Google Scholar]
- 12. Wang Z, Huang H.. Platelet factor-4 (CXCL4/PF-4): an angiostatic chemokine for cancer therapy. Cancer Lett 2013;331:147–153. [DOI] [PubMed] [Google Scholar]
- 13. Lapchak PH, Ioannou A, Rani P, Lieberman LA, Yoshiya K, Kannan L, Dalle Lucca JJ, Kowalska MA, Tsokos GC.. The role of platelet factor 4 in local and remote tissue damage in a mouse model of mesenteric ischemia/reperfusion injury. PLoS One 2012;7:e39934.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sachais BS, Turrentine T, Dawicki McKenna JM, Rux AH, Rader D, Kowalska MA.. Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE-/- mice. Thromb Haemost 2007;98:1108–1113. [PubMed] [Google Scholar]
- 15. Koenen RR, von Hundelshausen P, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, Kramp BK, Piccinini AM, Paludan SR, Kowalska MA, Kungl AJ, Hackeng TM, Mayo KH, Weber C.. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med 2009;15:97–103. [DOI] [PubMed] [Google Scholar]
- 16. von Hundelshausen P, Agten SM, Eckardt V, Blanchet X, Schmitt MM, Ippel H, Neideck C, Bidzhekov K, Leberzammer J, Wichapong K, Faussner A, Drechsler M, Grommes J, van Geffen JP, Li H, Ortega-Gomez A, Megens RT, Naumann R, Dijkgraaf I, Nicolaes GA, Doring Y, Soehnlein O, Lutgens E, Heemskerk JW, Koenen RR, Mayo KH, Hackeng TM, Weber C.. Chemokine interactome mapping enables tailored intervention in acute and chronic inflammation. Sci Transl Med 2017;9:eaah6650.. [DOI] [PubMed] [Google Scholar]
- 17. Pervushina O, Scheuerer B, Reiling N, Behnke L, Schroder JM, Kasper B, Brandt E, Bulfone-Paus S, Petersen F.. Platelet factor 4/CXCL4 induces phagocytosis and the generation of reactive oxygen metabolites in mononuclear phagocytes independently of Gi protein activation or intracellular calcium transients. J Immunol 2004;173:2060–2067. [DOI] [PubMed] [Google Scholar]
- 18. Wan E, Yeap XY, Dehn S, Terry R, Novak M, Zhang S, Iwata S, Han X, Homma S, Drosatos K, Lomasney J, Engman DM, Miller SD, Vaughan DE, Morrow JP, Kishore R, Thorp EB.. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res 2013;113:1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res 2012;110:159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kingery JR, Hamid T, Lewis RK, Ismahil MA, Bansal SS, Rokosh G, Townes TM, Ildstad ST, Jones SP, Prabhu SD.. Leukocyte iNOS is required for inflammation and pathological remodeling in ischemic heart failure. Basic Res Cardiol 2017;112:19.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ma Y, Halade GV, Zhang J, Ramirez TA, Levin D, Voorhees A, Jin YF, Han HC, Manicone AM, Lindsey ML.. Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation. Circ Res 2013;112:675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. DeLeon-Pennell KY, Iyer RP, Ma Y, Yabluchanskiy A, Zamilpa R, Chiao YA, Cannon P, Cates C, Flynn ER, Halade GV, de Castro Bras LE, Lindsey ML.. The Mouse Heart Attack Research Tool (mHART) 1.0 Database. Am J Physiol Heart Circ Physiol 2018;315:H522–H530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brooks HL, Lindsey ML.. Guidelines for authors and reviewers on antibody use in physiology studies. Am J Physiol Heart Circ Physiol 2018;314:H724–H732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR, Lindsey ML.. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol 2017;112:33.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Meschiari CA, Jung M, Iyer RP, Yabluchanskiy A, Toba H, Garrett MR, Lindsey ML.. Macrophage overexpression of matrix metalloproteinase-9 in aged mice improves diastolic physiology and cardiac wound healing after myocardial infarction. Am J Physiol Heart Circ Physiol 2018;314:H224–H235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, Flynn ER, Freeman TC, Saucerman JJ, Garrett MR, Ma Y, Harmancey R, Lindsey ML.. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res Cardiol 2018;113:26.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lindsey ML, Iyer RP, Jung M, DeLeon-Pennell KY, Ma Y.. Matrix metalloproteinases as input and output signals for post-myocardial infarction remodeling. J Mol Cell Cardiol 2016;91:134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lindsey ML, Bolli R, Canty JM Jr, Du XJ, Frangogiannis NG, Frantz S, Gourdie RG, Holmes JW, Jones SP, Kloner RA, Lefer DJ, Liao R, Murphy E, Ping P, Przyklenk K, Recchia FA, Schwartz Longacre L, Ripplinger CM, Van Eyk JE, Heusch G.. Guidelines for experimental models of myocardial ischemia and infarction. Am J Physiol Heart Circ Physiol 2018;314:H812–H838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De Jesus NM, Wang L, Lai J, Rigor RR, Francis Stuart SD, Bers DM, Lindsey ML, Ripplinger CM.. Antiarrhythmic effects of interleukin 1 inhibition after myocardial infarction. Heart Rhythm 2017;14:727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang Y, Guo T, Oda T, Chakraborty A, Chen L, Uchinoumi H, Knowlton AA, Fruen BR, Cornea RL, Meissner G, Bers DM.. Cardiac myocyte Z-line calmodulin is mainly RyR2-bound, and reduction is arrhythmogenic and occurs in heart failure. Circ Res 2014;114:295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lindsey ML, Kassiri Z, Virag JAI, de Castro Brás LE, Scherrer-Crosbie M.. Guidelines for measuring cardiac physiology in mice. Am J Physiol Heart Circ Physiol 2018;314:H733–H752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma Y, Chiao YA, Clark R, Flynn ER, Yabluchanskiy A, Ghasemi O, Zouein F, Lindsey ML, Jin YF.. Deriving a cardiac ageing signature to reveal MMP-9-dependent inflammatory signalling in senescence. Cardiovasc Res 2015;106:421–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT.. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 2009;55:611–622. [DOI] [PubMed] [Google Scholar]
- 34. DeLeon-Pennell KY, Tian Y, Zhang B, Cates CA, Iyer RP, Cannon P, Shah P, Aiyetan P, Halade GV, Ma Y, Flynn E, Zhang Z, Jin YF, Zhang H, Lindsey ML.. CD36 is a matrix metalloproteinase-9 substrate that stimulates neutrophil apoptosis and removal during cardiac remodeling. Circ Cardiovasc Genet 2016;9:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zamilpa R, Kanakia R, Cigarroa J, Dai Q, Escobar GP, Martinez H, Jimenez F, Ahuja SS, Lindsey ML.. CC chemokine receptor 5 deletion impairs macrophage activation and induces adverse remodeling following myocardial infarction. Am J Physiol Heart Circ Physiol 2011;300:H1418–H1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lindsey ML, Gray GA, Wood SK, Curran-Everett D.. Statistical considerations in reporting cardiovascular research. Am J Physiol Heart Circ Physiol 2018;315:H303–H313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blanchet X, Cesarek K, Brandt J, Herwald H, Teupser D, Kuchenhoff H, Karshovska E, Mause SF, Siess W, Wasmuth H, Soehnlein O, Koenen RR, Weber C, von Hundelshausen P.. Inflammatory role and prognostic value of platelet chemokines in acute coronary syndrome. Thromb Haemost 2014;112:1277–1287. [DOI] [PubMed] [Google Scholar]
- 38. Gleissner CA, Shaked I, Little KM, Ley K.. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J Immunol 2010;184:4810–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma Y, de Castro Bras LE, Toba H, Iyer RP, Hall ME, Winniford MD, Lange RA, Tyagi SC, Lindsey ML.. Myofibroblasts and the extracellular matrix network in post-myocardial infarction cardiac remodeling. Pflugers Arch 2014;466:1113–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van den Borne SW, Cleutjens JP, Hanemaaijer R, Creemers EE, Smits JF, Daemen MJ, Blankesteijn WM.. Increased matrix metalloproteinase-8 and -9 activity in patients with infarct rupture after myocardial infarction. Cardiovasc Pathol 2009;18:37–43. [DOI] [PubMed] [Google Scholar]
- 41. Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT.. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest 2000;106:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vrij AA, Rijken J, Van Wersch JW, Stockbrügger RW.. Platelet factor 4 and beta-thromboglobulin in inflammatory bowel disease and giant cell arteritis. Eur J Clin Invest 2000;30:188–194. [DOI] [PubMed] [Google Scholar]
- 43. Baltus T, von Hundelshausen P, Mause SF, Buhre W, Rossaint R, Weber C.. Differential and additive effects of platelet-derived chemokines on monocyte arrest on inflamed endothelium under flow conditions. J Leukoc Biol 2005;78:435–441. [DOI] [PubMed] [Google Scholar]
- 44. von Hundelshausen P, Petersen F, Brandt E.. Platelet-derived chemokines in vascular biology. Thromb Haemost 2007;97:704–713. [DOI] [PubMed] [Google Scholar]
- 45. Nahrendorf M, Pittet MJ, Swirski FK.. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation 2010;121:2437–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G, Kerkau T, Frantz S.. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 2012;125:1652–1663. [DOI] [PubMed] [Google Scholar]
- 47. Shi G, Field DJ, Long X, Mickelsen D, Ko KA, Ture S, Korshunov VA, Miano JM, Morrell CN.. Platelet factor 4 mediates vascular smooth muscle cell injury responses. Blood 2013;121:4417–4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hartwig H, Drechsler M, Lievens D, Kramp B, von Hundelshausen P, Lutgens E, Weber C, Doring Y, Soehnlein O.. Platelet-derived PF4 reduces neutrophil apoptosis following arterial occlusion. Thromb Haemost 2014;111:562–564. [DOI] [PubMed] [Google Scholar]
- 49. Bdeir K, Gollomp K, Stasiak M, Mei J, Papiewska-Pajak I, Zhao G, Worthen GS, Cines DB, Poncz M, Kowalska MA.. Platelet-specific chemokines contribute to the pathogenesis of acute lung injury. Am J Respir Cell Mol Biol 2017;56:261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med 2014;46:e99.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gleissner CA, Shaked I, Erbel C, Bockler D, Katus HA, Ley K.. CXCL4 downregulates the atheroprotective hemoglobin receptor CD163 in human macrophages. Circ Res 2010;106:203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bonen A, Campbell SE, Benton CR, Chabowski A, Coort SL, Han XX, Koonen DP, Glatz JF, Luiken JJ.. Regulation of fatty acid transport by fatty acid translocase/CD36. Proc Nutr Soc 2004;63:245–249. [DOI] [PubMed] [Google Scholar]
- 53. Imai T, Chantry D, Raport CJ, Wood CL, Nishimura M, Godiska R, Yoshie O, Gray PW.. Macrophage-derived chemokine is a functional ligand for the CC chemokine receptor 4. J Biol Chem 1998;273:1764–1768. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.