

Abstract
In nontypeable Haemophilus influenzae (NTHi), the oligopeptide-binding protein (OppA) serves as the substrate-binding protein (SBP) of the oligopeptide transport system responsible for the import of peptides. We solved the crystal structure of nthiOppA in complex with hydrophobic peptides of various sizes. Our novel hexapeptide complex demonstrates the flexibility of the nthiOppA-binding cavity to expand and accommodate the longer peptide while maintaining similar protein–peptide interactions of smaller peptide complexes. In addition to acquiring peptides from the host environment, as a heme auxotroph NTHi utilizes host hemoproteins as a source of essential iron. OppA is a member of the Cluster C SBP family, and unlike other SBP families, some members recognize two distinctly different substrates. DppA (dipeptide), MppA (murein tripeptide), and SapA (antimicrobial peptides) are Cluster C proteins known to also transport heme. We observed nthiOppA shares this heme-binding characteristic and established heme specificity and affinity by surface plasmon resonance (SPR) of the four Cluster C proteins in NTHi. Ligand-docking studies predicted a distinct heme-specific cleft in the binding pocket, and using SPR competition assays, we observed that heme does not directly compete with peptide in the substrate-binding pocket. Additionally, we identified that the individual nthiOppA domains differentially contribute to substrate binding, with one domain playing a dominant role in heme binding and the other in peptide binding. Our results demonstrate the multisubstrate specificity of nthiOppA and the role of NTHi Cluster C proteins in the heme-uptake pathway for this pathogen.
Keywords: heme, peptide transport, bacterial metabolism, ABC transporter, ligand-binding protein, Cluster C, oligopeptide-binding protein, opportunistic pathogen, PepT importer
Introduction
Nontypeable Haemophilus influenzae (NTHi)2 is a widespread Gram-negative commensal that colonizes the human nasopharynx. As an opportunistic pathogen, NTHi is one of the principal bacteria isolated from the middle ear during otitis media infections. NTHi infections can also exacerbate other upper and lower respiratory tract diseases and induce complications for patients with chronic obstructive pulmonary disease and cystic fibrosis (1–3). The host immune system limits access to essential nutrients and produces antimicrobial peptides to prevent infections. To circumvent host-mediated defenses, NTHi manages a complex network of transport proteins to scavenge nutrients from the host environment.
The oligopeptide (Opp) transport system is an ATP-binding cassette (ABC) transporter responsible for the uptake of peptides, supplying pathogens with essential nutrients as a source of carbon, nitrogen, and amino acids. These bacterial importers are composed of five subunits. In Gram-negative bacteria, the substrate-binding protein (SBP) binds the substrate in the periplasm and shuttles it to the transporter. Transmembrane domains (TMDs) form a dimer to create the translocation channel through the membrane. Energized by ATP binding and hydrolysis, a dimer of nucleotide-binding domains (NBDs) is coupled to the TMDs to drive conformational changes to import the substrate into the cell. Additionally, nutrient acquisition through the Opp system influences many cellular processes, including internalization of quorum-sensing peptides, biofilm production, modifying the cell surface, and antibiotic resistance (4–7).
OppA, the SBP of the Opp transport system, belongs to a group of structurally related SBPs involved in the uptake of a wide range of nutrients from nickel to peptides known as the Cluster C family. In Gram-negative bacteria, OppA has been shown to select for peptides between 3 and 5 amino acids with high affinity for tri- and tetrapeptides (8). Crystal structures of peptide-bound OppA from Salmonella typhimurium, Yersinia pestis, Escherichia coli, and Burkholderia pseudomallei show peptide specificity is independent of amino acid sequence of the peptides as protein–peptide interactions occur mainly through the peptide backbone (9–12). Gram-positive Lactococcus lactis OppA binds peptides 4–35 amino acids long with a preference for nonapeptides (13). Large binding cavities of oligopeptide homologs llOppA, Bacillus subtilis AppA, and Enterococcus faecalis PrgZ allow these proteins to fully enclose hepta- and nonapeptides in their substrate-binding pockets (14–16). OppA-mediated peptide transport is a necessary component of nutrient uptake and signaling pathways for survival and virulence of pathogens (17, 18).
One striking feature of the Cluster C family is that members play dual roles in nutrient uptake, importing their canonical substrates and heme. In E. coli, the Cluster C nickel SBP, NikA, binds heme in vivo and has been suggested to play a role as a heme chaperone in the periplasm (11). In addition to oppA, there are three other Cluster C family SBP genes encoded in the NTHi genome: hbpA (heme-binding protein, also known as GbpA) mediates import of GSH; sapA (sensitivity to antimicrobial peptides) provides antimicrobial peptide resistance; and NTHI0310 (19, 20). The predicted topology and high sequence similarities to Cluster C SBPs led us to identify NTHI0310 as a member of this family (Table S1), but its canonical substrate specificity is unknown. In H. influenzae, HbpA is necessary to use heme as an iron source during growth, and SapA is essential for NTHi survival in heme-starved growth conditions (21, 22). These in vivo studies highlight the overlapping functionality of some Cluster C proteins and their roles in the heme uptake network for this heme auxotroph.
Here, we probed peptide and heme substrate specificity of nthiOppA. We solved the structure of nthiOppA bound to a range of hydrophobic peptides and uncovered the peptide specificity of Gram-negative OppA extends beyond pentapeptides. In vivo studies have identified some Cluster C proteins are involved in the heme-uptake pathway, and we extended this study to compare the heme specificity and binding affinity of the NTHi Cluster C SBPs. In silico analysis of heme docking, coupled with substrate competition assays and individual domain analysis, allows us to propose how multisubstrate specificity may occur in nthiOppA. Our work highlights the multifunctional roles of Cluster C systems and the complexity of the heme transport network.
Results
Crystal structure of co-purified peptide nthiOppA complex
We expressed nthiOppA without the periplasmic signal sequence in E. coli and natively purified the protein using a C-terminal His tag. Crystallized nthiOppA was co-purified with bound endogenous peptide, indicated by additional electron density in the substrate-binding pocket. SBPs are frequently co-purified with their substrates, and OppA homologs have demonstrated a broad specificity for peptides (23, 24). The structure was solved by molecular replacement to a resolution of 1.85 Å with space group P21 21 21 and one monomer in the asymmetric unit. In general, SBPs have two structurally conserved globular domains consisting of a β-sheet that is flanked by α-helices (α/β-domains), and the five-strand sheet at the core of each domain is connected by two strands to the opposite α/β-domain. The substrate-binding pocket is formed at the interface of these domains. Specific to the Cluster C SBP family, domain I of nthiOppA is divided into two subdomains: domain IA is stitched together from residues 26 to 41, 210 to 290, and 510 to 541, and domain IB includes residues 42 to 209. Domain II spans residues 291 to 509 (Fig. 1A). The addition of domain IB makes Cluster C proteins larger than other SBPs in other clusters, and this allows for an expanded binding cavity to accommodate the larger substrates associated with this family of SBPs (25).
Figure 1.
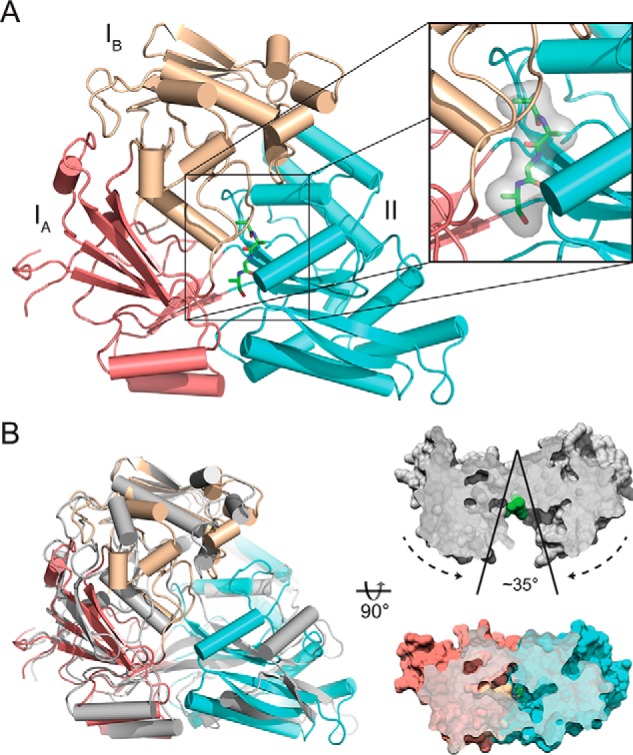
Crystal structure of nthiOppA in closed ligand-bound conformation. A, crystal structure of nthiOppA, colored by domain (IA, salmon; IB, wheat; II, cyan), bound to co-purified peptide in green. Inset, surface and stick representation of the co-purified peptide in substrate-binding pocket. B, cartoon representation of the ligand-bound structure of nthiOppA aligned with the open unbound ecOppA structure in gray (PDB code 3TCH), and surface representation of cross-sections of ecOppA and nthiOppA. Based on the alignment with nthiOppA, the green spheres in the open unbound ecOppA structure highlight the binding site where peptide interacts with the SBP to form the closed conformation.
When compared with open unbound ecOppA (RMSD = 4.42 Å), structural alignment of nthiOppA indicates a conformational change to the closed peptide-bound state (Fig. 1B). In the ligand-bound nthiOppA complex, domains I and II rotate toward the center of the protein to bury the peptide in the substrate-binding pocket. Interestingly, even though domains IA and IB are structurally independent, they rotate as a rigid domain in respect to domain II. The angle of rotation between the two domains is ∼35° from the open to the closed conformations. The co-purified peptide binds near the hinge region at the interface between the domains, and domain II provides a binding cleft with the majority of protein–peptide interactions for the bound peptide in the substrate-binding pocket.
Characterization of nthiOppA peptide interactions
Using a range of peptides with varying physical properties, we tested the specificity of nthiOppA. Peptides identified as Gram-negative or Gram-positive OppA substrates as well as peptides with an increase in hydrophobicity and length were used in a thermal shift assay. Peptides that increased the stability of nthiOppA (melting temperature, Tm) were identified as binding candidates (Fig. S2). The following six binding candidates raised the Tm between 2.5 and 6 °C: P1 (KKK); P2 (MGG); P3 (LGG); P4 (GIINTL); P5Long (YLGANGRGGGS); and P6Brady (bradykinin, RPPGFSPFR). P1KKK was one of the initial peptides discovered to bind OppA, and peptides P1KKK and P6Brady were first crystallized bound to seOppA and llOppA, respectively (9, 14, 26). Additionally, ecOppA has also been shown to have a preference for positively charged peptides with approximately half of co-purified peptides having at least one arginine, histidine, or lysine (11). The identified binding candidates indicate peptide specificity of nthiOppA is not limited to 3–5 amino acids peptides. nthiOppA can bind positive and hydrophobic peptides as well as longer peptides similar to Gram-positive OppA homologs.
The novel binding candidate peptides were co-crystallized with nthiOppA. Chemically denatured and refolded nthiOppA co-crystallized with P2MGG, P3LGG, and P4GIINTL in the same condition as natively purified nthiOppA with similar crystal morphology and diffraction quality. However, P5Long co-crystallization produced poorly formed and cracked crystals that reduced diffraction to 9 Å. For the endogenous peptide bound to the natively purified nthiOppA, a 4-residue peptide backbone was built and refined in the electron density. The observed electron density of the second residue of the co-purified peptide is large enough to fit a bulky side chain. However, the density for the side chains cannot be easily interpreted to confidently identify residue assignments, which is likely caused by a heterogeneous mixture of co-purified peptides bound to nthiOppA. Hydrophobic peptides P2MGG, P3LGG, and P4GIINTL have well-resolved electron density representing the bound peptides (Fig. S2). The overall structure of nthiOppA does not change dramatically between the different bound peptides with RMSD values from 0.07 to 0.33 Å compared with the co-purified peptide complex. While maintaining the protein–peptide interactions, the flexible binding cavity can accommodate longer peptides.
Broad substrate specificity of OppA is mainly regulated through hydrogen bonds to the peptide backbone, with few peptide side chain interactions (11, 27). In all of the nthiOppA complexes, the N-terminal end of the bound peptide is secured by a salt bridge (Fig. 2). In addition to the salt bridge, four hydrogen bonds secure the first residue of peptides in the substrate-binding pocket (Tyr-130, two from His-441, and Asp-443). A second salt bridge also contributes to the binding of the C-terminal ends of the tripeptides. For P2MGG, P3LGG, and the co-purified peptide, hydrogen bonds to both amino and carboxyl groups of the remaining residues secure the substrate in the binding pocket. P4GIINTL has the most peptide–backbone hydrogen bonds involving an additional eight residues: Glu-52, Val-54, Asn-388, and Asn-392; two from Arg-437, and Gly-439 and Tyr-509 (Fig. 2D). The P4GIINTL complex shows nthiOppA–peptide interactions occur with at least the first six residues of a peptide, providing a platform for nthiOppA to bind longer peptides such as PLong and PBrady. Like the tripeptide complexes, the amino and carbonyl groups of the first three P4GIINTL residues form hydrogen bonds in the binding cavity, and only the carbonyl groups of the remaining peptide residues hydrogen bond with nthiOppA. With extensive peptide backbone interactions, a salt bridge is not required to stabilize the charged C-terminal end of the hexapeptide.
Figure 2.

Protein–peptide backbone hydrogen bonds are independent of peptide side chains. The hydrogen bond interactions of P2MGG (A), P3LGG (B), co-purified peptide (C), and P4GIITNL (D) with nthiOppA are represented by orange dashed lines. The bound peptide is shown in green. Hydrogen bond lengths calculated in LigPlot+.
Flexible binding cavity accommodates bulky ligands
In the ligand-bound closed conformation, several Gram-positive OppA homologs have flexible substrate-binding pockets that vary in size. The binding cavities efPrgZ, bsAppA, and llOppA range in volume even when bound to long peptides, 1600, 2500, and 4900 Å3, respectively (16). Alignment of heptapeptide-bound efPrgZ with the P4GIINTL complex (RMSD = 1.30 Å) shows a conserved salt bridge and hydrogen bonds to the N-terminal residue of the peptide (Fig. S3). Similar to other Gram-positive OppA homologs, the P4GIINTL complex demonstrates the flexibility of the Gram-negative OppA-binding cavity by expanding to accommodate a larger substrate and maintaining the peptide interactions in the substrate-binding pocket. Comparing the P2MGG and the P4GIINTL complexes, rearrangement of backbone loops and side chains in the substrate-binding pocket widen the binding cavity to prevent steric clashes with the side chains of residues 4 and 5 of P4GIINTL. The side chains of Tyr-267 and His-441 adopt different rotamer conformations by rotating 89 and 88°, respectively, creating a larger binding pocket for the bound peptide (Fig. 3).
Figure 3.
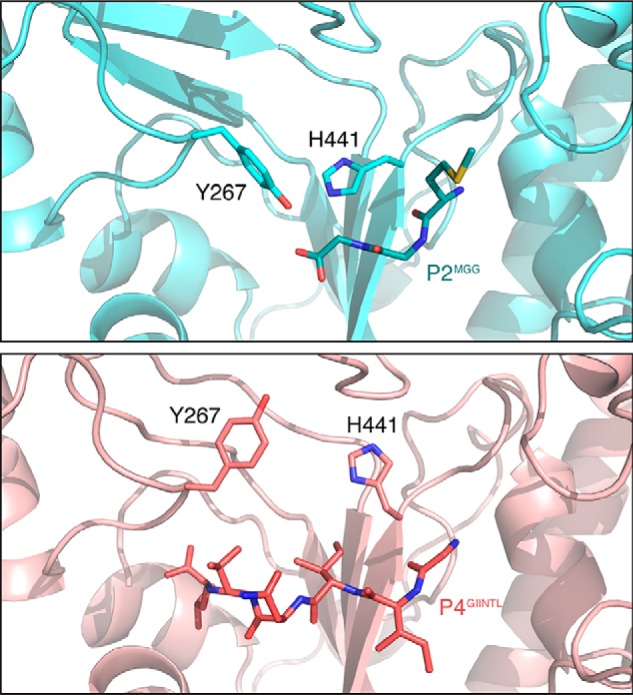
Flexible residues allow the substrate-binding pocket to expand and accommodate bulky hydrophobic side chains of bound peptide. Two binding pocket residues, Tyr-267 and His-441, adopt different rotamer conformations enlarging the binding cavity in the P4GIINTL complex. The P2MGG and P4GIINTL complexes are shown in cyan and salmon, respectively.
Heme specificity of Cluster C family in NTHi
The flexible binding cavity of nthiOppA and the established heme binding of other Cluster C SBPs led us to explore the ability of nthiOppA to bind heme. ABC transporters play a vital role in heme uptake in bacteria. To uncover how heme is shuttled to the inner membrane of this “heme-loving” pathogen, we looked at the conservation of heme ABC transport systems in NTHi. Homologs of other heme SBPs, including Yersinia enterocolitica HemT, Y. pestis HmuT, Shigella dysenteriae ShuT, Bordetella pertussis BhuT, Pseudomonas aeruginosa PhuT, E. coli ChuT, and Vibrio cholerae HutB have not been identified in the H. influenzae genome. NTHi Cluster C proteins provide the vital link in the uptake pathway between the OM receptors scavenging heme from the host and the ABC importers delivering the nutrient to the cytoplasm. We surveyed all of the identified NTHi Cluster C proteins. Despite the low sequence identity between the Cluster C SBP proteins in the NTHi genome (nthiHbpA (24.9% identity), nthiSapA (22.2% identity), and NTHI0310 (26.8% identity) compared with nthiOppA), these proteins have high sequence similarity (Fig. S1) and are predicted to have a similar overall structural topology.
Additional co-crystallization of nthiOppA with heme was attempted in the optimized crystallization condition and was screened for in a new crystallization condition, but no heme co-crystals were obtained. Therefore, we did in silico heme-binding studies of nthiOppA and two other solved Cluster C structures, Glaesserella (Haemophilus) parasuis HbpA and ecNikA. ROSIE ligand-docking program was used to predict the most likely interactions between the co-purified peptide-bound nthiOppA complex and heme (28). Representative top-ranked solutions for the SBPs show a similar location and ligand position of heme in each protein (Fig. 4). Based on the docking solutions, movement of backbone loops and side chains in the binding cavities of all three SBPs allows for the docking of heme near the canonical substrate-binding sites. The top-ranked solutions for nthiOppA indicate the substrate-binding pocket could be large enough to accommodate both heme and peptide (Fig. 4A). The docked heme demonstrates the substrate-binding pocket extends into domain I and is adjacent to the peptide-binding site (Fig. 6B). This suggests heme binding of nthiOppA does not conflict with peptide–protein interactions and the possibility of a heme-specific cleft in the substrate-binding pocket.
Figure 4.
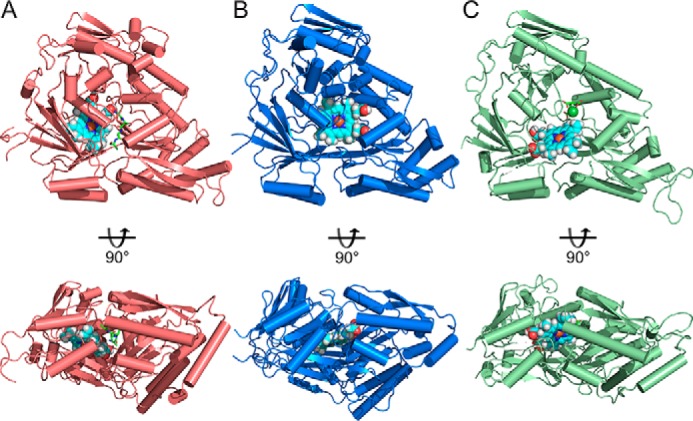
Comparing heme-docking studies of multiple Cluster C SBPs shows conserved heme-specific cleft in the substrate-binding pocket. Independent of canonical ligand size of these SBPs, structural conservation of Cluster C proteins creates flexible substrate-binding pockets large enough to accommodate heme binding. A, top-scoring model of heme docked in a heme-specific cleft of substrate-binding pocket of nthiOppA. The nthiOppA substrate-binding pocket is large enough to fit two substrates, heme and the co-purified peptide, shown in cyan and green, respectively. The docking model shows both ligands are fully enclosed and buried in the binding pocket. B, top-scoring model of gpHbpA (PDB code 3M8U) shows the docked heme in a relatively similar location in binding pocket. C, top-scoring heme docked model for ecNikA (PDB code 3DP8) also shows the substrate-binding pocket accommodates both heme and butane-1,2,4-tricarboxylate-chelated nickel, shown in green.
Figure 6.

Heme-specific binding cleft is independent of bound peptide in the nthiOppA substrate-binding pocket. Cutaway surface representations of the top-scoring ROSIE docking nthiOppA model show the heme-specific binding cleft does not overlap with the bound peptide. A, co-purified peptide discovered in the nthiOppA structure is buried in the peptide-binding site formed by a majority of hydrogen bonds with domain II. B, flexible binding cavity allows for a heme-specific cleft adjacent to the peptide-binding site. C–E, models of P1KKK (C), P2MGG (D), and P5Long (E) aligned with the heme-docking model. A competitive SPR assay was used to measure the ability of heme to bind nthiOppA in the presence of P1KKK (F), P2MGG (G), or P5Long (H). The sensograms display the response of peptide binding to nthiOppA, the combined injection of heme and peptide, and the theoretical sum model of the individual heme and peptide injections in green, blue, and black lines, respectively.
Previous studies have reported a wide range of heme equilibrium dissociation constants in the Cluster C SBPs. The heme affinity of ecNikA has previously been measured by tryptophan fluorescence quenching, KD value of 530 nm (29). Two other E. coli SBPs, DppA (dipeptide) and MppA (murein tripeptide), bind heme with estimated binding constants of 10 and 50 μm, respectively; a mutant strain lacking both of these SBPs was not able to use heme as an iron source during growth (30). The first identified Cluster C heme-binding protein, hiHbpA, was reported to have weak heme affinity with a KD value of 655 μm (19, 31). The equilibrium dissociation constants of ecDppA, ecMppA, and hiHbpA were calculated using native PAGE gel-shift assays. One major disadvantage of these assays is that the samples were not at equilibrium during electrophoresis (32). Heme-binding affinities of homologs for the other NTHi Cluster C SBPs have not previously been published. The limitations of the calculated heme affinity for some of the Cluster C SBPs led us to investigate the heme specificity and affinity of each of the NTHi SBPs.
To determine the shared heme-binding functionality of nthiOppA and other NTHi Cluster C proteins, we examined the heme specificity and binding affinity of the SBPs. The heme–protein binding kinetics of nthiHbpA, nthiOppA, nthiSapA, NTHI0310, and ecNikA were measured by SPR. The single-cycle kinetics method was used to measure heme binding of the immobilized SBPs, which included five serial injections with increasing concentrations of heme followed by an extended dissociation step. All four of the NTHi SBPs bind heme with variable affinity (Fig. 5). The heme affinity calculated by spectroscopic analysis of ecNikA provided a reference for the heme affinity measured by SPR. The SPR-calculated heme affinity of ecNikA, KD value of 526 nm, closely matches the previously reported KD value of 530 nm (29). Heme binds to nthiOppA with the highest affinity, KD value of 244 nm. The heme affinities of nthiHbpA and NTHI0310 are similar with KD values of 382 and 420 nm, respectively. nthiSapA showed a slightly lower heme binding with a KD value of 1.1 μm. The SPR sensograms for each SBP were fit to a two-state reaction model and indicate a 1:1 binding ratio of heme to SBP. The kinetic rate constants and equilibrium dissociation constant for each of the SBPs are summarized in Table 1. The differences in the heme affinities between the SBPs are largely explained by variability in the initial association rates. Despite their variation in their individual canonical substrates, the NTHi SBPs share specificity for heme.
Figure 5.

Surface plasmon resonance reveals NTHi SBPs bind heme. Single-cycle kinetic analysis of heme binding was performed using five analyte injections over immobilized NTHi SBPs. For the association phase, increasing concentrations of heme were injected in series followed by a final dissociation phase. Representative data in each sensogram is depicted by a red line and fit by a 1:1 two-state reaction model to determine kinetic constants and equilibrium dissociation constants, which is displayed by a dashed black line.
Table 1.
Kinetic and equilibrium constants of Cluster C SBPs
Standard deviation of triplicates is shown in parentheses.
| ka1 | kd1 | ka2 | kd2 | KD | |
|---|---|---|---|---|---|
| m−1 s−1 | s−1 | s−1 | s−1 | nm | |
| nthiOppA | 5.88 (0.21) × 104 | 3.98 (0.06) × 10−2 | 5.57 (0.08) × 10−3 | 3.14 (0.05) × 10−3 | 244 (11) |
| nthiHbpA | 3.46 (0.15) × 104 | 3.44 (0.31) × 10−2 | 4.65 (0.11) × 10−3 | 2.19 (0.07) × 10−3 | 382 (20) |
| nthiSapA | 1.77 (0.03) × 104 | 4.82 (0.14) × 10−2 | 5.07 (0.19) × 10−3 | 3.41 (0.07) × 10−3 | 1100 (24) |
| NTHI0310 | 4.21 (0.08) × 104 | 4.23 (0.16) × 10−2 | 5.42 (0.10) × 10−3 | 3.90 (0.06) × 10−3 | 420 (18) |
| ecNikA | 5.38 (0.46) × 104 | 7.66 (0.36) × 10−2 | 4.34 (0.28) × 10−2 | 2.52 (0.20) × 10−3 | 526 (76) |
Accommodating heme and peptide in nthiOppA substrate-binding pocket
To further elucidate the multisubstrate specificity of nthiOppA, we compared the heme and peptide interactions with the substrate-binding pocket. Using an SPR assay, we measured the competitive binding between heme and peptide by injecting either ligand alone or together over immobilized nthiOppA. The sum of the individual responses of heme and peptide was used to determine the theoretical response of both heme and peptide binding to the substrate pocket simultaneously. In the case of heme binding independent of peptide binding, we expect the observed SPR response of the combined ligands to correspond to the theoretical sum of the independent heme and the peptide responses. For competitive binding of heme, we expect a reduction of the observed SPR response of an injection with both ligands compared with the theoretical sum of the individual responses of heme and peptide. The SPR competition assay probes the availability of a heme-specific cleft in the presence of bound peptide in the substrate-binding pocket.
Using this experimental design, P1KKK, P2MGG, and P5Long were injected at a high concentration to load the peptide-specific site of the nthiOppA-binding pocket. The SPR sensograms of the individual peptide response and the combined heme and peptide response were collected for each peptide. The theoretical sum of the individual peptide and heme responses was calculated and compared with the observed combined heme and peptide response for each peptide (Fig. 6). The observed SPR response of the combined heme and P1KKK injection matched the theoretical sum of the responses for both ligands. For P1KKK, heme does not compete for binding at the peptide-specific binding site, and heme binding is independent of bound peptide (Fig. 6F). This is evidence of a heme-specific cleft in the substrate-binding pocket and corresponds to the heme-docking studies that predict bound peptide does not exclude heme binding. The combined heme and peptide injections of P2MGG and P5Long are lower than the theoretical sum of the individual responses. In these cases, the presence of peptide does limit but does not abolish heme binding (Fig. 6, G and H). In the ligand-bound closed conformation of P2MGG and P5Long complexes, steric hindrance of the bulky and rigid side chains or occlusion of the channel to the heme-specific binding cleft reduces heme binding. These competition assays show heme does not directly compete with peptide binding, and disruptions in heme and peptide binding are likely influenced by a heme-specific binding cleft in close proximity to the peptide-binding site.
Differential substrate binding of nthiOppA domains
To further expand our knowledge of nthiOppA substrate binding, we expressed and purified the individual domains of nthiOppA, nthiOppA1A1B and nthiOppA2 (Fig. 7, A and B). Using SPR, we determined nthiOppA1A1B has about 4-fold higher affinity for heme than nthiOppA2 with KD values of 577 nm and 2.46 μm, respectively (Fig. 7, C and D, and Table S3). nthiOppA1A1B binds heme with a similar affinity to nthiOppA. This corresponds with our heme-docking studies, which identified domain I as playing a major role in the formation of a heme-specific cleft in the binding cavity. An intrinsic tryptophan fluorescence quenching assay determined P4GIINTL binds nthiOppA2 with a KD value of 172 nm, and nthiOppA1A1B has a weaker affinity with a KD value of 11.7 μm (Fig. 7, E and F). Peptide binding of nthiOppA is largely mediated through interactions with domain II, and nthiOppA2 has a similar heme affinity as nthiOppA with a KD value of 754 nm (Fig. S4). Notably, there are two hydrogen bonds and a salt bridge between residues in domain II and the N-terminal residue of the bound peptide. Based on these binding studies, each domain plays a differential role in binding both substrates with domain I directing heme binding and domain II driving P4GIINTL binding of nthiOppA.
Figure 7.

nthiOppA1A1B and nthiOppA2 differentially bind heme and peptide substrates. Cartoon representations of individual nthiOppA domains, nthiOppA1A1B (A) and nthiOppA2 (B), are highlighted in salmon/wheat and cyan, respectively. C and D, single-cycle kinetic analysis of nthiOppA1A1B (C) and nthiOppA2 (D) heme binding. Each sensogram depicts representative data fit to a 1:1 two-state reaction model, shown in red and dashed black lines, respectively. The tryptophan fluorescence quenching of nthiOppA1A1B (E) and nthiOppA2 (F) upon P4GIINTL binding is shown. Representative data shown are fit to a one-site binding model.
Discussion
NTHi employs Cluster C SBPs to scavenge essential nutrients and adapt to rapidly changing microenvironments in the host. In Moraxella catarrhalis, oppA is necessary for invasion of human respiratory epithelial cells and persistence during infection (33). In addition to nutrient uptake, Gram-negative OppA plays a role in proteostasis of the periplasm. To aid in protein folding, ecOppA acts as a protein chaperone in the periplasm (34, 35). OppA can also assist in the recycling of misfolded periplasmic proteins by transporting protease-degraded peptides into the cytoplasm for reuse as a nutrient source.
Broadening our understanding of Gram-negative OppA peptide specificity is important for determining peptide utilization for essential nutrients and signaling pathways. Our data show nthiOppA bound to a longer peptide than previously observed in other Gram-negative OppA structures. The protein–peptide hydrogen bond interactions of the tripeptide-bound nthiOppA structures are maintained in the novel hexapeptide-bound complex and demonstrate peptide recognition of this SBP is independent of the length and amino acid composition of these peptides. The P4GIINTL nthiOppA complex illustrates the flexibility of the binding cavity that expands to accommodate the hexapeptide. Our findings bridge the peptide specificity of Gram-negative and Gram-positive OppA proteins and highlight the similarities of these SBPs.
During further study of the flexible binding cavity of nthiOppA, we identified heme as a novel substrate for this SBP. NTHi lacks the necessary enzymes for heme biosynthesis and has a strict growth requirement for heme as a source of iron to sustain aerobic respiration (36). This led us to survey all the Cluster C proteins in NTHi, and we identified that they all have specificity for heme with affinities ranging from 244 nm for nthiOppA to 1.1 μm for nthiSapA. Interestingly, the heme affinity of the individual SBP is not a determinant of the role these proteins play in the heme-uptake pathway. Considered a low-affinity heme–binding protein, nthiSapA plays a crucial role in the heme-uptake pathway for NTHi survival after heme starvation (22). Even in bacteria that employ high-affinity heme SBPs, such as HmuT, PhuT, and ShuT, there are functional overlapping heme transport systems. Deletion of the hmu locus in Y. pestis did not eliminate the ability of the mutant strain to utilize heme or colonize mice in the systemic infection model, indicating another heme transport system is sufficient for virulence in the mouse model (37). Functionally similar heme trafficking proteins in the cytoplasm, such as P. aeruginosa PhuS, have comparable heme affinity to Cluster C SBPs (38). These heme-binding proteins demonstrate heme affinity does not dictate functionality or limit the essential role of these proteins in the heme-uptake pathway for bacterial survival and pathogenesis.
NTHi Cluster C SBPs deliver substrate to three ABC transporters in the PepT (peptide/opine/nickel) family: gene clusters dpp, opp, and sap (Fig. 8). In vivo studies have demonstrated the Dpp and Sap importers are important for heme uptake. Deletion of components in the dpp or sap operons reduced the ability of the bacteria to utilize heme and recover after heme–iron starvation, respectively (19, 22, 44, 45). With more Cluster C SBP genes than PepT transporter gene clusters in NTHi, orphan SBPs have versatility in complex assembly within this family and share importers for substrate uptake. NTHi lacks the gene encoding dppA, and nthiHbpA (55% sequence identity to ecDppA) is an orphan SBP without an encoded transporter in its operon. In H. influenzae, HbpA recognizes the Dpp transporter and delivers substrate for import into the cell (19). EcMppA, another orphan SBP, delivers its peptide substrate to the Opp transporter, and both ecMppA and ecDppA are dependent on the Dpp transporter for heme uptake (30, 46). Similar to hiHbpA and ecMppA, orphan SBP NTHI0310 likely utilizes one of the PepT importers for substrate delivery. These functionally overlapping Cluster C SBPs are conserved in Haemophilus (Fig. S5).
Figure 8.
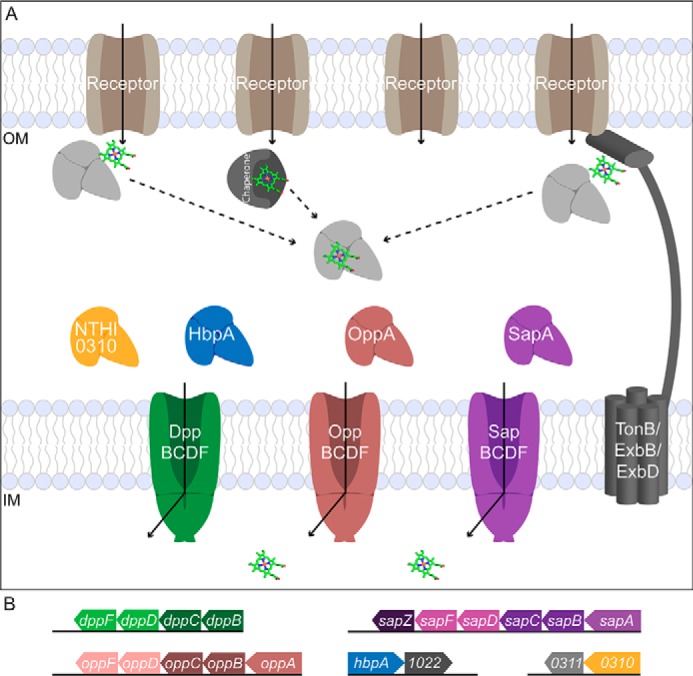
Multiple OM receptors and ABC transporters mediate heme uptake in NTHi. A, the human host has many iron/heme reservoirs, including hemoglobin, haptoglobin, myoglobin, and serum albumin. NTHi has many OM receptors (Hup, HgpABC, HxuC, and HemR) to scavenge heme, and each OM receptor recognizes a unique combination of host hemoproteins, hemophores, or free heme for utilization in the uptake pathway. The TonB/ExbB/ExbD complex provides energy to the OM receptors to transport the key nutrient across the membrane. SBPs acquire substrate from other components of the transport system, including metallochaperones, and the receptor/TonB complex (39–42) and deliver it to their cognate ABC transporter, powered by ATP hydrolysis, for import into the cell (adapted from Faraldo-Gomez and Sansom (43)). B, gene clusters of NTHi Cluster C SBPs and PepT importers. The opp and sap operons contain the SBP, two TMDs and two NBDs. The dpp operon includes the TMDs and NBDs without the corresponding dppA SBP. The remaining gene clusters include the orphan SBPs, hbpA and NTHI0310.
In the dynamic and perilous host environment, it is advantageous for bacteria to manage multiple heme SBPs to ensure the pathogen maintains access to the essential nutrient. Despite the canonical substrates of Cluster C SBPs ranging in diversity and size from a nickel ion to antimicrobial peptides, the unifying characteristic of these proteins has previously been their overall structure conserved from a common ancestor (47). Here, we have identified multisubstrate specificity for heme is a shared characteristic of NTHi Cluster C SBPs. This functional overlap between the Cluster C SBPs, particularly the previously unknown heme-binding capability of nthiOppA and NTHI0310, has made it a challenge to fully characterize the heme-uptake pathway. Further studies of the unique and essential role each SBP plays in the transport of heme need to be explored. Better understanding of the interplay between these multifunctional Cluster C SBPs will help us uncover how pathogens adapt and overcome host-mediated defenses.
Experimental procedures
Materials and methods
All peptides were solubilized according to the manufacturers' recommendations. Peptides P1KKK, P3LGG, and P6Brady (bradykinin, RPPGFSPFR) were purchased from Sigma. Custom peptides P2MGG, P4GIINTL, and P5Long (YLGANGRGGGS) were synthesized by GenScript. A 10 mm heme stock solution was prepared by dissolving hemin chloride (Strem Chemicals) in 100% DMSO, and the heme concentration was confirmed using the pyridine–hemochromagen method.
Bioinformatics analysis
The genome of NTHi 86-028NP was searched for Cluster C SBPs using BLAST queries. Four of these SBPs were identified in NTHi, HbpA, OppA, SapA, and a putative peptide–binding protein (NTHI0310). Percent identity and percent similarity comparisons between NTHi SBPs were calculated from pairwise sequence alignments (Table S1) generated by LALIGN using a BLOSUM50 matrix (EMBL-EBI). Alignment of NTHi SBPs was performed using ClustalW and superimposed with the secondary structure of nthiOppA in Jalview version 2 (48).
Expression vectors of SBPs
Expression constructs were amplified from the genomic DNA of the clinical strain of NTHi 86-028NP and E. coli strain K-12 substrain MG1655. Each construct was amplified without their predicted periplasmic signal sequence, nthiHbpA (residues 20–549), nthiOppA (residues 21–541), nthiSapA (residues 24–564), NTHI0310 (residues 24–514), and ecNikA (residues 23–524). The SBP constructs were cloned into the pET-21b vector using the NdeI and XhoI restriction sites to create constructs fused to a C-terminal His6 tag. Domain construct nthiOppA1A1B was created by site-directed mutagenesis; domain II was deleted and replaced with two glycine residues to fuse the C-terminal end of domain IA to domain IB (21–290 GG 510–541). nthiOppA2 (residues 291–509) was cloned using the same method as the other SBP constructs. All plasmids were verified by sequencing (ACGT Inc.).
Protein expression and purification
SBP proteins were expressed in E. coli BL21(DE3) grown in 1-liter cultures of Luria Broth media supplemented with 100 μg/ml ampicillin. Cells were grown at 37 °C to early log phase (OD600 of 0.4) and then cooled to 16 °C. After the incubator cooled, protein expression was induced (OD600 of 0.8) with 400 μm isopropyl 1-thio-β-d-galactopyranoside. Cells were cultured overnight and harvested by centrifugation at 5000 rpm and stored at −80 °C. Each step of the protein purification was carried out at 4 °C. Bacterial cells were resuspended in buffer A (25 mm HEPES, pH 8, 500 mm NaCl, 15 mm imidazole, pH 8) and lysed with an S-4000 sonicator (Misonix Sonicators). The cell debris was removed by centrifugation at 17,000 rpm for 1 h, and the soluble fraction was loaded on to an equilibrated nickel-nitrilotriacetic acid affinity chromatography column (ThermoFisher Scientific). The 5-ml column was rinsed with 10 column volumes of buffer A, followed by 5 column volumes of buffer B (buffer A, supplemented to 30 mm imidazole, pH 8). The SBPs were eluted with 8 column volumes of buffer C (buffer A, supplemented to 250 mm imidazole, pH 8). Eluted protein (determined by SDS-PAGE to be >95% pure) was dialyzed overnight in buffer D (25 mm HEPES, pH 7.5, 500 mm NaCl). The protein was applied to a HiLoad 16/600 Superdex 200 size-exclusion chromatography column (GE Healthcare). Buffers for nthiHbpA and NTHI0310 were supplemented with 5 mm 2-mercaptoethanol. Protein fractions were pooled and concentrated to 20 mg/ml. Proteins were stored in buffer D at −80 °C until needed.
For co-crystallization of nthiOppA with peptides, the protein was chemically denatured and refolded on the affinity column (see supporting Experimental procedures). The bacteria cells were resuspended, sonicated, and centrifuged as noted above. After the soluble fraction was applied to the affinity column and washed with 10 column volumes of buffer A, the protein was then denatured with 20 column volumes of denaturing buffer (6 m GdnHCl, 25 mm HEPES, 15 mm imidazole, pH 8). The column was rinsed with 10 column volumes of refolding buffers with decreasing concentrations of GdnHCl (buffer A supplemented with 3, 1.5, 1, and 0.5 m GdnHCl). To remove the remaining GdnHCl, the column was washed with 1 column volume of buffer A, and the protein was eluted with 8 column volumes of buffer C. The protein was dialyzed, applied to the Superdex column, and stored as mentioned above.
Protein crystallization
Crystallization of the co-purified peptide nthiOppA complex (25 mm HEPES, pH 7.5, 500 mm NaCl) was achieved by the vapor diffusion in sitting drops at 22 °C. The nthiOppA crystals were obtained with a 1:1 ratio of 10 mg/ml protein and reservoir solution containing 0.1 m sodium acetate at pH 4.6 and 2.4 m ammonium sulfate. Crystals usually appeared within 24 h. Co-crystallization of nthiOppA with peptides was achieved using a 1:10 refolded OppA to peptide mixture yielding final concentrations of 1 mm peptide and 6 mg/ml protein in binding buffer (25 mm HEPES, pH 7.5, 150 mm NaCl). The mixture was incubated on ice for 30 min. Co-crystals grew in the same reservoir solution and conditions. All crystals were briefly soaked in cryoprotectant consisting of reservoir solution supplemented with 15% v/v glycerol, harvested with a nylon loop, and flash-cooled in liquid nitrogen.
X-ray diffraction data collection and structure determination
Diffraction data were collected at the Advanced Photon Source (Argonne, IL) LS-CAT beamline 21-ID-D with an Eiger X 9M detector (DECTRIS AG). The diffraction data were integrated using XDS (49) and scaled with AIMLESS, from the CCP4 suite (50). The initial model for nthiOppA was determined using the molecular replacement pipeline Balbes (51), and model building was further improved with ARP/warp (52) from the on-line CCP4 platform. The output model was manually rebuilt over several cycles with Coot (53). Each peptide was built in the observed electron density, and the models were refined with REFMAC (54). Validation statistics of the final models were calculated with Molprobity (55). Details of data quality and structure refinement are summarized in Table 2. Also, see Table S2 for additional details of data quality and structure refinement for P1KKK, P5Long, and P6Brady peptides. Coordinate files have been deposited in the Protein Data Bank under the accession codes 6DQQ, 6DQR, 6DQT, 6DQU, 6DTF, 6DTG, and 6DTH. Structural figures, analysis of nthiOppA substrate-bound states, and sequence-independent structural alignments with RMSD calculations were performed in PyMOL version 2.0 (Schrödinger, LLC). Protein–peptide hydrogen bond lengths were calculated in LigPlot+ version 1.4 (56).
Table 2.
Data collection and refinement statistics
Data for the highest-resolution shell are given in parentheses. All structures were determined using single crystals.
| Statistics | OppA structures |
|||
|---|---|---|---|---|
| Co-purified + nthiOppA | P2MGG + nthiOppA | P3LGG + nthiOppA | P4GIINTL + nthiOppA | |
| Space group | P21 21 21 | P21 21 21 | P21 21 21 | P21 21 21 |
| Data collection | ||||
| Unit cell dimensions, Å | ||||
| Aa | 49.99 | 47.57 | 49.52 | 47.90 |
| Bb | 90.90 | 92.19 | 90.95 | 92.96 |
| Cc | 108.78 | 108.37 | 108.86 | 108.34 |
| Unit cell angles (°) | ||||
| Aα | 90.0 | 90.0 | 90.0 | 90.0 |
| Bβ | 90.0 | 90.0 | 90.0 | 90.0 |
| Cγ | 90.0 | 90.0 | 90.0 | 90.0 |
| Resolution (Å) | 46.67–1.85 | 35.75–2.08 | 36.63–1.95 | 35.88–1.65 |
| (1.89–1.85) | (2.14–2.08) | (2.00–1.95) | (1.68–1.65) | |
| Wavelength (Å) | 1.0782 | 1.0781 | 1.0781 | 1.0781 |
| Completeness (%) | 99.4 (99.5) | 99.1 (99.9) | 99.6 (99.8) | 99.9 (99.9) |
| Rmerge | 0.077 (0.538) | 0.080 (0.670) | 0.091 (0.665) | 0.059 (0.589) |
| Average I/σI | 18.6 (4.2) | 15.6 (3.1) | 15.3 (3.7) | 16.9 (3.0) |
| Redundancy | 13.3 | 11.1 | 10.9 | 9.0 |
| Total reflections | 572,217 | 323,020 | 397,113 | 527,954 |
| Unique reflections | 42,868 | 2,9098 | 36,506 | 58,988 |
| Refinement | ||||
| Rwork/Rfree | 0.1598/0.2069 | 0.1770/0.2344 | 0.1664/0.2048 | 0.1598/0.2069 |
| No. of atoms | ||||
| All atoms | 4586 | 4333 | 4349 | 4521 |
| Protein | 4169 | 4115 | 4157 | 4115 |
| Water | 375 | 184 | 153 | 357 |
| Average B-factor (Å2) | ||||
| All atoms | 33.3 | 49.3 | 37.1 | 32.1 |
| Protein | 32.6 | 49.1 | 36.9 | 31.4 |
| Water | 40.0 | 50.7 | 38.9 | 39.2 |
| RMSD | ||||
| Bond lengths (Å) | 0.017 | 0.013 | 0.010 | 0.017 |
| Bond angles (°) | 1.695 | 1.483 | 1.410 | 1.695 |
| Ramachandran statistics | ||||
| Favored (%) | 98.1 | 97.1 | 97.7 | 97.5 |
| Allowed (%) | 1.7 | 2.7 | 2.3 | 2.3 |
| Outliers (%) | 0.2 | 0.2 | 0.0 | 0.2 |
Thermal shift assay
Peptide binding candidates were identified using a thermal shift assay of nthiOppA. Reactions of 2 μg of refolded nthiOppA, 5× SYPRO Orange (ThermoFisher Scientific), and 1 mm peptide in binding buffer (25 mm HEPES, pH 7.5, 150 mm NaCl) were placed in a 384-well PCR plate. The plate was heated from 25 to 95 °C with a heating rate of 0.5 °C/min. The fluorescence intensity was measured with an excitation wavelength of 470 nm and emission wavelength of 580 nm. The thermal shift assay was performed using a Bio-Rad CFX384 Real-Time Detection System (Bio-Rad).
In silico heme-docking studies
Ligand docking of heme was conducted with ROSIE (Rosetta Online Server that Includes Everyone) ligand-docking protocol (28). Along with the structure of nthiOppA that we solved, we looked at other previously solved structures for gpHbpA (PDB code 3M8U) and ecNikA (PDB code 3DP8). The gpHbpA structure has 74% sequence identity to nthiHbpA. The solvent-free structure of each SBP was used as the template, and heme with a 2+ formal charge was used as the ligand. The substrate-binding pocket was probed with a 7 Å search radius starting at x = 38.0, y = 43.5, and z = 10.5, and the search radius was centered at the interior of the protein. For each protein, 200 structures were generated and ranked based on the calculated lowest interface energy. Of the 20 top-ranked ligand poses for nthiOppA, heme was docked in the same position in the substrate-binding pocket in 18 of the solutions. For hiHbpA and ecNikA there were 11 and 19 solutions with the same location, respectively. For the remaining solutions in the top 20, the heme molecule is overlapped with the most common ligand pose, but the porphyrin ring is slightly rotated. Top-scoring docking models were displayed using PyMOL.
Heme affinity determined by surface plasmon resonance
SBPs were coupled via a standard amine-coupling method in flow channels 2, 3, and 4 on a CM5 sensor chip (GE Healthcare); 1000–2000 RU of each SBP was immobilized. Flow channel 1 was designated the control channel. The surface of the chip was equilibrated in running buffer (25 mm HEPES, pH 7.5, 150 mm NaCl, 0.1% Tween 20, 2% DMSO) at a flow rate of 100 μl/min for at least 3 h. The data were obtained using single-cycle kinetic experiments in triplicate. For each replicate, five analyte (heme) injections were prepared by 2-fold serial dilution. The analyte samples were consecutively injected by increasing heme concentrations over all four channels to determine the binding constants of the SBPs. The association time of each analyte injection was 1 min, followed by a final 5-min dissociation step. SBP kinetic experiments were run at a flow rate of 40 μl/min at 25 °C. The sensor surface was regenerated after each experiment with two 30-s injections of running buffer, supplemented with 0.1% SDS, at a flow rate of 50 μl/min. All experiments were performed with the Biacore T200 instrument (GE Healthcare) according to the manufacturer's instructions. Kinetic rate constants and equilibrium dissociation constants were determined by fitting the data globally to the 1:1 two-state reaction model using the Biacore T200 evaluation software version 3.0 (GE Healthcare).
Surface plasmon resonance competition assay
After nthiOppA immobilization (3300 RU), the chip was equilibrated in running buffer as described above. For the competition assay, analytes were injected for 1 min at a flow rate of 40 μl/min at 25 °C. A 500 nm heme injection was used to calculate the heme response. Individual injections of P1KKK, P2MGG, and P5Long were measured at 250, 1.5, and 250 nm, respectively. Maintaining the concentration of each analyte, combined mixtures of heme and peptide were injected. The sensor surface was regenerated after each experiment with a 30-s injection of running buffer, supplemented with 0.1% SDS, at a flow rate of 50 μl/min. The response for each analyte was observed in triplicate. All experiments were performed with the Biacore T200 instrument (GE Healthcare) according to the manufacturer's instructions.
Circular dichroism (CD) spectroscopy
The nthiOppA domain samples were diluted to 16 μm in 5 mm Tris, pH 7.5, 15 mm NaCl. Far-UV CD spectra were collected using a 0.1-cm quartz cuvette from 260 to 190 nm in the step scan mode, with a 2-nm bandwidth, a 4-s response time, and a 1.0-nm step speed. Each spectrum is the accumulation of three scans. CD analysis was performed using a J-815 CD spectrometer (Jasco). The spectra show strong helical and β-sheet secondary structure characteristics indicating nthiOppA1A1B and nthiOppA2 are well-folded domains (Fig. S4).
Intrinsic tryptophan fluorescence quenching
Solutions of 100 nm nthiOppA, nthiOppA1A1B, and nthiOppA2 were prepared in reaction buffer (25 mm HEPES, pH 7.5, 150 mm NaCl) for steady-state fluorescence experiments. P4GIINTL was titrated into the protein solution in steps of 0.2 or 10 μm. After each titration step, samples were stirred for at least 5 min and then left to rest for at least 5 min at room temperature. Samples were excited at 295 nm, and the fluorescence maximum was observed at 329 nm. UV-grade polyacrylic cuvettes with 1-cm path length and excitation and emission slits of 1 mm were used for data collection. All fluorescence intensity experiments were performed using a PC1 photon-counting steady-state fluorometer (ISS). The percent quenching of tryptophan fluorescence at 329 nm versus the P4GIINTL concentration was fit to a single-site binding model using GraphPad Prism 6.0.
Author contributions
K. J. T. and H. W. P. conceptualization; K. J. T. data curation; K. J. T. and H. W. P. formal analysis; K. J. T. and H. W. P. validation; K. J. T. and H. W. P. visualization; K. J. T. and H. W. P. methodology; K. J. T. and H. W. P. writing-original draft; K. J. T. and H. W. P. writing-review and editing; H. W. P. resources; H. W. P. supervision; H. W. P. funding acquisition; H. W. P. investigation; H. W. P. project administration.
Supplementary Material
Acknowledgments
We thank Drs. Kevin Mason and Saemee Song for helpful discussions and critical reading of the manuscript. We especially thank the staff of Life Sciences Collaborative Access Team 21-ID at the Advanced Photon Source, Argonne National Laboratory. We thankfully acknowledge the use of the SPR instrument in the Biophysics Core at University of Illinois at Chicago and Dr. Hyun Lee. We also thank the Keck Biophysics Facility and Structural Biology Facility at Northwestern University for use of equipment.
This work was supported by the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust, National Science Foundation Grant MCB-1121872, and National Institutes of Health NIGMS Molecular Biophysics Training Program Grant T32 GM008382. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5, Tables S1–S3, and supporting Experimental procedures.
- NTHi
- nontypeable H. influenzae
- OM
- outer membrane
- SBP
- substrate-binding protein
- SPR
- surface plasmon resonance
- OPP
- oligopeptide
- ABC
- ATP-binding cassette
- TMD
- transmembrane domain
- NBD
- nucleotide-binding domain
- RMSD
- root-mean-square deviation
- PDB
- Protein Data Bank
- RU
- response unit
- GdnHCl
- guanidine HCl.
References
- 1. Turk D. C. (1984) The pathogenicity of Haemophilus influenzae. J. Med. Microbiol. 18, 1–16 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 2. Monsó E., Ruiz J., Rosell A., Manterola J., Fiz J., Morera J., and Ausina V. (1995) Bacterial infection in chronic obstructive pulmonary disease. A study of stable and exacerbated outpatients using the protected specimen brush. Am. J. Respir. Crit. Care Med. 152, 1316–1320 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 3. Rajan S., and Saiman L. (2002) Pulmonary infections in patients with cystic fibrosis. Semin. Respir. Infect. 17, 47–56 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 4. Gominet M., Slamti L., Gilois N., Rose M., and Lereclus D. (2001) Oligopeptide permease is required for expression of the Bacillus thuringiensis plcR regulon and for virulence. Mol. Microbiol. 40, 963–975 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 5. Lee E. M., Ahn S. H., Park J. H., Lee J. H., Ahn S. C., and Kong I. S. (2004) Identification of oligopeptide permease (opp) gene cluster in Vibrio fluvialis and characterization of biofilm production by oppA knockout mutation. FEMS Microbiol. Lett. 240, 21–30 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 6. Flores-Valdez M. A., Morris R. P., Laval F., Daffé M., and Schoolnik G. K. (2009) Mycobacterium tuberculosis modulates its cell surface via an oligopeptide permease (Opp) transport system. FASEB J. 23, 4091–4104 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mistry A., Warren M. S., Cusick J. K., Karkhoff-Schweizer R. R., Lomovskaya O., and Schweizer H. P. (2013) High-level pacidamycin resistance in Pseudomonas aeruginosa is mediated by an opp oligopeptide permease encoded by the opp-fabI operon. Antimicrob. Agents Chemother. 57, 5565–5571 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guyer C. A., Morgan D. G., and Staros J. V. (1986) Binding specificity of the periplasmic oligopeptide-binding protein from Escherichia coli. J. Bacteriol. 168, 775–779 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tame J. R., Murshudov G. N., Dodson E. J., Neil T. K., Dodson G. G., Higgins C. F., Wilkinson A. J., Neil T. K., Dodson G. G., Higgins C. F., and Wilkinson A. J. (1994) The structural basis of sequence-independent peptide binding by OppA protein. Science 264, 1578–1581 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 10. Tanabe M., Mirza O., Bertrand T., Atkins H. S., Titball R. W., Iwata S., Brown K. A., and Byrne B. (2007) Structures of OppA and PstS from Yersinia pestis indicate variability of interactions with transmembrane domains. Acta Crystallogr. D Biol. Crystallogr. 63, 1185–1193 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 11. Klepsch M. M., Kovermann M., Löw C., Balbach J., Permentier H. P., Fusetti F., de Gier J. W., Slotboom D. J., and Berntsson R. P. (2011) Escherichia coli peptide binding protein OppA has a preference for positively charged peptides. J. Mol. Biol. 414, 75–85 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 12. Lassaux P., Peri C., Ferrer-Navarro M., Gourlay L. J., Gori A., Conchillo-Solé O., Rinchai D., Lertmemongkolchai G., Longhi R., Daura X., Colombo G., and Bolognesi M. (2013) A structure-based strategy for epitope discovery in Burkholderia pseudomallei OppA antigen. Structure 21, 167–175 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 13. Detmers F. J. M., Lanfermeijer F. C., Abele R., Jack R. W., Tampe R., Konings W. N., and Poolman B. (2000) Combinatorial peptide libraries reveal the ligand-binding mechanism of the oligopeptide receptor OppA of Lactococcus lactis. Proc. Natl. Acad. Sci. U.S.A. 97, 12487–12492 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Berntsson R. P., Doeven M. K., Fusetti F., Duurkens R. H., Sengupta D., Marrink S.-J., Thunnissen A.-M., Poolman B., and Slotboom D.-J. (2009) The structural basis for peptide selection by the transport receptor OppA. EMBO J. 28, 1332–1340 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Levdikov V. M., Blagova E. V., Brannigan J. A., Wright L., Vagin A. A., and Wilkinson A. J. (2005) The structure of the oligopeptide-binding protein, AppA, from Bacillus subtilis in complex with a nonapeptide. J. Mol. Biol. 345, 879–892 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 16. Berntsson R. P., Schuurman-Wolters G. K., Dunny G., Slotboom D.-J., and Poolman B. (2012) Structure and mode of peptide binding of pheromone receptor PrgZ*. J. Biol. Chem. 287, 37165–37170 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang C. H., Lin C. Y., Luo Y. H., Tsai P. J., Lin Y. S., Lin M. T., Chuang W. J., Liu C. C., and Wu J. J. (2005) Effects of oligopeptide permease in group A streptococcal infection. Infect. Immun. 73, 2881–2890 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jones M. M., Johnson A., Koszelak-Rosenblum M., Kirkham C., Brauer A. L., Malkowski M. G., and Murphy T. F. (2014) Role of the oligopeptide permease ABC transporter of Moraxella catarrhalis in nutrient acquisition and persistence in the respiratory tract. Infect. Immun. 82, 4758–4766 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vergauwen B., Elegheert J., Dansercoer A., Devreese B., and Savvides S. N. (2010) Glutathione import in Haemophilus influenzae Rd is primed by the periplasmic heme-binding protein HbpA. Proc. Natl. Acad. Sci. U.S.A. 107, 13270–13275 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parra-Lopez C., Baer M. T., and Groisman E. A. (1993) Molecular genetic analysis of a locus required for resistance to antimicrobial peptides in Salmonella typhimurium. EMBO J. 12, 4053–4062 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morton D. J., Madore L. L., Smith A., Vanwagoner T. M., Seale T. W., Whitby P. W., and Stull T. L. (2005) The heme-binding lipoprotein (HbpA) of Haemophilus influenzae: role in heme utilization. FEMS Microbiol. Lett. 253, 193–199 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 22. Mason K. M., Raffel F. K., Ray W. C., and Bakaletz L. O. (2011) Heme utilization by nontypeable Haemophilus influenzae is essential and dependent on sap transporter function. J. Bacteriol. 193, 2527–2535 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Silhavy T. J., Szmelcman S., Boos W., and Schwartz M. (1975) On the significance of the retention of ligand by protein. Proc. Natl. Acad. Sci. U.S.A. 72, 2120–2124 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lanfermeijer F. C., Picon A., Konings W. N., and Poolman B. (1999) Kinetics and consequences of binding of nona- and dodecapeptides to the oligopeptide binding protein (OppA) of Lactococcus lactis. Biochemistry 38, 14440–14450 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 25. Berntsson R. P., Smits S. H., Schmitt L., Slotboom D. J., and Poolman B. (2010) A structural classification of substrate-binding proteins. FEBS Lett. 584, 2606–2617 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 26. Goodell E. W., and Higgins C. F. (1987) Uptake of cell wall peptides by Salmonella typhimurium and Escherichia coli. J. Bacteriol. 169, 3861–3865 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tame J. R., Dodson E. J., Murshudov G., Higgins C. F., and Wilkinson A. J. (1995) The crystal structures of the oligopeptide-binding protein OppA complexed with tripeptide and tetrapeptide ligands. Structure 3, 1395–1406 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 28. Lyskov S., Chou F. C., Conchúir S. Ó., Der B. S., Drew K., Kuroda D., Xu J., Weitzner B. D., Renfrew P. D., Sripakdeevong P., Borgo B., Havranek J. J., Kuhlman B., Kortemme T., Bonneau R., et al. (2013) Serverification of molecular modeling applications: the rosetta online server that includes everyone (ROSIE). PLoS ONE 8, e63906 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shepherd M., Heath M. D., and Poole R. K. (2007) NikA binds heme: a new role for an Escherichia coli periplasmic nickel-binding protein. Biochemistry 46, 5030–5037 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 30. Létoffé S., Delepelaire P., and Wandersman C. (2006) The housekeeping dipeptide permease is the Escherichia coli heme transporter and functions with two optional peptide binding proteins. Proc. Natl. Acad. Sci. U.S.A. 103, 12891–12896 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hanson M. S., and Hansen E. J. (1991) Molecular cloning, partial purification, and characterization of a haemin-binding lipoprotein from Haemophilus influenzae type-b. Mol. Microbiol. 5, 267–278 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 32. Hellman L. M., and Fried M. G. (2007) Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2, 1849–1861 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murphy T. F., Brauer A. L., Johnson A., and Kirkham C. (2016) ATP-binding cassette (ABC) transporters of the human respiratory tract pathogen, Moraxella catarrhalis: role in virulence. PLoS ONE 11, e0158689 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Richarme G., and Caldas T. D. (1997) Chaperone properties of the bacterial periplasmic substrate-binding proteins. J. Biol. Chem. 272, 15607–15612 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 35. Lennon C. W., Thamsen M., Friman E. T., Cacciaglia A., Sachsenhauser V., Sorgenfrei F. A., Wasik M. A., and Bardwell J. C. (2015) Folding optimization in vivo uncovers new chaperones. J. Mol. Biol. 427, 2983–2994 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Granick S., and Gilder H. (1946) The porphyrin requirements of Haemophilus influenzae and some functions of the vinyl and propionic acid side chains of heme. J. Gen. Physiol. 30, 1–13 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thompson J. M., Jones H. A., and Perry R. D. (1999) Molecular characterization of the hemin uptake locus (hmu) from Yersinia pestis and analysis of hmu mutants for hemin and hemoprotein utilization. Infect. Immun. 67, 3879–3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O'Neill M. J., Bhakta M. N., Fleming K. G., and Wilks A. (2012) Induced fit on heme binding to the Pseudomonas aeruginosa cytoplasmic protein (PhuS) drives interaction with heme oxygenase (HemO). Proc. Natl. Acad. Sci. U.S.A. 109, 5639–5644 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Handali M., Roychowdhury H., Neupane D. P., and Yukl E. T. (2015) AztD, a periplasmic zinc metallochaperone to an ATP-binding cassette (ABC) transporter system in Paracoccus denitrificans. J. Biol. Chem. 290, 29984–29992 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ilari A., Alaleona F., Tria G., Petrarca P., Battistoni A., Zamparelli C., Verzili D., Falconi M., and Chiancone E. (2014) The Salmonella enterica ZinT structure, zinc affinity and interaction with the high-affinity uptake protein ZnuA provide insight into the management of periplasmic zinc. Biochim. Biophys. Acta 1840, 535–544 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 41. Carter D. M., Miousse I. R., Gagnon J. N., Martinez E., Clements A, Lee J., Hancock M. A., Gagnon H., Pawelek P. D., and Coulton J. W. (2006) Interactions between TonB from Escherichia coli and the periplasmic protein FhuD. J. Biol. Chem. 281, 35413–35424 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 42. James K. J., Hancock M. A., Gagnon J. N., and Coulton J. W. (2009) TonB interacts with BtuF, the Escherichia coli periplasmic binding protein for cyanocobalamin. Biochemistry 48, 9212–9220 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 43. Faraldo-Gómez J. D., and Sansom M. S. (2003) Acquisition of siderophores in Gram-negative bacteria. Nat. Rev. Mol. Cell Biol. 4, 105–116 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 44. Morton D. J., Seale T. W., Vanwagoner T. M., Whitby P. W., and Stull T. L. (2009) The dppBCDF gene cluster of Haemophilus influenzae: Role in heme utilization. BMC Res. Notes 2, 166 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vogel A. R., Szelestey B. R., Raffel F. K., Sharpe S. W., Gearinger R. L., Justice S. S., and Mason K. M. (2012) SapF-mediated heme–iron utilization enhances persistence and coordinates biofilm architecture of Haemophilus. Front. Cell. Infect. Microbiol. 2, 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Park J. T., Raychaudhuri D., Li H., Normark S., and Mengin-Lecreulx D. (1998) MppA, a periplasmic binding protein essential for import of the bacterial cell wall peptide l-alanyl-γ-d-glutamyl-meso-diaminopimelate. J. Bacteriol. 180, 1215–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu L. F., and Mandrand-Berthelot M. A. (1995) A family of homologous substrate-binding proteins with a broad range of substrate specificity and dissimilar biological functions. Biochimie (Paris) 77, 744–750 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 48. Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., and Barton G. J. (2009) Jalview version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Long F., Vagin A. A., Young P., and Murshudov G. N. (2008) BALBES: a molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Langer G., Cohen S. X., Lamzin V. S., and Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Emsley P., and Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 54. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 55. Chen V. B., Arendall W. B. 3rd., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Laskowski R. A., and Swindells M. B. (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.