

Abstract
5-Aminoimidazole-4-carboxamide 1-β-d-ribofuranoside (AICAR, or acadesine) is a precursor of the monophosphate derivative 5-amino-4-imidazole carboxamide ribonucleoside 5′-phosphate (ZMP), an intermediate in de novo purine biosynthesis. AICAR proved to have promising anti-proliferative properties, although the molecular basis of its toxicity is poorly understood. To exert cytotoxicity, AICAR needs to be metabolized, but the AICAR-derived toxic metabolite was not identified. Here, we show that ZMP is the major toxic derivative of AICAR in yeast and establish that its metabolization to succinyl-ZMP, ZDP, or ZTP (di- and triphosphate derivatives of AICAR) strongly reduced its toxicity. Affinity chromatography identified 74 ZMP-binding proteins, including 41 that were found neither as AMP nor as AICAR or succinyl-ZMP binders. Overexpression of karyopherin-β Kap123, one of the ZMP-specific binders, partially rescued AICAR toxicity. Quantitative proteomic analyses revealed 57 proteins significantly less abundant on nuclei-enriched fractions from AICAR-fed cells, this effect being compensated by overexpression of KAP123 for 15 of them. These results reveal nuclear protein trafficking as a function affected by AICAR.
Keywords: purine, yeast genetics, yeast metabolism, nucleoside/nucleotide analogue, proteomics, drug metabolism, small molecule
Introduction
AICAR6 is a natural metabolite that can be taken up by nucleoside transporters in human cells (1) and has multiple promising pharmacological properties (2). Indeed, AICAR has proved in vivo anti-tumor effects in xenograft models (3–5). It is currently under clinical trial phase I/II to treat patients with chronic lymphocytic leukemia and has shown good safety and tolerability properties, although some side effects have been reported (6). Most of the described AICAR effects require its metabolic conversion to the monophosphate form ZMP by adenosine kinase (2). As an intermediate of the purine de novo pathway, ZMP is naturally present in cells at low concentration (7). ZMP and its precursor SZMP are important regulatory molecules that promote interactions between specific transcription factors and thereby directly couple the flux in the purine de novo pathway to transcription of the genes encoding the corresponding enzymes (8, 9). In addition, ZMP co-regulates purine synthesis to phosphate utilization through its interaction with common transcription factors (10). Strikingly, the regulatory role of ZMP is conserved in bacteria, although the riboswitch mechanism involved is totally different (11). Although under physiological conditions ZMP is present in the micromolar range (7), upon feeding of the riboside precursor AICAR, ZMP concentration can increase to milli molar levels (1), and, as a result, AICAR displayed cytotoxicity in yeast and mammalian cells (2), although the exact mechanisms involved are not known. The best documented pharmacological effect of ZMP is activation of the low-energy sensor AMP-activated protein kinase (AMPK) (12, 13). This well-characterized effect was attributed to the AMP-mimetic properties of ZMP (14) because of the structural similarity of these nucleotides (see Fig. 1A). However, the anti-proliferative effects of AICAR were shown to be largely AMPK-independent (1, 15), thus implying that other critical ZMP targets exist in the cell that have not yet been identified.
Figure 1.
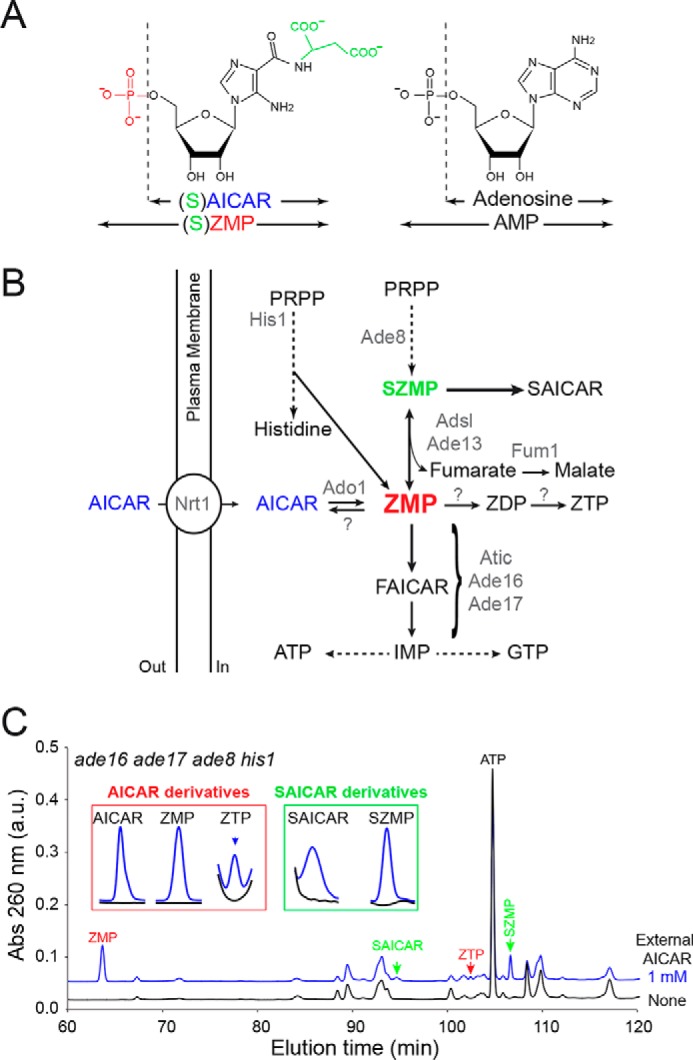
AICAR feeding of yeast cells results in accumulation of several AICAR-derived metabolites. A, chemical structure of ZMP and AMP derivatives. B, Adsl, adenylosuccinate lyase; Atic, AICAR transformylase IMP-cyclohydrolase; FAICAR, 5-formamido-4-imidazole carboxamide ribonucleoside 5′-phosphate; PRPP, 5-phosphoribosyl-1-pyrophosphate. Only the enzymes mentioned in the text are listed (in gray). Question marks correspond to enzymatic activities catalyzed by unidentified proteins prior to this work. C, accumulation of AICAR derivatives in yeast (ade8 ade16 ade17 his1; Y2950) fed (blue lines) or not (black lines) for 24 h with extracellular AICAR. Similar patterns were found in three independent metabolic extractions. Insets correspond to enlargement of chromatogram sections where AICAR and SAICAR derivatives were eluted.
Characterization of yeast mutants hypersensitive to AICAR revealed its uptake by the nicotinamide riboside transporter Nrt1 (see Fig. 1B) (1) and gave the first clues on the molecular basis of AICAR toxicity. Indeed, this approach established AICAR transport, ATP homeostasis, histone methylation, and ubiquitin metabolism as critical mechanisms underlying AICAR sensitivity (1, 16–18). Importantly, some of these mechanisms are conserved from yeast to human cells (16, 17). However, in these chemogenetic studies based on synthetic lethality, neither the toxic derivative of AICAR nor its protein target(s) were identified. Aiming to unveil the identity of the toxic derivative, our previous work established that intracellular AICAR and its succinyl-derivative SAICAR, which are the riboside forms of ZMP and SZMP, respectively, were not toxic for yeast cells (7). This situation is reminiscent of what happens in mammalian cells, in which the vast majority of AICAR effects are abolished by adenosine kinase inhibitors that block ZMP synthesis (19). Thus, for both yeast and mammalian cells, it was concluded that AICAR riboside had to be metabolically converted to the monophosphate form to become pharmacologically active and cytotoxic. Understanding how small molecules promote cytotoxicity is a complex task because small molecules are frequently metabolized to derivatives that may affect more than one biological process through interaction with multiple targets. This led us to ask two essential questions. First, is it ZMP itself and/or derived metabolites that are the toxic species? Second, which proteins are affected by the toxic metabolite?
In this work, we addressed these questions by combining yeast genetics, metabolic profiling, and proteomics to substantiate the assumption that ZMP itself is the toxic compound for yeast cells. Based on this result, we detected multiple ZMP-binding proteins and identified nuclear trafficking as one of the key functions affected by AICAR.
Results
AICAR treatment results in accumulation of ZMP, SZMP, and ZTP
Effects of AICAR feeding on yeast cells were evaluated by metabolic profiling of an ade16 ade17 ade8 his1 strain that is unable to metabolize ZMP to IMP (20) (Fig. 1B). As previously reported (1), in this genetic background, AICAR treatment caused massive ZMP accumulation (Fig. 1C). In addition, metabolic profile analysis revealed that AICAR treatment resulted in accumulation of SZMP, as well as of SAICAR, its riboside form (Fig. 1C). This is because in yeast, ZMP can be converted back to SZMP by adenylosuccinate lyase (Adsl), encoded by the ADE13 gene (21) (Fig. 1B). In turn, SZMP is dephosphorylated to SAICAR, which is nontoxic even at very high concentrations in yeast (7). We also observed that AICAR feeding led to accumulation of the triphosphate form, ZTP, whereas the diphosphate form ZDP was not detected (Fig. 1C). Finally, as expected, AICAR itself accumulated in yeast cells (Fig. 1C), but intracellular accumulation of this compound was previously demonstrated to be nontoxic (7).
Thus, AICAR treatment resulted in accumulation of ZMP, SZMP, and ZTP that could potentially affect cellular functions and result in toxicity. We thus used yeast genetics to identify the toxic derivative(s) of AICAR.
SZMP synthesis is not required for AICAR toxicity
We first asked whether SZMP could contribute to AICAR toxicity. The ade13 knockout mutation blocking SZMP synthesis from ZMP (Fig. 1B) was introduced in an ade16 ade17 ade8 his1 strain, and the resulting quintuple mutant strain was found to be highly sensitive to AICAR (Fig. 2A). As expected, the ade13 mutation abolished SZMP accumulation, whereas intracellular ZMP was higher than in the control strain (Fig. 2, B and C). We conclude that AICAR toxicity was not due to SZMP synthesis from ZMP but correlated with intracellular ZMP accumulation. These conclusions were strongly supported by analysis of a fumarase knockout mutant fum1, which accumulates fumarate (Fig. 2D) and thereby favors the reverse reaction from ZMP to SZMP by adenylosuccinate lyase Adsl (Ade13) (Fig. 1B), leading to increased SZMP and decreased ZMP levels (Fig. 2, E and F). In the corresponding mutant strain (fum1 ade16 ade17 ade8 his1), AICAR toxicity was abolished (Fig. 2G). From these results, we conclude that, in yeast, SZMP is not the metabolite that causes AICAR toxicity, but synthesis of SZMP rather contributes to its detoxification.
Figure 2.

SZMP is not required for AICAR toxicity but rather contributes to its detoxification in yeast. A–C, deletion of the ADE13 gene leads to increased ZMP accumulation and AICAR toxicity. A, yeast strains (Y2950 and Y10422) were grown overnight, serially diluted (1/10), and spotted on SDcasaWAU medium containing or lacking AICAR. The plates were imaged after 48 h of incubation at 30 °C. B and C, ZMP (B) and SZMP (C) relative content was determined on metabolic extracts from three independent cell cultures of each strain (Y2950 and Y10422) grown in SDcasaWAU medium and treated with AICAR (0.5 mm) for 24 h. Relative contents were set at 1 for the ADE13 control strain. D–G, favoring SZMP synthesis from ZMP lowers AICAR toxicity. D–F, metabolic analyses were done on three independent extractions from cells (Y2950 and Y3655) grown in SDcasaWAU medium and treated with AICAR (0.5 mm, 24 h). Relative content was set at 1 for the FUM1 control strain (Y2950). G, AICAR resistance was increased in a FUM1-deleted strain (Y3655) grown for 48 h in SDcasaWAU medium at 30 °C. B–F, errors bars and statistics correspond to standard deviation and Welch's unpaired t tests. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; UD, undetectable.
ZDP and ZTP are not toxic for yeast cells
To get a deeper insight into the mechanisms leading to AICAR toxicity, we performed an unbiased search for genes that suppress AICAR toxicity when overexpressed. A yeast genomic library carried on a multicopy vector was used to search for gene dosage suppressors of AICAR sensitivity in a yeast strain that can accumulate ZMP and derivatives when the riboside is provided (see “Experimental procedures” for details). The suppressor plasmids all carried an overlapping DNA region of chromosome XI containing four genes: two genes of unknown function (YKL023C-A and YKL023W), PAN3 encoding a subunit of the Pan2p-Pan3p poly(A)-RNase complex, and URA6, an essential gene encoding UMP kinase, which was reported to have significant AMP kinase activity (22). Of note, URA6 was previously identified as a suppressor of AICAR sensitivity hypomorphic allele of UBA1 but was not further characterized (18). Because ZMP is structurally close to AMP (Fig. 1A), we hypothesized that Ura6 could phosphorylate ZMP and thereby improve growth upon AICAR feeding. Indeed, overexpression of URA6 alone was sufficient to confer robust resistance to AICAR similarly to the initial suppressor plasmid (Fig. 3A). Metabolic profiling on yeast cells grown in the presence of AICAR revealed that overexpression of URA6 resulted in accumulation of ZDP and ZTP, both of which are hardly detectable in the control strain (Fig. 3, B and C). Noticeably, in the AICAR-resistant strain overexpressing URA6, ZMP was lower (Fig. 3D), whereas both ZDP and ZTP levels were higher than in the AICAR-sensitive control strain (Fig. 3, B and C). We conclude that ZMP, rather than its di- or triphosphate derivatives, is the toxic metabolite derived from AICAR. Accordingly, AICAR feeding had no effect on mutation rates in yeast (Fig. S1A) even under conditions (URA6 overexpression) resulting in high ZTP levels (Fig. S1B). Similarly, AICAR feeding did not affect other aspects of DNA metabolism such as mitotic recombination rate (Fig. S2) or telomere length (Fig. S3). Together, these results establish that AICAR toxicity is not associated with ZTP synthesis.
Figure 3.

The UMP kinase Ura6 promotes ZMP phosphorylation in yeast. A, overexpression of the URA6 gene strongly reduces ZMP toxicity. The ade3 ade16 ade17 ade8 his1 strain (Y8908) was transformed with either the suppressor plasmid isolated during the genetic screen (suppressor, P4979), a plasmid overexpressing URA6 gene (P4919), or the cognate empty vector as control (control; YEplac195). Transformants were grown overnight, serially diluted, and spotted on SDCasaWA medium containing, or lacking, AICAR. The plates were imaged after 2 days at 30 °C. B–D, effect of URA6 overexpression (p4919) on relative content of AICAR derivatives in the ade3 ade8 ade16 ade17 his1 strain (Y8908). Quantifications of metabolites were obtained from four independent extractions for each condition on strains grown in SDcasaWA medium for 24 h in the presence of 0.5 mm AICAR. Standard deviations are presented. Statistics correspond to Welch's t tests. ***, p < 0.001; ****, p < 0.0001. E–H, purified Ura6 (E) catalyzes ZDP, but not ZTP, synthesis in vitro. F–H, ZMP, ZDP, and ZTP were measured after a 15-min incubation at 30 °C with ZMP (1 mm) and ATP (2 mm) and His6–Ura6 either in total protein bacterial (Bact.) extracts or purified (E). Metabolite amounts were set at 1 for the amounts measured with bacterial extracts containing the empty vector (control). Errors correspond to standard deviation obtained from six independent measurements.
Notably, recombinant yeast Ura6 purified from Escherichia coli (Fig. 3E) could synthesize ZDP, but not ZTP, from ZMP in vitro (Fig. 3, F–H), as expected for a nucleoside monophosphate kinase. However, both ZTP and ZDP were found as reaction products when the reaction was run with total extracts from URA6-expressing bacteria (Fig. 3, G and H). Altogether, these results show that Ura6 can synthesize ZDP from ZMP and that another enzyme(s) can further phosphorylate it to ZTP in both yeast and bacteria. We established that yeast NDP-kinase Ynk1 was not required because ynk1 knockout had no effect on ZTP production (Fig. S4). Of note, purified yeast adenylate kinase Adk1, which was not found in the gene-dosage suppressor screening had no detectable ZMP-kinase activity in vitro.
Thus, our genetic and biochemical experiments in yeast demonstrate that ZMP, rather than SZMP, ZDP, or ZTP, is the toxic molecule derived from AICAR. The question arising then was: why is ZMP toxic?
Proteome-wide identification of ZMP binders
To identify potential effectors of ZMP toxicity, we isolated yeast proteins that interact with ZMP by an affinity chromatography approach (for details, see “Experimental procedures” and Ref. 10). Whole-cell protein extracts from six independent cultures were loaded on a ZMP-Sepharose resin. The eluted proteins (herein called ZMP binders) were identified by MS. A total of 74 proteins, representing ∼1.1% of the yeast proteome, were identified by at least two different peptides in at least four of six experiments (Fig. 4A and Table S1). Among them was the enzyme adenylosuccinate lyase (Ade13), a known ZMP binder (10), synthesizing ZMP from SZMP. However, identification of ZMP binders by this approach was not exhaustive because other ZMP-metabolizing enzymes (Ade16, Ade17, or Ura6) or previously identified binders (Pho2 or Pho4 (10)) were not found. Most interestingly, the ubiquitin-activating enzyme Uba1 was found as a ZMP binder, suggesting that it could be a direct target for ZMP and account for reported chemo-genetic effects of AICAR on the ubiquitin pathway (18). Analysis of the biological processes (Gene Ontology (GO) terms), associated with the proteins retained on the ZMP resin, showed a strong enrichment in purine nucleotide binding, unfolded protein binding, and translation factor activity (Fig. 4B). The best score was for “purine nucleotide binding” (p value 2.7 × 10−9), thus validating the affinity chromatography approach because ZMP is a precursor of purine nucleotides.
Figure 4.
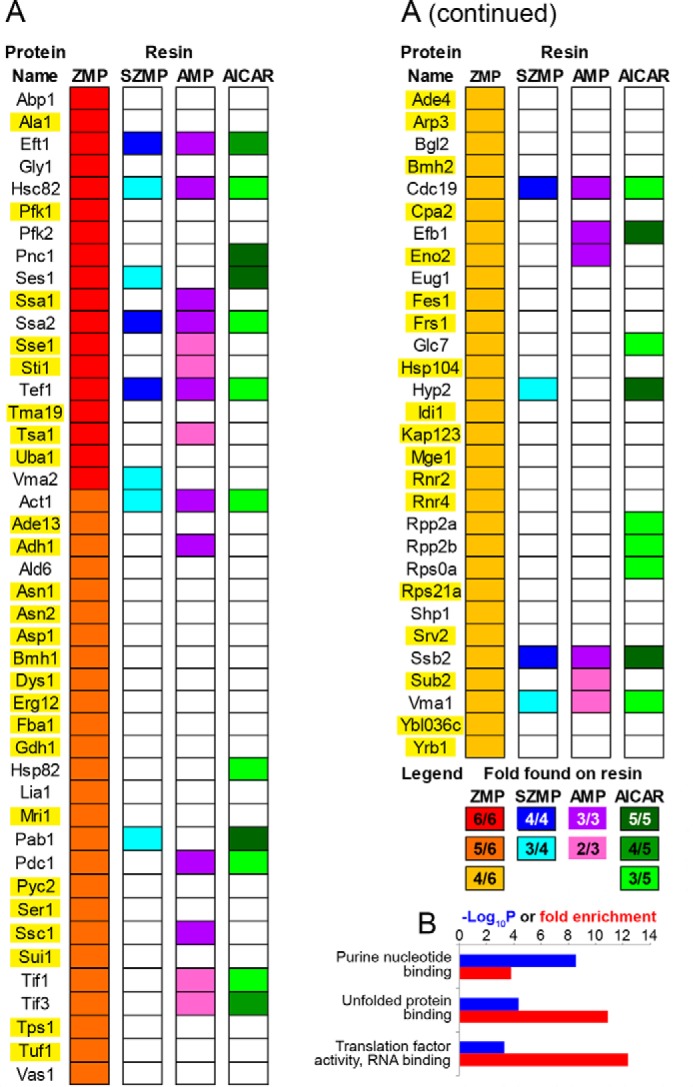
Proteome-wide identification of yeast ZMP binders by affinity chromatography. A, list of ZMP binders found at least four times in six affinity-chromatography experiments performed with independent samples. Proteins also found on other affinity resins are indicated. Complete lists are shown in Table S1. Genes highlighted in yellow were overexpressed, and their effects on AICAR sensitivity are shown in Fig. S7. B, biological process GO terms enrichment analysis for the 74 ZMP binders (http://www.geneontology.org/; please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site) (34, 35).
To evaluate the specificity of ZMP-binding proteins, a similar approach was applied to closely related molecules, namely AMP, SZMP, and AICAR. A list of proteins retained on AMP, SZMP, and AICAR resins is presented in Table S1, revealing enrichment for specific functions. The best scores for SZMP and AICAR corresponded to “structural constituent of the ribosome,” whereas for AMP it was “nucleotide binding” (Fig. S5). Among the 74 ZMP binders, 30 bound at least another compound tested (Fig. 4A). Interestingly, only eight proteins were found exclusively on ZMP and AMP columns, a binding pattern that would be expected for proteins echoing the AMP-mimetic effects of ZMP described for AMPK. Of note, the γ subunit of yeast AMPK (Snf4) was not retained on the columns as expected because yeast AMPK was found to be activated by ADP rather than AMP (23). Accordingly, a snf4 mutant did not show altered AICAR sensitivity (Fig. S6). Among the 74 ZMP binders, 44 were exclusive ZMP binders, strongly suggesting that ZMP effects are most likely not restricted to AMP mimicry.
Overexpression of ZMP binders reveals nuclear trafficking as a function affected by AICAR
We reasoned that the most likely effectors of ZMP toxicity in vivo are proteins that bind ZMP (and also possibly AMP) but not the nontoxic derivatives SZMP and AICAR. The genes encoding 44 such yeast proteins (highlighted in yellow in Fig. 4A) were overexpressed from multicopy plasmids, and their ability to affect resistance to AICAR in yeast cells was evaluated. These 44 proteins comprised 36 of the 44 exclusive ZMP binders, and all the 8 AMP/ZMP exclusive binders. Overexpression of most of these genes, including UBA1, had no or little effect (Fig. S7), but overexpression of KAP123 clearly improved the ability to grow in the presence of AICAR, as URA6 did (Fig. 5A). However, in contrast to URA6, overexpression of KAP123 did not lower intracellular ZMP (Fig. 5B), as expected for a ZMP target.
Figure 5.
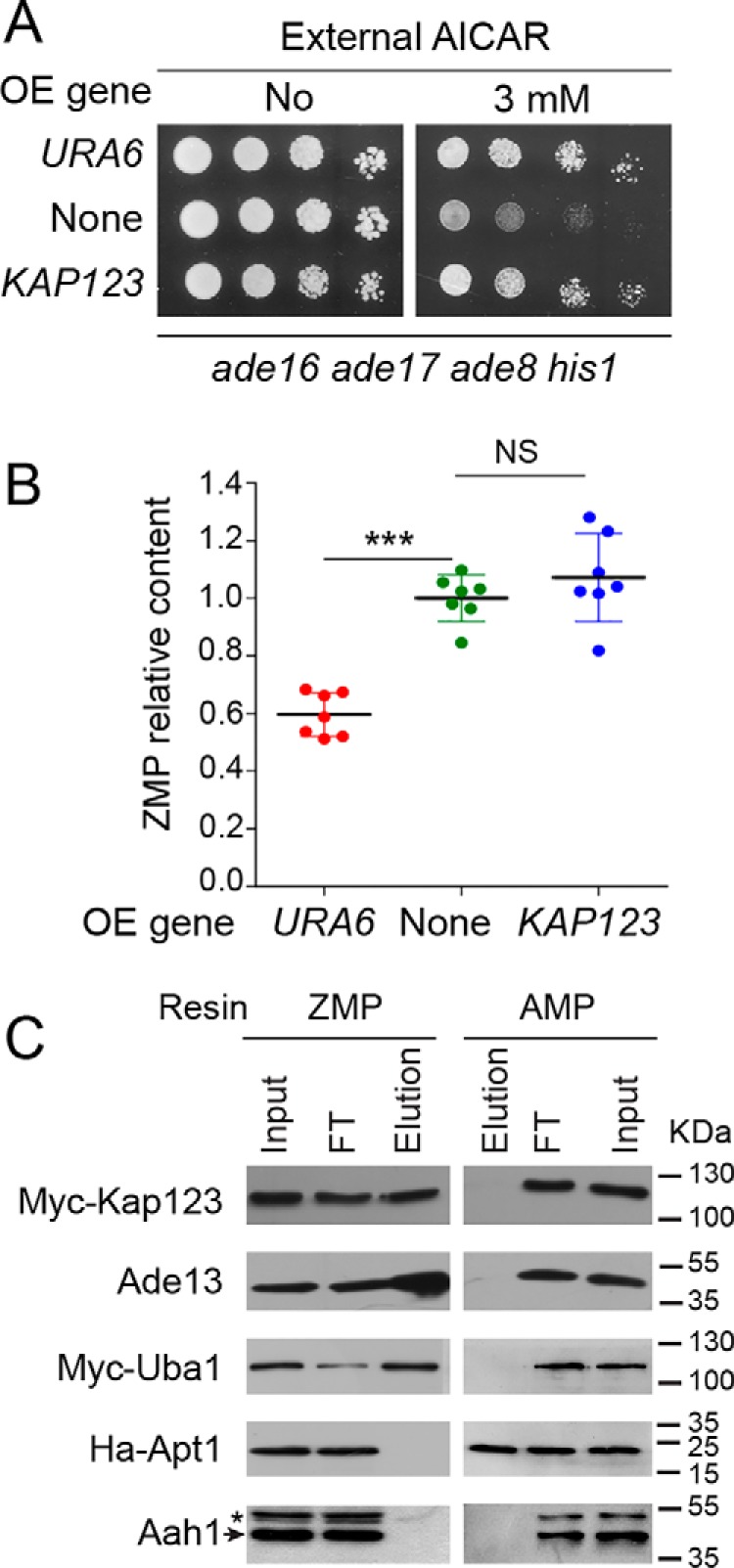
Overexpression of KAP123 enhances AICAR resistance without changing ZMP intracellular content. A, cells (ade8 ade16 ade17 his1; Y2950) were transformed with plasmids allowing overexpression (OE) of either URA6 (p4919) or KAP123 (p4983) or with the empty vector (YepLac195, none). Transformants were serially diluted and spotted on SDcasaWA medium. The plates were imaged after 2 days at 37 °C. B, ZMP relative content was determined on seven independent transformants from Fig. 5A and exponentially grown in SDcasaWA medium for 24 h in the presence of AICAR (3 mm). Errors bars and statistics correspond to standard deviation and Welch's t test. NS, nonsignificant; ***, p < 0.001. C, specific binding of Myc-tagged Kap123 and Uba1 proteins to the ZMP-affinity resin was confirmed by Western blotting. FT, flow through. Elution corresponds to the fraction eluted with the cognate specific nucleotide (5 mm) on each affinity resin.
We then confirmed the binding to ZMP of Kap123 (but also Uba1; see “Proteome-wide identification of ZMP binders”), using epitope-tagged versions of the proteins. Indeed, both Kap123 and Uba1 proteins were retained on the ZMP resin, but not on the AMP resin, as revealed by Western blotting analysis (Fig. 5C). In this experiment, Ade13 (adenylosuccinate lyase) and Apt1 (adenine phosphoribosyltransferase), for which ZMP and AMP are the corresponding reaction products, were used as positive controls for the ZMP and AMP resin, respectively (Fig. 5C). Aah1 (adenine deaminase) was used as a negative control for both resins (Fig. 5C).
Because Kap123 is an importin mediating the nuclear trafficking of several cargoes, including ribosomal proteins (24), we hypothesized that ZMP could affect nuclear transport. We first monitored nuclear localization of the Rpl25–NLS–GFP fusion (ribosomal 60S subunit protein L25 fused to green fluorescent protein via a nuclear localization signal), which is fully dependent on Kap123 (24). We found that it was not affected by AICAR (Fig. S8), indicating that AICAR does not primarily act through inhibition of Kap123, although overexpression of Kap123 can clearly bypass defects associated with AICAR treatment. The effect of AICAR on nuclear trafficking was then directly and globally assayed by comparing protein composition of nuclei-enriched extracts from yeast treated or not treated with AICAR by label-free quantitative analysis. Strikingly, 57 proteins were significantly less abundant (at least 2-fold) in the nuclei-enriched fraction following AICAR treatment (Fig. 6A and Table S2). Among these 57 proteins, 17 (highlighted in yellow in Table S2) were also detected in whole cell protein extracts by quantitative MS, and in all cases their abundance was not significantly changed in response to AICAR. We conclude that AICAR affects their nuclear localization rather than their overall abundance. GO-term analysis revealed that, among these 57 proteins, nuclear components and nuclear functions such as chromatin assembly and organization, rRNA metabolism, or transcription, were most significantly over-represented (Table S3).
Figure 6.
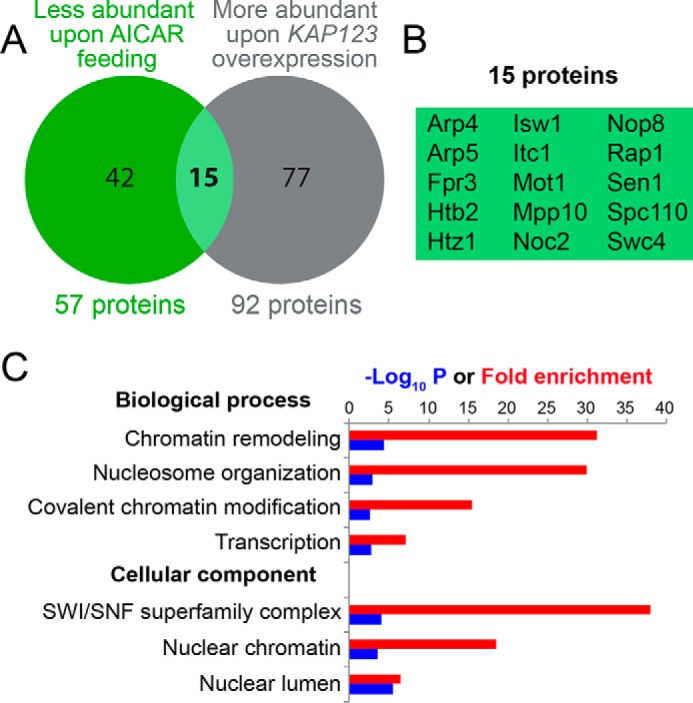
Nuclear protein content is altered by AICAR treatment in yeast. A, Venn diagram representation for proteins less abundant in the nuclei-enriched fraction after AICAR treatment (green circle) and proteins more abundant in nuclei-enriched fraction of cells overexpressing KAP123 in the presence of AICAR (gray circle). The entire lists of proteins that significantly changed in both conditions are presented in Tables S3 and S4. B, list of the 15 common proteins in the Venn diagram presented in Fig. 6A. C, GO term analysis (http://www.geneontology.org/; please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site) (34, 35)8 for the 15 proteins listed in Fig. 6B.
A parallel analysis, performed on yeast cells grown in the presence of AICAR and overexpressing or not KAP123, revealed 92 proteins that were significantly more abundant in the nuclei-enriched fraction when KAP123 was overexpressed (Table S4). Among those, proteins involved in chromatin and transcription-related processes were very significantly over-represented (Table S5). Most conspicuously, 15 proteins, less abundant in the nuclei-enriched fraction following AICAR treatment, were “restored” when KAP123 was overexpressed (Fig. 6B). This overlap is statistically highly significant (hypergeometric distribution: p = 2.16 × 10−5), pointing to a strong functional connection between AICAR feeding and KAP123 overexpression. Again, proteins involved in chromatin organization and transcription were overrepresented among these 15 proteins (Fig. 6C). This result shows that KAP123 overexpression can compensate for some of the effects that AICAR exerts on nuclear traffic and could in this way contribute to the suppression of AICAR-induced phenotypic effects. Taken together, these results reveal nuclear trafficking as a critical cellular function that is altered by AICAR and point to Kap123 as an important, although nonexclusive, effector protein.
Specificity of KAP123 and URA6 as dosage suppressors
In previous work, we identified several mutants that showed high sensitivity to AICAR because of increased uptake and accumulation (thi3, pdc2, and thi80 mutant strains) (1); impaired purine metabolism (aah1, ade13, and hpt1) (17); or interference with cell cycle progression (bre1, rad6, set1, and swd1) (16). Representative mutants among those were assayed to establish whether they could be phenotypically suppressed by URA6 or KAP123 overexpression. In fact, URA6 overexpression efficiently suppressed AICAR sensitivity of the set1 and thi3 mutants (Fig. 7, A and B) and had a weaker but reproducible effect on the aah1 mutant (Fig. 7C). By contrast, overexpression of KAP123 only rescued AICAR sensitivity of the set1 mutant (Fig. 7A) and not that of other mutants such as thi3 or aah1 (Fig. 7, B and C), indicating that increased sensitivity of the various mutants has distinct causes. Consistent with our previous findings (16), other mutants in the set1 pathway (bre1, swd1, and swd3) were also suppressed by KAP123 overexpression (Fig. S9). Overall, these results point to a central role for nuclear trafficking in AICAR sensitivity of yeast mutants affecting cell-cycle progression.
Figure 7.
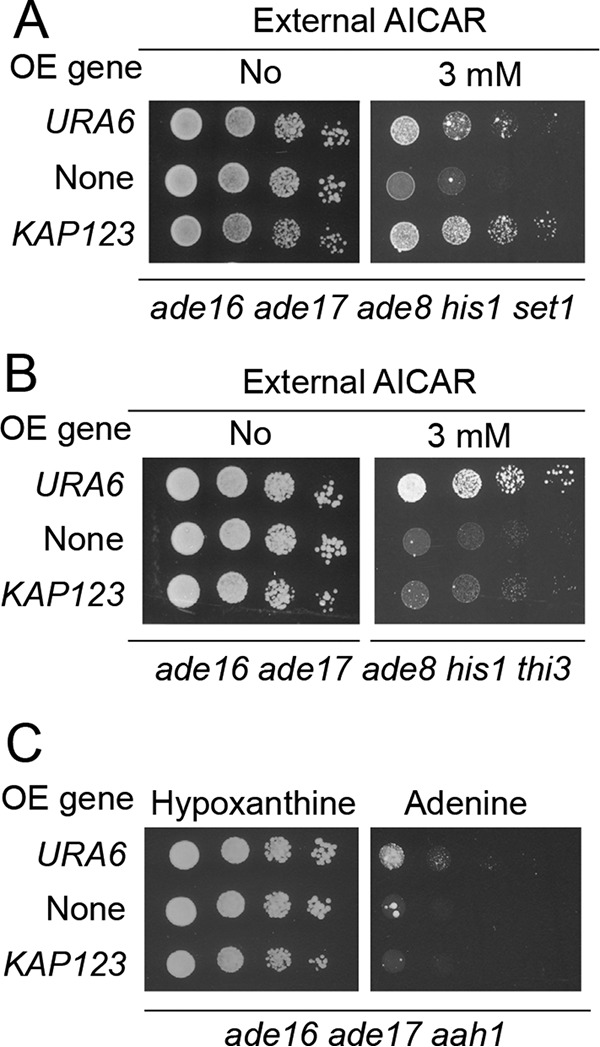
AICAR toxicity is alleviated by KAP123 overexpression specifically in the AICAR-sensitive set1 mutant affected in cell cycle progression. A–C, effect of URA6 and KAP123 overexpression on growth of AICAR-sensitive strains affected in cell-cycle progression (A), in AICAR uptake (B), and in purine metabolism (C). Mutant strains (Y9168, Y7321, and Y10846) were transformed with the empty plasmid (YepLac195, none) or with plasmids allowing overexpression (OE) of either URA6 (p4919) or KAP123 (p4983). Transformants were serially diluted and spotted on SDcasaWA medium ± AICAR (A and B) or SDcasaW medium supplemented with either hypoxanthine or adenine (C). The plates were imaged after 3 days at 37 °C.
Discussion
The purine biosynthesis metabolic intermediate ZMP is naturally present in eukaryotic cells at low concentration. It can accumulate when its riboside precursor AICAR is provided to the cell, but also in several metabolic diseases. Indeed, ZMP as well as ZDP and ZTP were found at very high concentrations (ZTP reaching even higher levels than ATP) in red blood cells of an AICAR transformylase IMP-cyclohydrolase–deficient patient lacking the enzyme that catalyzes the last two steps of the purine biosynthesis pathway (25) and was the best marker for hypoxanthine-guanine phospho-ribosyl-transferase deficiency in a large cohort of Lesch–Nyhan syndrome patients (26). The precise role of ZMP in the etiology of these diseases is unclear but should be addressed in connection with the fact that treating eukaryotic cells with AICAR at high concentration is toxic. In many studies, it was shown that the riboside precursor of ZMP had no biological effects, suggesting that ZMP itself, or a derivative, is the active compound. In our study, taking advantage of the power of yeast genetics, we established that AICAR toxicity tightly correlates with ZMP accumulation; lowering ZMP accumulation by stimulating its conversion to SZMP, ZDP, or ZTP derivatives decreased toxicity of the prodrug AICAR. Thus, increasing the levels of intracellular ZTP did not enhance toxicity of AICAR but rather decreased it. This suggests that AICAR toxicity is not due to massive incorporation of AICAR-nucleotides into nucleic acids. Accordingly, we found no effect of AICAR on mutation or recombination rate, as well as telomere length. These results are consistent with those obtained in previous work showing that yeast mutants displaying increased AICAR uptake became hypersensitive to this compound and that the sensitivity was associated with a 10-fold increase of the monophosphate (ZMP) concentration (1), whereas the di- and triphosphate forms (ZDP and ZTP) were hardly detectable.7 Altogether, these results reveal ZMP as the toxic compound in yeast cells treated with the prodrug AICAR.
Affinity chromatography allowed us to identify a spectrum of AMP- and ZMP-binding proteins from yeast, thus providing a new resource of nucleotide-binding proteins. Only 20 of the 81 AMP binders and 74 ZMP binders were retained on both AMP and ZMP resins, indicating that not all AMP binders are able to efficiently bind ZMP despite its well-established AMP-mimetic role best exemplified as an effector molecule acting on mammalian AMP-activated kinase (14). Symmetrically, most ZMP binders were not identified as AMP binders. Why do these proteins bind ZMP? It is possible that some of the identified proteins physiologically bind ZMP, as Ade13 does? It could also be that ZMP binding imitates physiological interaction of these proteins with another nucleotide. In any case, our results strongly suggest that some of the AICAR effects are not due to AMP-mimetic interactions of ZMP with target proteins. Thus, our data support the view that the toxic effects of ZMP are not due to its AMP-mimetic properties.
Overexpression of KAP123, which has been identified as an “exclusive ZMP binder,” significantly improved AICAR resistance, and this effect was enhanced in bre1, set1, swd1, and swd3 AICAR-sensitive mutants affecting histone methylation. Kap123 is a karyopherin-β involved in the nuclear entry of multiple proteins including ribosomal proteins and histones (24, 27). Strikingly, Srm1, the guanyl-nucleotide exchange factor for yeast Ran Gsp1p, was among the 57 proteins for which nuclear localization was impaired by AICAR. This protein, homologous to human RCC1, is critical for nuclear trafficking, and its mislocalization upon AICAR treatment could, in a cascading effect, contribute to mislocalization of several other proteins. Thus, these results point to AICAR affecting nuclear trafficking in a way that is at least partly Kap123-dependent.
Suppression of bre1, set1, and swd1 by Kap123 overexpression confirmed that nuclear trafficking contributes to the chemo-genetic effect of AICAR in several histone modification mutants, as proposed previously (16). It should be stressed that KAP123 overexpression only partially suppressed AICAR sensitivity and was not a suppressor of all AICAR-sensitive mutants (Fig. 7), implying that some AICAR effects most probably involve targets other than Kap123. Indeed, we had previously identified the ubiquitin pathway as an important target for ZMP toxicity (18), and remarkably, unfolded protein binding was the second enriched GO term among the ZMP binders (Fig. 4). In this work, Uba1, the ubiquitin-activating enzyme at the apex of the ubiquitin pathway, was identified as a ZMP binder, suggesting that it could be a direct target of ZMP. However, overexpression of UBA1 alone was not sufficient to increase the ability of yeast cells to cope with AICAR (Fig. S7), revealing a rather complex situation, as previously highlighted by our genetic analyses (18). Even at the level of nuclear trafficking, our finding of a partial overlap between KAP123 overexpression and AICAR effect on nuclear protein content suggests a complex interplay between ZMP and the nuclear trafficking machinery involving additional players. One such candidate is Yrb1, a Ran GTPase-binding protein involved in nuclear protein trafficking that was identified as one of the ZMP-specific binders in this work. However, overexpression of YRB1 had no major effect on growth in the presence of AICAR (Fig. S7).
This study illustrates that a molecule with anti-proliferative properties interacts with multiple proteins and, even on a single process such as nuclear trafficking, most probably acts through more than one effector. The polypharmacology paradigm proposed 10 years ago (28) underlines how combined effects of a drug on multiple targets can bypass phenotypic robustness while limiting toxicity. Importantly, acadesine (AICAR) was well-tolerated in clinical trials (6) and was found pharmacologically active in several animal-model studies (3–5, 15, 29). Understanding the diverse molecular bases of AICAR effects will give clues to its beneficial and adverse effects and could permit synthetic-lethality predictions, smart development of derived compounds, and drug repurposing (30).
Experimental procedures
Yeast media, yeast strains, and plasmids
SD is a synthetic minimal medium containing 0.5% ammonium sulfate, 0.17% yeast nitrogen base (BD-Difco, Franklin Lakes, NJ), 2% glucose. SDcasaW is SD medium supplemented with 0.2% casamino acids (catalog no. 233520; BD-Difco) and tryptophan (0.2 mm). When indicated, adenine (0.3 mm), hypoxanthine (0.3 mm), and/or uracil (0.3 mm) were added in SDcasaW medium, resulting in media termed SDcasaWA (+ adenine), SDcasaWHypox (+ hypoxanthine), SDcasaWU (+ uracil), and SDcasaWAU (+ adenine + uracil). Yeast strains are listed in Table S6 and belong to, or are derived from, a set of knockout mutant strains isogenic to BY4742 purchased from Euroscarf (Frankfurt, Germany). Multimutant strains were obtained by crossing, sporulation, and micromanipulation of meiosis progeny. The plasmids used in this study are described in Table S7. Cloning details for the unpublished plasmids are available upon request.
Yeast growth test
Yeast cells were resuspended in sterile water to 2 × 107 cells/ml and subjected to 1/10 serial dilutions. Drops (5 μl) of each dilution were spotted on freshly prepared plates and were incubated at indicated temperature for 48–72 h.
Isolation of multicopy suppressors of AICAR toxicity
To obtain multicopy suppressors of AICAR toxicity, an ade3 ade16 ade17 ade8 his1 strain (Y8908) was used because in such a strain both ade3 and ade16 ade17 mutations block AICAR transformylase IMP-cyclohydrolase activity and therefore neither ADE3, ADE16, nor ADE17 can be selected as a suppressor. This strain was transformed with a multicopy plasmid library (PFL44L backbone, generous gift from F. Lacroute), and transformants were selected on SDcasaWA medium after 48 h of incubation at 30 °C. The colonies were then transferred by replica plating on the same medium containing 5 mm AICAR and grown for 48 h at 37 °C. This screening temperature was chosen because in previous work we noticed that addition of AICAR was much more toxic for yeast cells at 37 °C than at 30 °C (1). Multicopy plasmids were extracted from six clones able to grow in the presence of AICAR and were sequenced. These plasmids contained a DNA fragment from the same region of chromosome 11. The plasmid (p4979) containing the 4.6-kbp fragment with chromosome coordinates 390,252–394,835 bp was further studied (see “Results”).
Metabolite extraction from yeast cells and separation by LC
Extraction of metabolites was performed in an ethanol with 10 mm HEPES, pH 7.2 (3/1), solution for 3 min at 80 °C for yeast as described (1). The extracts were then evaporated using a rotary evaporator (8 min, 65 °C), and the dried residues were resuspended in MilliQ water. Insoluble particles were removed by centrifugation (21,000 × g, 4 °C; 1 h). Metabolites were then separated on an ICS3000 chromatography station (Dionex, Sunnyvale, CA) using a carbopac PA1 column (250 × 2 mm; catalog no. 057178; Thermo Fisher Scientific) with a 0.25 ml/min flow rate. Elution of metabolites was achieved with a sodium acetate gradient in 50 mm NaOH as follows: elution was started at 50 mm sodium acetate for 2 min, rising up to 75 mm in 8 min, then to 100 mm in 25 min, and finally to 350 mm in 30 min, followed by a step at 350 mm for 5 min, rising to 500 mm in 10 min, and being kept at this sodium acetate concentration for 5 min, and finally raised to 800 mm within 10 min followed by a step at this concentration for 20 min. The resin was then equilibrated at 50 mm sodium acetate for 15 min before injection of a new sample. The peaks were identified by their retention time and their UV spectrum signature (Diode Array Detector Ultimate 3000 RS; Dionex), and when necessary by co-injection with standards. For each strain analyzed, normalization to cellular volume was done with a Multisizer 4 (Beckman Coulter). Peak quantifications were done at the following wavelengths: 230 nm for fumarate, 260 nm for ATP, and 269 nm for AICAR, SAICAR, ZMP, and SZMP. All metabolite extractions were performed on at least three to six independent yeast cell cultures.
Affinity chromatography
ZMP-binding proteins were identified by affinity chromatography using the ZMP-Sepharose resin described previously (10). Protein extracts were prepared from the BY4742 WT strain as described (10), except that elution of the ZMP binders was performed with ZMP (5 mm; catalog no. A611705; Toronto Research Chemicals) in buffer A (Na2HPO4/HCl, pH 7.2, 10 mm NaCl). Elution fractions were concentrated on a Nanosep 10K Omega filter, and protein concentration was measured. The ZMP binders were then identified using MS according to the procedure described in protein identification by LC-MS/MS and database search and results processing sections. A stringent cutoff was then applied, retaining only proteins identified with at least two peptides, and found at least four times in six independent experiments (Table S1). The ability of proteins to bind AMP, SZMP, or AICAR was also assayed using either a commercial AMP-agarose resin (catalog no. A1271; Merck) or SZMP- and AICAR-resins prepared as described for the ZMP resin (10). In each case, elution of interacting proteins was performed with 5 mm of the corresponding metabolite (AMP (catalog no. A1752; Merck), SZMP (lab collection, (8)), or AICAR (catalog no. A611700; Toronto Research Chemicals)) dissolved in buffer A.
Immunoblot
Western blots were hybridized with the following antibodies: anti-Myc (1/5,000; catalog no. MMS-150R; Covance), anti-Ade13 (1/15,000 (8)), anti-HA (1/10,000, catalog no. H9658; Sigma–Aldrich), and anti-Aah1 (1/10,000 (31)).
Protein identification by LC-MS/MS
Concentrated proteins from each affinity-chromatography elution fractions were solubilized in Laemmli sample buffer and subjected to SDS-PAGE to evaluate concentration and for cleaning purposes. After entering the resolving gel, separation was stopped. Following colloidal blue staining, the bands were cut out from the SDS-PAGE gel and subsequently cut into 1 × 1 mm gel pieces. Gel pieces were destained in 25 mmol/liter ammonium bicarbonate 50% (w/v), rinsed twice in ultrapure water, and shrunk in acetonitrile for 10 min. After acetonitrile removal, gel pieces were dried at room temperature, covered with trypsin solution (10 ng/μl in 40 mm NH4HCO3 and 10% acetonitrile), rehydrated at 4 °C for 10 min, and finally incubated overnight at 37 °C. Gel pieces were then incubated for 15 min in 40 mm NH4HCO3 and 10% acetonitrile at room temperature on a rotary shaker. The supernatant was collected, and an extraction solution (H2O, acetonitrile, HCOOH (47.5/47.5/5)) was added to the gel slices for 15 min. This extraction step was repeated twice. Supernatants were pooled and concentrated with a vacuum centrifuge to a final volume of 25 μl. Digests were finally acidified by addition of 1.5 μl of formic acid (5%, v/v) and stored at −20 °C. The peptide mixture was analyzed on an Ultimate 3000 nano-LC system (Dionex) coupled to either a LTQ, a LTQ-Orbitrap XL, or a Q-Exactive mass spectrometer (ThermoFinnigan, San Jose, CA). In all cases, 10 μl of peptide digests were loaded onto a C18 PepMapTM trap column (300-μm; 5-mm LC Packings) at a 30 μl/min flow rate. Peptides were eluted from the trap column and then loaded onto an analytical C18 Pep-Map column (75 mm × 15 cm; LC Packings) for 105 min with a 5–40% linear gradient of 80% acetonitrile with 0.1% formic acid in 5% acetonitrile with 0.1% formic acid at a 200 nl/min flow rate. The mass spectrometer operated in positive ion mode at a 1.8–2-kV needle voltage. Mass spectrometry acquisition parameters are given in Table S8.
Nuclei-enriched fractions preparation and label-free quantitative data analysis
Yeast cells (ade16 ade17 ade8 his1; Y8480) were transformed by the plasmid allowing KAP123 overexpression or by the cognate empty vector serving as negative control. Transformants exponentially grown in SDcasaWA were treated with 0 or 1 mm AICAR for 2 h. The cells were collected and used to prepare total protein extracts and in parallel to perform nuclei enrichment with a commercial kit (Abnova catalog no. KA3951). For each condition, the experiments were done in triplicate with samples obtained from independent cultures. LC-MS/MS data were acquired on a Q-Exactive mass spectrometer as described above and were subsequently imported in Progenesis LC-MS 4.0 (Non Linear Dynamics). Data processing included the following steps: (i) features detection, (ii) features alignment across the LC-MS/MS runs, (iii) volume integration for 2–6 charge-state ions, (iv) normalization on feature median ratio, (v) import of sequence information, (vi) analysis of variance test at peptide level and filtering for features (p < 0.05), (vii) calculation of protein abundance (sum of the volume of corresponding peptides), and (viii) analysis of variance test at protein level and filtering for features (p < 0.05). Proteins were grouped according to the parsimony principle so as to establish the minimal protein list covering all detected peptides. Noticeably, only nonconflicting features and unique peptides were considered for calculation at protein level. Quantitative data were considered for proteins quantified by a minimum of two peptides.
Database search and processing of proteomic results
The data were searched by SEQUEST through Proteome Discoverer 1.4 (Thermo Fisher Scientific Inc.) against the Saccharomyces cerevisiae Reference Proteome Set (Uniprot 2015-05; 6636 entries). Spectra from peptides higher than 5,000 Da or lower than 350 Da were rejected. The search parameters were as follows: mass accuracy of the monoisotopic peptide precursor and peptide fragments were as described in Table S8. Only b- and y-ions were considered for mass calculation. Oxidation of methionine (+16 Da) was considered as variable modification. Two missed trypsin cleavages were allowed. Peptide validation was performed using Percolator algorithm (32), and only “high confidence” peptides were retained corresponding to a 1% false positive rate at peptide level. Data for the affinity-chromatography and label-free quantitative proteomics were deposited and are available in PRIDE at the following links: identification of the S. cerevisiae ZMP binders (project accession number PXD007780) and effect of acadesine and overexpression of the karyopherin Kap123 on nuclear abundance of yeast proteins (project accession number PXD007779).
His6–Ura6 protein expression and purification
The His6–Ura6 protein was expressed in BL21 C41(DE3)pLysS bacteria (33) by transformation of the URA6-HIS6 plasmid (p5334). Transformants were grown overnight in LB medium + ampicillin, diluted in 500 ml of the same medium to A600 nm = 0.005 and grown for 4 h at 30 °C before isopropyl β-d-1-thiogalactopyranoside addition (2 mm). The cells were harvested by centrifugation (5,000 × g for 10 min at 4 °C) after 2 h 30 min at 30 °C, and the cell pellet was washed with 50 mm Tris-HCl, pH 7, 0.15 m KCl and frozen at −80 °C. The pellet was resuspended in 50 mm Tris-HCl, pH 7, 0.1 m KCl, 1 mm EDTA, 1 mm DTT containing a tablet of protease/phosphatase inhibitor (buffer B). The cells were disrupted by four cycles of 1 min sonication on ice, and the suspension was clarified by centrifugation (21,000 × g) for 30 min at 4 °C. The supernatant was dialyzed twice against buffer B without EDTA (buffer C) containing 10 mm imidazole and then loaded onto 1.5 ml of nickel–nitrilotriacetic acid resin (Qiagen). The resin was washed with 15 volumes of Buffer C containing 50 mm imidazole, and purified His6–Ura6 protein was then eluted with 250 mm imidazole in buffer C. The purified His6–Ura6 protein was concentrated (0.7 ml at 10 mg/ml), aliquoted and frozen at −80 °C.
Ura6 enzymatic activity
Ura6 enzymatic activity was measured on both total protein extracts from bacteria (containing the His6–Ura6 expressing plasmid (p5334) or the empty vector (pET15b; catalog no. 69661-3; Merck–Novagen)) and on the purified protein. In each case, measurements were carried out using initial rate conditions that were stable for at least 30 min at 30 °C. Enzymatic measurements were performed in 100 mm Tris-HCl, pH 7.5 buffer containing 100 mm KCl, 5 mm MgCl2, 2 mm ATP, and 1 mm ZMP. The reactions were started by addition of either the purified protein (at 1 μg/ml in enzymatic assay) or the total protein extracts from bacteria expressing or not His6–Ura6 (each at final concentration of 100 μg/ml in enzymatic assays). All enzymatic reactions were stopped by boiling (3 min at 80 °C) a 100-μl aliquot of the reaction mix in 900 μl of ethanol, 10 mm HEPES, pH 7 (4/1 v/v). Quantification of the reaction product(s) was done by LC under conditions described for intracellular metabolites determination. ZDP and ZTP standard solutions used for quantification were purchased from Jena Biosciences (catalog nos. NU-1167 and NU-1166, respectively).
Quantification and statistical analysis
All tests for significance were according to Welch's unpaired t tests, assuming bilateral distribution and unequal variances, by comparison with the relevant control as specified in legends of the figures and supplemental figures Experiments were in each case performed on at least three biologically independent samples (n ≥ 3), as detailed in each figure legend. Significance was defined by p values and indicated as *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; NS, nonsignificant (p > 0.05).
Author contributions
D. C. D., B. P., J. C., H. C. H., C. S.-M., D. L., M. K., M. B., and B. D.-F. conceptualization; D. C. D., B. P., J. C., H. C. H., C. S.-M., D. L., S. C., M. B., and B. D.-F. data curation; D. C. D., B. P., J. C., C. S.-M., S. C., M. B., and B. D.-F. formal analysis; D. C. D., B. P., J. C., C. S.-M., and B. D.-F. supervision; D. C. D., B. P., J. C., C. S.-M., and B. D.-F. funding acquisition; D. C. D., B. P., J. C., C. S.-M., and B. D.-F. validation; D. C. D., B. P., J. C., H. C. H., C. S.-M., D. L., S. C., and B. D.-F. investigation; D. C. D., B. P., J. C., H. C. H., C. S.-M., D. L., S. C., and B. D.-F. methodology; D. C. D., B. P., J. C., C. S.-M., M. K., and B. D.-F. writing-original draft; D. C. D., B. P., J. C., C. S.-M., M. K., and B. D.-F. writing-review and editing; M. K. resources.
Supplementary Material
Acknowledgment
We thank Ioanna Lemnian for help with hypergeometric distribution.
This work was supported by a Ligue contre le Cancer Dordogne 2016–2017 grant (to B. D.-F.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1–S8 and Figs. S1–S9.
The mass spectrometry proteomics data have been deposited to the Proteome Xchange Consortium via the PRIDE partner repository with the dataset identifiers PXD007780 and PXD007779.
J. Ceschin, B. Daignan-Fornier, and B. Pinson, unpublished results.
- AICAR
- 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside
- ZMP
- 5-amino-4-imidazole carboxamide ribonucleoside monophosphate
- SZMP
- succinyl-ZMP
- ZDP
- 5-amino-4-imidazole carboxamide ribonucleoside diphosphate
- ZTP
- 5-amino-4-imidazole carboxamide ribonucleoside triphosphate
- AMPK
- AMP-activated protein kinase
- SAICAR
- succinyl-AICAR
- GO
- Gene Ontology.
References
- 1. Ceschin J., Saint-Marc C., Laporte J., Labriet A., Philippe C., Moenner M., Daignan-Fornier B., and Pinson B. (2014) Identification of yeast and human 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) transporters. J. Biol. Chem. 289, 16844–16854 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Daignan-Fornier B., and Pinson B. (2012) 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranosyl 5′-monophosphate (AICAR), a highly conserved purine intermediate with multiple effects. Metabolites 2, 292–302 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guo D., Hildebrandt I. J., Prins R. M., Soto H., Mazzotta M. M., Dang J., Czernin J., Shyy J. Y., Watson A. D., Phelps M., Radu C. G., Cloughesy T. F., and Mischel P. S. (2009) The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc. Natl. Acad. Sci. U.S.A. 106, 12932–12937 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rattan R., Giri S., Singh A. K., and Singh I. (2005) 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J. Biol. Chem. 280, 39582–39593 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 5. Robert G., Ben Sahra I., Puissant A., Colosetti P., Belhacene N., Gounon P., Hofman P., Bost F., Cassuto J. P., and Auberger P. (2009) Acadesine kills chronic myelogenous leukemia (CML) cells through PKC-dependent induction of autophagic cell death. PLoS One 4, e7889 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Van Den Neste E., Cazin B., Janssens A., González-Barca E., Terol M. J., Levy V., Pérez de Oteyza J., Zachee P., Saunders A., de Frias M., and Campàs C. (2013) Acadesine for patients with relapsed/refractory chronic lymphocytic leukemia (CLL): a multicenter phase I/II study. Cancer Chemother. Pharmacol. 71, 581–591 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hürlimann H. C., Laloo B., Simon-Kayser B., Saint-Marc C., Coulpier F., Lemoine S., Daignan-Fornier B., and Pinson B. (2011) Physiological and toxic effects of purine intermediate 5-amino-4-imidazolecarboxamide ribonucleotide (AICAR) in yeast. J. Biol. Chem. 286, 30994–31002 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rébora K., Desmoucelles C., Borne F., Pinson B., and Daignan-Fornier B. (2001) Yeast AMP pathway genes respond to adenine through regulated synthesis of a metabolic intermediate. Mol. Cell Biol. 21, 7901–7912 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rébora K., Laloo B., and Daignan-Fornier B. (2005) Revisiting purine-histidine cross-pathway regulation in Saccharomyces cerevisiae: a central role for a small molecule. Genetics 170, 61–70 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pinson B., Vaur S., Sagot I., Coulpier F., Lemoine S., and Daignan-Fornier B. (2009) Metabolic intermediates selectively stimulate transcription factor interaction and modulate phosphate and purine pathways. Genes Dev. 23, 1399–1407 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim P. B., Nelson J. W., and Breaker R. R. (2015) An ancient riboswitch class in bacteria regulates purine biosynthesis and one-carbon metabolism. Mol. Cell 57, 317–328 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sullivan J. E., Brocklehurst K. J., Marley A. E., Carey F., Carling D., and Beri R. K. (1994) Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 353, 33–36 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 13. Sullivan J. E., Carey F., Carling D., and Beri R. K. (1994) Characterisation of 5′-AMP-activated protein kinase in human liver using specific peptide substrates and the effects of 5′-AMP analogues on enzyme activity. Biochem. Biophys. Res. Commun. 200, 1551–1556 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 14. Day P., Sharff A., Parra L., Cleasby A., Williams M., Hörer S., Nar H., Redemann N., Tickle I., and Yon J. (2007) Structure of a CBS-domain pair from the regulatory γ1 subunit of human AMPK in complex with AMP and ZMP. Acta Crystallogr. D Biol. Crystallogr. 63, 587–596 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 15. Liu X., Chhipa R. R., Pooya S., Wortman M., Yachyshin S., Chow L. M., Kumar A., Zhou X., Sun Y., Quinn B., McPherson C., Warnick R. E., Kendler A., Giri S., Poels J., et al. (2014) Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc. Natl. Acad. Sci. U.S.A. 111, E435–E444 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Albrecht D., Ceschin J., Dompierre J., Gueniot F., Pinson B., and Daignan-Fornier B. (2016) Chemo-genetic interactions between histone modification and the antiproliferation drug AICAR are conserved in yeast and humans. Genetics 204, 1447–1460 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ceschin J., Hürlimann H. C., Saint-Marc C., Albrecht D., Violo T., Moenner M., Daignan-Fornier B., and Pinson B. (2015) Disruption of nucleotide homeostasis by the antiproliferative drug 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside monophosphate (AICAR). J. Biol. Chem. 290, 23947–23959 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Albrecht D., Hürlimann H. C., Ceschin J., Saint-Marc C., Pinson B., and Daignan-Fornier B. (2018) Multiple chemo-genetic interactions between a toxic metabolite and the ubiquitin pathway in yeast. Curr. Genet. 64, 1275–1286 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 19. Nakamaru K., Matsumoto K., Taguchi T., Suefuji M., Murata Y., Igata M., Kawashima J., Kondo T., Motoshima H., Tsuruzoe K., Miyamura N., Toyonaga T., and Araki E. (2005) AICAR, an activator of AMP-activated protein kinase, down-regulates the insulin receptor expression in HepG2 cells. Biochem. Biophys. Res. Commun. 328, 449–454 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 20. Tibbetts A. S., and Appling D. R. (1997) Saccharomyces cerevisiae expresses two genes encoding isozymes of 5-aminoimidazole-4-carboxamide ribonucleotide transformylase. Arch. Biochem. Biophys. 340, 195–200 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 21. Guetsova M. L., Lecoq K., and Daignan-Fornier B. (1997) The isolation and characterization of Saccharomyces cerevisiae mutants that constitutively express purine biosynthetic genes. Genetics 147, 383–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schricker R., Magdolen V., Kaniak A., Wolf K., and Bandlow W. (1992) The adenylate kinase family in yeast: identification of URA6 as a multicopy suppressor of deficiency in major AMP kinase. Gene 122, 111–118 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 23. Mayer F. V., Heath R., Underwood E., Sanders M. J., Carmena D., McCartney R. R., Leiper F. C., Xiao B., Jing C., Walker P. A., Haire L. F., Ogrodowicz R., Martin S. R., Schmidt M. C., Gamblin S. J., et al. (2011) ADP regulates SNF1, the Saccharomyces cerevisiae homolog of AMP-activated protein kinase. Cell Metab. 14, 707–714 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schlenstedt G., Smirnova E., Deane R., Solsbacher J., Kutay U., Görlich D., Ponstingl H., and Bischoff F. R. (1997) Yrb4p, a yeast ran-GTP-binding protein involved in import of ribosomal protein L25 into the nucleus. EMBO J. 16, 6237–6249 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marie S., Heron B., Bitoun P., Timmerman T., Van Den Berghe G., and Vincent M. F. (2004) AICA-ribosiduria: a novel, neurologically devastating inborn error of purine biosynthesis caused by mutation of ATIC. Am. J. Hum. Genet. 74, 1276–1281 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ceballos-Picot I., Le Dantec A., Brassier A., Jaïs J. P., Ledroit M., Cahu J., Ea H. K., Daignan-Fornier B., and Pinson B. (2015) New biomarkers for early diagnosis of Lesch-Nyhan disease revealed by metabolic analysis on a large cohort of patients. Orphanet J. Rare Dis. 10, 7 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mosammaparast N., Guo Y., Shabanowitz J., Hunt D. F., and Pemberton L. F. (2002) Pathways mediating the nuclear import of histones H3 and H4 in yeast. J. Biol. Chem. 277, 862–868 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 28. Hopkins A. L. (2008) Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 4, 682–690 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 29. Tang Y. C., Williams B. R., Siegel J. J., and Amon A. (2011) Identification of aneuploidy-selective antiproliferation compounds. Cell 144, 499–512 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Medina-Franco J. L., Giulianotti M. A., Welmaker G. S., and Houghten R. A. (2013) Shifting from the single to the multitarget paradigm in drug discovery. Drug Disc. Today 18, 495–501 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Escusa S., Camblong J., Galan J. M., Pinson B., and Daignan-Fornier B. (2006) Proteasome- and SCF-dependent degradation of yeast adenine deaminase upon transition from proliferation to quiescence requires a new F-box protein named Saf1p. Mol. Microbiol. 60, 1014–1025 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 32. Käll L., Canterbury J. D., Weston J., Noble W. S., and MacCoss M. J. (2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 33. Dumon-Seignovert L., Cariot G., and Vuillard L. (2004) The toxicity of recombinant proteins in Escherichia coli: a comparison of overexpression in BL21(DE3), C41(DE3), and C43(DE3). Protein Expr. Purif. 37, 203–206 10.1074/jbc.RA117.001068 [DOI] [PubMed] [Google Scholar]
- 34. Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. The Gene Ontology Consortium (2017) Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 45, D331–D338 10.1074/jbc.RA117.001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.