

Abstract
Background:
Statins represent a class of medications widely prescribed to efficiently treat dyslipidemia. These drugs inhibit 3-βhydroxy 3β-methylglutaryl Coenzyme A reductase (HMGR), the rate-limiting enzyme of mevalonate (MVA) pathway. Besides cholesterol, MVA pathway leads to the production of several other compounds, which are essen-tial in the regulation of a plethora of biological activities, including in the central nervous system. For these reasons, statins are able to induce pleiotropic actions, and acquire increased interest as potential and novel modulators in brain processes, es-pecially during pathological conditions.
Objective:
The purpose of this review is to summarize and examine the current knowledge about pharmacokinetic and phar-macodynamic properties of statins in the brain. In addition, effects of statin on brain diseases are discussed providing the most up-to-date information.
Methods:
Relevant scientific information was identified from PubMed database using the following keywords: statins and brain, central nervous system, neurological diseases, neurodegeneration, brain tumors, mood, stroke.
Results:
315 scientific articles were selected and analyzed for the writing of this review article. Several papers highlighted that statin treatment is effective in preventing or ameliorating the symptomatology of a number of brain pathologies. Howev-er, other studies failed to demonstrate a neuroprotective effect.
Conclusion:
Even though considerable research studies suggest pivotal functional outcomes induced by statin therapy, addi-tional investigation is required to better determine the pharmacological effectiveness of statins in the brain, and support their clinical use in the management of different neuropathologies.
Keywords: Statins, brain, neurodegeneration, neurological disorders, mood, brain tumors
1. Introduction
Statins consist of a class of drugs at first identified as fungi metabolites, characterized by different lipophilicity and effectiveness [1]. They are potent inhibitors of mevalonate (MVA) pathway (Fig. 1). This pathway mainly produces cholesterol, but it is also responsible for the generation of other important end-products that play several physiological roles: Coenzyme Q (mitochondrial respiratory chain), farnesyl and geranylgeranyl moieties (protein post-translational modifications), isopentenyl tRNAs (RNA transcription), and dolichol (protein N-glycosylation) [2]. Statins competitively and reversibly inhibit the key enzyme of MVA pathway, the 3-βhydroxy 3β-methylglutaryl Coenzyme A reductase (HMGR, E.C. 1.1.1.34) by their lactone ring and side chains that bind them to the enzyme’s active site [1]. HMGR inhibition, and in turn cholesterol synthesis reduction, leads the cells to a homeostatic response that enhances the extra-cellular uptake of plasma LDL-cholesterol (LDL-C) [3]. In particular, low density lipoprotein receptors (LDLr) and other proteins involved in the regulation of cholesterol metabolism, as well as HMGR, are modulated depending on cellular sterol content by transcription factors named sterol regulatory element binding proteins (SREBPs). SREBPs are embedded into the endoplasmic reticulum (ER) where they are transcriptionally inactive. In the ER, SREBPs interact with the sterol sensor protein SREBP cleavage activating protein (SCAP) [4]. Thus, when intra-cellular sterol content is low, SCAP binds SREBPs and escorts them to the Golgi apparatus where they are proteolytically cleaved, releasing transcriptionally active fragments that migrate into the nucleus and induce the expression of target genes (e.g., LDLr, HMGR). On the contrary, when the amount of sterol increases, SCAP binds the insulin induced gene proteins (INSIGs), which block the SCAP/SREBP complex migration to Golgi apparatus, inhibiting the transcription of genes involved in lipid homeostasis [4]. This refined regulation is at the root of cholesterol homeostasis maintenance, and constitutes the mechanism by which statins exert their hypocholesterolemic activities.
Fig. (1).

The main steps of MVA pathway. MVA pathway, also known as cholesterol/isoprenoid biosynthetic pathway, is a pivotal metabolic pathway expressed in all mammalian cells. It leads to the production of several end-products, required for the proper functioning of cell physiology. HMGR represents the key and rate-limiting enzyme of the whole pathway, and is responsible for the conversion of HMG-CoA into MVA. HMGR is strongly inhibited by statins.
Lovastatin and simvastatin are administered as lactone pro-drug and are converted into their active hydroxy acids in the liver (the different hepatic statin metabolism is well reviewed in Marino et al., 2011) [5], whereas other statins are administered as active molecules [6]. Since sterol synthesis is high during the night, statins are more effective when taken in the evening [7]. Statins are mainly absorbed by the P-glycoprotein (P-gp) transporter in the gut [7-9] and bound to plasma proteins, mainly albumin, except pravastatin [6,10,11]. For this reason, circulating levels of free pravastatin are 10-fold higher than those of other statins [6,7,12]. Tissue distribution of statins is strongly influenced by the drug hydrophilicity [13]. In fact, lipophilic statins cross plasma membranes by passive diffusion, whilst hydrophilic ones activate carrier-mediated processes and are more selective for hepatic tissues, where the most part of cholesterol metabolism takes place [14-16]. Statins, beside their hypocholesterolemic action, exert both positive and negative pleiotropic activities in several body districts. For instance, statins can be responsible for the onset of different adverse effects, especially in skeletal muscles [17-19]. On the other hand, statin therapy has been associated to important beneficial outcomes in other organs including the brain, ranging from anti-inflammatory activity to reduction of brain atrophy and disability in multiple sclerosis. Depending on the physiopathological circumstances, it is uncertain whether these events are related to hepatic effects and/or whether they are independent of MVA pathway inhibition [20-28]. In this review we will summarize and discuss, providing up-to-date information, the current knowledge about the pleiotropic effects of statins in the brain. Pharmacokinetic and pharmacodynamic properties of statins will be analyzed, as well as their involvement as prospective pharmacological therapy for counteracting the onset and the progression of different neuropathological disorders.
2. End-products of mevalonate pathway in brain physiopathology
The effect of statins on MVA pathway inhibition has been well characterized in the liver, where a great part of cholesterol metabolism takes place [29]. However, this pathway is ubiquitously expressed in all mammalian cells, and several findings suggest that MVA pathway inhibition by statins can induce a plethora of pharmacological effects in extra-hepatic tissues [30,31]. For instance, MVA pathway end-products are particularly abundant in the central nervous system (CNS) and their presence at defined concentrations guarantees the correct functioning of different neuronal processes.
2.1. Cholesterol
Despite the brain constitutes 2% of the body weight, it contains about 25% of the total body cholesterol [32]. In almost all tissues, cholesterol need is not only assured by de novo biosynthesis, but also by intracellular uptake of lipoprotein-derived cholesterol from the circulation. On the contrary in the CNS, the blood brain barrier (BBB) hinders cholesterol uptake from the bloodstream, thus de novo synthesis represents the only source for practically all cholesterol present in this organ [33]. Essentially all (>99,5%) cholesterol in the CNS is present in an unesterified free form. This lipid is divided into two major pools: the former retains up to 70% of the brain cholesterol and represents a key constituent of myelin sheaths of oligodendrocytes; the latter is made up by plasma membranes of neurons and astrocytes [34]. Astrocytes are able to synthesize at least 2- to 3-fold more cholesterol than neurons. Cholesterol demand by neurons is very high, since this compound is required for a variety of neuronal processes, such as neurite formation and synapse activity [35]. Cholesterol in the brain presents a peculiar metabolic compartmentalization: for this reason, almost all neuronal cells depend on cholesterol synthetized by other cell types. A number of experimental findings indicate that astrocytes are the main brain cells responsible for the dispatch of cholesterol to neurons. A well-accepted model for cholesterol homeostasis in the CNS suggests that neurons strongly decrease or even interrupt their own cholesterol biosynthesis and import this molecule from astrocytes [36]. The hypothesis is supported by the fact that astrocytes efficiently secrete cholesterol through apolipoprotein (apo)E-rich lipoproteins [37]. ApoE is required for the lipoprotein endocytosis mediated by LDLr family members. Notably, the expression of LDLr family members is particularly relevant in neurons [36]. In addition to LDLr, ATP-binding cassette (ABC) transporters play a prominent function for cholesterol delivery from astrocytes to neurons. Indeed, ABCA1 is particularly expressed in astrocytes, and is involved in the transfer of intracellular cholesterol to the extracellular lipid-free apoA1 and apoE [38].
Cholesterol plays pivotal functions in CNS physiology [39]. It carries out structural roles in cellular membranes, as it modulates their fluidity and thickness, [40] and provides axonal electrical insulation for the conduction of rapid saltatory impulse [41] by restricting ion leakage through cholesterol-rich myelin membranes [42]. Furthermore, cholesterol is crucial for synapse formation, as it increases the number of synapses by enhancing their stability [32]. Different reports indicated that the biogenesis of organelles and structures belonging to the synaptic terminals requires a high cholesterol content. Indeed, synaptic vesicle membranes possess a relevant amount of cholesterol compared to other intracellular organelles [43], supporting the hypothesis that synaptic vesicle biogenesis depends on high cholesterol levels [36]. The remarkable need of cholesterol for vesicle membranes seems to be fundamental for maintaining a proper vesicle curvature and for the assembly of vesicle-specific proteins and lipids [44]. For instance, intracellular cholesterol levels are essential for the interaction between synaptobrevin and synaptophysin and, as a consequence, for the release of synaptic vesicles [45]. Experimental evidence has demonstrated that cholesterol is not only crucial in presynaptic terminals, but also for postsynaptic functions. For instance, a number of neurotransmitter receptors and other postsynaptic components are closely associated to cholesterol-rich lipid rafts, suggesting that an optimal cholesterol concentration is imperative for the structural and the functional organization of post-synaptic terminals [36]. Both reduction and enrichment of cholesterol hamper the activity of gamma aminobutyric acid A (GABAA) receptor [46]. Similarly, cholesterol depletion in hippocampal neurons destabilizes surface alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors and leads to progressive loss of dendritic spines and synapses [47]. The relevance of cholesterol in brain processes is reinforced by a growing body of clinical studies showing that alterations in cholesterol homoeostasis are associated to several brain pathological descriptions in the CNS, such as Alzheimer’s and Niemann-Pick type C disease [32,48,49]. More recently, independent research groups highlighted that cholesterol metabolism is impaired in brains and other tissues from mouse models of Rett syndrome (RTT), one of the most severe autism spectrum disorders [50-52]. Interestingly, other studies confirmed that perturbations in cholesterol homeostasis are also present in RTT patients [53-55].
2.2. Coenzyme Q, Prenylated Proteins and Dolichol
Besides cholesterol, other MVA end-products play essential roles in the brain. For instance, isoprenoids constitute the side chain of coenzyme Q (CoQ) [56], which assures ATP production in all mammalian cells including neurons. In mitochondria, CoQ is responsible for the electron transport during the oxidative phosphorylation. CoQ also preserves brain cells from central neurotoxic damages, acting as a powerful anti-oxidant and neuroprotective compound [57]. In addition, clinical evidence indicates that CoQ10 deficiency often results in neuropathological conditions, such as cerebellar ataxia, encephalomyopathy and multiple system atrophy [58-61].
Isoprenoids are also required for the post-translational modification of small GTPases. Protein prenylation consists in the covalent binding of farnesyl pyrophosphate (FPP) or geranylgeranyl pyrophosphate (GGPP) moieties to proteins. The attachment of a prenyl group is an essential prerequisite for the regulation of protein localization on cell membranes and, in turn, for key signaling cascades [62]. Ras is associated to a plethora of signal transduction pathways involved in the control of cell proliferation, morphology and gene transcription. Numerous literature shows that Ras/MAPK/ERK axis orchestrates synaptic connectivity mechanisms, thus modulating processes related to learning and memory and, in turn, influencing behavioral responses [63]. Ras proteins seem to be particularly implicated also in neural stem cell proliferation [64] and differentiation [65]. Furthermore, mutational activation of the Ras proto-oncogene represents the cause of several neurodevelopmental disorders, including neurofibromatosis type 1, Costello syndrome, and Noonan syndrome [66].
Among Rho GTPases, RhoA has been involved in important neurobiological functions. This small GTPase integrates extracellular and intracellular signals to coordinate elegant and specific modulations in gene transcription and actin cytoskeleton remodeling, thus regulating neurite outgrowth and synaptic connectivity [67,68]. Alterations in RhoA activity further sustain the essential role of this small GTPase in the brain. Indeed, RhoA dysfunctions have been associated to the establishment of mental retardation and psychiatric disorders [69]. In particular, KCTD13/Cul3/RhoA axis is essential for the control of brain size and synapse connectivity; this pathway infact results to be deeply deregulated in recurrent duplications and deletions of chromosome 16p11.2, which constitute a high risk factor for developing autism and schizophrenia [70]. Other research highlights that RhoA pathway is increased in neurological conditions such as Alzheimer’s and Huntington’s disease [71, 72], suggesting that a proper RhoA regulation is important for the homeostasis maintenance of brain functions.
Rab proteins are expressed in all eukaryotic cells and represent the greatest superfamily of small GTPases. Mammalian cells contain more than 50 Rab proteins, which are involved in the regulation of vesicle trafficking [73]. It is well known that, among Rab proteins, Rab3 carries out pivotal neurophysiological functions in neurotransmitter release at a late phase during the exocytosis of synaptic vesicles [74]. Once activated, Rab3 binds the synaptic vesicles through the prenyl moiety and forms complexes with effector proteins like rabphilin and RIM (Rab3-interacting molecule), thus promoting the recruitment and the docking of synaptic vesicles during Ca2+-induced exocytosis [73, 75]. Perturbations in Rab3 activity have been established in neuropathological conditions. For instance, alterations in Mss4 (mammalian suppressor of Sec4), a regulator of Rab3 activation, are strongly associated to deficits in neurotransmitter release and to the onset of neurodegenerative and psychological diseases in rodents such as depressive-like syndromes [76-78]. Notably, Rab3a interaction with rabphilin is abrogated by α-synuclein, representing a potential mechanism of impaired exocytosis and neurotransmitter release in Lewy´s Body diseases [79]. Mutations in the catalytic and non-catalytic domains of Rab3GAP, a Rab3 regulator, lead to Warburg-Micro syndrome and Martsolf syndrome, two inheritable forms of neurological diseases characterized by developmental abnormalities and intellectual disability [80, 81]. In addition, patients carrying mutations in GDI1 (Rab GDP-dissociator inhibitor) develop an x-linked non-specific mental retardation [82].
Even though few literature data are available, an interesting work demonstrated that a defect in dolichol metabolism is related to a novel syndrome of cerebellar ataxia [83], suggesting that also this MVA end-product could be implicated in brain development and function.
3. Statins in the brain: pharmacokinetic and pharmacodynamic aspects
It is now clear that brain processes can be directly influenced by statins, and few data reported that statin lactones and acids are able to cross the BBB. An interesting finding indicated that statins (simvastatin, lovastatin and pravastatin) can be detected in mouse cerebral cortex at 1, 3 and 6 hours following a single oral administration [84]. The detection levels of these compounds reflect their lipophilicity: simvastatin and lovastatin are administered as hydrophobic lactones that can readily cross the BBB; on the contrary, the acid form of pravastatin is highly hydrophilic, thus the permeability to the BBB is hindered. However, the capability of pravastatin (and other statins in the open acid form) to reach the CNS can be explained by active transport mechanisms. For instance, pravastatin was found to be a substrate for the organic anion transporter polypeptide 2 (OATP2) in the liver [85]. In addition, the acid forms of lovastatin, simvastatin and atorvastatin are also transported into cells by OATP2. Notably, OATP2 was found to be expressed at considerable levels in rat BBB and choroid plexus [86], suggesting this transporter as a pivotal player in the shuttling of acid statins from extra-cellular to intra-cellular compartments in the brain. Other evidence indicates that acid statins can also be transported by monocarboxylic acid transporters (MCT). MCT inhibition impairs the uptake of acid lovastatin in mesangial cells and MCT4 appears to be the most involved isoform in statin transport [87, 88]. MCT-mediated uptake of acid statins can be responsible for statin transport in the brain, as these transporters are expressed in neurons, astrocytes and BBB [89,90].
After a single dose administration, statins can be detected at significant levels in the brain [84], thus producing inhibitory effects on MVA production. It was reported that HMGR inhibition by statins significantly decreases cholesterol biosynthesis in rodent brains [84, 91] and, more recently, two-dimensional gel electrophoresis approaches provided evidence that statins also lead to a significant reduction in protein prenylation in this organ [92]. From these observations, it is not surprising that clinically relevant doses of statins can induce important biological effects in the brain.
Several reports indicated that statins may alter synaptic transmission by modulating the function of neurotransmitter receptors. For instance, cholesterol depletion by simvastatin impairs serotonin 3 (5-HT3) receptor function in neuronal cell line N1E-115 [93]. In addition, chronic mevastatin administration causes a decrease in the specific ligand binding and G-protein coupling to serotonin 1A (5-HT1A) receptor, without modifying the expression levels of the receptor [94]. Similarly, simvastatin administration does not affect the expression of the subunit 1 of N-methyl-D-aspartate (NMDA) receptor (NMDAR1), but hampers the association of NMDAR1 to lipid rafts, and protects primary neurons from NMDA-induced toxicity [95]. The effects of statins on neurotransmitter receptors are mediated, at least in part, by statin-dependent cholesterol depletion from cholesterol-rich membrane domains: notably, cholesterol replenishment is able to restore the modulations caused by the pharmacological treatment.
Recently, it has been shown that statins regulate cognition and memory pathways through different mechanisms (Fig. 2). HMGR inhibition by simvastatin leads to the enhancement of the consolidation phase of the memory process in the passive avoidance test. At molecular level, simvastatin treatment induces a significant reduction in RhoA prenylation in selected brain regions (i.e. hippocampus and amygdala) involved in the modulation of emotional memory. The reduction in prenylated RhoA determines an increase in Akt phosphorylation, which in turn leads to cAMP responsive element binding protein 1 (CREB1) activation [62]. The contribution of RhoA/Akt/CREB1 axis in the regulation of long-term memory mediated by statins is further strengthened by another independent study, showing that simvastatin promotes the upregulation of memory- and learning-related genes c-Fos and Egr-1, whose transcription is downstream of CREB1 activation [96]. Other evidence sustains that statins activate Akt not only through the inhibition of RhoA geranylgeranylation, but also by blocking protein farnesylation. Experiments conducted on CA1 region of mouse hippocampus showed that simvastatin is able to enhance long-term potentiation (LTP) by increasing Akt phosphorylation. However, this event is abolished upon farnesyl pyrophosphate replenishment. Accordingly, the specific inhibition of farnesylation mimics statin-related Akt activation during the LTP induction phase [97]. Aside from the direct inhibition of MVA production, an interesting work elegantly demonstrated that statins also affect memory by MVA pathway-independent actions. Notably, statins serve as ligands for peroxisome proliferator activated receptor α (PPARα), and the subsequent activation of this nuclear receptor enhances CREB transcription, driving the expression of neurotrophic factors such as brain derived neurotrophic factor (BDNF) and Neurotrophin-3 (NT-3) [98]. Other potential cellular mechanisms which may explain the functional outcomes of statins in cognitive processes are the regulation of neuronal differentiation and adult neurogenesis. It is postulated that defects during neurogenesis can be responsible for the onset of cognitive alterations and emotional disturbances in psychiatric disorders, neurological diseases and traumatic brain injury. Statins protect hippocampal neurons from traumatic brain injury, and ameliorate spatial learning in rats tested for Morris Water Maze task. The improvement in cognitive functions is associated to a reduction of neuronal loss in the non-neurogenic area of hippocampal CA3 area, and to an increased neurogenesis in the neurogenic region of the dentate gyrus [99]. A more recent experimental work elucidated the molecular mechanism exerted by statins in the induction of adult neurogenesis, highlighting that statin-dependent inhibition of MVA pathway is able to modulate hippocampal neurogenesis through the suppression of isoprenoid biosynthesis and, in turn, by upregulating Wnt signaling [100]. Statins control adult neurogenesis at multiple levels (Fig. 3): this class of compounds not only enhance the proliferation rate of adult neural stem cells, but also induce their differentiation [101]. In this context, several lines of evidence demonstrate that statins are potent inducers of neurite and axonal outgrowth. An interesting work showed that, among 50.400 small molecules screened for their ability to drive axon outgrowth, statins resulted to be the most effective [102]. Similarly, other reports highlighted that statins strongly support neuritogenesis in diverse experimental models of neuronal differentiation. The molecular mechanism underlying the statin-dependent induction of neurite outgrowth relies in the inhibition of RhoA prenylation and in the activation of Akt signal transduction [68, 103, 104].
Fig. (2).
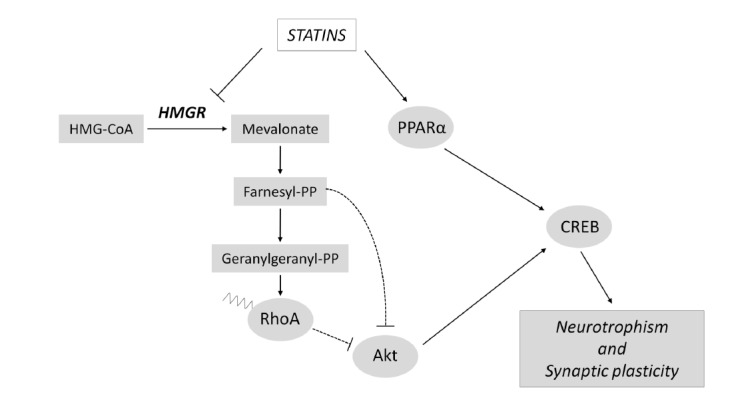
Statins exert neurotrophic actions and modulate synaptic plasticity. Statins are able to induce neurotrophism and synaptic plasticity through the activation of CREB. The molecular pathways by which statins activate CREB signaling are mediated by MVA pathway-dependent and independent mechanisms. For details see the main text.
Fig. (3).
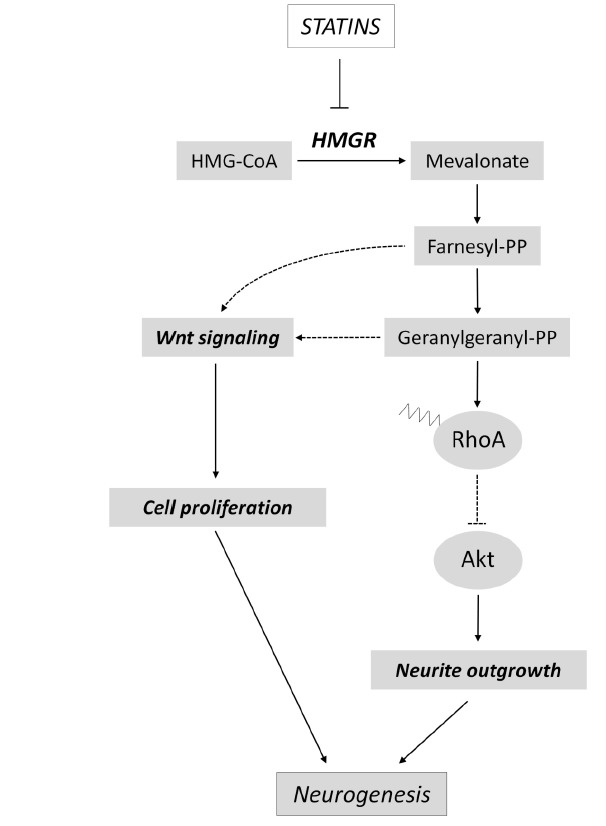
Statins activate neurogenesis. Statins influence adult neurogenesis at multiple levels. Indeed, this class of drugs can induce both proliferation and differentiation of neuronal precursor cells by modulating RhoA, Akt and Wnt signaling pathways.
Among their pleiotropic effects in brain physiopathology, statins also act as powerful anti-inflammatory agents (Fig. 4). The repression of Ras and Rac prenylation mediated by statins blunt the upregulation of pro-inflammatory molecules such as interleukin (IL) 1β, IL-6, IL-8, and tumor necrosis factor alpha (TNF-α) in numerous neuroinflammatory conditions, thus reducing the activation of astrocytes and microglia [98, 105-110]. The inhibition of Rac activity by the lack of prenylation is also at the root of statin-induced protection from oxidative stress, a common feature of several neurodegenerative conditions [111]. In particular, Rac plays pivotal functions in the activation of NADPH oxidase (NOX) for the generation of reactive oxygen species (ROS). The role of Rac consists in escorting the cytosolic components p67phox, p47phox and p40phox to the NOX2/p22phox complex, which is located on the membrane. Upon the interaction among these six proteins, NOX complex becomes active, and transfers an electron from NADPH to molecular oxygen producing superoxide [112]. Statins block Rac activity and effectively prevent the protein interaction and the formation of NOX complex, resulting in a decrease of ROS production (Fig. 4). The beneficial effect exerted by statins in oxidative stress conditions is now incontrovertible, and is clearly established in a plethora of neuropathological descriptions, ranging from ischemic brain injury to Alzheimer’s disease [113-117].
Fig. (4).

Statins reduce neuroinflammation and oxidative stress. Statins show strong anti-inflammatory and antioxidant properties in several physiopathological conditions. In particular, HMGR inhibition by statins determine the downregulation of protein prenylation, which in turn leads to the suppression of NOX activity and to the reduction of pro-inflammatory cytokines.
4. Statins and neurological disorders
The perturbation of cholesterol metabolism affects neural development and synaptogenesis, leading to synaptic dysfunction and, as a consequence, to disorders of nervous system [118-121]. Genetic syndromes often present a neurological component among their other symptoms. In some cases, this neurological phenotype may have an impairment of cholesterol balance as primary or secondary cause. For this reason, during the last decades the role of statins in the treatment of some genetic syndromes with neurological implication has attracted increasing interest.
4.1. Smith-Lemli-Opitz Syndrome
Smith-Lemli-Opitz syndrome (SLOS, OMIM: 270400) is an autosomal recessive syndrome characterized by multiple developmental abnormalities, like distinctive facial features, limb malformations, microcephaly, cleft palate, polydactyly and holoprosencephaly, as well as severe intellectual impairment [122]. Its birth prevalence is estimated to be approximately 1:20,000 to 1:40,000 live births [123, 124]. It is caused by mutations in 7-dehydrocholesterol reductase (DHCR7), the final enzyme of the cholesterol biosynthetic pathway, resulting not only in abnormally low amounts of cholesterol but also in the accumulation of its precursors 7-dehydrocholesterol (7-DHC) and 8-dehydrocholesterol (8-DHC) in plasma and cells of the brain [125].
Considering that both cholesterol deficiency and the excess of 7-DHC and 8-DHC seem to contribute to the pathogenesis of this disorder [126], the therapeutic goal for its treatment aims to increase cholesterol production and/or deposit, and to reduce the accumulation of potentially toxic cholesterol precursors 7-DHC and 8-DHC. The currently used treatment is based on feeding affected individuals with a cholesterol-rich diet [122]. Several reports have described multiple benefits of this treatment such as improved growth, decreased irritability, reduction in self-injurious behavior and tactile defensiveness, and increased sociability [127]. Dietary cholesterol supplementation not only increases plasma cholesterol in most individuals with SLOS [126,128,129], but also decreases 7-DHC and 8-DHC levels via feedback inhibition of HMGR [129, 130]. Nevertheless, this treatment does not allow to normalize the levels of the precursors [131]. Moreover, the neurological phenotype associated to SLOS does not improve by increasing the level of plasma cholesterol because plasma lipoproteins (and the cholesterol carried therein) do not cross the BBB or enter in the CNS [132].
For this reason, simvastatin administration has been suggested as a promising therapeutic avenue to address CNS dysfunctions and, as a consequence, to reduce 7-DHC levels. SLOS fibroblast cell lines with residual DHCR7 enzymatic activity treated with simvastatin increased fractional cholesterol synthesis and decreased 7-DHC levels [133]. Additionally, treatment with simvastatin in mouse models Dhcr7 T93M/T93M and Dhcr7 T93M/Δ3-5 showed decreased 7-DHC levels in both peripheral and brain tissues [134]. Furthermore, significantly reduced levels of the cholesterol precursors 7-DHC and 8-DHC were observed in 39 patients treated with cholesterol supplementation and simvastatin compared to a group who received only cholesterol supplementation. However, no anthropometric improvements were observed in the simvastatin-treated group [135]. Jira and collaborators revealed a rise in plasma cholesterol level and a decrease in the 7DHC/cholesterol ratio in two SLOS patients after long-term treatment with simvastatin [136]. Improvement in anthropometric measurements, including length, weight, and head circumference were also observed. Nonetheless, these observations were not reproduced in other studies [126,135]. Overall, further studies are necessary in order to confirm these results and evaluate possible side effects of this treatment.
4.2. Fragile X Syndrome
Fragile X Syndrome (FXS, OMIM: 300624) is an X-linked disorder which represents the most common inherited cause of intellectual disability (ID) and autism with an estimated frequency of 1/4,000 in males and 1/8,000 in females [137]. The patients show typical dysmorphic features like macrocephaly, narrow face with prominent ears, macroorchidism, and hyperextensible joints [138], while the cognitive phenotype is characterized by moderate to severe ID in males [139]. In addition, FXS individuals exhibit psychiatric problems such as attention-deficit hyperactivity disorder (ADHD), anxiety, compulsive behavior and aggressiveness [140]. FXS is caused by a cytosine-guanine-guanine (CGG) trinucleotide repeat expansion at the 5’-UTR region of the FMR1 gene, associated with the methylation of its promoter [141]. Consequently, FMR1 transcription is repressed, giving to the absence or a reduced expression level of the FMR1 protein, FMRP (fragile X mental retardation protein). This RNA binding protein acts as a translational repressor at the synapses [142] and its absence results in the formation of several thin and undeveloped dendrites causing impaired synaptic plasticity along with an increased protein synthesis rate [143].
Interestingly, many of the proteins upregulated in FXS lie downstream of the excessive ERK-mediated protein synthesis [144]. Indeed, the involvement of ERK1/2 was studied in Fmr1 KO mice, revealing an increased ERK activity [145, 146]. Recently, Osterweil and collaborators reported that the inhibition of ERK1/2 phosphorylation by specific drugs restored protein synthesis activity and attenuated audiogenic seizures, two relevant pathological features of the Fmr1 KO mouse [147]. Thus, the inhibition of the ERK1/2 pathway seems to be a promising therapeutic target for FXS patients. For this reason, lovastatin has been proposed as a possible treatment. It reduces ERK signaling pathway by decreasing the farnesylation-dependent membrane recruitment and activation of its upstream component Ras [148], as well as by decreasing lipid rafts’ cholesterol content, affecting membrane-dependent signaling interactions and dynamics [149]. Recently, it has been demonstrated that lovastatin can normalize the excessive protein synthesis and prevent mGluR-induced epileptogenesis in the Fmr1 KO hippocampus [150]. A similar effect was observed when lovastatin was administered to a mouse model of Neurofibromatosis [151]. These findings have stimulated human trials based on lovastatin treatment, and an open-label trial in children with FXS in Canada showed positive results [152]. This study suggests that lovastatin may have positive effects not only on behavioral symptoms but also on adaptive skills.
Until now, FXS patients are often treated with stimulants, anticonvulsants, antidepressants, selective serotonin reuptake inhibitors, alpha-receptor agonists, antipsychotics, and mood stabilizers [153]. As these treatments do not improve the progression of the disease and often cause important side effects, lovastatin may represent a new frontier in the treatment of FXS.
4.3. Rett Syndrome
Rett syndrome (RTT, OMIM: 312750) is an X-linked disorder characterized by progressive development of neurological and motor dysfunction. It affects almost exclusively females, with a frequency of approximately 1:10,000 live births [154]. After an apparent normal period of postnatal development, progressive neurological manifestations of disease occur, including loss of previously acquired speech and motor skills, stereotypic hand movements, difficulty walking, irregular breathing, and seizures [155]. Mutations in the X-linked gene, methyl CpG binding protein 2 (MeCP2), are the primary cause of RTT [156]. MeCP2 is involved in gene silencing through methylation-dependent remodeling of chromatin structure [157] and is able to repress gene transcription through the association with different co-repressors [154].
Recent findings demonstrated that cholesterol metabolism is perturbed in brains and livers of MeCP2-null male mice. Furthermore, statins (fluvastatin and lovastatin) have been reported to improve the systemic imbalance of lipid profile, ameliorate motor symptoms and confer increased longevity in MeCP2 mutant mice [50], suggesting that cholesterol homeostasis maintenance could be altered in RTT patients. A similar conclusion was reached by Segatto and collaborators, who showed that the protein network of cholesterol homeostasis maintenance is severely altered on plasma and cultured skin fibroblasts of RTT patients [54], providing an additional finding that highlights that cholesterol metabolism may be taken into account as a new therapeutic option for the treatment of RTT. However, a recent study revealed that lovastatin had no effect on motor performance and survival when the deletion of the MeCP2 allele is expressed on a different background strain, suggesting that the response to lovastatin is under the control of inherent genetic or other biological mechanisms specific to the effects of lovastatin on brain function [158]. Thus, the effectiveness of lovastatin in RTT may be limited to those patients who reproduce alterations in cholesterol homeostasis similar to those described in mice responding to statins [50]. In this context, additional studies are needed in order to clarify the results obtained and to elucidate the potential role that statins can play in the treatment of RTT pathology.
4.4. Autism and Epilepsy
Autism is one of the prevalent developmental disorders characterized by impaired language and communication, social relationship problems, and by restricted, repetitive and stereotyped behaviors and interests [159,160]. As the presence of an active neuroinflammatory process in the brain of autistic patients plays a role in the pathogenesis of the disorder [161], it may represent a target for therapeutic interventions. Increases in plasma levels of the pro-inflammatory cytokines IL-1β, IL-6, and IL-8 in the autism spectrum group have been reported [162]. For this reason, it seems reasonable to hypothesize that statins may play a role in the treatment of autism because of their anti-inflammatory activities. It has also been described that lovastatin reduces the mRNA expression of IL-1β, IL-6 and TNF-α in epileptic animals [105]. Moreover, other studies have suggested that statins can decrease seizure activity [163-165]. On the contrary, others have reported no effect [166] or an intensification of seizures [167].
Considering these findings and the high comorbidity between autism and epilepsy [168,169], it seems worthwhile to conduct additional experimental studies to evaluate the real feasibility of this possible novel treatment.
5. Statins and neurodegenerative diseases
As described above, statins are able to modulate a plethora of neuronal processes ranging from synaptic transmission to neuroinflammation. Since misbalances in these processes have been strongly associated to the pathogenesis of several brain diseases, it has seemed reasonable to consider statin treatment as a putative new therapeutic option for counteracting neurodegenerative disorders. In this section, we summarize the current knowledge about the neuroprotective action of this class of pharmacological compounds in the prevention and treatment of Alzheimer’s, Parkinson’s and Huntington’s diseases.
5.1. Alzheimer’s Disease
Alzheimer’s disease (AD), the most common form of dementia responsible for the cognitive impairment of 26.6 million people worldwide, is characterized by progressive neurodegeneration, particularly affecting cortical and hippocampal brain regions [170, 171]. AD brain is hallmarked microscopically by the combined presence of two types of abnormal structures, described over a century ago by Alois Alzheimer [172]. These include extracellular senile plaques composed of Amyloid β (Aβ) peptide and intraneuronal neurofibrillary tangles (NFTs) composed of hyperphosphorylated Tau protein. Impairment of cognitive and memory functions is associated with Aβ accumulation, inflammation, increased oxidative stress and lipid dysmetabolism [170].
Alteration in cholesterol/isoprenoid metabolism is considered one of the causative factors of the disease, there is indeed considerable attention being given to the association of AD and cholesterol homeostasis, particularly to the lipoprotein APOE, required for cholesterol transport [173]. Notably, the apoE4 allele of the APOE gene (APOE ε4), has been associated with elevated cholesterol levels and with increased risk of AD, having been established unequivocally as the most important susceptibility gene for Late Onset AD (LOAD) [174]. It has now been demonstrated that ApoE lipoproteins, besides binding to several cell-surface receptors to deliver lipids, also associate to hydrophobic peptide Aβ promoting its aggregation or decreasing the clearance of senile plaques [2]. These events are thought to initiate toxic pathways that lead to synaptic dysfunction and neurodegeneration in AD [175]. Neuropathological examination in LOAD suggests that APOE ε4 allele dosage is associated with increased Aβ, Aβ oligomers, and plaque accumulation in the brain [176-178] and that APOE receptors play an important role in modulating amyloid precursor protein (APP) trafficking and Aβ production [179].
Furthermore, it is worth to mention that emerging evidence indicates that isoprenoids/protein prenylation and small GTPases affect multiple aspects of AD, which may imply that prenylated proteins play important roles in the pathogenesis and in the development of the disease [180]. According to this observation, it has been demonstrated that statin-induced depletion of isoprenoids leads to reduced levels of protein prenylation and promotes non-amyloidogenic processing of APP and reduces the production of Aβ [92, 181-183]. Based on this evidence it seems reasonable and interesting to propose statins as neuroprotective agents in the prevention and treatment of AD. Studies on patients [184] and on animal models have shown that simvastatin or pravastatin reduce the levels of Aβ both in cerebrospinal fluid (CSF) and brain [185]. Atorvastatin has been proven to reduce Aβ through the reduction of amyloid precursor protein (APP) [186, 187], β-secretase (BACE1) [188] and oxidative stress in preclinical models of AD [113]. Furthermore, lovastatin decreases the production of components of senile plaques [189] and protects neurons in an induced Aβ-toxicity experimental model by reducing GSK-3β activity and enhancing Wnt signaling [190]. In a transgenic mouse model harboring a mutation for APP (APP-Tg model), atorvastatin and pitavastatin have been shown to exert neuroprotective action against neuroinflammatory cytokines (IL-1β, IL-6, TNF-α), reduce senile plaques and tau phosphorylation, and improve cognitive function [187]. The same group demonstrated that these statins prevent memory impairment and oxidative stress through the reduction of superoxide anion production occurring after intracerebroventricular injection of Aβ in an induced Aβ-toxicity preclinical model [191]. In the same model, simvastatin treatment improves cerebrovascular function and reduces glial activation as well as the number of Aβ plaque-associated dystrophic neurites [192]. Noteworthy, in AD patients, simvastatin stimulates the non-amyloidogenic pathway of APP [188] by increasing α-secretase activity [193]. Evidence suggests that the treatment with cholesterol-lowering drugs may have potential therapeutic benefit in AD in view of their effect on APOE4 carriers, for example treatment with simvastatin has been shown to reduce plasma levels of APOE [189,194]. Another study suggested that statin use might have a slightly stronger protective association for people who carry at least one APOE4 allele [195]. Likewise, one trial on AD patients suggested that atorvastatin might help preserve cognitive function for carriers of at least one APOE4 allele [196, 197]. Conversely, speaking of statins’ effects on brain health for APOE4 we also need to mention at least three observational studies reporting that APOE genotype had no effect on the relationship between statin use and dementia [197-199].
Although a plethora of preclinical and clinical studies suggest an overall neuroprotective effect, other evidence indicates inefficacy or detrimental roles of statin administration in AD [200]. For example, lovastatin and mevastatin alter microtubule system by increasing Tau phosphorylation and inducing axonal degeneration linked to the lack of cholesterol in cultured neurons [201]. In a human neuroblastoma cell line (SH-SY5Y) with induced Aβ toxicity, lovastatin increases ROS production, inhibition of the proteasome activity, microglia activation and TNF-α upregulation [200, 202]. Furthermore mitochondrial apoptotic pathway is induced in a time and dose-dependent manner, and is caused by the reduction of MVA after treatment with the same statin [203].
These conflicting results may be associated to divergences in the experimental design. Indeed, several variables can induce diverse outcomes, such as statin dose and timing of administration, statin lipophilicity, and age of the individuals enrolled in the clinical studies. Moreover, difference in the rate of basal cholesterol metabolism among the experimental models and human patients, as well as statin catabolism and their interaction with xenobiotic compounds may account for the ambiguous data present in literature [200].
5.2. Parkinson’s Disease
Parkinson disease (PD) is the second most common neurodegenerative disease after AD, characterized by classical motor and non-motor symptoms. Motor manifestations include bradykinesia, muscle rigidity, resting tremor, postural instability and gait disturbances, while non-motor symptoms may include depression, olfactory dysfunction, sleep disorders, psychiatric symptoms, pain, and cognitive impairment [204, 205]. The deterioration of motor functions observed in PD patients is predominantly attributable to the degeneration of dopaminergic neurons in the substantia nigra, showing that the nigrostriatal circuit is involved in neurodegeneration [206]. At cellular level, neuronal loss is accompanied by neurite degeneration and the presence of cytoplasmic inclusions known as Lewy bodies, involving α-synuclein aggregation [205].
During the last few years, different genes have been associated with the familiar form of PD, with altered mitochondrial homeostasis as one of the culprits of the pathology [207]. Indeed the disease-associated mutated genes, such as α-synuclein, DJ-1, PINK, parkin, and dominant mutations in LRRK2 are directly or indirectly associated with mitochondrial dysfunction in PD [208, 209]. Studies revealed that cell dysfunction in PD also involves biochemical factors, such as free radicals, mitochondrial dysfunction, excitotoxicity, inflammation and autophagy.
There is growing evidence that autophagic processes play important roles in neuronal degeneration and participate to regulated forms of cell death, particularly in large cells such as the dopaminergic neurons of substantia nigra [210, 211]. Autophagy is involved in the intracellular turnover of proteins and cell organelles and has an important role in regulating cell fate in response to stress [211, 212].
The dysregulation of the autophagic pathway in the brains of animal models and PD patients suggests a pivotal role for autophagy in the pathogenesis of the disease [213]. Notably, in PD, the autophagic pathway participates in the degradation of α-synuclein, as well as in the turnover of damaged mitochondria, a hallmark of the disease [210]. As a consequence of mitochondrial dysmetabolism, a striking increase of related-oxidative stress has been proven to occur.
Statins demonstrated neuroprotective effects through different mechanisms including anti-inflammatory actions, apoptosis regulation, and mitigation of oxidative damage [214]. Furthermore, statins can also show cytoprotective effects through the modulation of autophagy. Kang and collaborators (2016) hypothesized that statins may decrease rotenone-induced neurotoxicity in SH-SY5Y cells, an in vitro model of PD, by modulating the autophagy signaling pathway. In this study, the authors demonstrated that statin treatment up-regulates autophagic markers including Beclin-1 and AMP-activated protein kinase (AMPK). Specifically, treatment of SH-SY5Y with rotenone increased mTOR expression, while decreased Beclin-1 and AMPK levels, with consequent suppression of the autophagic mechanism. The co-administration of rosuvastatin restored the levels of autophagic markers, significantly increasing Beclin-1 and AMPK expression and decreasing mTOR levels. This study suggests that the mechanism of statin-mediated neuroprotection is associated with an increased autophagy. Thus, the neuroprotective effect of statins via autophagy modulation may provide a new therapeutic strategy for the treatment of PD [215].
Despite mounting observations in support of a protective role of statin treatment in various models of PD, some essential issues remain unsolved. Concerning PD clinical treatment with statins, a controversial scenario has been revealed. A 12-year follow-up of PD cases (338 women and 306 men) found that regular use of statins was associated with a modest reduction in PD risk [216]. A meta-analyses study demonstrated that all statins showed a non-significant inverse association with the risk of PD, except for pravastatin [206]. This effect is probably due to the weakly lipophilic properties of pravastatin resulting in its difficulty to cross the BBB [217]. Notably, atorvastatin, followed by simvastatin, being the most lipophilic, shows significantly larger protective effects [218, 219]. Wolozin and collaborators (2007), using a database (US Veterans Affairs database) containing diagnostic, medication and demographic information on 4.5 million subjects, compared the action of three statins (lovastatin, simvastatin and atorvastatin) to investigate whether some of these compounds might be associated with a reduction in the incidence of dementia and PD. Simvastatin was associated with a strong reduced incidence of both dementia and PD, while atorvastatin treatment led to a modest reduction in incidence of dementia and PD. Importantly, results were significant when age was included as a covariate, but not-significant when other comorbid diseases were included as covariates. Conversely, lovastatin was not associated with a reduction in the incidence of dementia [220]. Interestingly, a prospective study showed that statin use may be related to a higher risk of PD, whereas higher total cholesterol level may be associated with lower risk [221]. These findings are inconsistent with the results of many other studies, which indicate that statins protect against PD, as previously described. It is worth to mention that since HMGR is involved in the synthesis of CoQ10, the use of statins could result in a reduction of the serum coenzyme concentration. Given the role of CoQ as an essential carrier in the mitochondrial respiratory chain, its decrease could affect mitochondrial homeostasis increasing the risk of PD [216].
In conclusion, the current literature indicates that the relationship between statins and PD is not straightforward, and further research needs to be conducted in order to univocally validate the potential role of these drugs in PD therapy.
5.3. Huntington’s Disease
Hungtington’s disease (HD) is an inherited, autosomal dominant neurodegenerative disorder characterized by motor and psychiatric disturbances, as well as dementia. It is caused by a CAG repeat expansion in the gene coding for huntingtin (HTT). The possibility that changes in brain cholesterol homeostasis occur in HD has been suggested over the past several years [222]. Indeed, in HD patients and mouse models of HD, growing evidence has demonstrated early and long-lasting alterations in the cholesterol biosynthesis [223].
Whereas some studies on preclinical models show significant downregulation of cholesterol in the HD brain [224-226], there are others that, in spite of presenting progressive reduction of brain cholesterol biosynthesis, demonstrate constant steady-state levels of total cholesterol, suggesting compensatory mechanisms [227]. Conversely, other groups proposed that the accumulation of cholesterol in HD brain can result in neurotoxicity [228, 229]. According to this evidence, it was recently shown that in HD the decreased cholesterol synthesis is caused by altered cholesterol homeostasis and the dysregulation of cholesterol clearance is due to deficient expression of CYP46A1, the rate limiting enzyme for cholesterol degradation [223, 230].
The finding that cholesterol perturbations are involved in HD pathogenesis, led to the hypothesis that a pharmacological intervention on sterol metabolism could, at least in part, ameliorate the symptomatology of the disease. For example, the supplementation of CoQ, a compound synthetized from intermediary metabolites of MVA pathway, is being examined as a HD therapy [2, 231]. A surprising discovery was that simvastatin, a drug typically used to reduce cholesterol, also improved the synaptic phenotype in the R6/2 strain, a transgenic mouse model of HD [223]. Some studies postulate that cholesterol is accumulated in HD cell membranes [228], and simvastatin could enhance mobilization, redistribution, and/or efflux of accumulated cholesterol [232]. Moreover simvastatin has been also shown to increase membrane fluidity, exert anti-inflammatory effects, and promote BDNF production. This latter effect is important because BDNF reduces the enhanced spontaneous inhibitory postsynaptic currents (sIPSCs) observed in R6/2 mice [223]. In addition, there is evidence demonstrating that simvastatin can have neuroprotective effects in cell cultures expressing mutant huntingtin, which is in turn involved in the accumulation of cholesterol. As a matter of fact, the treatment with simvastatin reduces the content of ordered domains at the cell surface thus protecting cells against NMDA-mediated excitotoxicity [229].
6. Statins and brain tumors
Since early 90s, conflicting results have been reported about the role of statins in cancer prevention and treatment. From a biological point of view, statins are able to interfere in different processes of cancer cells, like progression through the cell cycle, resistance to apoptosis, angiogenesis and many others (for extensive review see Hindler et al., 2006) [233]. Indeed, MVA pathway blockade by statins leads to the down-regulation of several end-products, including cholesterol, dolichol, GGPP and FPP. Cancer cells are strongly dependent on cholesterol levels because of their high request for the neo-formation of membranes during rapid mitosis. De novo DNA synthesis, a key process for cell proliferation, is strongly dependent on the activity of dolichol-linked glycoproteins [234]. Furthermore, the relative abundance of GGPP and FPP could influence Ras activation and its anchoring to cell membranes, modulating pivotal cellular pathways (i.e. Akt, ERK) involved in proliferation and apoptosis. Several studies pointed out a positive effect of statins in preventing and treating different type of tumors such as breast cancer [235], prostate cancer [236], colorectal cancer [237], leukemia [238] and others [239-241]. Noteworthy, it has been demonstrated that statins could represent a therapeutic strategy for the treatment of Brain Cancers (BCs), alone or in combination with other chemotherapies. BCs, generally classified in gliomas and neuroblastoma, are usually characterized by poor prognosis and overall worsened survival. For instance, epidemiological data reveals that only 3% of glioma patients survives after 5 years since diagnosis [242]. Common therapies for BCs (chemo- or radio-therapy) are unable to improve this outcome, thus, alternative strategies are needed.
Glioma represents a large group of common brain tumors that comprises glioblastoma and astrocytoma [243]. Preclinical data assess that statins are able to strongly inhibit glioma growth and development through multiple mechanisms of action (Fig. 5). Indeed, it has been demonstrated that C6 cell line treated with simvastatin undergoes proliferation arrest and apoptosis induction via JNK-dependent phosphorylation of ATF-2 and c-jun [244]. Similar results were obtained by another group, which analyzed the effect of three different statins (mevastatin, fluvastatin and simvastatin) on C6 and U251MG cells. They found that statins, in particular fluvastatin, decrease the synthesis of geranylgeranyl pyrophosphate, a pivotal membrane-anchoring molecule of Ras protein. This led to the suppression of ERK1/2, Akt phosphorylation, and induction of apoptosis [245]. Statin-induced apoptosis in cancer cells can be also mediated by the enhanced expression of proapoptotic protein Bim, as observable in U87 and U251 glioblastoma cell lines [246]. A recent research demonstrated that atorvastatin inhibits proliferation and migration, and induces apoptosis in in vitro models of glioma (A172 cells) [247]. In addition, pitavastatin potentiates anti-tumor effects of low-dose irinotecan, a topoisomerase inhibitor, in glioblastoma cell lines. This effect is mediated by the inhibition of the multi-drug resistance protein (MDR-1), which is overexpressed in glioblastoma and is partly responsible for the resistance to chemotherapy [248]. Together with promising data in in vitro glioma models, statins have also shown to block cancer progression in murine models of brain tumors. For instance, simvastatin is able to reduce glioma cell growth into nude mice by its ability to inhibit MVA production [249]. Moreover, pitavastatin significantly reduced tumor size and tumor weight in a xenograft mouse model of glioblastoma multiforme [248]. Despite preclinical data strongly suggest a positive role of statins in the treatment of BCs, there is still lack of definitive evidence about their clinical utility, as different clinical studies with conflicting results were published. Larner and colleagues (1998) tested, for the first time, lovastatin as a potential drug against glioma in a phase II trial on 18 patients with either anaplastic glioma or glioblastoma multiforme. Patients entered into the clinical trial served for testing the safety of high-dose lovastatin with or without radiation. They demonstrated that lovastatin was well tolerated, but no significant inhibition of tumor progression was reported [250]. On the contrary, a case-control study about the correlation between statin administration and risk of glioma indicated a positive effect of statins on tumor incidence. 517 glioma patients and 400 controls were recruited and matched for age, ethnicity and education level. These two groups were stratified based on the time of statins use (<6 months, 7-24 months, 25-60 months, 60-120 months and >120 months). Simvastatin and lovastatin administration for more than 6 months showed a significant inverse correlation with risk of glioma, with an Odd Ratio (OR) of 0.49 and 0.47 respectively, and 95% Confidence Interval (CI) of 0.30-0.81 and 0.24-0.93, respectively. Furthermore, a significant inverse trend between duration of statin treatment and glioma risk was highlighted [251]. A nationwide population-based case–control study utilizing national registry data was conducted in Denmark to investigate the association between statin treatment and glioma incidence. This study compared 2656 cases of glioma and 18480 controls matched for age, diabetes, use of aspirin, selective Cox2 inhibitors, non-aspirin non-steroidal anti-inflammatory drugs and hormone replacement therapies. Notably, a significant inverse correlation between long-term statin therapy and risk of glioma was found in both men < 60 years old (OR = 0.40; 95% CI: 0.17–0.91) and > 60 years old (OR = 0.71; 95% CI: 0.49–1.03) and in women < 60 years old (OR = 0.28; 95% CI: 0.06–1.25). However, the statistical significance was only achieved with the use of lipophilic statins (OR = 0.69; 95% CI: 0.38–1.25), and not with the hydrophilic ones: the discrepancy probably reflects the hydrophobic properties of these compounds and, in turn, their capability to cross the BBB. Interestingly, long-term statin administration also reduced the risk of glioblastoma multiforme, the most common and highly aggressive type of glioma, by 21% [252]. Differently, a retrospective analysis of the follow-up of 284 glioblastoma multiforme patients indicated that the preoperative use of statins did not show any impact on progression free survival and overall survival of glioblastoma multiforme patients. Nevertheless, the authors of this study underlined some biases of their analysis, such as the small cohort analyzed, uncertainty about the effective dose, regularity of drug assumption and others [253]. The incoherent clinical results available in literature suggest the need of additional studies to assess the effective clinical relevance of statins in preventing and/or treating glioma.
Fig. (5).

Statins interfere with brain tumor growth at multiple levels. Statins lead to the alteration of both pro-survival and pro-apoptotic intracellular pathways, causing the inhibition of proliferation and the induction of apoptosis in brain cancer cells. Dashed lines indicate indirect and/or unknown signal transduction mechanisms. For details see the main text.
Neuroblastoma represents the most common solid tumor diagnosed in the first year of life. To date, only preclinical data about the effectiveness of statins in neuroblastoma treatment are available. Experimental data proved the efficacy of a wide concentration range (0-100 μM) of lovastatin in different neuroblastoma cell lines (SK-NSH, NUB-7, SMS-MSN, NBL-S, LAN-5, IMR-32, and GOTO), assessing their sensitivity to this drug. Lovastatin shows anti-tumor properties also in SH-SY5Y cells, a well-established in vitro model of neuroblastoma. In particular, lovastatin treatment leads to apoptosis through mitochondrial pathway [203]. Interestingly, simvastatin exerts anti-apoptotic effect on SH-SY5Y cells by stimulating Bcl-2 protein expression. The anti-apoptotic effect of simvastatin is independent from MVA pathway inhibition, but it is likely due to the stimulation of Endothelin-1 and Nuclear Factor of Activated T-cells 3 (NFATc3) [254]. Different dosage and diverse mechanism of action of the two statins could explain the opposite effect on the same cell line.
Data available in literature are still far from to definitely establish the real benefits of statins in the clinical treatment of BCs. Even though the majority of preclinical evidence sustains a beneficial role for statin treatment, there are still contrasting results about their efficacy in patients affected by BCs. For these reasons, further researches will be useful in order to fully understand the putative mechanisms of action of different statins in cancer cells. Moreover, additional clinical studies, both retrospective and prospective, could shed more light about the efficacy of statins in BCs.
7. Statins in mood and mental disorders
Several reports hint for a role of statins in the modulation of mood and affection. However, a direct correlation between these drugs and the onset of mood disturbances is still controversial. Indeed, MVA pathway end-products and lipoprotein levels seem to be associated with mood disorders, but statin efficacy, as well as the molecular mechanisms underlying their pleiotropic actions are not completely clarified.
7.1. Anxiety
A number of experimental groups independently demonstrated important roles for statins in anxiety behaviors. Chronic statin treatment (10 mg/kg atorvastatin; 10 mg/kg simvastatin; 30 mg/kg pravastatin) induces anxiolytic-like effects in rats tested in the open-field task (OF) [255, 256]. Statin-mediated behavioral changes are related, at least in part, to the modulation of NMDA receptors in different brain regions and by the release of striatal dopamine [255]. Cruz and colleagues assessed an interesting adjuvant effect of classical music on anxiolytic outcomes caused by simvastatin (10 mg/kg) in rats tested in Elevated Plus-Maze (EPM) and OF tasks [257]. Other research corroborates the previous findings, highlighting anxiolytic and antidepressant effects upon simvastatin administration in rats exposed to both standard diet (SD) and long-term high fat diet (HFD). HFD rats show increased anxiety and immobility, accompanied by morphological alterations, such as a reduction in the number of hippocampal pyramidal neurons. Importantly, simvastatin chronic administration determines anxiolytic activity during the EPM test and a normalization of anxiogenic effects induced by HFD [258, 259].
On the contrary, different effects were observed in guinea pigs who had undergone low dose statin treatment. In this preclinical model, simvastatin (1 mg/kg) and atorvastatin (0.5 mg/kg) do not show any effect on spatial memory and learning, but significantly increased anxiety [260]. Similarly, rats chronically treated with low dose of simvastatin (1,5 mg/kg) are less engaged in the active social investigation in the social interaction (SI) test, indicating the onset of anxiogenic behaviors following statin treatment. The effects are associated with a specific unbalance of Rab3 active fraction in the hippocampus and prefrontal cortex, two brain regions deeply involved in the modulation of emotional and anxiety processes [62].
The discrepancy among these preclinical data can be due not only to the different statin dosage used in each experimental design, but also to the different behavioral test used to estimate anxiety-like states. Even though both the EPM and the SI tests are two validated tasks to assess anxiety-like behaviors in rodents [261-263], they are used to evoke different anxiety states in laboratory rodents [264, 265]. The activation of diverse neurosignaling pathways may be at the root of this divergence [266-268], and could explain the different sensitivity to statin treatment in dependence on the test taken into consideration. Notably, the EPM test is able to induce a state of generalized anxiety in rodents, since it triggers an approach/avoidance conflict based on the motivation to explore the environment and an unconditioned fear of novel situations [268]. Instead, in the SI test, the considered parameter is represented by the time spent in active social interaction between a pair of rodents, and one animal influences the behavior of the other one [269]. In this context, statin treatment seems to decrease behaviors induced by generalized anxiety, whereas it is able to elicit social anxiety.
The use of statin as modulators of anxiety appears interesting also when anxiety status is a secondary symptom of another pathology. Chronic fatigue stress (CFS) is associated with mood disorders as anxiety and depression [270] and metabolic stress is probably involved. Atorvastatin (10 and 20 mg/kg) and fluvastatin (5 and 10 mg/kg), show a significant improvement of behavior, biochemical and mitochondrial alterations observed in rats exposed to chronic running wheel [271].
Differently from preclinical models, clinical data fail to identify effects of statin treatment in the modulation of anxiety. A scientific report performed on children with familiar hypercholesterolemia showed that statin administration has no effect on anxiety [272]. A study based on eight Italian regions database, indicated that there is no significant correlation between statin use and psychiatric adverse drug reactions (ADR), even though a strength relationship is found between statins and insomnia [273]. Similarly, no associations were found between pravastatin treatment and the development of psychiatric disorders in a military population [274]. Finally, treatment with rosuvastatin in JUPITER (Justification for the Use of Statins in Prevention) trial demonstrated no correlations with adverse psychiatric events [275].
7.2. Depression
The relationship between statins and depression has been extensively investigated. However, published findings are controversial and far from being univocal.
It has been described that statins reduce the risk of development of de novo depression phase, after 10-year follow up period [276]. Consistently, another study based on the United Kingdom General Practice Research Database (GPRD) demonstrated that statin treatment decreases the risk of depression in depressed patients and patients with suicidal behavior. However, the authors speculate that statin anti-depressant activity could be related to an indirect effect of these drugs, as it can be related to an improvement in quality of life of patients suffering from cardiovascular diseases [277]. Data analysis obtained from the National Health Insurance Research Database (NHIRD) in Taiwan, highlighted increased depression symptoms in hyperlipidemic patients. Notably, these mood disturbances were attenuated after statin treatment during a 4-year follow-up [278]. In accordance, preclinical data suggest an anti-depressant effect of statin treatment during modified forced swimming test (MFST). In particular, simvastatin reduces the immobility time and augments the swimming time in SD and HFD rats [258].
The onset of depression symptoms can be also associated to a traumatic brain injury (TBI), becoming a chronic pathology following hippocampal injury events [279, 280]. Hippocampal injury in humans and preclinical animal models easily leads to the rise of neuroinflammatory markers, such as TNF-α [281, 282] whose activation pathway may be involved in the development of depressive disorders [283, 284]. Simvastatin administration in TBI rats for 3 consecutive days determined a reduction in TNF-α activation and a decrease in depressive-like behaviors [108]. The anti-depressive effects of statins (atorvastatin, simvastatin and pravastatin) were also observed in WAG/Rij rats, a valuable animal model to study absence-type epilepsy, epileptogenesis and low-grade depression [256].
The molecular mechanism by which statins attenuate depressive symptoms could be related to the modulation of serotonergic pathway. Indeed, it is suggested that simvastatin counteracts depression by increasing tryptophan levels (and in turn serotonin) through the inhibition of indoleamine 2,3-dioxygenase (IDO) [285].
Despite these data, negative effects of statins in the treatment of depression have been described. In 1992, Leichleitner and collaborators reported the first case of depressive symptoms associated with paravastin treatment in four hypercholesterolemic patients [286]. Morales and colleagues confirmed these findings demonstrating a relationship between simvastatin treatment and the enhancement of depressive symptoms [287]. Accordingly, simvastatin induced depression in a crossover randomized controlled trial (RCT) [288].
7.3. Schizophrenia
Scientific evidence sustains that people suffering from mental illnesses, like schizophrenia, are more susceptible to develop cardiovascular disease (CVD) [289, 290]. Thus, treatment with hypocholesterolemic drugs like statins attracts interest as a potential therapeutic option for the management of these brain disorders. Schizophrenia is a mood disorder with an unknown etiology characterized, among other features, by a prominent inflammatory component [291-293]. Statin treatment has been proposed as a putative pharmacological approach in Schizophrenia because of its incontrovertible anti-inflammatory effect. A preliminary randomized double-blind placebo-controlled study failed to demonstrate an amelioration in mania symptoms in patients with schizophrenia treated with lovastatin. However, the authors assessed that the pharmacological interactions, the duration of the study and the small cohort may limit the evaluation of potential statin effects in schizophrenia [294]. On the other hand, a study on a military population showed that the risk of developing schizophrenia symptoms is higher in non-persistent statin patients, compared with persistent users [295].
8. Statins and stroke
In addition to their well-documented role in preventing cardiovascular diseases, increasing evidence suggests statin administration as one of the major pharmacological approaches for the treatment and prevention of ischemic stroke (for extensive review, see Zhao et al., 2014) [296].
Several meta-analysis and clinical trials demonstrated that statins positively influence the outcomes in patients affected by stroke. Statin therapy, especially at stroke onset, is associated with consistent beneficial effects [297-299]. Indeed, statin treatment before and during ischemic stroke hospitalization is predictive of improved life expectancy, whereas its discontinuation is associated with a significant poor prognosis [300]. In Long-term Intervention with Pravastatin in Ischaemic Disease (LIPID) study, the incidence of stroke was significantly lower (19% reduction in risk) in patients who received pravastatin [301]. The randomized-controlled Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) trial indicated that atorvastatin was effective in reducing stroke by 50%, as only 12 events were observed in the atorvastatin group if compared to 24 cases in the placebo group [302]. Furthermore, a meta-analysis of 13 trials based on statin administration highlighted a decrease in the relative risk for stroke of 15% in primary prevention, and of 25% in secondary prevention. The relative risk reduction for stroke was comparable with simvastatin and pravastatin [303]. Interestingly, it was shown that both low and high statin doses are effective in ameliorating stroke severity in accordance with the NIH Stroke Scale score [304].
It is suggested that statins successfully counteract the incidence of stroke through cholesterol-independent mechanisms. Indeed, a number of reports failed to demonstrate protection from stroke with lipid-lowering drugs different from statins. It is postulated that a great part of pleiotropic statins’ systemic effects on the vasculature can explain their outcomes in stroke, including improvement of endothelial function, anti-inflammatory activities, induction of angiogenesis, antioxidant properties and anti-thrombotic actions. Several lines of evidence suggest that statins ameliorate endothelial function during stroke by upregulating endothelial nitric oxide synthase (eNOS). Simvastatin treatment reduces infarct size, improves cerebral blood flow and stabilizes neurological function in a mouse model of stroke. Importantly, these effects are absent in eNOS-deficient mice [305]. Statin-induced modulation of eNOS depends, at least in part, on the inhibition of RhoA prenylation, which leads to the activation of PI3K/Akt/eNOS pathway and to the stabilization of eNOS mRNA [306, 307]. A recent study provides evidence for the contribution of neuronal nitric oxide synthase (nNOS) in neuronal apoptosis during stroke. Ischemia/reperfusion suppresses Akt/nNOS pathway, leading to the activation of JNK3 and induction of apoptosis. In this context, atorvastatin pre-treatment prevents the reduction in Akt phosphorylation mediated by ischemic damage and promotes survival in hippocampal neurons [308]. Statin-mediated neuroprotection in stroke may be also exerted by immunomodulatory actions. The release of Matrix MetalloProteinases (MMPs) by astrocytes and microglia are strongly linked to neuroinflammation and BBB disruption [309, 310]. Notably, both simvastatin and atorvastatin efficiently block MMP dysregulations induced by recombinant human tissue Plasminogen Activator (rht-PA) [311, 312]. It is well described that statins suppress the expression of inflammatory biomarkers such as c-rective protein, IL-1, IL-6, IL-12, TNF-α and Interferon gamma (IFN-γ) [296]. In addition, statins inhibit signaling pathways involved in ischemia-induced inflammation by acting on NF-kB transcription factor [313]. Similarly to neuroinflammation, ROS production is thought to participate to ischemic injury by disrupting the structure and/or function of proteins, lipid and nucleic acids. Statin treatment reduces the infarct volume following middle cerebral artery occlusion, and this event is accompanied by a significant decrease in NADPH oxidase-dependent superoxide production [314].
Despite the majority of literature data highlight the involvement of cholesterol-independent actions in the amelioration of stroke outcomes, recent data suggest that statins can be beneficial, at least in part, through their lipid-lowering properties. Indeed, a meta-analysis demonstrated that lower stroke incidence is positively correlated to cholesterol reduction, and that all lipid-lowering interventions are effective in decreasing stroke risk [315]. Even though the molecular mechanisms are not still well established, the role of statins in the prevention of ischemic stroke is well described, and some of them (i.e. atorvastatin) are currently used in the pharmacological management of ischemic cerebrovascular diseases [308]. Furthermore, an ongoing clinical trial is aimed at better defining the beneficial effect of statins on neurological protection in stroke (ASSORT, ClinicalTrials.gov identifier: NCT02549846).
Conclusion
Statins represent an important class of medications commonly prescribed to reduce hypercholesterolemia. The biological activity of statins is achieved by their capability to inhibit HMGR, the key enzyme of MVA pathway. The effect of statins on MVA pathway inhibition has been extensively studied in the liver, where the major part of cholesterologenesis takes place. However, this pathway is ubiquitously activated in almost all eukaryotic cells, and numerous experimental evidences indicate that MVA inhibition by statins may lead to several pharmacological actions also in extra-hepatic tissues. In this context, MVA pathway end-products appear to be abundant in the CNS, and their maintenance at defined concentrations assures the proper functioning of a variety of neurobiological processes.
Several lines of evidence demonstrate that the production of MVA pathway end-products can be altered during different neuropathological states, suggesting that statin use may be useful as a new therapeutic avenue for the treatment of diverse neurological impairments. Clinical and preclinical research indicates that statins ameliorate the symptomatology of a number of brain pathologies, such as SLOS, FXS and autism-like disorders. Additional findings highlight statin efficacy in the treatment of brain tumors, as well as in the therapy of neurodegeneration, and mood disorders and stroke. Despite promising data, other studies failed to demonstrate a neuroprotective effect of statin administration, raising important questions about the concrete pharmacological efficacy of these drugs in counteracting brain pathologies. The discrepancy observed among different studies can be explained by several factors. For instance, preclinical experimental models could not adequately reproduce the human pathology. In other cases, statin dosage and administration route can differently affect the biosynthesis of MVA pathway end-products, thus leading to different biological responses. Concerning clinical studies, a variety of factors such as statin type, duration of the trial, sample size, population heterogeneity and genetic background can contribute to the collection of contrasting experimental outcomes observed in literature.
In conclusion, even though considerable research suggests important neuropharmacological effects exerted by statin administration, additional studies are required to better define the biological activity of statins in the brain, and support their clinical use in the treatment of different neuropathologies.
Acknowledgements
The authors thank Dr Elena Fico (Department of Biotechnological and Applied Clinical Sciences, University of L’Aquila, Italy) and Dr Rachel Price (Department of Systems Medicine, University of Rome “Tor Vergata”, Rome, Italy) for the helpful comments and for the support during the editing of the manuscript.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no conflict of interest, financial or otherwise.
References
- 1.Sirtori C.R. The pharmacology of statins. Pharmacol. Res. 2014;88:3–11. doi: 10.1016/j.phrs.2014.03.002. [http://dx.doi.org/10.1016/j.phrs.2014.03.002]. [DOI] [PubMed] [Google Scholar]
- 2.Segatto M., Leboffe L., Trapani L., Pallottini V. Cholesterol homeostasis failure in the brain: implications for synaptic dysfunction and cognitive decline. Curr. Med. Chem. 2014;21(24):2788–2802. doi: 10.2174/0929867321666140303142902. [http://dx.doi.org/10.2174/0929867321666140303142902]. [DOI] [PubMed] [Google Scholar]
- 3.Trapani L., Segatto M., Pallottini V. Regulation and deregulation of cholesterol homeostasis: The liver as a metabolic “power station”. World J. Hepatol. 2012;4(6):184–190. doi: 10.4254/wjh.v4.i6.184. [http://dx.doi.org/ 10.4254/wjh.v4.i6.184]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Espenshade P.J., Hughes A.L. Regulation of sterol synthesis in eukaryotes. Annu. Rev. Genet. 2007;41:401–427. doi: 10.1146/annurev.genet.41.110306.130315. [http://dx.doi. org/10.1146/annurev.genet.41.110306.130315]. [DOI] [PubMed] [Google Scholar]
- 5.Marino M., di Masi A., Trezza V., Pallottini V., Polticelli F., Ascenzi P. Xenosensors CAR and PXR at work: impact on statin metabolism. Curr. Drug Metab. 2011;12(3):300–311. doi: 10.2174/138920011795101859. [http://dx. doi.org/10.2174/138920011795101859]. [DOI] [PubMed] [Google Scholar]
- 6.Corsini A., Maggi F.M., Catapano A.L. Pharmacology of competitive inhibitors of HMG-CoA reductase. Pharmacol. Res. 1995;31(1):9–27. doi: 10.1016/1043-6618(95)80042-5. [http://dx.doi.org/10.1016/1043-6618(95)80042-5]. [DOI] [PubMed] [Google Scholar]
- 7.Neuvonen P.J. Drug interactions with HMG-CoA reductase inhibitors (statins): the importance of CYP enzymes, transporters and pharmacogenetics. Curr. Opin. Investig. Drugs. 2010;11(3):323–332. [PubMed] [Google Scholar]
- 8.Neuvonen P.J., Niemi M., Backman J.T. Drug interactions with lipid-lowering drugs: mechanisms and clinical relevance. Clin. Pharmacol. Ther. 2006;80(6):565–581. doi: 10.1016/j.clpt.2006.09.003. [http://dx.doi.org/10.1016/ j.clpt.2006.09.003]. [DOI] [PubMed] [Google Scholar]
- 9.Lennernas H., Fager G. Pharmacodynamics and pharmacokinetics of the HMG-CoA reductase inhibitors. Similarities and differences. Clin. Pharmacokinet. 1997;32(5):403–425. doi: 10.2165/00003088-199732050-00005. [http://dx.doi.org/10. 2165/00003088-199732050-00005]. [DOI] [PubMed] [Google Scholar]
- 10.Williams D., Feely J. Pharmacokinetic-pharmacodynamic drug interactions with HMG-CoA reductase inhibitors. Clin. Pharmacokinet. 2002;41(5):343–370. doi: 10.2165/00003088-200241050-00003. [http://dx.doi.org/10.2165/00003088-200241050-00003]. [DOI] [PubMed] [Google Scholar]
- 11.Christians U., Jacobsen W., Floren L.C. Metabolism and drug interactions of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors in transplant patients: are the statins mechanistically similar? Pharmacol. Ther. 1998;80(1):1–34. doi: 10.1016/s0163-7258(98)00016-3. [http://dx.doi.org/10. 1016/S0163-7258(98)00016-3]. [DOI] [PubMed] [Google Scholar]
- 12.Quion J.A., Jones P.H. Clinical pharmacokinetics of pravastatin. Clin. Pharmacokinet. 1994;27(2):94–103. doi: 10.2165/00003088-199427020-00002. [http://dx.doi.org/10. 2165/00003088-199427020-00002]. [DOI] [PubMed] [Google Scholar]
- 13.Hamelin B.A., Turgeon J. Hydrophilicity/lipophilicity: relevance for the pharmacology and clinical effects of HMG-CoA reductase inhibitors. Trends Pharmacol. Sci. 1998;19(1):26–37. doi: 10.1016/s0165-6147(97)01147-4. [http://dx. doi.org/10.1016/S0165-6147(97)01147-4]. [DOI] [PubMed] [Google Scholar]
- 14.Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam. Clin. Pharmacol. 2005;19(1):117–125. doi: 10.1111/j.1472-8206.2004.00299.x. [http://dx.doi.org/10.1111/j.1472-8206.2004.00299.x]. [DOI] [PubMed] [Google Scholar]
- 15.Kivisto K.T., Niemi M. Influence of drug transporter polymorphisms on pravastatin pharmacokinetics in humans. Pharm. Res. 2007;24(2):239–247. doi: 10.1007/s11095-006-9159-2. [http://dx.doi.org/10.1007/s11095-006-9159-2]. [DOI] [PubMed] [Google Scholar]
- 16.Kitamura S., Maeda K., Wang Y., Sugiyama Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab. Dispos. 2008;36(10):2014–2023. doi: 10.1124/dmd.108.021410. [http://dx.doi.org/ 10.1124/dmd.108.021410]. [DOI] [PubMed] [Google Scholar]
- 17.Trapani L., Melli L., Segatto M., Trezza V., Campolongo P., Jozwiak A., Swiezewska E., Pucillo L.P., Moreno S., Fanelli F., Linari M., Pallottini V. Effects of myosin heavy chain (MHC) plasticity induced by HMGCoA-reductase inhibition on skeletal muscle functions. FASEB J. 2011;25(11):4037–4047. doi: 10.1096/fj.11-184218. [http://dx. doi.org/10.1096/fj.11-184218]. [DOI] [PubMed] [Google Scholar]
- 18.Trapani L., Segatto M., Ascenzi P., Pallottini V. Potential role of nonstatin cholesterol lowering agents. IUBMB Life. 2011;63(11):964–971. doi: 10.1002/iub.522. [http://dx.doi.org/10.1002/iub.522]. [DOI] [PubMed] [Google Scholar]
- 19.Trapani L., Segatto M., La Rosa P., Fanelli F., Moreno S., Marino M., Pallottini V. 3-hydroxy 3-methylglutaryl coenzyme A reductase inhibition impairs muscle regeneration. J. Cell. Biochem. 2012;113(6):2057–2063. doi: 10.1002/jcb.24077. [http://dx.doi.org/10.1002/jcb.24077]. [DOI] [PubMed] [Google Scholar]
- 20.Tonelli M., Moye L., Sacks F.M., Cole T., Curhan G.C. Effect of pravastatin on loss of renal function in people with moderate chronic renal insufficiency and cardiovascular disease. J. Am. Soc. Nephrol. 2003;14(6):1605–1613. doi: 10.1097/01.asn.0000068461.45784.2f. [http://dx.doi.org/10.1097/01. ASN.0000068461.45784.2F]. [DOI] [PubMed] [Google Scholar]
- 21.Vidt D.G., Cressman M.D., Harris S., Pears J.S., Hutchinson H.G. Rosuvastatin-induced arrest in progression of renal disease. Cardiology. 2004;102(1):52–60. doi: 10.1159/000077704. [http://dx.doi.org/10.1159/ 000077704]. [DOI] [PubMed] [Google Scholar]
- 22.Chopra V., Rogers M.A., Buist M., Govindan S., Lindenauer P.K., Saint S., Flanders S.A. Is statin use associated with reduced mortality after pneumonia? A systematic review and meta-analysis. Am. J. Med. 2012;125(11):1111–1123. doi: 10.1016/j.amjmed.2012.04.011. [http://dx.doi.org/10.1016/ j.amjmed.2012.04.011]. [DOI] [PubMed] [Google Scholar]
- 23.Novack V., MacFadyen J., Malhotra A., Almog Y., Glynn R.J., Ridker P.M. The effect of rosuvastatin on incident pneumonia: results from the JUPITER trial. CMAJ. 2012;184(7):E367–E372. doi: 10.1503/cmaj.111017. [http://dx.doi.org/10.1503/cmaj.111017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chataway J., Schuerer N., Alsanousi A., Chan D., MacManus D., Hunter K., Anderson V., Bangham C.R., Clegg S., Nielsen C., Fox N.C., Wilkie D., Nicholas J.M., Calder V.L., Greenwood J., Frost C., Nicholas R. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet. 2014;383(9936):2213–2221. doi: 10.1016/S0140-6736(13)62242-4. [http://dx.doi. org/10.1016/S0140-6736(13)62242-4]. [DOI] [PubMed] [Google Scholar]
- 25.Preiss D., Tikkanen M.J., Welsh P., Ford I., Lovato L.C., Elam M.B., LaRosa J.C., DeMicco D.A., Colhoun H.M., Goldenberg I., Murphy M.J., MacDonald T.M., Pedersen T.R., Keech A.C., Ridker P.M., Kjekshus J., Sattar N., McMurray J.J. Lipid-modifying therapies and risk of pancreatitis: a meta-analysis. JAMA. 2012;308(8):804–811. doi: 10.1001/jama.2012.8439. [http://dx.doi.org/10.1001/jama. 2012.8439]. [DOI] [PubMed] [Google Scholar]
- 26.El-Sisi A.A., Hegazy S.K., Salem K.A. AbdElkawy, K. S. Atorvastatin improves erectile dysfunction in patients initially irresponsive to Sildenafil by the activation of endothelial nitric oxide synthase. Int. J. Impot. Res. 2013;25(4):143–148. doi: 10.1038/ijir.2012.46. [http://dx.doi. org/10.1038/ijir.2012.46]. [DOI] [PubMed] [Google Scholar]
- 27.Subramanian S., Emami H., Vucic E., Singh P., Vijayakumar J., Fifer K.M., Alon A., Shankar S.S., Farkouh M., Rudd J.H., Fayad Z.A., Van Dyke T.E., Tawakol A. High-dose atorvastatin reduces periodontal inflammation: a novel pleiotropic effect of statins. J. Am. Coll. Cardiol. 2013;62(25):2382–2391. doi: 10.1016/j.jacc.2013.08.1627. [http://dx.doi.org/10.1016/j.jacc.2013.08.1627]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cartocci V., Servadio M., Trezza V., Pallottini V. Can cholesterol metabolism modulation affect brain function and behavior? J. Cell. Physiol. 2017;232(2):281–286. doi: 10.1002/jcp.25488. [http://dx.doi.org/10.1002/ jcp.25488]. [DOI] [PubMed] [Google Scholar]
- 29.Horton J.D. Sterol regulatory element-binding proteins: transcriptional activators of lipid synthesis. Biochem. Soc. Trans. 2002;30(Pt 6):1091–1095. doi: 10.1042/bst0301091. [http://dx.doi.org/10.1042/bst0301091]. [DOI] [PubMed] [Google Scholar]
- 30.Segatto M., Trapani L., Marino M., Pallottini V. Age- and sex-related differences in extra-hepatic low-density lipoprotein receptor. J. Cell. Physiol. 2011;226(10):2610–2616. doi: 10.1002/jcp.22607. [http://dx.doi.org/ 10.1002/jcp.22607]. [DOI] [PubMed] [Google Scholar]
- 31.Segatto M., Trapani L., Lecis C., Pallottini V. Regulation of cholesterol biosynthetic pathway in different regions of the rat central nervous system. Acta Physiol. (Oxf.) 2012;206(1):62–71. doi: 10.1111/j.1748-1716.2012.02450.x. [http://dx.doi.org/10.1111/j.1748-1716.2012.02450.x]. [DOI] [PubMed] [Google Scholar]
- 32.Pfrieger F.W. Role of cholesterol in synapse formation and function. Biochim. Biophys. Acta. 2003;1610(2):271–280. doi: 10.1016/s0005-2736(03)00024-5. [http://dx. doi.org/10.1016/S0005-2736(03)00024-5]. [DOI] [PubMed] [Google Scholar]
- 33.Bjorkhem I., Meaney S. Brain cholesterol: long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 2004;24(5):806–815. doi: 10.1161/01.ATV.0000120374.59826.1b. [http://dx.doi.org/10.1161/01.ATV.0000120374.59826.1b]. [DOI] [PubMed] [Google Scholar]
- 34.Snipes G.J., Orfali W. Common themes in peripheral neuropathy disease genes. Cell Biol. Int. 1998;22(11-12):815–835. doi: 10.1006/cbir.1998.0389. [http://dx. doi.org/10.1006/cbir.1998.0389]. [DOI] [PubMed] [Google Scholar]
- 35.Goritz C., Mauch D.H., Pfrieger F.W. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol. Cell. Neurosci. 2005;29(2):190–201. doi: 10.1016/j.mcn.2005.02.006. [http://dx.doi.org/10.1016/ j.mcn.2005.02.006]. [DOI] [PubMed] [Google Scholar]
- 36.Pfrieger F.W. Outsourcing in the brain: do neurons depend on cholesterol delivery by astrocytes? BioEssays. 2003;25(1):72–78. doi: 10.1002/bies.10195. [http://dx.doi.org/10.1002/bies.10195]. [DOI] [PubMed] [Google Scholar]
- 37.Shanmugaratnam J., Berg E., Kimerer L., Johnson R.J., Amaratunga A., Schreiber B.M., Fine R.E. Retinal muller glia secrete apolipoproteins E and J which are efficiently assembled into lipoprotein particles. Brain Res. Mol. Brain Res. 1997;50(1-2):113–120. doi: 10.1016/s0169-328x(97)00176-9. [http://dx.doi.org/10.1016/S0169-328X(97)00176-9]. [DOI] [PubMed] [Google Scholar]
- 38.Oram J.F., Heinecke J.W. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol. Rev. 2005;85(4):1343–1372. doi: 10.1152/physrev.00005.2005. [http://dx.doi.org/ 10.1152/physrev.00005.2005]. [DOI] [PubMed] [Google Scholar]
- 39.Segatto M., Di Giovanni A., Marino M., Pallottini V. Analysis of the protein network of cholesterol homeostasis in different brain regions: an age and sex dependent perspective. J. Cell. Physiol. 2013;228(7):1561–1567. doi: 10.1002/jcp.24315. [http://dx.doi.org/10.1002/jcp.24315]. [DOI] [PubMed] [Google Scholar]
- 40.Ohvo-Rekila H., Ramstedt B., Leppimaki P., Slotte J.P. Cholesterol interactions with phospholipids in membranes. Prog. Lipid Res. 2002;41(1):66–97. doi: 10.1016/s0163-7827(01)00020-0. [http://dx.doi.org/10.1016/S0163-7827 (01)00020-0]. [DOI] [PubMed] [Google Scholar]
- 41.Saher G., Brugger B., Lappe-Siefke C., Mobius W., Tozawa R., Wehr M.C., Wieland F., Ishibashi S., Nave K.A. High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 2005;8(4):468–475. doi: 10.1038/nn1426. [http://dx.doi.org/10.1038/nn1426]. [DOI] [PubMed] [Google Scholar]
- 42.Haines T.H. Do sterols reduce proton and sodium leaks through lipid bilayers? Prog. Lipid Res. 2001;40(4):299–324. doi: 10.1016/s0163-7827(01)00009-1. [http://dx. doi.org/10.1016/S0163-7827(01)00009-1]. [DOI] [PubMed] [Google Scholar]
- 43.Deutsch J.W., Kelly R.B. Lipids of synaptic vesicles: relevance to the mechanism of membrane fusion. Biochemistry. 1981;20(2):378–385. doi: 10.1021/bi00505a024. [http://dx.doi.org/10.1021/bi00505a024]. [DOI] [PubMed] [Google Scholar]
- 44.Huttner W.B., Zimmerberg J. Implications of lipid microdomains for membrane curvature, budding and fission. Curr. Opin. Cell Biol. 2001;13(4):478–484. doi: 10.1016/s0955-0674(00)00239-8. [http://dx.doi.org/10.1016/S0955-0674 (00)00239-8]. [DOI] [PubMed] [Google Scholar]
- 45.Mitter D., Reisinger C., Hinz B., Hollmann S., Yelamanchili S.V., Treiber-Held S., Ohm T.G., Herrmann A., Ahnert-Hilger G. The synaptophysin/synaptobrevin interaction critically depends on the cholesterol content. J. Neurochem. 2003;84(1):35–42. doi: 10.1046/j.1471-4159.2003.01258.x. [http://dx.doi.org/10.1046/j.1471-4159.2003.01258.x]. [DOI] [PubMed] [Google Scholar]
- 46.Sooksawate T., Simmonds M.A. Effects of membrane cholesterol on the sensitivity of the GABA(A) receptor to GABA in acutely dissociated rat hippocampal neurones. Neuropharmacology. 2001;40(2):178–184. doi: 10.1016/s0028-3908(00)00159-3. [http://dx.doi.org/10.1016/S0028-3908(00)00159-3]. [DOI] [PubMed] [Google Scholar]
- 47.Hering H., Lin C.C., Sheng M. Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J. Neurosci. 2003;23(8):3262–3271. doi: 10.1523/JNEUROSCI.23-08-03262.2003. [http://dx.doi.org/10.1523/ JNEUROSCI.23-08-03262.2003]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zipp F., Waiczies S., Aktas O., Neuhaus O., Hemmer B., Schraven B., Nitsch R., Hartung H.P. Impact of HMG-CoA reductase inhibition on brain pathology. Trends Pharmacol. Sci. 2007;28(7):342–349. doi: 10.1016/j.tips.2007.05.001. [http://dx.doi.org/10.1016/j.tips.2007.05.001]. [DOI] [PubMed] [Google Scholar]
- 49.Lecis C., Segatto M. Cholesterol Homeostasis Imbalance and Brain Functioning: Neuro-logical Disorders and Behavioral Consequences. J. Neurol. Neurol. Disord. 2014;1(1):101. [Google Scholar]
- 50.Buchovecky C.M., Turley S.D., Brown H.M., Kyle S.M., McDonald J.G., Liu B., Pieper A.A., Huang W., Katz D.M., Russell D.W., Shendure J., Justice M.J. A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat. Genet. 2013;45(9):1013–1020. doi: 10.1038/ng.2714. [http://dx.doi.org/10. 1038/ng.2714]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kyle S.M., Saha P.K., Brown H.M., Chan L.C., Justice M.J. MeCP2 co-ordinates liver lipid metabolism with the NCoR1/ HDAC3 corepressor complex. Hum. Mol. Genet. 2016;25(14):3029–3041. doi: 10.1093/hmg/ddw156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lopez A.M., Chuang J.C., Posey K.S., Turley S.D. ‘Corrigenda to “Suppression of brain cholesterol synthesis in male Mecp2-deficient mice is age dependent and not accompanied by a concurrent change in the rate of fatty acid synthesis” [Brain Res. 1654 (2017) 77-84]’. Brain Res. 2017;1657:383. doi: 10.1016/j.brainres.2016.10.021. [http://dx.doi.org/10. 1016/j.brainres.2016.12.016]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sticozzi C., Belmonte G., Pecorelli A., Cervellati F., Leoncini S., Signorini C., Ciccoli L., De Felice C., Hayek J., Valacchi G. Scavenger receptor B1 post-translational modifications in Rett syndrome. FEBS Lett. 2013;587(14):2199–2204. doi: 10.1016/j.febslet.2013.05.042. [http://dx.doi.org/ 10.1016/j.febslet.2013.05.042]. [DOI] [PubMed] [Google Scholar]
- 54.Segatto M., Trapani L., Di Tunno I., Sticozzi C., Valacchi G., Hayek J., Pallottini V. Cholesterol metabolism is altered in Rett syndrome: a study on plasma and primary cultured fibroblasts derived from patients. PLoS One. 2014;9(8):e104834. doi: 10.1371/journal.pone.0104834. [http://dx. doi.org/10.1371/journal.pone.0104834]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pecorelli A., Belmonte G., Meloni I., Cervellati F., Gardi C., Sticozzi C., De Felice C., Signorini C., Cortelazzo A., Leoncini S., Ciccoli L., Renieri A., Jay Forman H., Hayek J., Valacchi G. Alteration of serum lipid profile, SRB1 loss, and impaired Nrf2 activation in CDKL5 disorder. Free Radic. Biol. Med. 2015;86:156–165. doi: 10.1016/j.freeradbiomed.2015.05.010. [http://dx.doi.org/10.1016/j.freeradbiomed.2015.05.010]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matthews R.T., Yang L., Browne S., Baik M., Beal M.F. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. USA. 1998;95(15):8892–8897. doi: 10.1073/pnas.95.15.8892. [http://dx.doi.org/10.1073/pnas.95. 15.8892]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Young A.J., Johnson S., Steffens D.C., Doraiswamy P.M. Coenzyme Q10: a review of its promise as a neuroprotectant. CNS Spectr. 2007;12(1):62–68. doi: 10.1017/s1092852900020538. [http://dx.doi.org/10.1017/S10928529 00020538]. [DOI] [PubMed] [Google Scholar]
- 58.Quinzii C.M., Hirano M. Coenzyme Q and mitochondrial disease. Dev. Disabil. Res. Rev. 2010;16(2):183–188. doi: 10.1002/ddrr.108. [http://dx.doi.org/10. 1002/ddrr.108]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barca E., Kleiner G., Tang G., Ziosi M., Tadesse S., Masliah E., Louis E.D., Faust P., Kang U.J., Torres J., Cortes E.P., Vonsattel J.P., Kuo S.H., Quinzii C.M. Decreased coenzyme Q10 levels in multiple system atrophy cerebellum. J. Neuropathol. Exp. Neurol. 2016;75(7):663–672. doi: 10.1093/jnen/nlw037. [http://dx.doi.org/10.1093/ jnen/nlw037]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schottlaender L.V., Bettencourt C., Kiely A.P., Chalasani A., Neergheen V., Holton J.L., Hargreaves I., Houlden H. Coenzyme Q10 levels are decreased in the cerebellum of multiple-system atrophy patients. PLoS One. 2016;11(2):e0149557. doi: 10.1371/journal.pone.0149557. [http://dx.doi.org/10.1371/journal.pone.0149557]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stefely J.A., Licitra F., Laredj L., Reidenbach A.G., Kemmerer Z.A., Grangeray A., Jaeg-Ehret T., Minogue C.E., Ulbrich A., Hutchins P.D., Wilkerson E.M., Ruan Z., Aydin D., Hebert A.S., Guo X., Freiberger E.C., Reutenauer L., Jochem A., Chergova M., Johnson I.E., Lohman D.C., Rush M.J., Kwiecien N.W., Singh P.K., Schlagowski A.I., Floyd B.J., Forsman U., Sindelar P.J., Westphall M.S., Pierrel F., Zoll J., Dal Peraro M., Kannan N., Bingman C.A., Coon J.J., Isope P., Puccio H., Pagliarini D.J. Cerebellar ataxia and coenzyme Q deficiency through loss of unorthodox kinase activity. Mol. Cell. 2016;63(4):608–620. doi: 10.1016/j.molcel.2016.06.030. [http://dx.doi.org/10.1016/j.molcel.2016.06.030]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Segatto M., Manduca A., Lecis C., Rosso P., Jozwiak A., Swiezewska E., Moreno S., Trezza V., Pallottini V. Simvastatin treatment highlights a new role for the isoprenoid/cholesterol biosynthetic pathway in the modulation of emotional reactivity and cognitive performance in rats. Neuropsychopharmacology. 2014;39(4):841–854. doi: 10.1038/npp.2013.284. [http://dx.doi.org/10.1038/npp.2013.284]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mazzucchelli C., Brambilla R. Ras-related and MAPK signalling in neuronal plasticity and memory formation. Cell. Mol. Life Sci. 2000;57(4):604–611. doi: 10.1007/PL00000722. [http://dx.doi.org/10.1007/PL00000722]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bender R.H., Haigis K.M., Gutmann D.H. Activated k-ras, but not h-ras or N-ras, regulates brain neural stem cell proliferation in a raf/rb-dependent manner. Stem Cells. 2015;33(6):1998–2010. doi: 10.1002/stem.1990. [http://dx.doi.org/10.1002/stem.1990]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park J.C., Jeong W.J., Kim M.Y., Min D., Choi K.Y. Retinoic-acid-mediated HRas stabilization induces neuronal differentiation of neural stem cells during brain development. J. Cell Sci. 2016;129(15):2997–3007. doi: 10.1242/jcs.184366. [http://dx.doi.org/10.1242/jcs.184366]. [DOI] [PubMed] [Google Scholar]
- 66.Tidyman W.E., Rauen K.A. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 2009;19(3):230–236. doi: 10.1016/j.gde.2009.04.001. [http://dx.doi.org/10.1016/j.gde. 2009.04.001]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lingor P., Teusch N., Schwarz K., Mueller R., Mack H., Bahr M., Mueller B.K. Inhibition of Rho kinase (ROCK) increases neurite outgrowth on chondroitin sulphate proteoglycan in vitro and axonal regeneration in the adult optic nerve in vivo. J. Neurochem. 2007;103(1):181–189. doi: 10.1111/j.1471-4159.2007.04756.x. [DOI] [PubMed] [Google Scholar]
- 68.Cartocci V., Segatto M., Di Tunno I., Leone S., Pfrieger F.W., Pallottini V. Modulation of the isoprenoid/cholesterol biosynthetic pathway during neuronal differentiation In Vitro. J. Cell. Biochem. 2016;117(9):2036–2044. doi: 10.1002/jcb.25500. [http://dx.doi.org/10.1002/jcb.25500]. [DOI] [PubMed] [Google Scholar]
- 69.Ramakers G.J. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 2002;25(4):191–199. doi: 10.1016/s0166-2236(00)02118-4. [http://dx.doi.org/10.1016/S0166-2236(00)02118-4]. [DOI] [PubMed] [Google Scholar]
- 70.Lin G.N., Corominas R., Lemmens I., Yang X., Tavernier J., Hill D.E., Vidal M., Sebat J., Iakoucheva L.M. Spatiotemporal 16p11.2 protein network implicates cortical late mid-fetal brain development and KCTD13-Cul3-RhoA pathway in psychiatric diseases. Neuron. 2015;85(4):742–754. doi: 10.1016/j.neuron.2015.01.010. [http://dx.doi.org/10.1016/ j.neuron.2015.01.010]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Narayanan K.L., Chopra V., Rosas H.D., Malarick K., Hersch S. Rho kinase pathway alterations in the brain and leukocytes in Huntington’s Disease. Mol. Neurobiol. 2016;53(4):2132–2140. doi: 10.1007/s12035-015-9147-9. [http://dx.doi.org/10.1007/s12035-015-9147-9]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Henderson B.W., Gentry E.G., Rush T., Troncoso J.C., Thambisetty M., Montine T.J., Herskowitz J.H. Rho-associated protein kinase 1 (ROCK1) is increased in Alzheimer’s disease and ROCK1 depletion reduces amyloid-beta levels in brain. J. Neurochem. 2016;138(4):525–531. doi: 10.1111/jnc.13688. [http://dx.doi.org/10.1111/jnc.13688]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takai Y., Sasaki T., Matozaki T. Small GTP-binding proteins. Physiol. Rev. 2001;81(1):153–208. doi: 10.1152/physrev.2001.81.1.153. [http://dx.doi.org/10.1152/physrev. 2001.81.1.153]. [DOI] [PubMed] [Google Scholar]
- 74.Geppert M., Sudhof T.C. RAB3 and synaptotagmin: the yin and yang of synaptic membrane fusion. Annu. Rev. Neurosci. 1998;21:75–95. doi: 10.1146/annurev.neuro.21.1.75. [http://dx.doi.org/10.1146/annurev.neuro.21.1.75]. [DOI] [PubMed] [Google Scholar]
- 75.Cheng Y., Wang J., Wang Y., Ding M. Synaptotagmin 1 directs repetitive release by coupling vesicle exocytosis to the Rab3 cycle. eLife. 2015;4:1–19. doi: 10.7554/eLife.05118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Andriamampandry C., Muller C., Schmidt-Mutter C., Gobaille S., Spedding M., Aunis D., Maitre M. Mss4 gene is up-regulated in rat brain after chronic treatment with antidepressant and down-regulated when rats are anhedonic. Mol. Pharmacol. 2002;62(6):1332–1338. doi: 10.1124/mol.62.6.1332. [http://dx.doi.org/10.1124/mol.62.6.1332]. [DOI] [PubMed] [Google Scholar]
- 77.Baskys A., Bayazitov I., Zhu E., Fang L., Wang R. Rab-mediated endocytosis: linking neurodegeneration, neuroprotection, and synaptic plasticity? Ann. N. Y. Acad. Sci. 2007;1122:313–329. doi: 10.1196/annals.1403.023. [http://dx.doi.org/10.1196/annals.1403.023]. [DOI] [PubMed] [Google Scholar]
- 78.Blaveri E., Kelly F., Mallei A., Harris K., Taylor A., Reid J., Razzoli M., Carboni L., Piubelli C., Musazzi L., Racagni G., Mathe A., Popoli M., Domenici E., Bates S. Expression profiling of a genetic animal model of depression reveals novel molecular pathways underlying depressive-like behaviours. PLoS One. 2010;5(9):e12596. doi: 10.1371/journal.pone.0012596. [http://dx.doi.org/10.1371/journal.pone.0012596]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dalfo E., Barrachina M., Rosa J.L., Ambrosio S., Ferrer I. Abnormal alpha-synuclein interactions with rab3a and rabphilin in diffuse Lewy body disease. Neurobiol. Dis. 2004;16(1):92–97. doi: 10.1016/j.nbd.2004.01.001. [http://dx.doi.org/10.1016/j.nbd.2004.01.001]. [DOI] [PubMed] [Google Scholar]
- 80.Aligianis I.A., Johnson C.A., Gissen P., Chen D., Hampshire D., Hoffmann K., Maina E.N., Morgan N.V., Tee L., Morton J., Ainsworth J.R., Horn D., Rosser E., Cole T.R., Stolte-Dijkstra I., Fieggen K., Clayton-Smith J., Megarbane A., Shield J.P., Newbury-Ecob R., Dobyns W.B., Graham J.M., Jr, Kjaer K.W., Warburg M., Bond J., Trembath R.C., Harris L.W., Takai Y., Mundlos S., Tannahill D., Woods C.G., Maher E.R. Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat. Genet. 2005;37(3):221–223. doi: 10.1038/ng1517. [http://dx.doi.org/10. 1038/ng1517]. [DOI] [PubMed] [Google Scholar]
- 81.Aligianis I.A., Morgan N.V., Mione M., Johnson C.A., Rosser E., Hennekam R.C., Adams G., Trembath R.C., Pilz D.T., Stoodley N., Moore A.T., Wilson S., Maher E.R. Mutation in Rab3 GTPase-activating protein (RAB3GAP) noncatalytic subunit in a kindred with Martsolf syndrome. Am. J. Hum. Genet. 2006;78(4):702–707. doi: 10.1086/502681. [http://dx.doi.org/10.1086/502681]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.D’Adamo P., Menegon A., Lo Nigro C., Grasso M., Gulisano M., Tamanini F., Bienvenu T., Gedeon A.K., Oostra B., Wu S.K., Tandon A., Valtorta F., Balch W.E., Chelly J., Toniolo D. Mutations in GDI1 are responsible for X-linked non-specific mental retardation. Nat. Genet. 1998;19(2):134–139. doi: 10.1038/487. [http://dx.doi. org/10.1038/487]. [DOI] [PubMed] [Google Scholar]
- 83.Morava E., Wevers R.A., Cantagrel V., Hoefsloot L.H., Al-Gazali L., Schoots J., van Rooij A., Huijben K., van Ravenswaaij-Arts C.M., Jongmans M.C., Sykut-Cegielska J., Hoffmann G.F., Bluemel P., Adamowicz M., van Reeuwijk J., Ng B.G., Bergman J.E., van Bokhoven H., Korner C., Babovic-Vuksanovic D., Willemsen M.A., Gleeson J.G., Lehle L., de Brouwer A.P., Lefeber D.J. A novel cerebello-ocular syndrome with abnormal glycosylation due to abnormalities in dolichol metabolism. Brain. 2011;133(11):3210–3220. doi: 10.1093/brain/awq261. [http://dx.doi.org/ 10.1093/brain/awq261]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Johnson-Anuna L.N., Eckert G.P., Keller J.H., Igbavboa U., Franke C., Fechner T., Schubert-Zsilavecz M., Karas M., Muller W.E., Wood W.G. Chronic administration of statins alters multiple gene expression patterns in mouse cerebral cortex. J. Pharmacol. Exp. Ther. 2005;312(2):786–793. doi: 10.1124/jpet.104.075028. [http://dx.doi.org/ 10.1124/jpet.104.075028]. [DOI] [PubMed] [Google Scholar]
- 85.Hsiang B., Zhu Y., Wang Z., Wu Y., Sasseville V., Yang W.P., Kirchgessner T.G. A novel human hepatic organic anion transporting polypeptide (OATP2). Identification of a liver-specific human organic anion transporting polypeptide and identification of rat and human hydroxymethylglutaryl-CoA reductase inhibitor transporters. J. Biol. Chem. 1999;274(52):37161–37168. doi: 10.1074/jbc.274.52.37161. [http://dx.doi.org/ 10.1074/jbc.274.52.37161]. [DOI] [PubMed] [Google Scholar]
- 86.Lee G., Dallas S., Hong M., Bendayan R. Drug transporters in the central nervous system: brain barriers and brain parenchyma considerations. Pharmacol. Rev. 2001;53(4):569–596. [http://dx. doi.org/10.1146/annurev.pharmtox.41.1.569]. [PubMed] [Google Scholar]
- 87.Nagasawa K., Nagai K., Sumitani Y., Moriya Y., Muraki Y., Takara K., Ohnishi N., Yokoyama T., Fujimoto S. Monocarboxylate transporter mediates uptake of lovastatin acid in rat cultured mesangial cells. J. Pharm. Sci. 2002;91(12):2605–2613. doi: 10.1002/jps.10246. [http://dx.doi.org/10.1002/jps.10246]. [DOI] [PubMed] [Google Scholar]
- 88.Vijay N., Morris M.E. Role of monocarboxylate transporters in drug delivery to the brain. Curr. Pharm. Des. 2014;20(10):1487–1498. doi: 10.2174/13816128113199990462. [http://dx.doi.org/10.2174/13816128113199990462]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tsuji A., Saheki A., Tamai I., Terasaki T. Transport mechanism of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors at the blood-brain barrier. J. Pharmacol. Exp. Ther. 1993;267(3):1085–1090. [PubMed] [Google Scholar]
- 90.Pierre K., Pellerin L. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J. Neurochem. 2005;94(1):1–14. doi: 10.1111/j.1471-4159.2005.03168.x. [http://dx.doi.org/10.1111/j.1471-4159. 2005.03168.x]. [DOI] [PubMed] [Google Scholar]
- 91.Lutjohann D., Stroick M., Bertsch T., Kuhl S., Lindenthal B., Thelen K., Andersson U., Bjorkhem I., Bergmann Kv K., Fassbender K. High doses of simvastatin, pravastatin, and cholesterol reduce brain cholesterol synthesis in guinea pigs. Steroids. 2004;69(6):431–438. doi: 10.1016/j.steroids.2004.03.012. [http://dx.doi.org/10.1016/j.steroids.2004.03.012]. [DOI] [PubMed] [Google Scholar]
- 92.Ostrowski S.M., Johnson K., Siefert M., Shank S., Sironi L., Wolozin B., Landreth G.E., Ziady A.G. Simvastatin inhibits protein isoprenylation in the brain. Neuroscience. 2016;329:264–274. doi: 10.1016/j.neuroscience.2016.04.053. [http://dx.doi.org/10.1016/j.neuroscience.2016.04.053]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nothdurfter C., Tanasic S., Di Benedetto B., Rammes G., Wagner E.M., Kirmeier T., Ganal V., Kessler J.S., Rein T., Holsboer F., Rupprecht R. Impact of lipid raft integrity on 5-HT3 receptor function and its modulation by antidepressants. Neuropsychopharmacology. 2010;35(7):1510–1519. doi: 10.1038/npp.2010.20. [http://dx.doi.org/10. 1038/npp.2010.20]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shrivastava S., Pucadyil T.J., Paila Y.D., Ganguly S., Chattopadhyay A. Chronic cholesterol depletion using statin impairs the function and dynamics of human serotonin(1A) receptors. Biochemistry. 2010;49(26):5426–5435. doi: 10.1021/bi100276b. [http://dx.doi.org/10.1021/ bi100276b]. [DOI] [PubMed] [Google Scholar]
- 95.Ponce J., de la Ossa N.P., Hurtado O., Millan M., Arenillas J.F., Davalos A., Gasull T. Simvastatin reduces the association of NMDA receptors to lipid rafts: a cholesterol-mediated effect in neuroprotection. Stroke. 2008;39(4):1269–1275. doi: 10.1161/STROKEAHA.107.498923. [http://dx.doi.org/ 10.1161/STROKEAHA.107.498923]. [DOI] [PubMed] [Google Scholar]
- 96.Tong X.K., Lecrux C., Rosa-Neto P., Hamel E. Age-dependent rescue by simvastatin of Alzheimer’s disease cerebrovascular and memory deficits. J. Neurosci. 2012;32(14):4705–4715. doi: 10.1523/JNEUROSCI.0169-12.2012. [http://dx. doi.org/10.1523/JNEUROSCI.0169-12.2012]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mans R.A., McMahon L.L., Li L. Simvastatin-mediated enhancement of long-term potentiation is driven by farnesyl-pyrophosphate depletion and inhibition of farnesylation. Neuroscience. 2012;202:1–9. doi: 10.1016/j.neuroscience.2011.12.007. [http://dx.doi.org/10.1016/j.neuroscience. 2011.12.007]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Roy A., Jana M., Kundu M., Corbett G.T., Rangaswamy S.B., Mishra R.K., Luan C.H., Gonzalez F.J., Pahan K. HMG-CoA reductasei bind to PPARalpha to upregulate neurotrophin Expression in the brain and improve memory in mice. Cell Metab. 2015;22(2):253–265. doi: 10.1016/j.cmet.2015.05.022. [http://dx.doi.org/10.1016/j.cmet.2015.05.022]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lu D., Qu C., Goussev A., Jiang H., Lu C., Schallert T., Mahmood A., Chen J., Li Y., Chopp M. Statins increase neurogenesis in the dentate gyrus, reduce delayed neuronal death in the hippocampal CA3 region, and improve spatial learning in rat after traumatic brain injury. J. Neurotrauma. 2007;24(7):1132–1146. doi: 10.1089/neu.2007.0288. [http://dx.doi.org/10.1089/neu.2007.0288]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Robin N.C., Agoston Z., Biechele T.L., James R.G., Berndt J.D., Moon R.T. Simvastatin promotes adult hippocampal neurogenesis by enhancing Wnt/beta-catenin signaling. Stem Cell Reports. 2013;2(1):9–17. doi: 10.1016/j.stemcr.2013.11.002. [http://dx.doi.org/10.1016/j.stemcr.2013.11. 002]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wu H., Lu D., Jiang H., Xiong Y., Qu C., Li B., Mahmood A., Zhou D., Chopp M. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J. Neurotrauma. 2008;25(2):130–139. doi: 10.1089/neu.2007.0369. [http://dx.doi.org/10.1089/neu.2007.0369]. [DOI] [PubMed] [Google Scholar]
- 102.Li H., Kuwajima T., Oakley D., Nikulina E., Hou J., Yang W.S., Lowry E.R., Lamas N.J., Amoroso M.W., Croft G.F., Hosur R., Wichterle H., Sebti S., Filbin M.T., Stockwell B., Henderson C.E. Protein prenylation constitutes an endogenous brake on axonal growth. Cell Reports. 2016;16(2):545–558. doi: 10.1016/j.celrep.2016.06.013. [http://dx.doi.org/10.1016/j.celrep.2016.06.013]. [DOI] [PubMed] [Google Scholar]
- 103.Jin Y., Sui H.J., Dong Y., Ding Q., Qu W.H., Yu S.X., Jin Y.X. Atorvastatin enhances neurite outgrowth in cortical neurons in vitro via up-regulating the Akt/mTOR and Akt/GSK-3beta signaling pathways. Acta Pharmacol. Sin. 2012;33(7):861–872. doi: 10.1038/aps.2012.59. [http:// dx.doi.org/10.1038/aps.2012.59]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pooler A.M., Xi S.C., Wurtman R.J. The 3-hydroxy-3-methylglutaryl co-enzyme A reductase inhibitor pravastatin enhances neurite outgrowth in hippocampal neurons. J. Neurochem. 2006;97(3):716–723. doi: 10.1111/j.1471-4159.2006.03763.x. [http://dx.doi.org/10.1111/j.1471-4159.2006. 03763.x]. [DOI] [PubMed] [Google Scholar]
- 105.Gouveia T.L., Scorza F.A., Iha H.A., Frangiotti M.I., Perosa S.R., Cavalheiro E.A., Silva J.A., Jr, Feliciano R.S., de Almeida A.C., Naffah-Mazzacoratti M.G. Lovastatin decreases the synthesis of inflammatory mediators during epileptogenesis in the hippocampus of rats submitted to pilocarpine-induced epilepsy. Epilepsy Behav. 2014;36:68–73. doi: 10.1016/j.yebeh.2014.04.009. [http://dx.doi.org/10.1016/j.yebeh.2014. 04.009]. [DOI] [PubMed] [Google Scholar]
- 106.Griffin J.M., Kho D., Graham E.S., Nicholson L.F., O’Carroll S.J. Statins inhibit fibrillary beta-amyloid induced inflammation in a Model of the human blood brain barrier. PLoS One. 2016;11(6):e0157483. doi: 10.1371/journal.pone.0157483. [http://dx.doi.org/10.1371/journal.pone.0157483]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Reis P.A., Alexandre P.C., D’Avila J.C., Siqueira L.D., Antunes B., Estato V., Tibirica E.V., Verdonk F., Sharshar T., Chretien F., Castro-Faria-Neto H.C., Bozza F.A. Statins prevent cognitive impairment after sepsis by reverting neuroinflammation, and microcirculatory/endothelial dysfunction. Brain Behav. Immun. 2017;60:293–303. doi: 10.1016/j.bbi.2016.11.006. [http://dx.doi.org/10.1016/j.bbi.2016.11.006]. [DOI] [PubMed] [Google Scholar]
- 108.Lim S.W., Shiue Y.L., Liao J.C., Wee H.Y., Wang C.C., Chio C.C., Chang C.H., Hu C.Y., Kuo J.R. Simvastatin therapy in the acute stage of traumatic brain injury attenuates brain trauma-induced depression-Like behavior in rats by reducing neuroinflammation in the Hippocampus. Neurocrit. Care. 2017;26(1):122–132. doi: 10.1007/s12028-016-0290-6. [http://dx.doi.org/10.1007/s12028-016-0290-6]. [DOI] [PubMed] [Google Scholar]
- 109.Chu L.W., Chen J.Y., Wu P.C., Wu B.N. Atorvastatin prevents neuroinflammation in chronic constriction injury rats through nuclear NFkappaB downregulation in the dorsal root ganglion and spinal cord. ACS Chem. Neurosci. 2015;6(6):889–898. doi: 10.1021/acschemneuro.5b00032. [http://dx. doi.org/10.1021/acschemneuro.5b00032]. [DOI] [PubMed] [Google Scholar]
- 110.Yan J., Sun J., Huang L., Fu Q., Du G. Simvastatin prevents neuroinflammation by inhibiting N-methyl-D-aspartic acid receptor 1 in 6-hydroxydopamine-treated PC12 cells. J. Neurosci. Res. 2014;92(5):634–640. doi: 10.1002/jnr.23329. [http://dx.doi.org/10.1002/jnr.23329]. [DOI] [PubMed] [Google Scholar]
- 111.Ma M.W., Wang J., Zhang Q., Wang R., Dhandapani K.M., Vadlamudi R.K., Brann D.W. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017;12(1):7. doi: 10.1186/s13024-017-0150-7. [http://dx.doi.org/10.1186/s13024-017-0150-7]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kwok J.M., Ma C.C., Ma S. Recent development in the effects of statins on cardiovascular disease through Rac1 and NADPH oxidase. Vascul. Pharmacol. 2013;58(1-2):21–30. doi: 10.1016/j.vph.2012.10.003. [http://dx.doi.org/ 10.1016/j.vph.2012.10.003]. [DOI] [PubMed] [Google Scholar]
- 113.Barone E., Cenini G., Di Domenico F., Martin S., Sultana R., Mancuso C., Murphy M.P., Head E., Butterfield D.A. Long-term high-dose atorvastatin decreases brain oxidative and nitrosative stress in a preclinical model of Alzheimer disease: a novel mechanism of action. Pharmacol. Res. 2011;63(3):172–180. doi: 10.1016/j.phrs.2010.12.007. [http:// dx.doi.org/10.1016/j.phrs.2010.12.007]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Catalao C.H., Santos-Junior N.N., da Costa L.H., Souza A.O., Alberici L.C., Rocha M.J. Brain oxidative stress during experimental sepsis is attenuated by simvastatin administration. Mol. Neurobiol. 2017;54(9):7008–7018. doi: 10.1007/s12035-016-0218-3. [DOI] [PubMed] [Google Scholar]
- 115.Butterfield D.A., Barone E., Di Domenico F., Cenini G., Sultana R., Murphy M.P., Mancuso C., Head E. Atorvastatin treatment in a dog preclinical model of Alzheimer’s disease leads to up-regulation of haem oxygenase-1 and is associated with reduced oxidative stress in brain. Int. J. Neuropsychopharmacol. 2012;15(7):981–987. doi: 10.1017/S1461145711001118. [http://dx.doi.org/10.1017/S1461145711001118]. [DOI] [PubMed] [Google Scholar]
- 116.Tong X.K., Nicolakakis N., Fernandes P., Ongali B., Brouillette J., Quirion R., Hamel E. Simvastatin improves cerebrovascular function and counters soluble amyloid-beta, inflammation and oxidative stress in aged APP mice. Neurobiol. Dis. 2009;35(3):406–414. doi: 10.1016/j.nbd.2009.06.003. [http://dx.doi.org/10.1016/j.nbd.2009.06.003]. [DOI] [PubMed] [Google Scholar]
- 117.Hayashi T., Hamakawa K., Nagotani S., Jin G., Li F., Deguchi K., Sehara Y., Zhang H., Nagano I., Shoji M., Abe K. HMG CoA reductase inhibitors reduce ischemic brain injury of Wistar rats through decreasing oxidative stress on neurons. Brain Res. 2005;1037(1-2):52–58. doi: 10.1016/j.brainres.2004.12.051. [http://dx.doi.org/10.1016/j.brainres.2004. 12.051]. [DOI] [PubMed] [Google Scholar]
- 118.Simons K., Ehehalt R. Cholesterol, lipid rafts, and disease. J. Clin. Invest. 2002;110(5):597–603. doi: 10.1172/JCI16390. [http://dx.doi.org/10.1172/ JCI0216390]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Linetti A., Fratangeli A., Taverna E., Valnegri P., Francolini M., Cappello V., Matteoli M., Passafaro M., Rosa P. Cholesterol reduction impairs exocytosis of synaptic vesicles. J. Cell Sci. 2010;123(Pt 4):595–605. doi: 10.1242/jcs.060681. [http://dx.doi.org/10.1242/jcs.060681]. [DOI] [PubMed] [Google Scholar]
- 120.Pani A., Mandas A., Dessi S. Cholesterol, Alzheimer’s disease, prion disorders: a menage a trois? Curr. Drug Targets. 2010;11(8):1018–1031. doi: 10.2174/138945010791591386. [http://dx.doi.org/10.2174/138945010791591386]. [DOI] [PubMed] [Google Scholar]
- 121.Karasinska J.M., Hayden M.R. Cholesterol metabolism in Huntington disease. Nat. Rev. Neurol. 2011;7(10):561–572. doi: 10.1038/nrneurol.2011.132. [http://dx.doi.org/10.1038/nrneurol.2011.132]. [DOI] [PubMed] [Google Scholar]
- 122.Porter F.D., Herman G.E. Malformation syndromes caused by disorders of cholesterol synthesis. J. Lipid Res. 2011;52(1):6–34. doi: 10.1194/jlr.R009548. [http://dx.doi.org/10.1194/jlr.R009548]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tint G.S., Irons M., Elias E.R., Batta A.K., Frieden R., Chen T.S., Salen G. Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N. Engl. J. Med. 1994;330(2):107–113. doi: 10.1056/NEJM199401133300205. [http://dx.doi.org/10.1056/NEJM199401133300205]. [DOI] [PubMed] [Google Scholar]
- 124.Nowaczyk M.J., Zeesman S., Waye J.S., Douketis J.D. Incidence of Smith-Lemli-Opitz syndrome in Canada: results of three-year population surveillance. J. Pediatr. 2004;145(4):530–535. doi: 10.1016/j.jpeds.2004.06.045. [http://dx.doi.org/10.1016/j.jpeds.2004.06.045]. [DOI] [PubMed] [Google Scholar]
- 125.Vance J.E. Dysregulation of cholesterol balance in the brain: contribution to neurodegenerative diseases. Dis. Model. Mech. 2012;5(6):746–755. doi: 10.1242/dmm.010124. [http://dx.doi.org/10.1242/dmm.010124]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chan Y.M., Merkens L.S., Connor W.E., Roullet J.B., Penfield J.A., Jordan J.M., Steiner R.D., Jones P.J. Effects of dietary cholesterol and simvastatin on cholesterol synthesis in Smith-Lemli-Opitz syndrome. Pediatr. Res. 2009;65(6):681–685. doi: 10.1203/PDR.0b013e31819ea4eb. [http://dx.doi.org/10.1203/PDR.0b013e31819ea4eb]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Porter F.D. Human malformation syndromes due to inborn errors of cholesterol synthesis. Curr. Opin. Pediatr. 2003;15(6):607–613. doi: 10.1097/00008480-200312000-00011. [http://dx.doi.org/10.1097/00008480-200312000-00011]. [DOI] [PubMed] [Google Scholar]
- 128.Elias E.R., Irons M.B., Hurley A.D., Tint G.S., Salen G. Clinical effects of cholesterol supplementation in six patients with the Smith-Lemli-Opitz syndrome (SLOS). Am. J. Med. Genet. 1997;68(3):305–310. doi: 10.1002/(sici)1096-8628(19970131)68:3<305::aid-ajmg11>3.0.co;2-x. [http://dx.doi.org/10.1002/(SICI)1096-8628(19970131) 68:3<305:AID-AJMG11>3.0.CO;2-X]. [DOI] [PubMed] [Google Scholar]
- 129.Linck L.M., Lin D.S., Flavell D., Connor W.E., Steiner R.D. Cholesterol supplementation with egg yolk increases plasma cholesterol and decreases plasma 7-dehydrocholesterol in Smith-Lemli-Opitz syndrome. Am. J. Med. Genet. 2000;93(5):360–365. doi: 10.1002/1096-8628(20000828)93:5<360::aid-ajmg4>3.0.co;2-p. [http://dx.doi.org/10.1002/1096-8628(20000828)93:5<360:AID-AJMG4>3.0.CO;2-P]. [DOI] [PubMed] [Google Scholar]
- 130.Pappu A.S., Steiner R.D., Connor S.L., Flavell D.P., Lin D.S., Hatcher L., Illingworth D.R., Connor W.E. Feedback inhibition of the cholesterol biosynthetic pathway in patients with Smith-Lemli-Opitz syndrome as demonstrated by urinary mevalonate excretion. J. Lipid Res. 2002;43(10):1661–1669. doi: 10.1194/jlr.m200163-jlr200. [http://dx.doi.org/ 10.1194/jlr.M200163-JLR200]. [DOI] [PubMed] [Google Scholar]
- 131.Merkens L.S., Connor W.E., Linck L.M., Lin D.S., Flavell D.P., Steiner R.D. Effects of dietary cholesterol on plasma lipoproteins in smith-Lemli-Opitz syndrome. Pediatr. Res. 2004;56(5):726–732. doi: 10.1203/01.PDR.0000141522.14177.4F. [http://dx.doi.org/10.1203/01.PDR.0000141522.14177.4F]. [DOI] [PubMed] [Google Scholar]
- 132.Dietschy J.M., Turley S.D. Cholesterol metabolism in the brain. Curr. Opin. Lipidol. 2001;12(2):105–112. doi: 10.1097/00041433-200104000-00003. [http://dx.doi.org/ 10.1097/00041433-200104000-00003]. [DOI] [PubMed] [Google Scholar]
- 133.Wassif C.A., Krakowiak P.A., Wright B.S., Gewandter J.S., Sterner A.L., Javitt N., Yergey A.L., Porter F.D. Residual cholesterol synthesis and simvastatin induction of cholesterol synthesis in Smith-Lemli-Opitz syndrome fibroblasts. Mol. Genet. Metab. 2005;85(2):96–107. doi: 10.1016/j.ymgme.2004.12.009. [http://dx.doi.org/10.1016/j.ymgme.2004.12. 009]. [DOI] [PubMed] [Google Scholar]
- 134.Correa-Cerro L.S., Wassif C.A., Kratz L., Miller G.F., Munasinghe J.P., Grinberg A., Fliesler S.J., Porter F.D. Development and characterization of a hypomorphic Smith-Lemli-Opitz syndrome mouse model and efficacy of simvastatin therapy. Hum. Mol. Genet. 2006;15(6):839–851. doi: 10.1093/hmg/ddl003. [http://dx.doi.org/10.1093/hmg/ ddl003]. [DOI] [PubMed] [Google Scholar]
- 135.Haas D., Garbade S.F., Vohwinkel C., Muschol N., Trefz F.K., Penzien J.M., Zschocke J., Hoffmann G.F., Burgard P. Effects of cholesterol and simvastatin treatment in patients with Smith-Lemli-Opitz syndrome (SLOS). J. Inherit. Metab. Dis. 2007;30(3):375–387. doi: 10.1007/s10545-007-0537-7. [http://dx.doi.org/10.1007/s10545-007-0537-7]. [DOI] [PubMed] [Google Scholar]
- 136.Jira P.E., Wevers R.A., de Jong J., Rubio-Gozalbo E., Janssen-Zijlstra F.S., van Heyst A.F., Sengers R.C., Smeitink J.A. Simvastatin. A new therapeutic approach for Smith-Lemli-Opitz syndrome. J. Lipid Res. 2000;41(8):1339–1346. [PubMed] [Google Scholar]
- 137.Peprah E. Fragile X syndrome: the FMR1 CGG repeat distribution among world populations. Ann. Hum. Genet. 2012;76(2):178–191. doi: 10.1111/j.1469-1809.2011.00694.x. [http://dx.doi.org/10.1111/j.1469-1809.2011.00694.x]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gallagher A., Hallahan B. Fragile X-associated disorders: a clinical overview. J. Neurol. 2012;259(3):401–413. doi: 10.1007/s00415-011-6161-3. [http://dx.doi.org/ 10.1007/s00415-011-6161-3]. [DOI] [PubMed] [Google Scholar]
- 139.Hessl D., Nguyen D.V., Green C., Chavez A., Tassone F., Hagerman R.J., Senturk D., Schneider A., Lightbody A., Reiss A.L., Hall S. A solution to limitations of cognitive testing in children with intellectual disabilities: the case of fragile X syndrome. J. Neurodev. Disord. 2009;1(1):33–45. doi: 10.1007/s11689-008-9001-8. [http://dx.doi.org/10.1007/ s11689-008-9001-8]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wadell P.M., Hagerman R.J., Hessl D.R., Fragile X. Syndrome: Psychiatric Manifestations, Assessment and Emerging Therapies. Curr. Psychiatry Rev. 2013;9(1):53–58. doi: 10.2174/157340013805289644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Verkerk A.J., Pieretti M., Sutcliffe J.S., Fu Y.H., Kuhl D.P., Pizzuti A., Reiner O., Richards S., Victoria M.F., Zhang F.P. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65(5):905–914. doi: 10.1016/0092-8674(91)90397-h. [http://dx.doi.org/ 10.1016/0092-8674(91)90397-H]. [DOI] [PubMed] [Google Scholar]
- 142.Corbin F., Bouillon M., Fortin A., Morin S., Rousseau F., Khandjian E.W. The fragile X mental retardation protein is associated with poly(A)+ mRNA in actively translating polyribosomes. Hum. Mol. Genet. 1997;6(9):1465–1472. doi: 10.1093/hmg/6.9.1465. [http://dx.doi.org/10. 1093/hmg/6.9.1465]. [DOI] [PubMed] [Google Scholar]
- 143.Irwin S.A., Patel B., Idupulapati M., Harris J.B., Crisostomo R.A., Larsen B.P., Kooy F., Willems P.J., Cras P., Kozlowski P.B., Swain R.A., Weiler I.J., Greenough W.T. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am. J. Med. Genet. 2001;98(2):161–167. doi: 10.1002/1096-8628(20010115)98:2<161::aid-ajmg1025>3.0.co;2-b. [http://dx.doi.org/10.1002/ 1096-8628(20010115)98:2<161:AID-AJMG1025>3.0.CO;2-B]. [DOI] [PubMed] [Google Scholar]
- 144.Bhakar A.L., Dolen G., Bear M.F. The pathophysiology of fragile X (and what it teaches us about synapses). Annu. Rev. Neurosci. 2012;35:417–443. doi: 10.1146/annurev-neuro-060909-153138. [http://dx.doi.org/10.1146/annurev-neuro-060909-153138]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hou L., Antion M.D., Hu D., Spencer C.M., Paylor R., Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51(4):441–454. doi: 10.1016/j.neuron.2006.07.005. [http://dx.doi.org/10. 1016/j.neuron.2006.07.005]. [DOI] [PubMed] [Google Scholar]
- 146.Price T.J., Rashid M.H., Millecamps M., Sanoja R., Entrena J.M., Cervero F. Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J. Neurosci. 2007;27(51):13958–13967. doi: 10.1523/JNEUROSCI.4383-07.2007. [http://dx.doi. org/10.1523/JNEUROSCI.4383-07.2007]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Osterweil E.K., Krueger D.D., Reinhold K., Bear M.F. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci. 2010;30(46):15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010. [http://dx.doi.org/10. 1523/JNEUROSCI.3888-10.2010]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cerezo-Guisado M.I., Garcia-Roman N., Garcia-Marin L.J., Alvarez-Barrientos A., Bragado M.J., Lorenzo M.J. Lovastatin inhibits the extracellular-signal-regulated kinase pathway in immortalized rat brain neuroblasts. Biochem. J. 2007;401(1):175–183. doi: 10.1042/BJ20060731. [http://dx.doi.org/10.1042/BJ20060731]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kumari R., Castillo C., Francesconi A. Agonist-dependent signaling by group I metabotropic glutamate receptors is regulated by association with lipid domains. J. Biol. Chem. 2013;288(44):32004–32019. doi: 10.1074/jbc.M113.475863. [http://dx.doi.org/10.1074/jbc.M113.475863]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Osterweil E.K., Chuang S.C., Chubykin A.A., Sidorov M., Bianchi R., Wong R.K., Bear M.F. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron. 2013;77(2):243–250. doi: 10.1016/j.neuron.2012.01.034. [http://dx.doi. org/10.1016/j.neuron.2012.01.034]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Li W., Cui Y., Kushner S.A., Brown R.A., Jentsch J.D., Frankland P.W., Cannon T.D., Silva A.J. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr. Biol. 2005;15(21):1961–1967. doi: 10.1016/j.cub.2005.09.043. [http://dx.doi.org/10.1016/j.cub.2005.09.043]. [DOI] [PubMed] [Google Scholar]
- 152.Caku A., Pellerin D., Bouvier P., Riou E., Corbin F. Effect of lovastatin on behavior in children and adults with fragile X syndrome: an open-label study. Am. J. Med. Genet. A. 2014;164A(11):2834–2842. doi: 10.1002/ajmg.a.36750. [http://dx.doi.org/10.1002/ajmg.a.36750]. [DOI] [PubMed] [Google Scholar]
- 153.Bailey D.B., Jr, Raspa M., Bishop E., Olmsted M., Mallya U.G., Berry-Kravis E. Medication utilization for targeted symptoms in children and adults with fragile X syndrome: US survey. J. Dev. Behav. Pediatr. 2012;33(1):62–69. doi: 10.1097/DBP.0b013e318236c0e1. [http://dx.doi.org/10. 1097/DBP.0b013e318236c0e1]. [DOI] [PubMed] [Google Scholar]
- 154.Patankar J.V. Cholesterol metabolism is a potential therapeutic target for Rett syndrome. Clin. Genet. 2014;85(3):229–230. doi: 10.1111/cge.12284. [http://dx.doi.org/10.1111/cge.12284]. [DOI] [PubMed] [Google Scholar]
- 155.Chahrour M., Jung S.Y., Shaw C., Zhou X., Wong S.T., Qin J., Zoghbi H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320(5880):1224–1229. doi: 10.1126/science.1153252. [http://dx.doi.org/10.1126/science.1153252]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Amir R.E., Van den Veyver I.B., Wan M., Tran C.Q., Francke U., Zoghbi H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23(2):185–188. doi: 10.1038/13810. [http://dx.doi.org/10.1038/13810]. [DOI] [PubMed] [Google Scholar]
- 157.Macdonald J.L., Verster A., Berndt A., Roskams A.J. MBD2 and MeCP2 regulate distinct transitions in the stage-specific differentiation of olfactory receptor neurons. Mol. Cell. Neurosci. 2010;44(1):55–67. doi: 10.1016/j.mcn.2010.02.003. [http://dx.doi.org/10.1016/j.mcn.2010.02.003]. [DOI] [PubMed] [Google Scholar]
- 158.Villani C., Sacchetti G., Bagnati R., Passoni A., Fusco F., Carli M., Invernizzi R.W. Lovastatin fails to improve motor performance and survival in methyl-CpG-binding protein2-null mice. eLife. 2018;5:1–14. doi: 10.7554/eLife.22409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zoghbi H.Y., Bear M.F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 2012;4(3) doi: 10.1101/cshperspect.a009886. [http://dx.doi. org/10.1101/cshperspect.a009886]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Huguet G., Ey E., Bourgeron T. The genetic landscapes of autism spectrum disorders. Annu. Rev. Genomics Hum. Genet. 2013;14:191–213. doi: 10.1146/annurev-genom-091212-153431. [http://dx.doi.org/10.1146/annurev-genom-091212-153431]. [DOI] [PubMed] [Google Scholar]
- 161.Vargas D.L., Nascimbene C., Krishnan C., Zimmerman A.W., Pardo C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005;57(1):67–81. doi: 10.1002/ana.20315. [http://dx.doi.org/10.1002/ana.20315]. [DOI] [PubMed] [Google Scholar]
- 162.Molloy C.A., Morrow A.L., Meinzen-Derr J., Schleifer K., Dienger K., Manning-Courtney P., Altaye M., Wills-Karp M. Elevated cytokine levels in children with autism spectrum disorder. J. Neuroimmunol. 2006;172(1-2):198–205. doi: 10.1016/j.jneuroim.2005.11.007. [http://dx.doi.org/10. 1016/j.jneuroim.2005.11.007]. [DOI] [PubMed] [Google Scholar]
- 163.Lee J.K., Won J.S., Singh A.K., Singh I. Statin inhibits kainic acid-induced seizure and associated inflammation and hippocampal cell death. Neurosci. Lett. 2008;440(3):260–264. doi: 10.1016/j.neulet.2008.05.112. [http://dx.doi. org/10.1016/j.neulet.2008.05.112]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ramirez C., Tercero I., Pineda A., Burgos J.S. Simvastatin is the statin that most efficiently protects against kainate-induced excitotoxicity and memory impairment. J. Alzheimers Dis. 2011;24(1):161–174. doi: 10.3233/JAD-2010-101653. [http://dx.doi.org/10.3233/JAD-2010-101653]. [DOI] [PubMed] [Google Scholar]
- 165.Xie C., Sun J., Qiao W., Lu D., Wei L., Na M., Song Y., Hou X., Lin Z. Administration of simvastatin after kainic acid-induced status epilepticus restrains chronic temporal lobe epilepsy. PLoS One. 2011;6(9):e24966. doi: 10.1371/journal.pone.0024966. [http://dx.doi.org/10.1371/journal.pone. 0024966]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.van Vliet E.A., Holtman L., Aronica E., Schmitz L.J., Wadman W.J., Gorter J.A. Atorvastatin treatment during epileptogenesis in a rat model for temporal lobe epilepsy. Epilepsia. 2011;52(7):1319–1330. doi: 10.1111/j.1528-1167.2011.03073.x. [http://dx.doi.org/10.1111/j.1528-1167.2011.03073.x]. [DOI] [PubMed] [Google Scholar]
- 167.Serbanescu I., Ryan M.A., Shukla R., Cortez M.A., Snead O.C., III, Cunnane S.C. Lovastatin exacerbates atypical absence seizures with only minimal effects on brain sterols. J. Lipid Res. 2004;45(11):2038–2043. doi: 10.1194/jlr.M400097-JLR200. [http://dx.doi.org/10.1194/jlr.M400097-JLR200]. [DOI] [PubMed] [Google Scholar]
- 168.Canitano R. Epilepsy in autism spectrum disorders. Eur. Child Adolesc. Psychiatry. 2007;16(1):61–66. doi: 10.1007/s00787-006-0563-2. [http://dx.doi.org/10. 1007/s00787-006-0563-2]. [DOI] [PubMed] [Google Scholar]
- 169.Pintaudi M., Calevo M.G., Vignoli A., Parodi E., Aiello F., Baglietto M.G., Hayek Y., Buoni S., Renieri A., Russo S., Cogliati F., Giordano L., Canevini M., Veneselli E. Epilepsy in Rett syndrome: clinical and genetic features. Epilepsy Behav. 2010;19(3):296–300. doi: 10.1016/j.yebeh.2010.06.051. [http://dx.doi.org/10.1016/j.yebeh.2010.06.051]. [DOI] [PubMed] [Google Scholar]
- 170.Querfurth H.W., LaFerla F.M. Alzheimer’s disease. N. Engl. J. Med. 2010;362(4):329–344. doi: 10.1056/NEJMra0909142. [http://dx.doi.org/10.1056/NEJMra 0909142]. [DOI] [PubMed] [Google Scholar]
- 171.Bonda D.J., Wang X., Lee H.G., Smith M.A., Perry G., Zhu X. Neuronal failure in Alzheimer’s disease: a view through the oxidative stress looking-glass. Neurosci. Bull. 2014;30(2):243–252. doi: 10.1007/s12264-013-1424-x. [http://dx.doi.org/10.1007/s12264-013-1424-x]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Alzheimer A. Uber eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrife Psychiatrie. 1907;64:146–148. [Google Scholar]
- 173.Wood W.G., Igbavboa U., Eckert G.P., Johnson-Anuna L.N., Muller W.E. Is hypercholesterolemia a risk factor for Alzheimer’s disease? Mol. Neurobiol. 2005;31(1-3):185–192. doi: 10.1385/MN:31:1-3:185. [http://dx.doi. org/10.1385/MN:31:1-3:185]. [DOI] [PubMed] [Google Scholar]
- 174.Yu J.T., Tan L., Hardy J. Apolipoprotein E in Alzheimer’s disease: an update. Annu. Rev. Neurosci. 2014;37:79–100. doi: 10.1146/annurev-neuro-071013-014300. [http://dx.doi.org/10.1146/annurev-neuro-071013-014300]. [DOI] [PubMed] [Google Scholar]
- 175.Liu C.C., Kanekiyo T., Xu H., Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 2013;9(2):106–118. doi: 10.1038/nrneurol.2012.263. [http://dx.doi.org/10.1038/nrneurol.2012.263]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Christensen D.Z., Schneider-Axmann T., Lucassen P.J., Bayer T.A., Wirths O. Accumulation of intraneuronal Abeta correlates with ApoE4 genotype. Acta Neuropathol. 2010;119(5):555–566. doi: 10.1007/s00401-010-0666-1. [http://dx.doi.org/10.1007/s00401-010-0666-1]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hashimoto T., Serrano-Pozo A., Hori Y., Adams K.W., Takeda S., Banerji A.O., Mitani A., Joyner D., Thyssen D.H., Bacskai B.J., Frosch M.P., Spires-Jones T.L., Finn M.B., Holtzman D.M., Hyman B.T. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid beta peptide. J. Neurosci. 2012;32(43):15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012. [http://dx.doi.org/10.1523/ JNEUROSCI.1542-12.2012]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Koffie R.M., Hashimoto T., Tai H.C., Kay K.R., Serrano-Pozo A., Joyner D., Hou S., Kopeikina K.J., Frosch M.P., Lee V.M. [DOI] [PMC free article] [PubMed]; Holtzman D.M., Hyman B.T., Spires-Jones T.L. Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-beta. Brain. 2012;135(Pt 7):2155–2168. doi: 10.1093/brain/aws127. [http://dx.doi.org/10.1093/brain/aws127]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009;10(5):333–344. doi: 10.1038/nrn2620. [http://dx.doi.org/10.1038/nrn2620]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hottman D.A., Li L. Protein prenylation and synaptic plasticity: implications for Alzheimer’s disease. Mol. Neurobiol. 2014;50(1):177–185. doi: 10.1007/s12035-013-8627-z. [http://dx.doi.org/10.1007/s12035-013-8627-z]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Pedrini S., Carter T.L., Prendergast G., Petanceska S., Ehrlich M.E., Gandy S. Modulation of statin-activated shedding of Alzheimer APP ectodomain by ROCK. PLoS Med. 2005;2(1):e18. doi: 10.1371/journal.pmed.0020018. [http://dx.doi.org/10.1371/journal.pmed.0020018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Cole S.L., Grudzien A., Manhart I.O., Kelly B.L., Oakley H., Vassar R. Statins cause intracellular accumulation of amyloid precursor protein, beta-secretase-cleaved fragments, and amyloid beta-peptide via an isoprenoid-dependent mechanism. J. Biol. Chem. 2005;280(19):18755–18770. doi: 10.1074/jbc.M413895200. [http://dx.doi.org/10.1074/ jbc.M413895200]. [DOI] [PubMed] [Google Scholar]
- 183.Ostrowski S.M., Wilkinson B.L., Golde T.E., Landreth G. Statins reduce amyloid-beta production through inhibition of protein isoprenylation. J. Biol. Chem. 2007;282(37):26832–26844. doi: 10.1074/jbc.M702640200. [http://dx.doi.org/10.1074/jbc.M702640200]. [DOI] [PubMed] [Google Scholar]
- 184.Simons M., Schwarzler F., Lutjohann D., von Bergmann K., Beyreuther K., Dichgans J., Wormstall H., Hartmann T., Schulz J.B. Treatment with simvastatin in normocholesterolemic patients with Alzheimer’s disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann. Neurol. 2002;52(3):346–350. doi: 10.1002/ana.10292. [http://dx.doi.org/10.1002/ana.10292]. [DOI] [PubMed] [Google Scholar]
- 185.Fassbender K., Simons M., Bergmann C., Stroick M., Lutjohann D., Keller P., Runz H., Kuhl S., Bertsch T., von Bergmann K., Hennerici M., Beyreuther K., Hartmann T. Simvastatin strongly reduces levels of Alzheimer’s disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA. 2001;98(10):5856–5861. doi: 10.1073/pnas.081620098. [http://dx.doi.org/10.1073/ pnas.081620098]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tamboli I.Y., Barth E., Christian L., Siepmann M., Kumar S., Singh S., Tolksdorf K., Heneka M.T., Lutjohann D., Wunderlich P., Walter J. Statins promote the degradation of extracellular amyloid {beta}-peptide by microglia via stimulation of exosome-associated insulin-degrading enzyme (IDE) secretion. J. Biol. Chem. 2010;285(48):37405–37414. doi: 10.1074/jbc.M110.149468. [http://dx.doi.org/10.1074/ jbc.M110.149468]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kurata T., Miyazaki K., Kozuki M., Panin V.L., Morimoto N., Ohta Y., Nagai M., Ikeda Y., Matsuura T., Abe K. Atorvastatin and pitavastatin improve cognitive function and reduce senile plaque and phosphorylated tau in aged APP mice. Brain Res. 2011;1371:161–170. doi: 10.1016/j.brainres.2010.11.067. [http://dx.doi.org/10.1016/j.brainres.2010.11.067]. [DOI] [PubMed] [Google Scholar]
- 188.Murphy M.P., Morales J., Beckett T.L., Astarita G., Piomelli D., Weidner A., Studzinski C.M., Dowling A.L., Wang X., Levine H., III, Kryscio R.J., Lin Y., Barrett E., Head E. Changes in cognition and amyloid-beta processing with long term cholesterol reduction using atorvastatin in aged dogs. J. Alzheimers Dis. 2010;22(1):135–150. doi: 10.3233/JAD-2010-100639. [http://dx.doi.org/10.3233/JAD-2010-100639]. [DOI] [PubMed] [Google Scholar]
- 189.SjÃgren M.; Blennow, K. The link between cholesterol and Alzheimer’s disease. World J. Biol. Psychiatry. 2005;6(2):85–97. doi: 10.1080/15622970510029795. [http://dx.doi.org/10.1080/15622970510029795]. [DOI] [PubMed] [Google Scholar]
- 190.Salins P., Shawesh S., He Y., Dibrov A., Kashour T., Arthur G., Amara F. Lovastatin protects human neurons against Abeta-induced toxicity and causes activation of beta-catenin-TCF/LEF signaling. Neurosci. Lett. 2007;412(3):211–216. doi: 10.1016/j.neulet.2006.07.045. [http://dx.doi. org/10.1016/j.neulet.2006.07.045]. [DOI] [PubMed] [Google Scholar]
- 191.Kurata T., Miyazaki K., Morimoto N., Kawai H., Ohta Y., Ikeda Y., Abe K. Atorvastatin and pitavastatin reduce oxidative stress and improve IR/LDL-R signals in Alzheimer’s disease. Neurol. Res. 2013;35(2):193–205. doi: 10.1179/1743132812Y.0000000127. [http://dx.doi.org/10.1179/ 1743132812Y.0000000127]. [DOI] [PubMed] [Google Scholar]
- 192.Zhang Y.Y., Fan Y.C., Wang M., Wang D., Li X.H. Atorvastatin attenuates the production of IL-1beta, IL-6, and TNF-alpha in the hippocampus of an amyloid beta1-42-induced rat model of Alzheimer’s disease. Clin. Interv. Aging. 2013;8:103–110. doi: 10.2147/CIA.S40405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hoglund K., Thelen K.M., Syversen S., Sjogren M., von Bergmann K., Wallin A., Vanmechelen E., Vanderstichele H., Lutjohann D., Blennow K. The effect of simvastatin treatment on the amyloid precursor protein and brain cholesterol metabolism in patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2005;19(5-6):256–265. doi: 10.1159/000084550. [http://dx.doi.org/10.1159/000084550]. [DOI] [PubMed] [Google Scholar]
- 194.Malfitano A.M., Marasco G., Proto M.C., Laezza C., Gazzerro P., Bifulco M. Statins in neurological disorders: an overview and update. Pharmacol. Res. 2014;88:74–83. doi: 10.1016/j.phrs.2014.06.007. [http://dx.doi.org/10. 1016/j.phrs.2014.06.007]. [DOI] [PubMed] [Google Scholar]
- 195.Li G., Higdon R., Kukull W.A., Peskind E., Van Valen Moore K., Tsuang D., van Belle G., McCormick W., Bowen J.D., Teri L., Schellenberg G.D., Larson E.B. Statin therapy and risk of dementia in the elderly: a community-based prospective cohort study. Neurology. 2004;63(9):1624–1628. doi: 10.1212/01.wnl.0000142963.90204.58. [http://dx.doi.org/10.1212/ 01.WNL.0000142963.90204.58]. [DOI] [PubMed] [Google Scholar]
- 196.Sparks D.L., Sabbagh M.N., Connor D.J., Lopez J., Launer L.J., Browne P., Wasser D., Johnson-Traver S., Lochhead J., Ziolwolski C. Atorvastatin for the treatment of mild to moderate Alzheimer disease: preliminary results. Arch. Neurol. 2005;62(5):753–757. doi: 10.1001/archneur.62.5.753. [http://dx.doi.org/10.1001/archneur.62.5.753]. [DOI] [PubMed] [Google Scholar]
- 197.Dufouil C., Richard F., Fievet N., Dartigues J.F., Ritchie K., Tzourio C., Amouyel P., Alperovitch A. APOE genotype, cholesterol level, lipid-lowering treatment, and dementia: the Three-City Study. Neurology. 2005;64(9):1531–1538. doi: 10.1212/01.WNL.0000160114.42643.31. [http://dx.doi.org/10. 1212/01.WNL.0000160114.42643.31]. [DOI] [PubMed] [Google Scholar]
- 198.Sparks D.L., Kryscio R.J., Sabbagh M.N., Connor D.J., Sparks L.M., Liebsack C. Reduced risk of incident AD with elective statin use in a clinical trial cohort. Curr. Alzheimer Res. 2008;5(4):416–421. doi: 10.2174/156720508785132316. [http://dx.doi.org/10.2174/156720508785132316]. [DOI] [PubMed] [Google Scholar]
- 199.Li G., Shofer J.B., Rhew I.C., Kukull W.A., Peskind E.R., McCormick W., Bowen J.D., Schellenberg G.D., Crane P.K., Breitner J.C., Larson E.B. Age-varying association between statin use and incident Alzheimer’s disease. J. Am. Geriatr. Soc. 2010;58(7):1311–1317. doi: 10.1111/j.1532-5415.2010.02906.x. [http://dx.doi.org/10.1111/j.1532-5415.2010. 02906.x]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Mendoza-Oliva A., Zepeda A., Arias C. The complex actions of statins in brain and their relevance for Alzheimer’s disease treatment: an analytical review. Curr. Alzheimer Res. 2014;11(9):817–833. [PubMed] [Google Scholar]
- 201.Fan Q.W., Yu W., Senda T., Yanagisawa K., Michikawa M. Cholesterol-dependent modulation of tau phosphorylation in cultured neurons. J. Neurochem. 2001;76(2):391–400. doi: 10.1046/j.1471-4159.2001.00063.x. [http://dx.doi. org/10.1046/j.1471-4159.2001.00063.x]. [DOI] [PubMed] [Google Scholar]
- 202.Mendoza-Oliva A., Ferrera P., Arias C. Interplay Between Cholesterol and Homocysteine in the Exacerbation of Amyloid-beta Toxicity in Human Neuroblastoma Cells. CNS Neurol. Disord. Drug Targets. 2013 doi: 10.2174/18715273113129990083. [http://dx.doi.org/10.2174/187152731131299 90083]. [DOI] [PubMed] [Google Scholar]
- 203.Marcuzzi A., Tricarico P.M., Piscianz E., Kleiner G., Vecchi Brumatti L., Crovella S. Lovastatin induces apoptosis through the mitochondrial pathway in an undifferentiated SH-SY5Y neuroblastoma cell line. Cell Death Dis. 2013;4:e585. doi: 10.1038/cddis.2013.112. [http://dx.doi.org/ 10.1038/cddis.2013.112]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Saeed U., Compagnone J., Aviv R.I., Strafella A.P., Black S.E., Lang A.E., Masellis M. Imaging biomarkers in Parkinson’s disease and Parkinsonian syndromes: current and emerging concepts. Transl. Neurodegener. 2017;6:8. doi: 10.1186/s40035-017-0076-6. [http://dx.doi.org/10.1186/s40035-017-0076-6]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kalia L.V., Lang A.E. Parkinson’s disease. Lancet. 2015;386(9996):896–912. doi: 10.1016/S0140-6736(14)61393-3. [http://dx.doi.org/10.1016/S0140-6736(14) 61393-3]. [DOI] [PubMed] [Google Scholar]
- 206.Bai S., Song Y., Huang X., Peng L., Jia J., Liu Y., Lu H. Statin use and the risk of parkinson’s disease: An updated meta-analysis. PLoS One. 2016;11(3):e0152564. doi: 10.1371/journal.pone.0152564. [http://dx.doi.org/10.1371/journal. pone.0152564]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Corti O., Lesage S., Brice A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol. Rev. 2011;91(4):1161–1218. doi: 10.1152/physrev.00022.2010. [http://dx.doi.org/10.1152/physrev.00022. 2010]. [DOI] [PubMed] [Google Scholar]
- 208.Giannoccaro M.P., La Morgia C., Rizzo G., Carelli V. Mitochondrial DNA and primary mitochondrial dysfunction in Parkinson’s disease. Mov. Disord. 2017;32(3):346–363. doi: 10.1002/mds.26966. [http://dx.doi. org/10.1002/mds.26966]. [DOI] [PubMed] [Google Scholar]
- 209.Ng C.H., Guan M.S., Koh C., Ouyang X., Yu F., Tan E.K., O’Neill S.P., Zhang X., Chung J., Lim K.L. AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in Drosophila models of Parkinson’s disease. J. Neurosci. 2012;32(41):14311–14317. doi: 10.1523/JNEUROSCI.0499-12.2012. [http://dx.doi.org/10.1523/JNEUROSCI. 0499-12.2012]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Cheung Z.H., Ip N.Y. The emerging role of autophagy in Parkinson’s disease. Mol. Brain. 2009;2:29. doi: 10.1186/1756-6606-2-29. [http://dx.doi.org/10.1186/ 1756-6606-2-29]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120(2):159–162. doi: 10.1016/j.cell.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 212.Glick D., Barth S., Macleod K.F. Autophagy: cellular and molecular mechanisms. J. Pathol. 2010;221(1):3–12. doi: 10.1002/path.2697. [http://dx. doi.org/10.1002/path.2697]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lynch-Day M.A., Mao K., Wang K., Zhao M., Klionsky D.J. The role of autophagy in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012;2(4):a009357. doi: 10.1101/cshperspect.a009357. [http://dx.doi.org/10.1101/ cshperspect.a009357]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Li Q., Zhuang Q.K., Yang J.N., Zhang Y.Y. Statins excert neuroprotection on cerebral ischemia independent of their lipid-lowering action: the potential molecular mechanisms. Eur. Rev. Med. Pharmacol. Sci. 2014;18(8):1113–1126. [PubMed] [Google Scholar]
- 215.Kang S.Y., Lee S.B., Kim H.J., Kim H.T., Yang H.O., Jang W. Autophagic modulation by rosuvastatin prevents rotenone-induced neurotoxicity in an in vitro model of Parkinson’s disease. Neurosci. Lett. 2017;642:20–26. doi: 10.1016/j.neulet.2017.01.063. [http://dx.doi.org/10.1016/j.neulet.2017.01. 063]. [DOI] [PubMed] [Google Scholar]
- 216.Gao X., Simon K.C., Schwarzschild M.A., Ascherio A. Prospective study of statin use and risk of Parkinson disease. Arch. Neurol. 2012;69(3):380–384. doi: 10.1001/archneurol.2011.1060. [http://dx.doi.org/10.1001/archneurol.2011. 1060]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Illingworth D.R., Crouse J.R., III, Hunninghake D.B., Davidson M.H., Escobar I.D., Stalenhoef A.F., Paragh G., Ma P.T., Liu M., Melino M.R., O’Grady L., Mercuri M., Mitchel Y.B. A comparison of simvastatin and atorvastatin up to maximal recommended doses in a large multicenter randomized clinical trial. Curr. Med. Res. Opin. 2001;17(1):43–50. [http://dx.doi.org/10.1185/ 0300799039117026]. [PubMed] [Google Scholar]
- 218.van der Most P.J., Dolga A.M., Nijholt I.M., Luiten P.G., Eisel U.L. Statins: mechanisms of neuroprotection. Prog. Neurobiol. 2009;88(1):64–75. doi: 10.1016/j.pneurobio.2009.02.002. [http://dx.doi.org/10.1016/j.pneurobio.2009.02. 002]. [DOI] [PubMed] [Google Scholar]
- 219.Sierra S., Ramos M.C., Molina P., Esteo C., Vazquez J.A., Burgos J.S. Statins as neuroprotectants: a comparative in vitro study of lipophilicity, blood-brain-barrier penetration, lowering of brain cholesterol, and decrease of neuron cell death. J. Alzheimers Dis. 2011;23(2):307–318. doi: 10.3233/JAD-2010-101179. [http://dx.doi.org/10.3233/JAD-2010-101179]. [DOI] [PubMed] [Google Scholar]
- 220.Wolozin B., Wang S.W., Li N.C., Lee A., Lee T.A., Kazis L.E. Simvastatin is associated with a reduced incidence of dementia and Parkinson’s disease. BMC Med. 2007;5:20. doi: 10.1186/1741-7015-5-20. [http://dx.doi.org/10. 1186/1741-7015-5-20]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Huang X., Alonso A., Guo X., Umbach D.M., Lichtenstein M.L., Ballantyne C.M., Mailman R.B., Mosley T.H., Chen H. Statins, plasma cholesterol, and risk of Parkinson’s disease: a prospective study. Mov. Disord. 2015;30(4):552–559. doi: 10.1002/mds.26152. [http://dx.doi. org/10.1002/mds.26152]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Valenza M., Cattaneo E. Emerging roles for cholesterol in Huntington’s disease. Trends Neurosci. 2011;34(9):474–486. doi: 10.1016/j.tins.2011.06.005. [http://dx.doi.org/10.1016/j.tins.2011.06.005]. [DOI] [PubMed] [Google Scholar]
- 223.Chen J.Y., Tran C., Hwang L., Deng G., Jung M.E., Faull K.F., Levine M.S., Cepeda C. Partial Amelioration of Peripheral and Central Symptoms of Huntington’s Disease via Modulation of Lipid Metabolism. J. Huntingtons Dis. 2016;5(1):65–81. doi: 10.3233/JHD-150181. [http:// dx.doi.org/10.3233/JHD-150181]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Valenza M., Leoni V., Karasinska J.M., Petricca L., Fan J., Carroll J., Pouladi M.A., Fossale E., Nguyen H.P., Riess O., MacDonald M., Wellington C., DiDonato S., Hayden M., Cattaneo E. Cholesterol defect is marked across multiple rodent models of Huntington’s disease and is manifest in astrocytes. J. Neurosci. 2010;30(32):10844–10850. doi: 10.1523/JNEUROSCI.0917-10.2010. [http://dx.doi.org/10.1523/JNEUROSCI. 0917-10.2010]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Valenza M., Marullo M., Di Paolo E., Cesana E., Zuccato C., Biella G., Cattaneo E. Disruption of astrocyte-neuron cholesterol cross talk affects neuronal function in Huntington’s disease. Cell Death Differ. 2015;22(4):690–702. doi: 10.1038/cdd.2014.162. [http://dx.doi.org/10.1038/cdd. 2014.162]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Shankaran M., Di Paolo E., Leoni V., Caccia C., Ferrari Bardile C., Mohammed H., Di Donato S., Kwak S., Marchionini D., Turner S., Cattaneo E., Valenza M. Early and brain region-specific decrease of de novo cholesterol biosynthesis in Huntington’s disease: A cross-validation study in Q175 knock-in mice. Neurobiol. Dis. 2017;98:66–76. doi: 10.1016/j.nbd.2016.11.013. [http://dx.doi.org/10.1016/ j.nbd.2016.11.013]. [DOI] [PubMed] [Google Scholar]
- 227.Valenza M., Leoni V., Tarditi A., Mariotti C., Bjorkhem I., Di Donato S., Cattaneo E. Progressive dysfunction of the cholesterol biosynthesis pathway in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 2007;28(1):133–142. doi: 10.1016/j.nbd.2007.07.004. [http://dx.doi. org/10.1016/j.nbd.2007.07.004]. [DOI] [PubMed] [Google Scholar]
- 228.Trushina E., Singh R.D., Dyer R.B., Cao S., Shah V.H., Parton R.G., Pagano R.E., McMurray C.T. Mutant huntingtin inhibits clathrin-independent endocytosis and causes accumulation of cholesterol in vitro and in vivo. Hum. Mol. Genet. 2006;15(24):3578–3591. doi: 10.1093/hmg/ddl434. [http://dx.doi.org/10.1093/hmg/ddl434]. [DOI] [PubMed] [Google Scholar]
- 229.del Toro D., Xifro X., Pol A., Humbert S., Saudou F., Canals J.M., Alberch J. Altered cholesterol homeostasis contributes to enhanced excitotoxicity in Huntington’s disease. J. Neurochem. 2010;115(1):153–167. doi: 10.1111/j.1471-4159.2010.06912.x. [http://dx.doi.org/10.1111/j.1471-4159. 2010.06912.x]. [DOI] [PubMed] [Google Scholar]
- 230.Boussicault L., Alves S., Lamaziere A., Planques A., Heck N., Moumne L., Despres G., Bolte S., Hu A., Pages C., Galvan L., Piguet F., Aubourg P., Cartier N., Caboche J., Betuing S. CYP46A1, the rate-limiting enzyme for cholesterol degradation, is neuroprotective in Huntington’s disease. Brain. 2016;139(Pt 3):953–970. doi: 10.1093/brain/awv384. [http://dx.doi.org/10.1093/brain/awv384]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Valenza M., Cattaneo E. Cholesterol dysfunction in neurodegenerative diseases: is Huntington’s disease in the list? Prog. Neurobiol. 2006;80(4):165–176. doi: 10.1016/j.pneurobio.2006.09.005. [http://dx.doi.org/10.1016/j.pneurobio. 2006.09.005]. [DOI] [PubMed] [Google Scholar]
- 232.Burns M.P., Igbavboa U., Wang L., Wood W.G., Duff K. Cholesterol distribution, not total levels, correlate with altered amyloid precursor protein processing in statin-treated mice. Neuromol. Med. 2006;8(3):319–328. doi: 10.1385/nmm:8:3:319. [http://dx.doi.org/10.1385/NMM:8:3:319]. [DOI] [PubMed] [Google Scholar]
- 233.Hindler K., Cleeland C.S., Rivera E., Collard C.D. The role of statins in cancer therapy. Oncologist. 2006;11(3):306–315. doi: 10.1634/theoncologist.11-3-306. [http://dx.doi.org/10.1634/theoncologist.11-3-306]. [DOI] [PubMed] [Google Scholar]
- 234.Wejde J., Hjertman M., Carlberg M., Egestad B., Griffiths W.J., Sjovall J., Larsson O. Dolichol-like lipids with stimulatory effect on DNA synthesis: substrates for protein dolichylation? J. Cell. Biochem. 1998;71(4):502–514. [http://dx.doi.org/10.1002/ (SICI)1097-4644(19981215)71:4<502:AID-JCB5>3.0.CO;2-P]. [PubMed] [Google Scholar]
- 235.Murtola T.J., Visvanathan K., Artama M., Vainio H., Pukkala E. Statin use and breast cancer survival: a nationwide cohort study from Finland. PLoS One. 2014;9(10):e110231. doi: 10.1371/journal.pone.0110231. [http://dx.doi.org/ 10.1371/journal.pone.0110231]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Song C., Park S., Park J., Shim M., Kim A., Jeong I.G., Hong J.H., Kim C.S., Ahn H. Statin use after radical prostatectomy reduces biochemical recurrence in men with prostate cancer. Prostate. 2015;75(2):211–217. doi: 10.1002/pros.22907. [http://dx.doi.org/10.1002/pros.22907]. [DOI] [PubMed] [Google Scholar]
- 237.Wei T.T., Lin Y.T., Chen W.S., Luo P., Lin Y.C., Shun C.T., Lin Y.H., Chen J.B., Chen N.W., Fang J.M., Wu M.S., Yang K.C., Chang L.C., Tai K.Y., Liang J.T., Chen C.C. Dual targeting of 3-Hydroxy-3-methylglutaryl coenzyme a reductase and histone deacetylase as a therapy for colorectal cancer. EBioMedicine. 2016;10:124–136. doi: 10.1016/j.ebiom.2016.07.019. [http://dx.doi.org/10.1016/j.ebiom.2016.07.019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Minden M.D., Dimitroulakos J., Nohynek D., Penn L.Z. Lovastatin induced control of blast cell growth in an elderly patient with acute myeloblastic leukemia. Leuk. Lymphoma. 2001;40(5-6):659–662. doi: 10.3109/10428190109097663. [http://dx.doi.org/10.3109/10428190109097663]. [DOI] [PubMed] [Google Scholar]
- 239.Kawata S., Yamasaki E., Nagase T., Inui Y., Ito N., Matsuda Y., Inada M., Tamura S., Noda S., Imai Y., Matsuzawa Y. Effect of pravastatin on survival in patients with advanced hepatocellular carcinoma. A randomized controlled trial. Br. J. Cancer. 2001;84(7):886–891. doi: 10.1054/bjoc.2000.1716. [http://dx.doi.org/10.1054/bjoc.2000.1716]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Kim W.S., Kim M.M., Choi H.J., Yoon S.S., Lee M.H., Park K., Park C.H., Kang W.K. Phase II study of high-dose lovastatin in patients with advanced gastric adenocarcinoma. Invest. New Drugs. 2001;19(1):81–83. doi: 10.1023/a:1006481423298. [http://dx.doi.org/10.1023/A:1006481423298]. [DOI] [PubMed] [Google Scholar]
- 241.Knox J.J., Siu L.L., Chen E., Dimitroulakos J., Kamel-Reid S., Moore M.J., Chin S., Irish J., LaFramboise S., Oza A.M. A Phase I trial of prolonged administration of lovastatin in patients with recurrent or metastatic squamous cell carcinoma of the head and neck or of the cervix. Eur. J. Cancer. 2005;41(4):523–530. doi: 10.1016/j.ejca.2004.12.013. [http://dx.doi.org/10.1016/j.ejca.2004.12.013]. [DOI] [PubMed] [Google Scholar]
- 242.Ohgaki H. Epidemiology of brain tumors. Methods Mol. Biol. 2009;472:323–342. doi: 10.1007/978-1-60327-492-0_14. [http://dx.doi.org/10.1007/978-1-60327-492-0_14]. [DOI] [PubMed] [Google Scholar]
- 243.Appin C.L., Brat D.J. Molecular pathways in gliomagenesis and their relevance to neuropathologic diagnosis. Adv. Anat. Pathol. 2015;22(1):50–58. doi: 10.1097/PAP.0000000000000048. [http://dx.doi.org/10.1097/PAP.00000000000000 48]. [DOI] [PubMed] [Google Scholar]
- 244.Koyuturk M., Ersoz M., Altiok N. Simvastatin induces proliferation inhibition and apoptosis in C6 glioma cells via c-jun N-terminal kinase. Neurosci. Lett. 2004;370(2-3):212–217. doi: 10.1016/j.neulet.2004.08.020. [http:// dx.doi.org/10.1016/j.neulet.2004.08.020]. [DOI] [PubMed] [Google Scholar]
- 245.Yanae M., Tsubaki M., Satou T., Itoh T., Imano M., Yamazoe Y., Nishida S. Statin-induced apoptosis via the suppression of ERK1/2 and Akt activation by inhibition of the geranylgeranyl-pyrophosphate biosynthesis in glioblastoma. J. Exp. Clin. Cancer Res. 2011;30:74. doi: 10.1186/1756-9966-30-74. [http://dx.doi.org/10.1186/1756-9966-30-74]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Jiang Z., Zheng X., Lytle R.A., Higashikubo R., Rich K.M. Lovastatin-induced up-regulation of the BH3-only protein, Bim, and cell death in glioblastoma cells. J. Neurochem. 2004;89(1):168–178. doi: 10.1111/j.1471-4159.2004.02319.x. [http://dx.doi.org/10.1111/j.1471-4159.2004.02319.x]. [DOI] [PubMed] [Google Scholar]
- 247.Oliveira K.A., Dal-Cim T., Lopes F.G., Ludka F.K., Nedel C.B., Tasca C.I. Atorvastatin promotes cytotoxicity and reduces migration and proliferation of human A172 glioma cells. Mol. Neurobiol. 2018;55:1509–1523. doi: 10.1007/s12035-017-0423-8. [DOI] [PubMed] [Google Scholar]
- 248.Jiang P., Mukthavaram R., Chao Y., Bharati I.S., Fogal V., Pastorino S., Cong X., Nomura N., Gallagher M., Abbasi T., Vali S., Pingle S.C., Makale M., Kesari S. Novel anti-glioblastoma agents and therapeutic combinations identified from a collection of FDA approved drugs. J. Transl. Med. 2014;12:13. doi: 10.1186/1479-5876-12-13. [http://dx.doi.org/10.1186/1479-5876-12-13]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kikuchi T., Nagata Y., Abe T. In vitro and in vivo antiproliferative effects of simvastatin, an HMG-CoA reductase inhibitor, on human glioma cells. J. Neurooncol. 1997;34(3):233–239. doi: 10.1023/a:1005753523949. [http:// dx.doi.org/10.1023/A:1005753523949]. [DOI] [PubMed] [Google Scholar]
- 250.Larner J., Jane J., Laws E., Packer R., Myers C., Shaffrey M. A phase I-II trial of lovastatin for anaplastic astrocytoma and glioblastoma multiforme. Am. J. Clin. Oncol. 1998;21(6):579–583. doi: 10.1097/00000421-199812000-00010. [http://dx.doi.org/10.1097/00000421-199812000-00010]. [DOI] [PubMed] [Google Scholar]
- 251.Ferris J.S., McCoy L., Neugut A.I., Wrensch M., Lai R. HMG CoA reductase inhibitors, NSAIDs and risk of glioma. Int. J. Cancer. 2012;131(6):E1031–E1037. doi: 10.1002/ijc.27536. [http://dx.doi.org/10.1002/ijc.27536]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Gaist D., Andersen L., Hallas J., Sorensen H.T., Schroder H.D., Friis S. Use of statins and risk of glioma: a nationwide case-control study in Denmark. Br. J. Cancer. 2013;108(3):715–720. doi: 10.1038/bjc.2012.536. [http://dx.doi.org/10.1038/bjc.2012.536]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Bhavsar S., Hagan K., Arunkumar R., Potylchansky Y., Grasu R., Dang A., Carlson R., Cowels C., Arnold B., Rahlfs T.F., Lipski I., Walsh C., Nguyen A.T., Feng L., Cata J.P. Preoperative statin use is not associated with improvement in survival after glioblastoma surgery. J. Clin. Neurosci. 2016;31:176–180. doi: 10.1016/j.jocn.2016.03.010. [http://dx.doi.org/10.1016/j.jocn.2016.03.010]. [DOI] [PubMed] [Google Scholar]
- 254.Butterick T.A., Igbavboa U., Eckert G.P., Sun G.Y., Weisman G.A., Muller W.E., Wood W.G. Simvastatin stimulates production of the antiapoptotic protein Bcl-2 via endothelin-1 and NFATc3 in SH-SY5Y cells. Mol. Neurobiol. 2010;41(2-3):384–391. doi: 10.1007/s12035-010-8122-8. [http://dx.doi.org/10.1007/s12035-010-8122-8]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Wang H.X., Gao W.J. Cell type-specific development of NMDA receptors in the interneurons of rat prefrontal cortex. Neuropsychopharmacology. 2009;34(8):2028–2040. doi: 10.1038/npp.2009.20. [http://dx.doi.org/10.1038/ npp.2009.20]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Citraro R., Chimirri S., Aiello R., Gallelli L., Trimboli F., Britti D., De Sarro G., Russo E. Protective effects of some statins on epileptogenesis and depressive-like behavior in WAG/Rij rats, a genetic animal model of absence epilepsy. Epilepsia. 2014;55(8):1284–1291. doi: 10.1111/epi.12686. [http://dx.doi.org/10.1111/epi.12686]. [DOI] [PubMed] [Google Scholar]
- 257.Niehues da Cruz J.l., Delwing de Lima D., Delwing Dal Magro D.b., Geraldo Pereira da Cruz J. The power of classic music to reduce anxiety in rats treated with simvastatin. Basic Clin. Neurosci. 2011;2(4):5–11. [Google Scholar]
- 258.Can O.D., Ulupinar E., Ozkay U.D., Yegin B., Ozturk Y. The effect of simvastatin treatment on behavioral parameters, cognitive performance, and hippocampal morphology in rats fed a standard or a high-fat diet. Behav. Pharmacol. 2012;23(5-6):582–592. doi: 10.1097/FBP.0b013e328356c3f2. [http://dx.doi.org/10.1097/FBP.0b013e328356c3f2]. [DOI] [PubMed] [Google Scholar]
- 259.Zanoli P., Rivasi M., Zavatti M., Brusiani F., Baraldi M. New insight in the neuropharmacological activity of Humulus lupulus L. J. Ethnopharmacol. 2005;102(1):102–106. doi: 10.1016/j.jep.2005.05.040. [http://dx.doi.org/10. 1016/j.jep.2005.05.040]. [DOI] [PubMed] [Google Scholar]
- 260.Maggo S., Clark D., Ashton J.C. The effect of statins on performance in the Morris water maze in guinea pig. Eur. J. Pharmacol. 2012;674(2-3):287–293. doi: 10.1016/j.ejphar.2011.11.006. [http://dx.doi.org/10.1016/j.ejphar. 2011.11.006]. [DOI] [PubMed] [Google Scholar]
- 261.File S.E. Naloxone reduces social and exploratory activity in the rat. Psychopharmacology (Berl.) 1980;71(1):41–44. doi: 10.1007/BF00433250. [http://dx. doi.org/10.1007/BF00433250]. [DOI] [PubMed] [Google Scholar]
- 262.File S.E., Seth P. A review of 25 years of the social interaction test. Eur. J. Pharmacol. 2003;463(1-3):35–53. doi: 10.1016/s0014-2999(03)01273-1. [http://dx.doi.org/ 10.1016/S0014-2999(03)01273-1]. [DOI] [PubMed] [Google Scholar]
- 263.Pellow S., File S.E. Anxiolytic and anxiogenic drug effects on exploratory activity in an elevated plus-maze: a novel test of anxiety in the rat. Pharmacol. Biochem. Behav. 1986;24(3):525–529. doi: 10.1016/0091-3057(86)90552-6. [http://dx.doi.org/10.1016/0091-3057(86)90552-6]. [DOI] [PubMed] [Google Scholar]
- 264.Gonzalez L.E., Andrews N., File S.E. 5-HT1A and benzodiazepine receptors in the basolateral amygdala modulate anxiety in the social interaction test, but not in the elevated plus-maze. Brain Res. 1996;732(1-2):145–153. doi: 10.1016/0006-8993(96)00517-3. [http://dx.doi.org/10.1016/0006-8993(96)00517-3]. [DOI] [PubMed] [Google Scholar]
- 265.File S. E. Usefulness of animal models with newer anxiolytics. 1992. [DOI] [PubMed]
- 266.Cheeta S., Kenny P.J., File S.E. Hippocampal and septal injections of nicotine and 8-OH-DPAT distinguish among different animal tests of anxiety. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2000;24(7):1053–1067. doi: 10.1016/s0278-5846(00)00129-9. [http://dx.doi.org/10.1016/S0278-5846(00)00129-9]. [DOI] [PubMed] [Google Scholar]
- 267.File S.E., Cheeta S., Kenny P.J. Neurobiological mechanisms by which nicotine mediates different types of anxiety. Eur. J. Pharmacol. 2000;393(1-3):231–236. doi: 10.1016/s0014-2999(99)00889-4. [http://dx.doi.org/10.1016/S0014-2999(99)00889-4]. [DOI] [PubMed] [Google Scholar]
- 268.File S.E., Lippa A.S., Beer B., Lippa M.T. Animal tests of anxiety. 2004. [DOI] [PubMed] [Google Scholar]
- 269.File S.E., Hyde J.R. Can social interaction be used to measure anxiety? Br. J. Pharmacol. 1978;62(1):19–24. doi: 10.1111/j.1476-5381.1978.tb07001.x. [http://dx.doi.org/ 10.1111/j.1476-5381.1978.tb07001.x]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Heim C., Wagner D., Maloney E., Papanicolaou D.A., Solomon L., Jones J.F., Unger E.R., Reeves W.C. Early adverse experience and risk for chronic fatigue syndrome: results from a population-based study. Arch. Gen. Psychiatry. 2006;63(11):1258–1266. doi: 10.1001/archpsyc.63.11.1258. [http://dx.doi.org/10.1001/archpsyc.63.11.1258]. [DOI] [PubMed] [Google Scholar]
- 271.Kumar A., Vashist A., Kumar P., Kalonia H., Mishra J. Protective effect of HMG CoA reductase inhibitors against running wheel activity induced fatigue, anxiety like behavior, oxidative stress and mitochondrial dysfunction in mice. Pharmacol. Rep. 2012;64(6):1326–1336. doi: 10.1016/s1734-1140(12)70930-1. [http://dx.doi.org/10.1016/S1734-1140(12)70930-1]. [DOI] [PubMed] [Google Scholar]
- 272.Tonstad S. Children and statins. Acta Paediatr. 2003;92(9):1001–1002. doi: 10.1080/08035250310004900. [http://dx.doi.org/10.1111/j.1651-2227.2003.tb02565.x]. [DOI] [PubMed] [Google Scholar]
- 273.Tuccori M., Lapi F., Testi A., Coli D., Moretti U., Vannacci A., Motola D., Salvo F., Rivolta A.L., Blandizzi C., Mugelli A., Del Tacca M. Statin-associated psychiatric adverse events: a case/non-case evaluation of an Italian database of spontaneous adverse drug reaction reporting. Drug Saf. 2008;31(12):1115–1123. doi: 10.2165/0002018-200831120-00007. [http://dx.doi.org/10.2165/0002018-200831120-00007]. [DOI] [PubMed] [Google Scholar]
- 274.Mansi I., Frei C.R., Pugh M.J., Mortensen E.M. Psychologic disorders and statin use: a propensity score-matched analysis. Pharmacotherapy. 2013;33(6):615–626. doi: 10.1002/phar.1272. [http://dx.doi.org/10. 1002/phar.1272]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Hsia J., MacFadyen J.G., Monyak J., Ridker P.M. Cardiovascular event reduction and adverse events among subjects attaining low-density lipoprotein cholesterol <50 mg/dl with rosuvastatin. The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). J. Am. Coll. Cardiol. 2011;57(16):1666–1675. doi: 10.1016/j.jacc.2010.09.082. [http://dx.doi.org/10.1016/ j.jacc.2010.09.082]. [DOI] [PubMed] [Google Scholar]
- 276.Pasco J.A., Nicholson G.C., Williams L.J., Jacka F.N., Henry M.J., Kotowicz M.A., Schneider H.G., Leonard B.E., Berk M. Association of high-sensitivity C-reactive protein with de novo major depression. Br. J. Psychiatry. 2010;197(5):372–377. doi: 10.1192/bjp.bp.109.076430. [http:// dx.doi.org/10.1192/bjp.bp.109.076430]. [DOI] [PubMed] [Google Scholar]
- 277.Yang C.C., Jick S.S., Jick H. Lipid-lowering drugs and the risk of depression and suicidal behavior. Arch. Intern. Med. 2003;163(16):1926–1932. doi: 10.1001/archinte.163.16.1926. [http://dx.doi.org/10.1001/archinte.163.16. 1926]. [DOI] [PubMed] [Google Scholar]
- 278.Chuang C.S., Yang T.Y., Muo C.H., Su H.L., Sung F.C. ; Kao C.H. Hyperlipidemia, statin use and the risk of developing depression: a nationwide retrospective cohort study. Gen. Hosp. Psychiatry. 2014;36(5):497–501. doi: 10.1016/j.genhosppsych.2014.05.008. [http://dx.doi.org/10.1016/ j.genhosppsych.2014.05.008]. [DOI] [PubMed] [Google Scholar]
- 279.LeDoux J.E. Emotion circuits in the brain. Annu. Rev. Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [http://dx.doi.org/10.1146/annurev.neuro.23.1.155]. [DOI] [PubMed] [Google Scholar]
- 280.Zaloshnja E., Miller T., Langlois J.A., Selassie A.W. Prevalence of long-term disability from traumatic brain injury in the civilian population of the United States, 2005. J. Head Trauma Rehabil. 2008;23(6):394–400. doi: 10.1097/01.HTR.0000341435.52004.ac. [http://dx.doi.org/10.1097/01.HTR.0000341435. 52004.ac]. [DOI] [PubMed] [Google Scholar]
- 281.Allan S.M., Harrison D.C., Read S., Collins B., Parsons A.A., Philpott K., Rothwell N.J. Selective increases in cytokine expression in the rat brain in response to striatal injection of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate and interleukin-1. Brain Res. Mol. Brain Res. 2001;93(2):180–189. doi: 10.1016/s0169-328x(01)00211-x. [http://dx.doi. org/10.1016/S0169-328X(01)00211-X]. [DOI] [PubMed] [Google Scholar]
- 282.Zhang Q., Raoof M., Chen Y., Sumi Y., Sursal T., Junger W., Brohi K., Itagaki K., Hauser C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107. doi: 10.1038/nature08780. [http://dx.doi.org/10.1038/nature08780]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Miller A.H., Maletic V., Raison C.L. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol. Psychiatry. 2009;65(9):732–741. doi: 10.1016/j.biopsych.2008.11.029. [http://dx.doi. org/10.1016/j.biopsych.2008.11.029]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Wajant H., Henkler F., Scheurich P. The TNF-receptor-associated factor family: scaffold molecules for cytokine receptors, kinases and their regulators. Cell. Signal. 2001;13(6):389–400. doi: 10.1016/s0898-6568(01)00160-7. [http://dx.doi.org/10.1016/S0898-6568(01)00160-7]. [DOI] [PubMed] [Google Scholar]
- 285.Kilic F.S., Ozatik Y., Kaygisiz B., Baydemir C., Erol K. Acute antidepressant and anxiolytic effects of simvastatin and its mechanisms in rats. Neurosciences (Riyadh) 2012;17(1):39–43. [PubMed] [Google Scholar]
- 286.Lechleitner M., Hoppichler F., Konwalinka G., Patsch J.R., Braunsteiner H. Depressive symptoms in hypercholesterolaemic patients treated with pravastatin. Lancet. 1992;340(8824):910. doi: 10.1016/0140-6736(92)93318-h. [http://dx.doi.org/10.1016/0140-6736(92)93318-H]. [DOI] [PubMed] [Google Scholar]
- 287.Morales K., Wittink M., Datto C., DiFilippo S., Cary M., TenHave T., Katz I.R. Simvastatin causes changes in affective processes in elderly volunteers. J. Am. Geriatr. Soc. 2006;54(1):70–76. doi: 10.1111/j.1532-5415.2005.00542.x. [http://dx.doi.org/10.1111/j.1532-5415.2005.00542.x]. [DOI] [PubMed] [Google Scholar]
- 288.Hyyppa M.T., Kronholm E., Virtanen A., Leino A., Jula A. Does simvastatin affect mood and steroid hormone levels in hypercholesterolemic men? A randomized double-blind trial. Psychoneuroendocrinology. 2003;28(2):181–194. doi: 10.1016/s0306-4530(02)00014-8. [http://dx.doi.org/10.1016/ S0306-4530(02)00014-8]. [DOI] [PubMed] [Google Scholar]
- 289.Osborn D.P., Nazareth I., King M.B. Physical activity, dietary habits and Coronary Heart Disease risk factor knowledge amongst people with severe mental illness: a cross sectional comparative study in primary care. Soc. Psychiatry Psychiatr. Epidemiol. 2007;42(10):787–793. doi: 10.1007/s00127-007-0247-3. [http://dx.doi.org/10.1007/s00127-007-0247-3]. [DOI] [PubMed] [Google Scholar]
- 290.Laursen T.M., Munk-Olsen T., Gasse C. Chronic somatic comorbidity and excess mortality due to natural causes in persons with schizophrenia or bipolar affective disorder. PLoS One. 2011;6(9):1–7. doi: 10.1371/journal.pone.0024597. [http://dx.doi.org/10.1371/journal.pone.0024597]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Berk M., Copolov D., Dean O., Lu K., Jeavons S., Schapkaitz I., Anderson-Hunt M., Judd F., Katz F., Katz P., Ording-Jespersen S., Little J., Conus P., Cuenod M., Do K.Q., Bush A.I. N-acetyl cysteine as a glutathione precursor for schizophrenia--a double-blind, randomized, placebo-controlled trial. Biol. Psychiatry. 2008;64(5):361–368. doi: 10.1016/j.biopsych.2008.03.004. [http://dx.doi.org/10.1016/j.biopsych. 2008.03.004]. [DOI] [PubMed] [Google Scholar]
- 292.Chang S.H., Chiang S.Y., Chiu C.C., Tsai C.C., Tsai H.H., Huang C.Y., Hsu T.C., Tzang B.S. Expression of anti-cardiolipin antibodies and inflammatory associated factors in patients with schizophrenia. Psychiatry Res. 2011;187(3):341–346. doi: 10.1016/j.psychres.2010.04.049. [http://dx. doi.org/10.1016/j.psychres.2010.04.049]. [DOI] [PubMed] [Google Scholar]
- 293.Song X.Q., Lv L.X., Li W.Q., Hao Y.H., Zhao J.P. The interaction of nuclear factor-kappa B and cytokines is associated with schizophrenia. Biol. Psychiatry. 2009;65(6):481–488. doi: 10.1016/j.biopsych.2008.10.018. [http://dx. doi.org/10.1016/j.biopsych.2008.10.018]. [DOI] [PubMed] [Google Scholar]
- 294.Ghanizadeh A., Dehbozorgi S., Omrani S.M., Rezaei Z. Minocycline as add-on treatment decreases the negative symptoms of schizophrenia; a randomized placebo-controlled clinical trial. Recent Pat. Inflamm. Allergy Drug Discov. 2014;8(3):211–215. doi: 10.2174/1872213x08666141029123524. [http://dx.doi.org/10. 2174/1872213X08666141029123524]. [DOI] [PubMed] [Google Scholar]
- 295.Lilly S.M., Mortensen E.M., Frei C.R., Pugh M.J., Mansi I.A. Comparison of the risk of psychological and cognitive disorders between persistent and nonpersistent statin users. Am. J. Cardiol. 2014;114(7):1035–1039. doi: 10.1016/j.amjcard.2014.07.010. [http://dx.doi.org/10.1016/j.amjcard.2014. 07.010]. [DOI] [PubMed] [Google Scholar]
- 296.Zhao J., Zhang X., Dong L., Wen Y., Cui L. The many roles of statins in ischemic stroke. Curr. Neuropharmacol. 2014;12(6):564–574. doi: 10.2174/1570159X12666140923210929. [http://dx.doi.org/10.2174/1570159X12666140923210929]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Ni Chroinin D., Asplund K., Asberg S., Callaly E., Cuadrado-Godia E., Diez-Tejedor E., Di Napoli M., Engelter S.T., Furie K.L., Giannopoulos S., Gotto A.M., Jr, Hannon N., Jonsson F., Kapral M.K., Marti-Fabregas J., Martinez-Sanchez P., Milionis H.J., Montaner J., Muscari A., Pikija S., Probstfield J., Rost N.S., Thrift A.G., Vemmos K., Kelly P.J. Statin therapy and outcome after ischemic stroke: systematic review and meta-analysis of observational studies and randomized trials. Stroke. 2013;44(2):448–456. doi: 10.1161/STROKEAHA.112.668277. [http://dx.doi.org/10.1161/STROKEAHA.112.668277]. [DOI] [PubMed] [Google Scholar]
- 298.Merwick A., Albers G.W., Arsava E.M., Ay H., Calvet D., Coutts S.B., Cucchiara B.L., Demchuk A.M., Giles M.F., Mas J.L., Olivot J.M., Purroy F., Rothwell P.M., Saver J.L., Sharma V.K., Tsivgoulis G., Kelly P.J. Reduction in early stroke risk in carotid stenosis with transient ischemic attack associated with statin treatment. Stroke. 2013;44(10):2814–2820. doi: 10.1161/STROKEAHA.113.001576. [http://dx.doi.org/10. 1161/STROKEAHA.113.001576]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Al-Khaled M., Matthis C., Eggers J. Statin treatment in patients with acute ischemic stroke. Int. J. Stroke. 2014;9(5):597–601. doi: 10.1111/ijs.12256. [http://dx.doi.org/10.1111/ijs.12256]. [DOI] [PubMed] [Google Scholar]
- 300.Flint A.C., Kamel H., Navi B.B., Rao V.A., Faigeles B.S., Conell C., Klingman J.G., Sidney S., Hills N.K., Sorel M. ; Cullen S.P., Johnston S.C. Statin use during ischemic stroke hospitalization is strongly associated with improved poststroke survival. Stroke. 2012;43(1):147–154. doi: 10.1161/STROKEAHA.111.627729. [http://dx.doi.org/10.1161/ STROKEAHA.111.627729]. [DOI] [PubMed] [Google Scholar]
- 301.Tonkin A., Simes R., Sharpe N., Thomson A. The long term intervention with pravastatin in ischaemic disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N. Engl. J. Med. 1998;339(19):1349–1357. doi: 10.1056/NEJM199811053391902. [http://dx.doi.org/10.1056/NEJM199811053391902]. [DOI] [PubMed] [Google Scholar]
- 302.Schwartz G.G., Olsson A.G., Ezekowitz M.D., Ganz P., Oliver M.F., Waters D., Zeiher A., Chaitman B.R., Leslie S., Stern T. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA. 2001;285(13):1711–1718. doi: 10.1001/jama.285.13.1711. [http://dx.doi.org/10.1001/ jama.285.13.1711]. [DOI] [PubMed] [Google Scholar]
- 303.Sirol M., Bouzamondo A., Sanchez P., Lechat P. Does statin therapy reduce the risk of stroke? A meta-analysis. Ann. Med. Interne (Paris) 2001;152(3):188–193. [PubMed] [Google Scholar]
- 304.Martinez-Sanchez P., Fuentes B., Martinez-Martinez M., Ruiz-Ares G., Fernandez-Travieso J., Sanz-Cuesta B.E., Cuellar-Gamboa L., Diaz-Dominguez E., Diez-Tejedor E. Treatment with statins and ischemic stroke severity: does the dose matter? Neurology. 2013;80(19):1800–1805. doi: 10.1212/WNL.0b013e3182918d38. [http://dx.doi.org/10.1212/WNL. 0b013e3182918d38]. [DOI] [PubMed] [Google Scholar]
- 305.Endres M., Laufs U., Huang Z., Nakamura T., Huang P., Moskowitz M.A., Liao J.K. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA. 1998;95(15):8880–8885. doi: 10.1073/pnas.95.15.8880. [http://dx.doi.org/10.1073/pnas.95.15.8880]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Kureishi Y., Luo Z., Shiojima I., Bialik A., Fulton D., Lefer D.J., Sessa W.C., Walsh K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat. Med. 2000;6(9):1004–1010. doi: 10.1038/79510. [http://dx.doi.org/10.1038/79510]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Laufs U., Liao J.K. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J. Biol. Chem. 1998;273(37):24266–24271. doi: 10.1074/jbc.273.37.24266. [http://dx.doi.org/10.1074/ jbc.273.37.24266]. [DOI] [PubMed] [Google Scholar]
- 308.Shao S., Xu M., Zhou J., Ge X., Chen G., Guo L., Luo L., Li K., Zhu Z., Zhang F. Atorvastatin attenuates ischemia/ reperfusion-induced hippocampal neurons injury Via Akt-nNOS-JNK signaling pathway. Cell. Mol. Neurobiol. 2017;37(4):753–762. doi: 10.1007/s10571-016-0412-x. [http://dx.doi.org/10.1007/s10571-016-0412-x]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Gasche Y., Copin J.C., Sugawara T., Fujimura M., Chan P.H. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2001;21(12):1393–1400. doi: 10.1097/00004647-200112000-00003. [http://dx.doi.org/10.1097/00004647-200112000-00003]. [DOI] [PubMed] [Google Scholar]
- 310.Woo M.S., Park J.S., Choi I.Y., Kim W.K., Kim H.S. Inhibition of MMP-3 or -9 suppresses lipopolysaccharide-induced expression of proinflammatory cytokines and iNOS in microglia. J. Neurochem. 2008;106(2):770–780. doi: 10.1111/j.1471-4159.2008.05430.x. [http://dx.doi.org/10.1111/j.1471-4159.2008.05430.x]. [DOI] [PubMed] [Google Scholar]
- 311.Liu X.S., Zhang Z.G., Zhang L., Morris D.C., Kapke A., Lu M., Chopp M. Atorvastatin downregulates tissue plasminogen activator-aggravated genes mediating coagulation and vascular permeability in single cerebral endothelial cells captured by laser microdissection. J. Cereb. Blood Flow Metab. 2006;26(6):787–796. doi: 10.1038/sj.jcbfm.9600227. [http://dx.doi.org/10.1038/sj.jcbfm.9600227]. [DOI] [PubMed] [Google Scholar]
- 312.Wang S., Lee S.R., Guo S.Z., Kim W.J., Montaner J. ; Wang X., Lo E.H. Reduction of tissue plasminogen activator-induced matrix metalloproteinase-9 by simvastatin in astrocytes. Stroke. 2006;37(7):1910–1912. doi: 10.1161/01.STR.0000226923.48905.39. [http://dx.doi.org/10.1161/01.STR. 0000226923.48905.39]. [DOI] [PubMed] [Google Scholar]
- 313.Sironi L., Banfi C., Brioschi M., Gelosa P., Guerrini U., Nobili E., Gianella A., Paoletti R., Tremoli E., Cimino M. Activation of NF-kB and ERK1/2 after permanent focal ischemia is abolished by simvastatin treatment. Neurobiol. Dis. 2006;22(2):445–451. doi: 10.1016/j.nbd.2005.12.004. [http://dx.doi.org/10.1016/j.nbd.2005.12.004]. [DOI] [PubMed] [Google Scholar]
- 314.Hong H., Zeng J.S., Kreulen D.L., Kaufman D.I., Chen A.F. Atorvastatin protects against cerebral infarction via inhibition of NADPH oxidase-derived superoxide in ischemic stroke. Am. J. Physiol. Heart Circ. Physiol. 2006;291(5):H2210–H2215. doi: 10.1152/ajpheart.01270.2005. [http://dx.doi.org/10.1152/ajpheart.01270.2005]. [DOI] [PubMed] [Google Scholar]
- 315.De Caterina R., Salvatore T., Marchioli R. Cholesterol-lowering interventions and stroke: Insights from IMPROVE-IT. Atherosclerosis. 2016;248:216–218. doi: 10.1016/j.atherosclerosis.2016.03.024. [http://dx.doi.org/10.1016/j.atherosclerosis. 2016.03.024]. [DOI] [PubMed] [Google Scholar]