

Abstract
Although accurate identification of Mycobacterium tuberculosis is the gold standard for tuberculosis (TB) diagnosis, there have been several reports of false-positive results. After identifying a sudden increase in extensively drug-resistant TB, false-positive mycobacterial culture tests were suspected, and we contacted the supranational reference center for molecular typing. In silico genotyping tests showed that isolates from all five patients had an identical genotype pattern, and all harbored the same Beijing strain based on sequence-based phylogenic analysis and drug-resistant single nucleotide polymorphisms (SNPs) analysis. We also used whole genome sequencing (WGS) to compare the SNPs of all isolates with a reference genome, and all were identical. We adapted WGS to efficiently detect false-positive MTB culture tests.
Keywords: Drug resistance, molecular epidemiology, molecular typing, single nucleotide polymorphism
In South Korea with intermediate tuberculosis (TB) burden, many laboratory institutions often face a high burden of work related to mycobacterial tests. Although accurate identification of Mycobacterium tuberculosis (MTB) complex is the gold standard for TB diagnosis, it is labor intensive and time-consuming and not always 100% accurate. There are several studies reporting false-positive MTB culture despite advances in microbiological tests.[1] If clinicians do not suspect them and perform further investigation, false-positive cultures of MTB can go unrecognized. Many molecular typing quality control tests have been developed for TB laboratories to detect false-positive cultures and cross-contamination.[2] In recent years, whole genome sequencing (WGS) has been used for MTB genotyping.[3] False-positive mycobacterial culture tests were retrospectively suspected among patients whose specimens yielded MTB with the same drug-resistant profile in our institution. We herein report the application of WGS to determine whether these isolates were identical strains.
Case Report
Patient P1 (supposed index case) was a 23-year-old female who was diagnosed with multidrug-resistant TB 2 years ago. Her acid-fast bacilli (AFB) culture was positive for MTB (S1–S3), which was later found to have an extensively drug-resistant profile through culture-based drug susceptibility test (DST) [Table 1 and Figure 1]. Patient P2 was a 75-year-old female who visited for a household contact investigation. Despite normal chest X-ray, a sputum AFB culture test revealed positive for MTB (S4). Patient P3, an 81-year-old female with hemoptysis, was initially diagnosed with bronchiectasis combined with pneumonia. Her AFB culture test using a bronchial aspirate specimen was positive for MTB (S7). Patient P4, an 81-year-old male with lung cancer, was diagnosed with drug-sensitive pulmonary TB. After 1 month of anti-TB treatment, a follow-up AFB culture test was performed and was positive for MTB (S6). Patient P5, a 74-year-old male with leukemia, was diagnosed with drug-sensitive pulmonary TB. After completing standard anti-TB treatment, a follow-up AFB culture test was performed on the last day, which was positive for MTB (S7).
Table 1.
List of specimens requested and their culture results
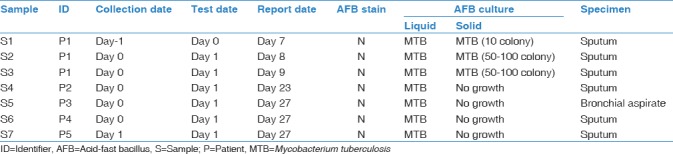
Figure 1.

Timeline of collecting samples, performing tests, and reporting test results, ID=Identifier; P=Patient; S=Sample; C=Collection of samples; T=Tests performed; R=Report of test results; D=Day; D0=Date of collecting samples, which revealed false-positive results; D1=Date of performing tests of samples, which revealed false-positive results
After P1's diagnosis of extensively drug-resistant TB, the rest of the patients (P2–P5) were subsequently reported to have the same resistance profile. Because of the sudden increase in extensively drug-resistant TB incidence, we suspected false-positive results and contacted the supranational reference center regarding the further investigation to investigate the origins of these MTB isolates.
To investigate epidemiological links between the isolates, in silico genotyping was performed using the total genotyping solution for TB (TGS-TB) website,[4] which showed that all five isolates had identical genotype patterns [Figure 2]. For WGS, genomic DNA extracted from individual isolates was sequenced using the Illumina MiSeq (Illumina, San Diego, CA, USA). A genomic DNA library was prepared using the Nextera XT Library Preparation Kit, and sequence data were produced using MiSeq Reagent Kit v3. The Qubit dsDNA High Sensitivity Assay Kit was used to quantify libraries for optimal cluster density (Thermo Fisher Scientific, CA, USA). Coverage of the H37Rv reference genome ranged from 98.70% to 98.87%. The average depth was 107.5. Sequence-based phylogenic analysis and DST were performed on all isolates using the using the online TB-Profiler tool.[5] Raw sequence (FASTQ format) was inputted to the website, which identified lineage-specific regions of difference (RD) and drug-resistant single nucleotide polymorphisms (SNPs). All isolates contained deletions in RD105, RD207, and RD181 according to sequence-based phylogenic analysis and represented Lineage 2_East Asian Beijing. SNP analysis demonstrated that all isolates contained the same nine SNPs related to drug resistance and had identical resistance patterns. We also compared the SNPs of all isolates with the H37Rv reference genome. All isolates contained 1572 SNPs compared with the reference genome, which were identical. Therefore, we confirmed that these five isolates were identical strains.
Figure 2.

The results of in silico genotyping. (a) In silico detection of IS6110 insertion site on five isolates (blue = forward; red = reverse). (b) In silico spoligotyping. (c) In silico 43 loci mycobacterial interspersed repetitive units-variable number of tandem repeats
Based on the results of genotypic tests, anti-TB treatments were discontinued in patients P2 and P3 [Table 2]. Since patient P4 was initially diagnosed with drug-sensitive TB, the physician-in-charge did not change the treatment regimen, and he successfully completed treatment. Patient P5 died of leukemia.
Table 2.
Summaries of patient’s medical records

Discussion
In this study, a WGS-based genotyping test was applied to accurately identify false-positive mycobacterial culture results. Although clinicians were suspicious when the results were reported, there was no evidence to confirm their false-positive nature. However, after culture-based DST results revealed the same extensively drug-resistant profile in five different patients in a single month, clinicians became alarmed, and an investigation was conducted using molecular strain-typing tests.
Molecular MTB typing has been used for various epidemiologic purposes, such as community outbreak investigation and identification of culture cross-contamination.[3] We used four different molecular typing methods: IS6110-RFLP, spoligotyping, mycobacterial interspersed repetitive units-variable number of tandem repeats (MIRU-VNTR) typing, and WGS. For MTB genetic fingerprinting, the gold standard is insertion sequence IS6110-RFLP.[6] The IS6110-RFLP method is highly discriminatory and reproducible, and its profiles are stable over time. Its main limitation is its low discriminatory power in isolates presenting five or fewer IS6110 copies, and thus, the use of additional typing methods such as spoligotyping and MIRU-VNTR is usually required to improve discrimination.[7] In recent years, WGS has also been used for MTB genotyping. WGS not only provides higher resolution than any other genotyping methods during outbreak examination but also it has the great advantage of simultaneously acquiring additional information, including drug-resistant patterns.[8] WGS has higher discriminatory power than classical genotyping and shows a better correlation with the spatial distribution of cases and contact tracing data.[9] Although the cost of WGS has rapidly declined, it is still too expensive to use as a routine diagnostic tool. In addition, WGS data interpretation and bioinformatics analysis require skilled experts, as commercialized WGS-based diagnostic tools have not been developed for MTB.
Several factors can result in false-positive mycobacterial culture tests, including contamination of medical sampling instruments, lack of skilled technicians, reagent contamination, equipment failure, and aerosol creation.[10] We interviewed health-care professionals in the mycobacteriology laboratory; however, we were unable to determine an obvious cause for the observed false-positive culture results.
We used WGS to efficiently detect false-positive mycobacterial culture tests. The results suggest that the potential for false-positive results in mycobacterial tests should be recognized and that new and advanced methods such as WGS can help prevent erroneous diagnosis and unneeded TB treatment.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
The study was conducted by the amended Declaration of Helsinki. The Institutional Review Board of Chungbuk National University Hospital approved the study protocol and waived the need for informed consent because no patients were at risk (IRB No 2018-02-005).
References
- 1.Schoch OD, Pfyffer GE, Buhl D, Paky A. False-positive Mycobacterium tuberculosis culture revealed by restriction fragment length polymorphism analysis. Infection. 2003;31:189–91. doi: 10.1007/s15010-002-3060-7. [DOI] [PubMed] [Google Scholar]
- 2.Martínez M, García de Viedma D, Alonso M, Andrés S, Bouza E, Cabezas T, et al. Impact of laboratory cross-contamination on molecular epidemiology studies of tuberculosis. J Clin Microbiol. 2006;44:2967–9. doi: 10.1128/JCM.00754-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ei PW, Aung WW, Lee JS, Choi GE, Chang CL. Molecular strain typing of Mycobacterium tuberculosis: A review of frequently used methods. J Korean Med Sci. 2016;31:1673–83. doi: 10.3346/jkms.2016.31.11.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sekizuka T, Yamashita A, Murase Y, Iwamoto T, Mitarai S, Kato S, et al. TGS-TB: Total genotyping solution for Mycobacterium tuberculosis using short-read whole-genome sequencing. PLoS One. 2015;10:e0142951. doi: 10.1371/journal.pone.0142951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coll F, McNerney R, Preston MD, Guerra-Assunção JA, Warry A, Hill-Cawthorne G, et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. 2015;7:51. doi: 10.1186/s13073-015-0164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci U S A. 2002;99:3684–9. doi: 10.1073/pnas.052548299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cowan LS, Mosher L, Diem L, Massey JP, Crawford JT. Variable-number tandem repeat typing of Mycobacterium tuberculosis isolates with low copy numbers of IS6110 by using mycobacterial interspersed repetitive units. J Clin Microbiol. 2002;40:1592–602. doi: 10.1128/JCM.40.5.1592-1602.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohl TA, Diel R, Harmsen D, Rothgänger J, Walter KM, Merker M, et al. Whole-genome-based Mycobacterium tuberculosis surveillance: A standardized, portable, and expandable approach. J Clin Microbiol. 2014;52:2479–86. doi: 10.1128/JCM.00567-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roetzer A, Diel R, Kohl TA, Rückert C, Nübel U, Blom J, et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: A longitudinal molecular epidemiological study. PLoS Med. 2013;10:e1001387. doi: 10.1371/journal.pmed.1001387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fitzpatrick L, Braden C, Cronin W, English J, Campbell E, Valway S, et al. Investigation of laboratory cross-contamination of Mycobacterium tuberculosis cultures. Clin Infect Dis. 2004;38:e52–4. doi: 10.1086/382076. [DOI] [PubMed] [Google Scholar]