

Abstract
Small-angle X-ray scattering (SAXS) is a powerful reemerging biophysical technique that can be used to directly analyze many properties related to the size and shape of a macromolecule in solution. For example, the radius of gyration and maximum diameter of a macromolecule can be readily extracted from SAXS data, as can information regarding how well folded a protein is. Similarly, the molecular weight of macromolecular complexes can be directly determined from the complex’s scattering profile, providing insight into the oligomeric state and stoichiometry of the assembly. Furthermore, recently developed procedures for ab initio shape determination can provide low-resolution (~20 Å) molecular envelopes of proteins/ complexes in their native state. In conjunction with high-resolution structural data, more sophisticated analysis of SAXS data can help address questions regarding conformational change, molecular flexibility, and populations of states within molecular ensembles. Because SAXS samples are easy to prepare and SAXS data is relatively easy to collect, the technique holds great promise for investigating the structure of macromolecules and their assemblies as well as monitoring and modeling their conformational changes. Here we describe typical steps in SAXS sample preparation and data collection and analysis and provide examples of SAXS analysis to investigate the structure and function of dengue virus NS3 and NS5.
Keywords: Small-angle X-ray scattering, Ab initio shape determination, Radius of gyration, Pair-wise distribution function
1. Introduction
Although small-angle X-ray scattering (SAXS) has only recently begun to garner the attention of nonspecialists, SAXS has a long history. In 1939, Guinier described the measurement of the radius of gyration of particles in solution from their diffuse X-ray scattering [1]. At the time, small-angle techniques provided a direct method for estimating the size of biological macromolecules, yet widespread application of the technique never gained momentum. The subsequent emergence of X-ray crystallography, electron microscopy, and NMR probably overshadowed the fledgling SAXS technique. Despite the considerable successes of these other structural methods, SAXS analysis is undergoing a renaissance. There are several reasons for this reemergence, but perhaps the most important are the growing availability of adequate X-ray sources to collect useful SAXS data and sufficient computational power to fully analyze SAXS data. Another important impetus for the rapid growth of SAXS is the number of recently developed methodologies for extracting structural information.
There are several advantages of SAXS analysis compared to other structural techniques such as X-ray crystallography and NMR. First, protein solutions can be directly analyzed with relatively few restrictions regarding buffer composition, salts, or pH, and thus protein molecules can be investigated in their native state in solution. Second, large protein molecules or virus particles (~500 Å in diameter) that are off-limits to NMR and X-ray crystallography can be studied by SAXS to determine their molecular shape and size. On the other hand, unlike electron microscopy, there is no lower size limit for analyzing structure via SAXS. Hence, SAXS analysis is compatible with virtually the entire range of molecular analyses. Thirdly, relatively small amounts of protein are required for SAXS analysis compared to analytical ultracentrifugation, NMR, or crystallography. Lastly, experimental setup and initial data analysis are fairly straightforward, as long as the protein solution is homogeneous and monodisperse.
In a SAXS experiment, a protein solution is exposed to X-rays, and the diffuse scattering at very small angles from the incident beam is recorded, typically on a 2-D area detector. This region of the scattered radiation contains low-resolution structural information and corresponds to data that would mostly be blocked by the beam stop in an X-ray crystallographic experiment. In contrast to atomic resolution structural techniques like NMR or X-ray crystallography, the high-resolution limit of a typical SAXS experiment is between 10 and 20 Å, and thus SAXS analysis only provides information regarding the global molecular shape of the object. From SAXS data, we can determine many parameters related to protein size such as molecular mass, radius of gyration, hydrated volume, and maximum diameter of the molecule. Additionally, it is possible to construct a low-resolution, 3-D molecular envelope using ab initio shape determination programs. Furthermore, SAXS is useful for evaluating different 3-D models of a protein; SAXS scattering curves can be calculated from a protein model and compared to the experimental scattering curves to validate the model. Similarly, SAXS has been used to evaluate crystal structures of multi-domain proteins that show two or more conformations. In this review, we will focus on the types of structural information that can be extracted from experimental SAXS data as well as the practical considerations that go into conducting a SAXS experiment.
2. Materials
2.1. Instrumentation
SAXS data collection requires a monochromatic X-ray source and a compatible detector system. SAXS data are usually collected either at synchrotron radiation sources or using an inhouse X-ray generator coupled with a specialized detector/camera for recording small-angle scattering data. Currently, SAXSess (Anton Paar), BioSAXS-1000 (Rigaku), and MICROPix (Bruker AXS) instruments are commercially available for collecting SAXS data on biological samples.
2.2. Protein and Buffer Solutions
Prepare protein solutions for SAXS analysis. Protein solutions need to be pure and monodisperse (see Note 1). A typical protocol involves purifying his-tagged proteins via metal-affinity and size-exclusion chromatography. Several protein concentrations between 1 and 10 mg/mL are initially used for data acquisition to determine the optimal concentration for the final data collection and analysis. Depending on the sample holder of the instrument, between 20 and 80 μL of protein solution is typically needed for each measurement.
Prepare buffer solution. Buffer solutions should contain the same exact components in the same concentrations as the protein solution, but without the protein present. Although we are only interested in scattering from our protein, scattering measured from a protein solution is the sum of scattering from the protein and the buffer. Scattering from the protein is calculated by subtraction of buffer scattering from protein solution scattering. It is thus essential that the buffer solution exactly matches the buffer present in the protein solution. It is recommended that the buffer solution is obtained from the protein solution by either buffer exchange or dialysis rather than separately preparing a buffer with the same composition. For example, the protein solution can be concentrated or buffer exchanged using an Amicon Ultra Centrifugal Filter Unit (Millipore) and the filtrate used as a “buffer” solution. Most of buffer conditions are compatible with SAXS data collection, provided there is an adequate difference in electron density between the buffer and the macromolecule of interest.
Minimize radiation damage. Radiation damage is a significant source of data deterioration during SAXS data collection. In order to reduce radiation damage during data collection, free radical scavengers such as ~5 % glycerol or 1–5 mM DTT can be added to protein and buffer solutions (see Note 2).
3. Methods
3.1. Data Collection
Plan experiments. SAXS can be measured either at synchrotron sources or using an in-house SAXS instrument. Typical data collection for an individual sample takes 5–10 s at a synchrotron source and 1–4 h using currently available in-house X-ray generators. Temperature-controlled chambers can also be used for long data collection times to preserve sample integrity.
Collect scattering data for both protein and buffer solutions. Protein and buffer solutions are placed in a capillary tube and irradiated with the X-ray beam. The same capillary tube should be used for each solution to eliminate scattering differences arising from different capillary tubes. The scattering intensities I(q) are recorded as a function of the scattering vector q (q = 4πsin θ/λ, where 2θ is the angle between the incident and scattered radiation, and λ is the radiation wavelength, generally around 0.1 nm). Typical high q values for SAXS measurement are 0.2–0.3 Å−1, which correspond to 30–20 Å resolution (Fig. 1a).
Integrate scattering intensities at each q value. Because the particles are randomly distributed (i.e., their positions and orientations are thus uncorrelated), the scattering intensity from a protein solution is continuous and isotropic (Fig. 1a) and is proportional to the scattering from a single protein molecule averaged over all orientations [2]. Thus, the scattering intensities are radially (circularly) averaged and integrated as onedimensional (1-D) function, I(q) vs. q, using the SAXS software package provided by either the beamline or SAXS camera manufacturer (Fig. 1b).
Calculate protein-only scattering. Scattering intensities are measured from both protein and buffer solutions. Scattering intensities contributed only from protein molecules are obtained by subtracting the scattering intensities of the buffer solution from those of the protein solution at each q value (Fig. 1b). This corrected intensity for protein molecules is then used for further analysis (see Note 3).
Check for radiation damage. Radiation damage can be significant in SAXS data collection, particularly at high flux synchrotron sources, even though scattering data are acquired within 10 s. To determine whether the protein solution suffered from radiation damage, short irradiation (i.e., 0.5 s) of a protein sample can be measured before and after the data collection, and their scattering curves compared. Using a home X-ray source, data collection can take up to several hours. Scattering data can be collected at set time intervals, compared for signs of radiation damage, and then merged with previously collected data (Fig. 1c).
Repeat the above data collection procedure at several protein concentrations. Since the scattering intensity is proportional to the amount of scattering objects (electrons for X-ray radiation), higher concentrations of protein solutions will have higher scattering intensities, which increases the signal to noise ratio. However, concentrated solutions may exhibit “interference” between neighboring molecules, and thus SAXS data are collected in several different protein concentrations to determine whether there are concentration-dependent effects on the scattering profile. Protein concentrations that show protein aggregation or interparticle effects are then omitted from subsequent data analysis (see Note 4).
Fig. 1.
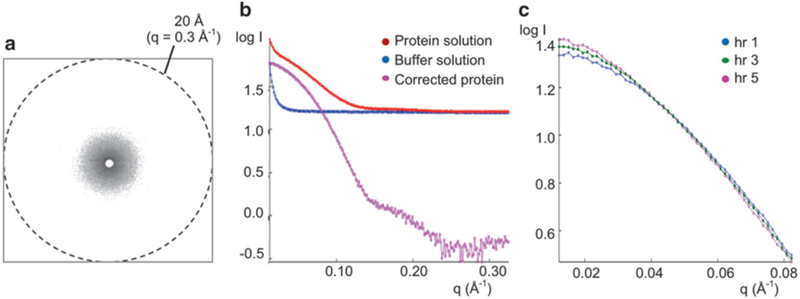
Small-angle X-ray scattering data collection and processing. (a) Typical X-ray scattering pattern of a protein solution. The scattering intensities are radially averaged to yield a 1-D scattering curve of I(q) vs. q. (b) 1-D SAXS scattering curves for a protein solution, a buffer solution, and corrected protein. The SAXS data are collected for protein and buffer solutions. The scattering intensity of the buffer solution is then subtracted from that of the protein solution to obtain the corrected scattering intensity for the target protein. This corrected curve is then used for data analysis. (c) Example of radiation damage. Protein samples were exposed to X-rays for five consecutive hours and data recorded at 1 h intervals. Only the first, third, and fifth hours are shown for clarity. Increased exposure times led to an increase in log I(q) at the lowest q regions, indicative of protein aggregation due to radiation damage
3.2. Primary Data Analysis
Perform initial data analysis. Primary data analysis can be carried out using the program PRIMUS from the ATSAS Suite [3]. All software can be downloaded from http://www.embl-hamburg.de/biosaxs/atsas-online/. The PRIMUS program is used to carry out simple operations on scattering profiles such as buffer subtraction, data averaging, merging, and extrapolation to zero concentrations.
- Examine the quality of the data first by calculating a Guinier plot, ln[I(q)] vs. q2. The scattering curve at low angles near q = 0 follows the Guinier approximation
where I(0) is the forward scattering intensity (also called the extrapolated scattering intensity at zero angle) and RG is the radius of gyration. Radius of gyration is defined as the root mean square average of the distance of the electrons from the center of the particle. Linearity of the Guinier plot near q = 0 indicates non-aggregated and monodisperse samples (Fig. 2a). For protein samples that obey the Guinier approximation, the radius of gyration is calculated from the slope of the line. If the Guinier plot does not follow a straight line at low angles, the protein is either heterogeneous, aggregated, or there are interparticle repulsions. The data should be discarded for further data analysis unless these behaviors are explicitly accounted for (see Note 4). Calculate Kratky plot. A Kratky plot, I(q)-q2 vs. q, can provide an indication as to whether the protein of interest is properly folded in solution. As shown in the example in Fig. 2b, folded, partially folded, and unfolded proteins have characteristic Kratky plots [4, 6]. Globular proteins will have a bell-shaped curve. In contrast, extended, semi-stiff polymers such as random coil peptides yield a curve like the unfolded protein shown in Fig. 2b. Partially folded proteins would have a curve that has characteristics of both folded and unfolded proteins. In addition, it is possible to determine whether multiple domains within a protein are arranged as a single unit or whether the domains are structurally independent and connected by a flexible linker. If the two domains are in a fixed arrangement as a single unit, a bell-shaped curve would be obtained. If two domains are connected by a flexible linker, the plot will have a broad multi-peak profile.
Fig. 2.
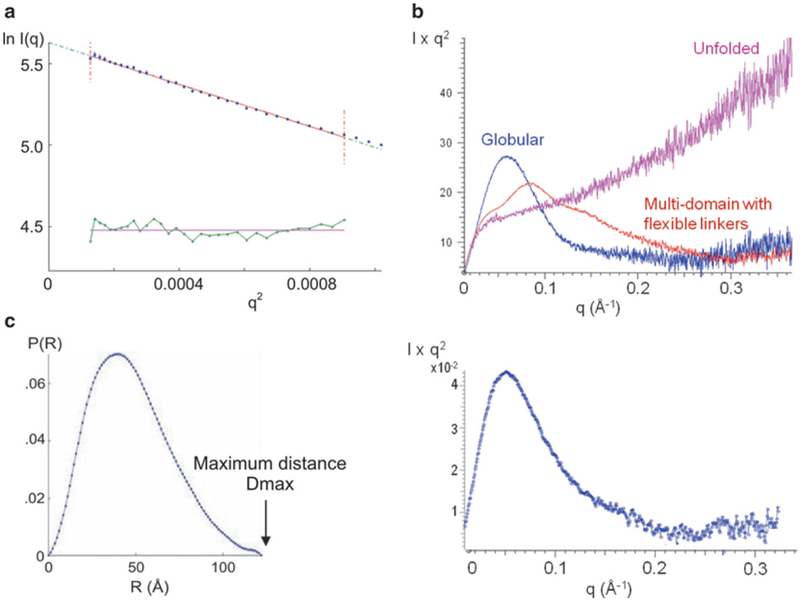
SAXS data analysis. (a) Guinier plot, ln[I(q)] vs. q2. The fit of the Guinier equation is shown in red. The bottom green line shows the residual. The radius of gyration RG is calculated from the slope of the Guinier plot. (B) Kratky plot, I(q)q2 vs. q. Top. Examples of a globular protein, an unfolded protein, and a multi-domain protein with flexible linkers are shown (from [4]). Bottom: The Kratky plot for this particular protein (dengue NS5) shows a profile typical of a globular protein [5]. (c) Pair-wise distance distribution function P(r). The radius of gyration RG calculated from the P(r) for the dengue NS5 is 35.6 ± 1 Å, which is in a good agreement with the value of 35.1 ± 1 Å obtained from the Guinier plot [5]. The shape of the P(r) for the protein has an asymmetric peak with a tailing profile for the higher distances, indicative of a rather elongated shape
3.3. Protein Size and Shape Analysis
Determine Porod invariant. The volume and molecular weight of a protein can be estimated from the small-angle X-ray scattering data via the Porod invariant, which can be calculated using the program PRIMUS. The hydrated volume VP of the particle is computed from the Porod invariant and is used to estimate the molecular mass of a globular protein [3]. The hydrated volume in cubic nanometers (nm3) is empirically found to be between 1.5 and 2 times the molecular mass in kilodaltons (kDa) [6].
- Determine forward scattering intensity I(0). The forward scattering intensity I(0) measured at zero angle (q=0) on an absolute scale is equal to the square of the number of electrons in the scattering object and is thus proportional to the molecular mass. Although I(0) cannot be experimentally measured since it is coincident with the direct beam and hence blocked by the beam stop, I(0) can be determined by extrapolation of the scattering curve to q=0. However, this approach still requires that I(0) is on an absolute scale, a condition that is not always practical to meet. In practice, the apparent molecular mass is often determined from the I(0) of a set of standard proteins using the formula
where MM, I(0), and c are the molecular mass, the forward scattering intensity, and the concentration of the protein of interest (subscript p) or the standard protein (subscript st), respectively [7]. Lysozyme (14.3 kDa), bovine serum albumin (66.2 kDa), and glucose isomerase (172 kDa) are often used as standard proteins. Determine pair-wise distance distribution function, P(r). The pair-wise distance distribution function P(r) describes the probability of finding an electron in the macromolecule separated by distance r from another electron in the particle. In theory, the P(r) can be directly obtained via Fourier transformation of the scattering intensity as a function of q. In practice, the P(r) is calculated from the scattering pattern via indirect Fourier inversion of the scattering intensity I(q), which can be accomplished using the program GNOM [8]. The boundary constraints of P(r) = 0 at r = 0 and at the maximum linear dimension, Dmax, are applied to P(r). This real space representation of the scattering intensity provides information about the particle shape and the maximum dimension Dmax (Fig. 2c). For example, globular proteins display a bell-shaped curve with maximum at about Dmax/2. Elongated molecules have skewed distributions with a maximum at small distances corresponding to the radius of the cross section and a tailing profile for the longer distances. Proteins consisting of wellseparated subunits would have multiple maxima, with the first maximum corresponding to the intra-subunit distances and the others corresponding to the distances between subunits. Radius of gyration RG can also be determined by P(r) function and should be compared to the RG determined from the Guinier plot. In some ways, the RG obtained from the P(r) function may be more reliable than that obtained via the Guinier approximation since all the data is used, whereas only low-resolution data contributes to Guinier analysis.
3.4. Ab Initio Shape Calculations
Perform initial shape determination. Although a unique three-dimensional (3-D) structure cannot be retrieved from the one-dimensional (1-D) scattering curve, it is possible to determine approximate molecular shapes, or envelopes, of macromolecules that are consistent with the scattering data. The programs GASBOR and DAMMIN can be used for ab initio molecular shape determination [2, 9]. Both programs use an ensemble of dummy residues (GASBOR) or dummy atoms (DAMMIN) placed in a volume, of which the radius is Dmax/2. Scattering curves from different arrangements of these dummy residues or atoms are calculated and compared to the experimental SAXS data (Fig. 3a). The agreement between a resulting model and the data is determined using the discrepancy χ2, defined according to Konarev et al. [3]. Since many 3-D shapes can fit a 1-D scattering curve equally well (χ2 < 2.0), an averaged shape from multiple runs is believed to be a better representative of a protein molecule. Ten to twenty independent calculations are thus performed with or without molecular symmetry imposed (Fig. 3a). The program DAMAVER is used to align the multiple molecular envelopes, select the most typical ones, and build an averaged model [10] (Fig. 3b). If an atomic structure or model structures are available, they can be superimposed to the calculated envelope by programs such as SUPCOMB [11] or Situs [12] to evaluate their agreement (see Note 5).
Fig. 3.
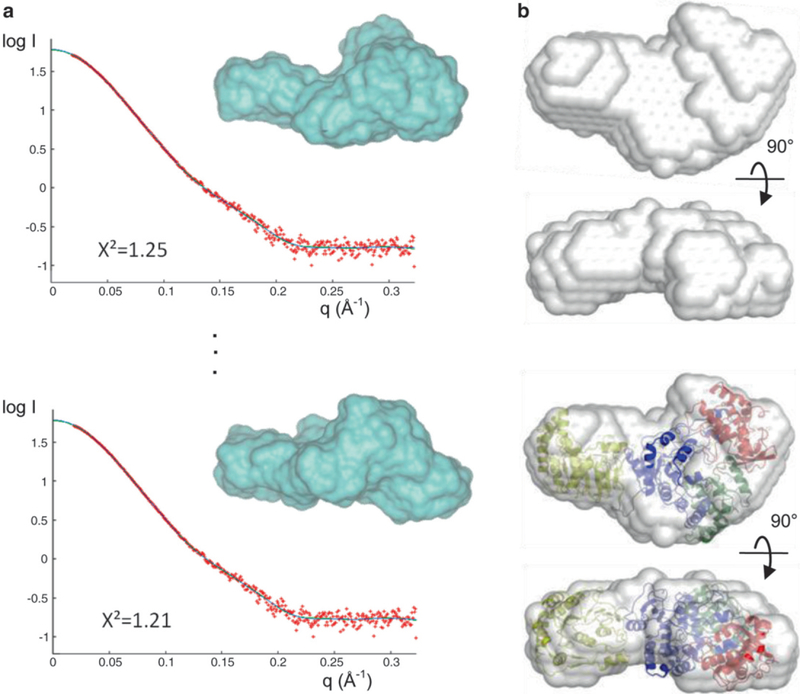
Applications of SAXS. (a) De novo shape determination using GASBOR. Ten individual runs generate ten different models (black lines) that fit the experimental scattering curve (red) well with X2 values between 1.2 and 1.4; two of the ten curves and de novo models (blue surface) are shown. (b) Two views of an averaged molecular envelope obtained from (a). In this example, the protein consists of two domains whose independent crystal structures are known. These structures were fitted into the SAXS envelope using SUPCOMB to generate a full-length model of the protein (bottom)
3.5. Building Multicomponent Assemblies from Their Partial Structures
Model multicomponent assemblies. If individual domain structures are known but not the full-length protein structure, SAXS data can be used to build a full-length protein model from the known subunits [5, 13]. Similarly, multicomponent assemblies can be constructed from the structures of individual components and SAXS data for the entire assembly. Both SASREF and BUNCH programs minimize the discrepancy between the calculated SAXS curves of the assembled model and the experimental scattering data [14]. The SASREF program uses a simulated annealing protocol to construct an interconnected ensemble of known subunits without steric clashes. The BUNCH program combines a rigid-body modeling for the known subunits and ab initio modeling for the regions of unknown structure and thus can be more useful if the structures of the protein subunits are incomplete (see Note 6).
3.6. Comparison of Different 3-D Structures with Solution SAXS Data
-
Compare SAXS data with other structural information. SAXS is particularly useful for verification of other structural or modeling data, e.g., whether a crystal structure or a 3-D model likely exists in solution. It is important to realize that it is not necessary to calculate an ab initio molecular envelope in order to compare X-ray structures to SAXS data. It is more appropriate to calculate a scattering curve from a structure for direct comparison with SAXS data. A SAXS profile can be calculated from the 3-D model (PDB coordinates) after adding a hydration shell using the program CRYSOL [15]. The agreement between the calculated SAXS profile of the 3-D model and the experimental SAXS data is determined using the discrepancy χ2. The lower χ2 indicates a better fit. Agreement would indicate that the crystallized structure (or model) represents the conformational state of the protein in solution. Disagreement might indicate that crystal packing forces have trapped your protein in a nonbiological conformation.
This approach has been used to study the flavivirus NS5 and NS3 proteins. Flavivirus NS5 consists of an N-terminal methyltransferase and a C-terminal RNA polymerase domain. Although structures of the two separate domains have been determined, there is no crystal structure for the full-length protein. Hence, various models of the full-length protein have been proposed, which differ in the relative arrangement of the two domains. Comparing scattering curves calculated from the different models to experimental SAXS data indicated which model more likely resembles the structure in solution [5]. Similarly, flavivirus NS3 consists of an N-terminal protease domain and a C-terminal helicase domain. The crystal structures of the full-length NS3 from dengue and Murray Valley encephalitis viruses have been determined in two conformations, which differ in the relative arrangements of the two domains [16, 17]. In order to determine whether NS3 protein exists in either or both conformations in solution, SAXS curves are calculated from the crystal structures and compared to the solution SAXS data [16, 17]. Both structures agree well with the solution SAXS data. Taken together with the crystallographic data, it is concluded that the conformational flexibility of the linker between the two domains exists and both NS3 conformations are likely present in the solution.
3.7. Modeling Multiple Conformational Ensembles
When two or more conformations of the protein are expected to exist in the solution, the relative abundance of each population can be estimated using the program EOM (ensemble optimization method) [18]. The program calculates a pool of potential conformations that are randomly generated (N > 1,000). Then subsets of the potential conformations (N = 50) are selected to fit the experimental SAXS data. By comparing the profile of the selected conformations to the random conformations, one can also address the protein flexibility.
4. Notes
Monodispersity (identical molecules) of protein samples can be addressed by a combination of analytical methods such as dynamic light scattering and/or analytical ultracentrifugation.
Radiation damage in data collected at synchrotron sources is frequently observed. We found that the addition of radical scavengers such as 1–5 % glycerol, 1–5 mM DTT, or 1–2 mM TCEP in protein solutions reduces the secondary radiation damage. The use of lower molecular weight salts can help reduce primary radiation since lighter atoms have a smaller cross-sectional scattering area.
The scattering intensity difference between protein and buffer solutions is small, especially at high q ranges where intensities are low. Thus, accurate background (buffer) subtraction is essential to obtain accurate measurements of the protein scattering.
SAXS data should be collected at a range of protein concentrations (typically 1–10 mg/mL) to determine the optimal concentration for SAXS data analysis. Overlaying the protein samples at different concentrations in the Guinier region should indicate whether there are concentration-dependent effects such as aggregation or interparticle repulsion.
SAXS-based ab initio shape calculations assume uniform particle density. Thus, shape calculations could be problematic for a multicomponent system such as protein–nucleic acid and protein–lipid complexes, in which each component has different average electron densities.
In some cases, the low-resolution ab initio envelope does not allow accurate positioning of individual subunits. Thus, model building is greatly facilitated if a priori information regarding the structure or assembly is available.
Acknowledgments
This work is supported by NIH grant AI087856 to KHC and GM095516 to MCM. We thank Dr. Mark White and Dr. Cecile Bussetta for providing SAXS examples [19] and for helpful discussions.
References
- 1.Guinier A (1939) La diffraction des rayons X aux tres petits angles; application a l’etude de phenomenes ultramicroscopiques. Ann Phys (Paris) 12:161–237 [Google Scholar]
- 2.Svergun DI, Petoukhov MV, Koch MHJ (2001) Determination of domain structure of proteins from X-ray solution scattering. Biophys J 80(6):2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr 36:1277–1282 [Google Scholar]
- 4.Putnam CD, Hammel M, Hura GL, Tainer JA (2007) X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys 40(3):191–285 [DOI] [PubMed] [Google Scholar]
- 5.Bussetta C, Choi KH (2012) Dengue virus nonstructural protein 5 adopts multiple conformations in solution. Biochemistry 51(30):5921–5931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mertens HD, Svergun DI (2010) Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J Struct Biol 172(1):128–141 [DOI] [PubMed] [Google Scholar]
- 7.Mylonas E, Svergun DI (2007) Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J Appl Crystallogr 40:S245–S249 [Google Scholar]
- 8.Svergun DI (1992) Determination of the Regularization Parameter in Indirect-Transform Methods Using Perceptual Criteria. J Appl Crystallogr 25:495–503 [Google Scholar]
- 9.Svergun DI (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing (vol 76, p 2879, 1999). Biophys J 77(5):2896–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Volkov VV, Svergun DI (2003) Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr 36:860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kozin MB, Svergun DI (2001) Automated matching of high- and low-resolution structural models. J Appl Crystallogr 34:33–41 [Google Scholar]
- 12.Wriggers W (2010) Using Situs for the integration of multi-resolution structures. Biophys Rev 2(1):21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mastrangelo E, Milani M, Bollati M, Selisko B, Peyrane F, Pandini V, Sorrentino G, Canard B, Konarev PV, Svergun DI, de Lamballerie X, Coutard B, Khromykh AA, Bolognesi M (2007) Crystal structure and activity of Kunjin virus NS3 helicase; protease and helicase domain assembly in the full length NS3 protein. J Mol Biol 372(2):444–455 [DOI] [PubMed] [Google Scholar]
- 14.Petoukhov MV, Svergun DI (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J 89(2):1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Svergun D, Barberato C, Koch MHJ (1995) CRYSOL—a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr 28:768–773 [Google Scholar]
- 16.Luo D, Xu T, Hunke C, Gruber G, Vasudevan SG, Lescar J (2008) Crystal structure of the NS3 protease-helicase from dengue virus. J Virol 82(1):173–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Assenberg R, Mastrangelo E, Walter TS, Verma A, Milani M, Owens RJ, Stuart DI, Grimes JM, Mancini EJ (2009) Crystal structure of a novel conformational state of the flavivirus NS3 protein: implications for polyprotein processing and viral replication. J Virol 83(24):12895–12906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI (2007) Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc 129(17):5656–5664 [DOI] [PubMed] [Google Scholar]
- 19.Zheng J, Gay DC, Demeler B, White MA, Keatinge-Clay AT (2012) Divergence of multimodular polyketide synthases revealed by a didomain structure. Nat Chem Biol 8(7):615–621 [DOI] [PMC free article] [PubMed] [Google Scholar]