

Abstract
Apoptosis is crucial for the normal development of the nervous system, whereas neurons in the adult CNS are relatively resistant to this form of cell death. However, under pathological conditions, upregulation of death receptor family ligands, such as tumour necrosis factor (TNF), can sensitize cells in the CNS to apoptosis and a form of regulated necrotic cell death known as necroptosis that is mediated by receptor-interacting protein kinase 1 (RIPK1), RIPK3 and mixed lineage kinase domain-like protein (MLKL). Necroptosis promotes cell death and neuroinflammation to mediate pathogenesis in several neurodegenerative diseases, including multiple sclerosis, amyotrophic lateral sclerosis, Parkinson disease and Alzheimer disease. In this Review, we outline the evidence implicating necroptosis in these neurological diseases, and suggest that targeting RIPK1 might help to inhibit multiple cell death pathways and ameliorate neuroinflammation.
Introduction
Apoptosis and necroptosis are two distinct regulated cell death mechanisms. Apoptosis is mediated by caspases and involved in mediating neural development 1. Necroptosis is a form of regulated necrotic cell death that can be activated under apoptosis-deficient conditions2. Although different forms of cell death characterized by distinct morphological features have long been appreciated, the definitive identification of necroptosis and its implications in neurological diseases only began to be elucidated about a decade ago, in a study that used small-molecule inhibitors of tumour necrosis factor (TNF)-induced necrosis under apoptosis-deficient conditions2,3. Intensive research on necroptosis in the past decade has led to the elucidation of the molecular mechanisms that underlie the activation of this cell death modality and its pathophysiological importance in human diseases4,5.
Necroptosis can be triggered by the activation of death receptors [G] (DRs) such as TNF receptor 1 (TNFR1). Under certain conditions, TNFR1 activation leads to the activation of receptor-interacting protein kinase 1 (RIPK1), and the RIPK1-kinase activity-dependent formation of a RIPK1–RIPK3–mixed lineage kinase-like protein (MLKL) complex (also known as complex IIb) (Fig. 1). Here, the phosphorylation of MLKL by RIPK3 ultimately leads to necroptosis via disruption of the plasma membrane and cell lysis6. In addition, the activation of RIPK1 may alternatively lead to apoptosis or neuroinflammation, depending on the cell type and context 4.
Fig. 1 |. TNF signalling-mediated apoptosis and necroptosis.
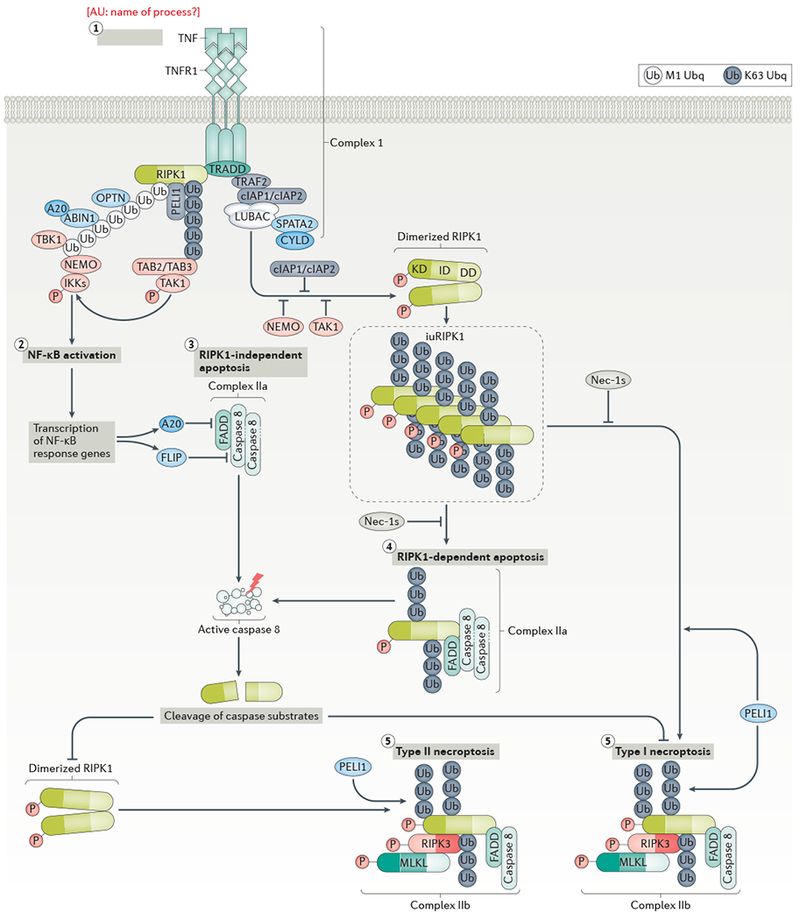
a | The binding of tumour necrosis factor (TNF) to TNF receptor 1 (TNFR1) leads to receptor trimerization and activation. TNFR1-associated death domain (DD) protein (TRADD) and receptor-interacting protein kinase 1 (RIPK1) are recruited to the intracellular DD of activated TNFR1 via their own DDs to initate the formation of complex I. In complex I, TRADD recruits the E3 ubiquitin ligases cellular inhibitor of apoptosis 1 (cIAP1) and cIAP2, which in turn add K63 ubiquitin chains to complex I members, including to the K377 residue of RIPK1. The linear ubiquitylation assembly complex (LUBAC) is recruited by binding with K63 ubiquitin chains in complex I and performs linear (M1) ubiquitylation of RIPK1. Deubiquitinating enzyme CYLD and its adaptor protein SPATA2 are recruited together with the LUBAC to complex I. Transforming-growth-factor-β-activated kinase 1 (TAK1) is recruited to K63 ubiquitin chains on RIPK1 via TAK1-binding protein 2 (TAB2) or TAB3. The nuclear factor-κB (NF-κB) essential modulator (NEMO)–IκB kinase (IKK) complex and the kinase TBK1 are recruited to complex I by binding with M1 ubiquitin chains on RIPK1. M1-ubiquitin binding proteins ABIN-1 and OPTN are also recruited to complex I. ABIN-1 in turn recruits ubiquitin editing enzyme A20 to complex I. b | Under normal conditions, activated IKKs and TAK1 lead to NF-κB activation and transcription of pro-inflammatory and pro-survival genes, such as those encoding FLICE-inhibitory protein (FLIP) and A20, which suppress the activation of caspase-8 and RIPK1, respectively. c | When NF-κB activation is inhibited, a cytosolic complex named complex IIa can form to mediate the activation of caspase-8 and subsequent cleavage of RIPK1 and RIPK1-independent apoptosis. d | The cleavage of RIPK1 at the C-terminus of its kinase domain (after residue D324) by caspase 8 provides an important mechanism by which apoptosis, both RIPK1-dependent and RIPK1-independent, may pre-empt the activation of RIPK1 and necroptosis. When caspase-8 activation is blocked, dimerization of RIPK1 via its C-terminal DD leads to RIPK1 activation and formation of complex IIb, which includes FADD, caspase-8, RIPK1, RIPK3 and MLKL.. RIPK1 activity is required for the formation of complex IIb. PELI1 mediates K63 ubiquitination of RIPK1 in complex IIb which promotes the activation of RIPK3 and MLKL to mediate type II necroptosis. e | Alternatively, when suppression of RIPK1 activity in complex I fails under pathological conditions, RIPK1 may mediate RIPK1-dependent apoptosis or type I necroptosis. Specifically, under cIAP1/2, NEMO-, IKK- or TAK1-deficient conditions, an activated, detergent-insoluble ubiquitinated RIPK1 species (iuRIPK1) may form following complex I to accelerate RIPK1 dimerization and subsequent formation of complex II and RIPK1-dependent apoptosis or type I necroptosis. f | When caspase-8 activation is blocked, the formation of complex IIb, which includes RIPK1, RIPK3, and MLKL, can lead to type I necroptosis. High levels of activated RIPK1 in complex I and the formation of iuRIPK1 distinguish type I necroptosis from type II necroptosis.Inhibition of RIPK1 kinase by Nec-1s blocks RIPK1 activation to prevent RDA and necroptosis.
In this Review, we provide an overview of the importance of RIPK1 signalling in necroptosis and neuroinflammation in CNS diseases. We briefly describe the role of apoptosis in the developing nervous system and how the adult CNS exhibits reduced sensitivity to intrinsic pathways of apoptosis. We explain how, under pathological conditions, DR-mediated extrinsic cell death [G] pathways may become activated and overcome the resistance of the adult CNS to intrinsic apoptosis [G], thus promoting both extrinsic apoptosis and necroptosis. Whereas the intrinsic apoptosis pathway is required in normal development, necroptosis is a common deleterious mediator of neurodegeneration. We discuss the role of necroptosis in various neurological diseases and examine how RIPK1 and necroptosis may be activated in specific CNS diseases. Finally, we evaluate evidence suggesting that RIPK1 activity may also contribute to CNS pathology by promoting neuroinflammation, and describe the underlying mechanisms. Overall, the activity of RIPK1 has emerged as a central therapeutic target of drug candidates currently being clinically tested for the treatment of neurological diseases 7,8.
Apoptosis in the developing CNS
An evolutionarily conserved apoptosis programme.
Elegant genetic studies in the nematode Caenorhabditis elegans revealed the first insights into the mechanisms of programmed cell death9: loss-of-function mutations in the ced-3 and ced-4 genes prevented all apoptosis during nematode development. Strikingly, most of the 131 cells that die during C. elegans development are in the neuronal lineage10; thus, in the developing C. elegans nervous system, apoptosis has a major role in eliminating superfluous neurons and refining neuronal connections. Caspases, which are the homologues of CED-3 in mammals11, mediate neuronal apoptosis in vertebrates, as demonstrated by the finding that, in the chicken dorsal root ganglion, expression of a viral inhibitor of caspases inhibits neuronal death induced by trophic factor deprivation, a model of developmental neuronal death12. Whereas CED-3 controls all of the developmental apoptosis in C. elegans, mammals express 12 different caspases that can regulate apoptosis as well as certain non-apoptotic functions13.
Caspases are synthesized as catalytically inactive pro-enzymes consisting of a pro-domain, a large subunit and a small subunit13,14. They may be activated by cell-intrinsic pathways, such as by DNA damage, or by cell-extrinsic pathways — that is, in response to ligands that activate death receptors. In the intrinsic pathway of apoptosis, caspase-9 is activated following mitochondrial damage primarily by binding to the cytochrome c–apoptotic protease-activating factor 1 (APAF1) complex, or apoptosome15. In the extrinsic pathway of apoptosis, caspases that have a death-effector domain, such as caspase-8, are activated by interacting with signalling complexes downstream of DRs such as TNFR1 and FAS (also known as CD95)16. In either case, the activation of initiator caspases by intrinsic or extrinsic pathways leads to further activation of downstream effector caspases, such as caspase-3 and caspase-717. Once activated, effector caspases cleave hundreds of cellular substrates, leading to cellular demise18.
Apoptosis during nervous system development in mammals.
In the nervous system, the intrinsic apoptosis pathway is important for the proper formation of the CNS. Neurons generated during embryonic and early postnatal development are highly sensitive to apoptosis. The survival of immature neurons depends upon trophic factors such as nerve growth factor (NGF) or brain-derived neurotrophic factor (BDNF) provided by their targeting sites19. Trophic factors protect developing neurons by suppressing the mitochondrial pathway of apoptosis, which is mediated by the translocation of BAX from the cytosol to mitochondria, followed by the loss of mitochondrial membrane integrity and caspase activation via apoptosome formation20. This formation of apoptosome promotes the activation of caspase-9 and the downstream caspases such as caspase-3 that are involved in mediating development cell death. Both Casp-3−/− mice 21 and Casp-9−/− mice22 show similar severe defects in developmental neuronal apoptosis, with marked expansion of the periventricular zone and ectopic cell masses in the CNS, and perinatal lethality. After the proper formation of the CNS, multiple caspases, as well as members of the BCL-2 family proteins, which serve as regulators of mitochondrial apoptosis, are downregulated in the CNS, in part explaining the relative resistance of mature neurons to apoptosis23,24.
Mature neurons show increased resistance to intrinsic apoptosis.
Dependence on trophic factors for survival is largely lost once neurons mature (by postnatal day 28 in mice)25. Trophic factor withdrawal from cultures of mature sympathetic neurons does not induce apoptosis20. Further, adult brain cells are largely refractory to intrinsic apoptosis induced by mitochondrial damage 26. This resistance to apoptosis may be an adaptive mechanism to ensure the long-term survival of mature neurons for many decades, as in the human brain. The resistance of mature neurons to apoptosis may be mediated by mechanisms both upstream and downstream of mitochondria in the intrinsic apoptosis pathway 27–29.
Overcoming apoptosis resistance of mature neurons.
The increased resistance of the adult nervous system to intrinsic pathway of apoptosis can be overcome under pathological conditions by the activation of the extrinsic apoptosis pathway via the upregulation of the cognate ligands of multiple DRs such as TNF, FAS ligand (FASL) and TNF-related apoptosis-inducing ligand (TRAIL). These DR ligands may be upregulated in the context of chronic neurodegenerative diseases such as Alzheimer disease (AD), Parkinson disease (PD), amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS)30–36, to mediate both cell death and inflammation37.
Whereas the loss of TNFR1 is beneficial in multiple animal models of neurodegenerative diseases, inhibiting more than one DR may have increased benefit. For example, inhibiting both TNF and FASL, through genetic mutations or neutralizing antibodies, is more efficacious in reducing infarct size in the rodent middle cerebral artery occlusion [G] (MCAo) model of stroke than blocking TNF or FASL alone38. Similarly, in the experimental autoimmune encephalomyelitis [G] (EAE) mouse model of MS, oligodendrocyte loss can be more effectively blocked by inactivating both FAS and TNFR1 than by inhibiting either alone39. In addition, in cultured slices of human brain tissue, TRAIL robustly induced apoptosis of neurons, oligodendrocytes, microglia and astrocytes40. TRAIL expression is upregulated in the cerebral cortex of individuals with AD, often near plaques of amyloid-β (Aβ)41. In chronic and acute human MS lesions, and in mouse brains after induction of EAE, TRAIL is upregulated, predominantly by activated microglia and invading immune cells42.
Thus, whereas intrinsic apoptosis pathway is necessary for normal development, the extrinsic apoptosis pathway may serve as a sensor of extracellular inflammatory signalling in the adult nervous system. Moreover, several ligands of the DR family, including TNF, FASL and TRAIL, may cooperate to overcome the resistance of mature neurons to apoptosis by promoting cell death through the extrinsic apoptosis pathway, and thus collectively contribute to pathogenesis in neurological disorders. The possible activation of multiple DR family members under disease conditions suggests that therapeutic strategies that can target multiple DRs simultaneously may be more promising than those targeting a single DR.
Necroptosis in neurological diseases
By acting on DRs of various CNS cell types such as neurons and oligodendrocytes, TNF, FASL and TRAIL can induce not only extrinsic apoptosis, but also necroptosis through downstream involvement of RIPK1, RIPK3 and MLKL (Fig. 1). Necroptosis and RIPK1 have been implicated in many major human CNS diseases (Table 1). RIPK3-null mice and mice carrying various RIPK1 kinase-dead knock-in mutations are normal in development and adult life, demonstrating that unlike apoptosis, necroptosis is not involved in mediating normal development or adult homeostasis. Strikingly, however, these mutant mice do show resistance in multiple animal models of human inflammatory and neurological diseases (Table 2).
Table 1 |.
Targeting RIPK1 and necroptosis in neurodegenerative diseases.
| Disease | Comment | Refs |
|---|---|---|
| ALS | RIPK1 inhibition or RIPK3 deficiency blocked oligodendrocyte death, microglial inflammation and axonal degeneration in Optn−/− mice | 75 |
| RIPK1 inhibition by Nec-1s or RIPK3 deficiency blocked oligodendrocyte death, microglial inflammation and axonal degeneration in SOD1G93A mice | 75 | |
| Nec-1 blocked the death of human ESC-derived motor neurons in co-cultures with iPSC-derived astrocytes derived from individuals with primary familial or sporadic ALS | 110 | |
| ALS/FTD | Reduced myeloid TAK1 expression in Tbk1+/− mice promoted all the key hallmarks of ALS/FTD, including neuroinflammation, TDP43 aggregation, axonal degeneration, neuronal loss and behaviour deficits; all these effects were blocked upon inhibition of RIPK1 | 79 |
| MS | Cortical lesions in human MS brain samples showed reduced caspase 8 activation, along with increased activation of RIPK1, RIPK3, and MLKL. In the EAE and cuprizone models of MS, and in vitro, necroptosis mediated TNF-induced degeneration of oligodendrocytes, and RIPK1 inhibition protected against oligodendrocyte death | 92 |
| Compound 22 attenuated disease progression in the mouse EAE model of MS | 105 | |
| AD | Necroptosis markers were observed in human AD brains and correlated positively with Braak stage, and negatively with brain mass and cognition. Gene sets regulated by AD and RIPK1 show substantial overlap | 124 |
| RIPK1 was shown to be activated in human AD pathological samples. RIPK1 inhibition, using Nec-1s or the RIPK1 D138N kinase-dead mutation, reduced Aβ burden, levels of inflammatory cytokines and memory deficits in a mouse model of AD. RIPK1 inhibition promoted microglial degradation of Aβ in vitro, possibly in part by suppressing the expression of cystatin F | 125 | |
| Stroke | Administration of Nec-1 or its analogues up to 6 hours after the initiation of MCAo in mice dose-dependently reduced infarct volume and improved neurological score | 2 |
| Pre-treatment with Nec-1 before MCAo protected against OPC death, white-matter injury and loss of neurological function, and inhibited necroptotic kinases in the SVZ and corpus callosum | 159 | |
| Nec-1 reduced inflammatory response and improved cognitive function in BCAS mouse model of chronic ischaemic stroke | 160 | |
| Nec-1 reduced neurovascular injury (including cell death, BBB opening and oedema development) after collagenase-induced ICH in mice | 161 | |
| Traumatic brain injury | Nec-1 reduced histopathology and improved functional outcome after CCI in mice | 162 |
| Traumatic SCI | Nec-1 reduced cell death and tissue damage induced by traumatic SCI | 163 |
| Glaucoma | RIC protected retinal ganglion cells against ischaemic injury in a rat glaucoma model | 164 |
| Reinal ischaemia | Nec-1 protected the thickness and histoarchitecture of the inner retina and improved function after retinal ischaemia | 165 |
| TNF and RIPK1 were increased in the retina ganglion cell layer after IR injury. Nec-1 reduced retinal cell loss in IR-injured retinas | 166 | |
| Nec-1 reduced retinal damage and inflammation after IR injury | 167 | |
| Retinal detachment | Nec-1 attenuated retinal histopathological damage and plasma membrane breakdown in outer retinal layers and improved function after retinal detachment in rats | 168 |
| Age-related macular degeneration | In the degenerative mouse retina, RIPK3 levels were increased. RIPK3 loss protects retinal histoarchitecture and the RPE and prevents inflammation | 169 |
| Gaucher disease | RIPK3 deficiency increased survival and motor function in a mouse model of Gaucher disease | 114 |
| Niemann–Pick disease, type C1 | Nec-1 delayed cerebellar Purkinje cell loss, progression of neurological symptoms, and death in Npc1-mutant mice | 170 |
| PD | Nec-1s improved survival of OPA1-mutant human iPSC-derived neurons in vitro. Nec-1s attenuated MPTP-induced dopaminergic neuronal loss | 112 |
Aβ, amyloid-β; AD, Alzheimer disease; ALS, amyotrophic lateral sclerosis; ALS/FTD, comoribid ALS and frontotemporal dementia; BBB, blood–brain barrier; BCAS, bilateral carotid artery stenosis; CCI, controlled cortical impact; EAE, experimental autoimmune encephalitis; ESC, embryonic stem cell; ICH, intracerebral haemorrhage; iPSC, induced pluripotent stem cell; IR, ischaemia–reperfusion; MCAo, middle cerebral artery occlusion; MLKL; mixed lineage kinase domain-like protein; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; MS, multiple sclerosis; Nec-1, necrostation-1; Nec-1s, necrostation 1s; NPC1, Niemann–Pick C1 protein; OPA1, optic atrophy 1; OPC, oligodendrocyte progenitor cell; OPTN, optineurin; PD, Parkinson disease; RIC, RIPK1-inhibitory compound; RIPK1, receptor-interacting protein kinase 1; RPE, retinal pigmented epithelium; SCI, spinal cord injury; SODG93A mice, mice expressing the G93A variant of superoxide dismutase 1; SVZ, subventricular zone; TAK1, transforming-growth-factor-β-activated kinase 1; TBK1, kinase TBK1; TDP43, TAR DNA-binding protein 43; TNF, tumour necrosis factor.
Table 2 |.
Effects of null and kinase-dead mutations in Ripk1, Ripk3 and Mlkl in the mouse CNS.
| Genotype | Viable | Comments | Refs |
|---|---|---|---|
| Ripk1D138N kinase-dead knock-in mutation | Yes | Protects against TNF-induced SIRS | 151,171 |
| Ameliorates axonal pathology in Optn−/− mice | 75 | ||
| Protects against neuroinflammation, TDP43 aggregation, axonal degeneration, neuronal loss and behavior deficits in Tbk1+/−,Tak1ΔM/+ mice. | 79 | ||
| Protects against neuronal loss following intracerebral haemorrhage | 172 | ||
| Reduces Aβ burden, inflammation, and memory deficits in APP/PS1 mice | 125 | ||
| Protects systemic inflammation in A20−/− mice | 173 | ||
| Protects kidney ischaemia–reperfusion injury | 173 | ||
| Ripk1K45A kinase-dead knock-in mutation | Yes | Protects against TNF-induced SIRS | 174 |
| Reduces inflammation in SHARPIN-deficient mice | 70 | ||
| Protects against neuronal loss following intracerebral haemorrhage | 172 | ||
| Ripk1K584R knock-in mutation (disrupts DD homodimerization to block RIPK1 activation) | Yes | Protects against TNF-induced SIRS | 87 |
| Ripk3D161N kinase-dead knock-in mutation | No | Mice with catalytically inactive RIPK3 kinase die at around embryonic day 11.5 with abnormal yolk sac vasculature | 171 |
| Ripk3−/− | Yes | Protects against TNF-induced SIRS | 153 |
| Ameliorates axonal pathology in Optn−/− mice | 75 | ||
| Ameliorates axonal pathology in SODG93A mice | 75 | ||
| Protects against spinal cord injury | 175 | ||
| Protects against demyelination following cuprizone treatment | 92 | ||
| Protects against traumatic brain injury | 176 | ||
| Prevents necrosis and inflammation in poly(I:C)-induced retinal degeneration | 169 | ||
| No effect in MCAo stroke model | 173 | ||
| Protects against photoreceptor death after retinal detachment | 177 | ||
| Protects against Gaucher disease model | 114 | ||
| Mlkl−/− | Yes | No effect on hypothermia induced by 300 μg TNF per kg body weight; some resistance to hypothermia induced by 500 μg TNF per kg body weight | 173 |
DD, death domain; MCAo, middle cerebral artery occlusion; OPTN, optineurin; poly(I:C), polyinosinic:polycytidylic acid; RIPK1, receptor-interacting protein kinase 1; SHARPIN, shank-associated RH domain-interacting protein; SIRS, systemic inflammatory response syndrome; TAK1, transforming-growth-factor-β-activated kinase 1; TBK1, kinase TBK1; TDP43, TAR DNA-binding protein 43; TNF, tumour necrosis factor.
In the mature nervous system, RIPK1 kinase-dependent necroptosis serves as a major executioner of cell death in response to extracellular inflammatory signalling. Necroptosis can be experimentally activated under apoptosis-deficient conditions. Whereas developmental apoptosis is mostly immune silent [G], necroptosis elicits a strong inflammatory response that can drastically alter the local tissue environment and mediate the pathogenesis of CNS diseases 43.
Necroptosis induction.
The intracellular domains of TNFR1, FAS, and TRAIL receptor 1 (TRAILR1) and TRAILR2 (also known as DR4 and DR5) all include a death domain [G] (DD) and, following stimulation by their respective cognate ligands, TNF, FASL and TRAIL, recruit RIPK1 by homotypic binding of their DD to that of RIPK144. Several small-molecule RIPK1 inhibitors have been developed to target necroptosis activated by multiple members of the DR family (Box 1).
Box 1 |. Development of small-molecule inhibitors of RIPK1.
Receptor-interacting protein kinase 1 (RIPK1) is an important therapeutic target for the treatment of neurodegenerative diseases. Inactivation of RIPK1 by various genetic mutations, including D138N, K45A and ΔG26F27, leads to strong resistance to TNF and no other gross phenotype in mouse models151–154. Furthermore, RIPK1 provides a pharmacologically ‘druggable’ target, as its kinase domain presents a hydrophobic pocket that is highly amenable for specific small-molecule inhibitors155,156.
The first small-molecule inhibitors of RIPK1, including necrostatin-1 (Nec-1), and necrostatin-4 (Nec-4), were isolated in a cell-based high-throughput chemical screen for inhibitors of caspase-independent necrosis2,3. An improved analogue of necrostatin-1 (Nec-1), named Nec-1s (also known as 7-Cl-O-Nec-1) is a highly specific inhibitor of both human and murine RIPK1 with excellent blood–brain barrier (BBB) permeability and reasonable oral availability126. Because Nec-1 and Nec-1s can effectively inhibit both murine and human RIPK1, they have been used widely to investigate the role of RIPK1 and necroptosis in various cellular and animal models of human diseases.
Nec-1s binds to the hydrophobic pocket between the N-lobe and C-lobe of the RIPK1 kinase domain outside of the ATP-binding site. Through interactions with highly conserved amino acids in the activation loop and the surrounding structural elements, this binding stabilizes RIPK1 in an inactive conformation, known as the DFG-out conformation [G]3. Thus, Nec-1s is an excellent example of a type II kinase inhibitor [G].
GSK2982772 is a highly specific and potent inhibitor of human RIPK1 that was identified and optimized from hits of DNA-encoded libraries [G] 157. GSK2982772 binds the same allosteric lipophilic pocket at the back of the ATP binding site of the RIPK1 kinase domain as does Nec-4. GSK2982772 demonstrates a species selectivity for inhibition of primate RIPK1 compared with non-primate RIPK1 and is not BBB-permeable157,158. GSK2982772 has been advanced into phase IIa clinical trials for the treatment of non-neurological disorders (namely, psoriasis, rheumatoid arthritis and ulcerative colitis).
Denali Therapeutics is advancing a BBB-permeable RIPK1 inhibitor (DNL747) into phase I trials in Alzheimer disease and amyotrophic lateral sclerosis and has another BBB-permeable RIPK1 inhibitor (DNL788) in preclinical development7. In addition, Takeda has reported the development of 7-oxo-2,4,5,7-tetrahydro-6H-pyrazolo[3,4-c]pyridine derivatives, including compound 22, as a novel class of potent, orally available and BBB-permeable RIPK1 inhibitors105. Compound 22 attenuated disease progression in the mouse experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis (MS)105.
Although both FASL and TRAIL can activate necroptosis, their pro-inflammatory activities are considerably weaker than that of TNF45. Thus, our discussion here will focus on the downstream pathways activated by TNF, one of the most extensively studied neuroinflammatory cytokines involved in neurodegenerative diseases. TNF can interact with two receptors, TNFR1, which is expressed most prominently by microglia, astrocytes, oligodendrocytes and endothelial cells, and TNFR2, which is predominantly expressed by microglia46. Neuronal expression of TNFR1 and TNFR2 is very low under normal conditions47,48. Unlike TNFR1, TNFR2 does not contain the intracellular DD necessary for the formation of complex I (also known as TNF-receptor signalling complex (TNF-RSC))49 involved in mediating cell death responses; therefore, our discussion here will focus on the signalling pathways mediated by TNFR1 (Fig. 1).
Stimulation of TNFR1 by TNF leads to the formation of the membrane-associated, intracellular complex I, through the recruitment of two DD-containing proteins, RIPK1 and TNFR1-associated DD protein (TRADD), by homotypic interactions with the TNFR1 DD50. In complex I, RIPK1 is subject to several types of ubiquitin modifications, including Ml, K11, K48 and K63 ubiquitylation, and which play a critical role in modulating the activation of RIPK14 (Fig. 2).
Fig. 2 |. Post-translational modifications regulate the activation of RIPK1.
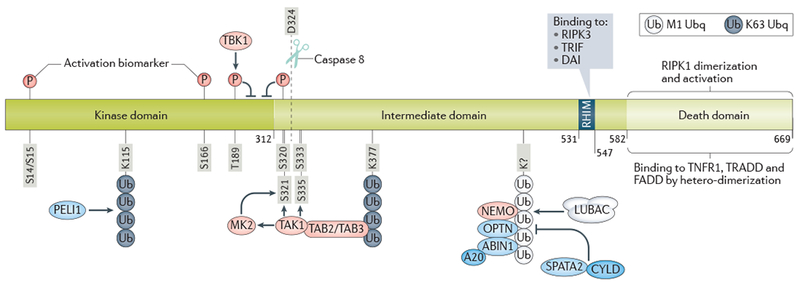
The domain structure of receptor-interacting protein 1 (RIPK1) consists of an N-terminal kinase domain, an intermediate domain and a C-terminal death domain (DD). The N-terminal kinase domain is crucial for RIPK1-dependent apoptosis and necroptosis. The cleavage of RIPK1 at D324 by caspase-8 blocks the activation of RIPK1 by separating the kinase domain from the C-terminal DD, which is critical for mediating RIPK1 activation by dimerization. Autophosphorylation of Ser14/15 and Ser166 of RIPK1 are biomarkers of its kinase activation. K115 in activated RIPK1 is ubiquitylated by the E3 ubiquitin ligase pellino 1 (PELI1). The kinase TBK1 phosphorylates T189 RIPK1 to suppress its activation. The intermediate domain of RIPK1 is involved in regulating nuclear factor-κB (NF-κB) signalling. RIPK1 can be ubiquitylated on multiple residues within this domain; one of these sites, Lys377, is a crucial residue that can be ubiquitylated by E3 ubiquitin ligases such as cellular inhibitor of apoptosis 1 (cIAP1) or cIAP2 and parkin. The Lys377 residue also serves as a binding hub for downstream signalling, including the NF-κB signalling pathway, by recruiting K63 ubiquitin binding proteins TAK1-binding protein 2 (TAB2) or TAB3, which in turn promote the activation of transforming-growth-factor-β-activated kinase 1 (TAK1). Activated TAK1 mediates multiple inhibitory phosphorylations of RIPK1 directly and indirectly by promoting the activation of IκB kinases (IKKs) and MAPK-activated protein kinase 2 (MK2). TBK1 mediates inhibitory phosphorylation of RIPK1 on T189 to block its substrate recognition in autophosphorylation-mediated activation. The receptor-interacting protein homotypic interaction motif (RHIM) within the intermediate domain of RIPK1 mediates homotypic binding with the RHIM of RIPK3 to promote downstream signalling in necroptosis and inflammation. The C-terminal DD of RIPK1 mediates heterodimerization with the death domains of TNFR1, TRADD and FADD, as well as homodimerization with itself to mediate the activation of N-terminal kinase domain. M1 ubiquitylation of RIPK1 by the linear ubiquitylation assembly complex (LUBAC) suppresses RIPK1 activation by enabling the recruitment of multiple UBAN-containing ubiquitin-binding proteins, including NEMO, ABIN1 and OPTN, which promote the activation of IKKs (NEMO), recruit A20 (ABIN1) or regulate K48 ubiquitylation of RIPK1 (OPTN).
The activation of RIPK1 represents a key signalling event in the TNFR1 pathway and dictates downstream cell death or survival (Fig. 1). RIPK1 activation can lead to cell death by driving the formation of a RIPK1–TRADD-FAS-associated DD protein (FADD)–caspase 8 complex (complex IIa), which triggers caspase activation and leads to RIPK1-dependent apoptosis [G] (RDA)6. By contrast, when caspase activity is deficient, RIPK1 activation leads to necroptosis through the formation of a RIPK1–RIPK3–MLKL complex (complex IIb), in which phosphorylation of MLKL by RIPK3 triggers disruption of the cellular membrane and cell lysis (Fig. 3)51–54.
Fig. 3 |. Execution of necroptosis.
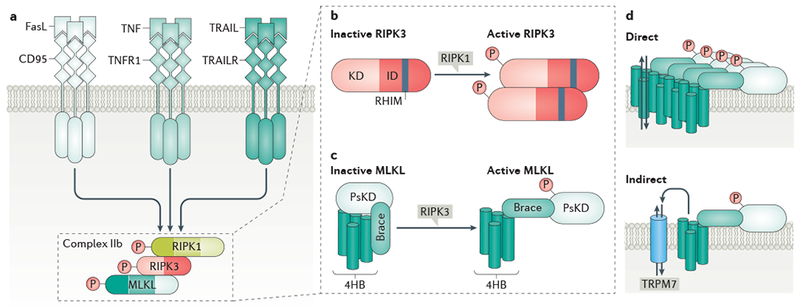
a | Activation of death receptors such as FAS, tumour necrosis factor (TNF) receptor 1 (TNFR1) or TNF-related apoptosis-inducing ligand receptors (TRAILRs) by their respective ligands can lead to the RIPK1 dimerization and activation, which in turn promotes the formation of a RIPK1–RIPK3–MLKL complex, referred to as complex IIb, and activation of RIPK3 and MLKL. b | RIPK3 consists of an N-terminal kinase domain and a C-terminal intermediate domain containing a RHIM motif that mediates binding to RIPK1. Dimerization or oligomerization of RIPK3, through RIPK1 kinase-dependent complex IIb formation, allows transphosphorylation of RIPK3 at T231/S232 in mouse RIPK3 and S227 in human RIPK3, and RIPK3 activation. c | MLKL contains a N-terminal 4-helix bundle (4HB), a brace region and a C-terminal pseudo-kinase domain (PsKD). Phosphorylation of the PsKD of MLKL by RIPK3, at S345 in mouse MLKL or T357/S358 in human MLKL, results in a conformational change of MLKL to reveal phosphatidylinositol phosphate (PIP) binding sites. d | Phosphorylated MLKL binds directly to PIPs and cardiolipin in the plasma membrane or cellular organelle membranes, leading to membrane disruption and cell lysis. The precise mechanism is unclear, but this may occur directly through MLKL oligomerization and pore formation, or indirectly, through activation of transporters or ion channels, such as transient receptor potential cation channel M7 (TRPM7).
Regulation of RIPK1 activation.
The extensive post-translational modifications of RIPK1 in complex I, including ubiquitylation, deubiquitylation and phosphorylation (Fig. 2), play a crucial role in deciding if the cell will live or die and how it might die. Under certain conditions, e.g. with cIAP1/2 deficiency or TAK1 deficiency, activated, ubiquitylated RIPK1 in complex I can transition to form an insoluble and highly ubiquitylated intermediate complex called iuRIPK1 that can in turn form complex IIa or complex IIb and drive cell death (Fig. 1) 55.
Under conditions when cells do not die, the activation of TNFR1 and recruitment of TRADD into complex I leads to activation of the transcription factor NF-κB, which in turn drives transcription of NF-κB response genes. In complex I, TRADD recruits the adaptor proteins TNFR-associated factor 2 (TRAF2) or TRAF5 and the E3 ubiquitin ligases cellular inhibitor of apoptosis 1 (cIAP1) or cIAP2, which then catalyse the K63 ubiquitylation of RIPK156. K63 ubiquitin chains on RIPK1 promote the recruitment and activation of transforming-growth-factor-β-activated kinase 1 (TAK1), which activates IκB kinases (IKKs) to promote the activation of nuclear factor-κB (NF-κB) pathway, leading to transcription of pro-survival factors, such as FLICE-inhibitory protein (FLIP), and pro-inflammatory cytokines57. Recent evidence suggests that TAK1 can suppress RIPK1 activation either directly through inhibitory phosphorylation, or indirectly, via activation of downstream kinases including MAPK-activated protein kinase 2 (MK2) and the IKKs 58–61. Thus, TAK1 mediates important pro-survival responses by both promoting NF-κB-mediated gene transcription and suppressing RIPK1 activation.
The presence of cIAP1/2 and the K63 ubiquitylation of RIPK1 in complex I also mediates the recruitment of the linear (M1) ubiquitylation assembly complex LUBAC. LUBAC is composed of haem-oxidized IRP2 ubiquitin ligase 1 (HOIL1), the catalytic subunit HOIL1-interacting protein (HOIP) and shank-associated RH domain-interacting protein (SHARPIN), as well as the deubiqitinating enzyme CYLD and its adaptor spermatogenesis-associated 2 (SPATA2), which can disassemble M1 ubiquitylation62–68. The activation of RIPK1 is negatively modulated by its M1 ubiquitylation status65,69,70. Three ubiquitin-binding proteins that bind predominantly M1 ubiquitin chains, including optineurin (OPTN), NF-κB essential modulator (NEMO) and A20-binding inhibitor of NF-κB activation 1 (ABIN1), all negatively regulate RIPK1 activation71–77. For example, recruitment of NEMO into complex I leads to activation of the IKK complex, which, like TAK1 activation, leads to both NF-κB activation and the suppression of RIPK1 activation via inhibitory phosphorylation59,78. In addition, M1 ubiquitylation of RIPK1 in complex I also mediates the recruitment of the kinase TBK1, which leads to phosphorylation of RIPK1 T189, a site important for RIPK1 substrate recognition; thus, phosphorylation of T189 RIPK1 by TBK1 blocks the transphosphorylation of RIPK1 itself, preventing its activation 79.
Recruitment of ABIN1 into complex I facilitates the recruitment of A20 (also known as TNFAIP3), a ubiquitin-editing enzyme that inhibits the activation of RIPK176,80–82 (Fig. 1). A20 contains an OTU (ovarian tumour) domain that functions as a K63 deubiquitinase of RIPK182–84. A20 deficiency thus promotes necroptosis by activating RIPK1 and its pro-necroptotic partner RIPK3. ABIN1 deficiency also sensitizes to necroptosis: Abin1−/− mice die during embryonic development (E18.5) owing to massive liver degeneration that can be blocked by the kinase-dead RIPK1 D138N mutation and by RIPK3 deficiency76. Interestingly, a genome-wide association study (GWAS) linked ABIN1 with both ALS and schizophrenia85. Thus, ABIN1 deficiency might promote the activation of RIPK1 in ALS and possibly also in schizophrenia.
Defective activation of caspase-8 in brain disorders.
Caspase inhibition is a prerequisite for susceptibility to necroptosis in most cell types2. In several cell lines, caspase inhibition alone promotes the activation of RIPK1 and thus autocrine production of proinflammatory cytokines and necroptosis43. Caspase-8 is the most important caspase that regulates necroptosis: it inhibits necroptosis by proteolytically cleaving RIPK1 after Asp324 (Fig. 2), separating the N-terminal kinase domain from the C-terminal part of the molecule86 and preventing RIPK1 kinase activation through dimerization via the C-terminal DD87.
Genetic ablation of caspase-8 or its adaptor protein FADD results in defective vascular and cardiac development and mouse embryonic lethality at E10.5 88,89; however, Casp-8−/− mice and Fadd−/− mice are fully viable when bred on necroptosis-deficient Ripk3−/− or Mlkl−/− backgrounds90,91. These results indicate that caspase-8 and FADD block necroptosis during normal development.
Caspase-8 inactivation may provide an important mechanism for sensitization to necroptosis in human CNS diseases. Tissue samples from cortical lesions of MS patients showed reduced levels of the active 18 kDa caspase-8 subunit and correspondingly elevated levels of full-length caspase-8 and the partially processed p43 subunit, as well as reduced caspase-8 activity92, implying defective caspase-8 activation. The expression of cellular FLICE-like inhibitory protein long form (cFLIPl), an inhibitor of caspase-8, was also substantially elevated in MS samples92. In the CNS, cFLIPl is expressed in many cell types, including microglia, oligodendrocyte precursor cells, and mature oligodendrocytes, but to a low extent in neurons under normal conditions48. Increased levels of cFLIPl may allow for activation of necroptosis in MS, as cFLIPl promotes the cleavage of pro-caspase-8 to yield the p43 subunit, but prevents the full activation of caspase-892. Similarly, in the EAE model of MS, levels of full-length caspase-8 and c-FLIPl are increased, whereas levels of the active 18 kDa caspase-8 are decreased92. cFLIPl expression can be induced by NF-κB93; thus, the ongoing inflammatory response in MS human spinal cords and brains and in mice after induction of EAE might promote cFLIPl expression via the NF-κB pathway, and thus facilitate necroptosis by blocking caspase-8 activation.
Caspase-8 deficiency may also be involved in AD. Two CASP8 variants, p.K148R and p.I298V, were identified as risk alleles of AD in a GWAS94. Both the K148R and the I1298V variants show a reduced ability to activate caspase-3, a downstream substrate of caspase-8. It will be interesting to examine whether the K148R mutation may affect the activation of caspase-8 by blocking a ubiquitylation site on the enzyme, as caspase-8 is subject to regulation by multiple E3 ubiquitin ligases95.
Necroptosis of oligodendrocytes and axonal degeneration.
Pathological axonal degeneration is frequently observed in patients with neurodegenerative disorders such as ALS, MS, AD and PD, and contributes significantly to neurological disability 96–98. Axonal degeneration may be acutely induced by direct nerve transection in a process known as Wallerian degeneration [G] 99–101. In chronic neurodegenerative disorders such as MS and ALS, axons may undergo so-called ‘Wallerian-like’ degeneration without affecting cell bodies, similar to that of Wallerian degeneration96. Increasing evidence suggests that the degeneration and dysfunction of oligodendrocytes is an important mechanism involved in driving axonal degeneration, such as Wallerian-like degeneration, in neurodegenerative diseases 102.
Focal inflammatory demyelination and axonal degeneration in the white matter of the brain and spinal cord are key pathological features and the cause of permanent disability for individuals with MS. Inhibiting oligodendrocyte death and promoting their ability to remyelinate are major therapeutic goals for treating MS103. Whereas apoptosis of oligodendrocytes occurs in the developing mouse CNS104, necroptosis of mature primary rodent oligodendrocytes can occur in vitro response to TNF that is inhibited by the RIPK1 inhibitor Nec-1s (also named 7N-1) or RIPK3 deficiency92. Thus, oligodendrocytes may be intrinsically ‘hard-wired’ for necroptosis when stimulated by TNF, even without exogenous caspase inhibitors. In the cuprizone model [G] of MS, oligodendrocytes undergo rapid degeneration, with concomitant microglial activation and behavioural deficit92. RIPK1 inhibition or RIPK3 deficiency reduces motor deficits, myelin loss and microglial activation in the corpus callosum of mice treated with cuprizone92. Similarly, the inhibition of RIPK1 by Nec-1s or compound 22 (another RIPK1 inhibitor developed by Takeda), can protect against demyelination and oligodendrocyte degeneration in EAE animals92,105.
ALS affects motor neurons, which have the longest axons of any neurons in the body. Growing evidence suggests that axonal degeneration in ALS may begin early, well before the onset of clinical symptoms106,107, and oligodendrocyte degeneration is a prevalent pathological feature in ALS97. One study75 demonstrated a RIPK1-dependent axonal pathology in mice lacking OPTN (one of the ubiquitin-binding proteins that inhibit RIPK1). These animals exhibited a marked reduction in the number of motor axons and a considerable amount of abnormal myelination in the white matter of the ventrolateral spinal cord, without the loss of motor neuron cell bodies. Moreover, these mice also showed more large-diameter axons and fewer total axons in the ventrolateral white matter, remniscent of the degeneration and swelling of motor neuron axons in the spinal cords of individuals with early-stage ALS108. The motor axons of Optn−/− mice also show ultrastructural evidence of immature myelination with decompaction [G], Thus, OPTN deficiency leads to the selective axonal degeneration of motor neurons without affecting cell bodies, providing an animal model of Wallerian-like axonal degeneration.
Notably, the axonal degeneration phenotype of Optn−/− mice can be reproduced by the conditional knockout of Optn specifically from oligodendrocytes75. Compared with WT oligodendrocytes, Optn−/− oligodendrocytes are more sensitive to TNF–induced necroptosis, but can be protected by Nec-1s, RIPK1 D138N kinase-dead mutation or RIPK3 deficiency75. Similarly, RIPK1 inhibition and RIPK3 deficiency block axonal degeneration and neural dysfunction in the Optn−/− mice. Thus, the loss of OPTN induces the activation of RIPK1- and RIPK3-mediated necroptosis of oligodendrocytes to promote Wallerian-like axonal degeneration.
Extensive degeneration of oligodendrocytes has also been demonstrated in SOD1G93A mice, another model of ALS in which mice express a mutant form of superoxide dismutase 1 (SOD1)109. RIPK1 inhibition and RIPK3 deficiency inhibited axonal myelination defects and delayed the onset of motor dysfunction in SOD1G93A mice75. Thus, necroptosis contributes to the degeneration of oligodendrocytes and motor neuron axons in SOD1G93A mice. The involvement of necroptosis in ALS may not be limited only to familial ALS: several markers of necroptosis, including levels of insoluble RIPK1, RIPK3, MLKL and phospho-MLKL and increased Ser14/15-phosphorylated RIPK1, were found to be increased in post-mortem spinal cord samples from individuals with sporadic ALS75.
Although the studies above highlight the role of necroptosis in the degeneration of oligodendrocytes to promote axonal degeneration in a non-cell-autonomous manner, necroptosis may also contribute directly to motor neuron cell death. In a humanized in vitro model of ALS, astrocytes derived from individuals with sporadic ALS, but not control individuals, killed human embryonic stem cell-derived motor neurons by promoting necroptosis110.
Mitochondrial and lysosomal defects promote necroptosis.
PD is characterized by progressive loss of domaminergic neurons in the substantia nigra, and a central role for mitochondrial dysfunction in the etiology of the disorder has been long established111. Recent studies implicate necroptosis in PD and potentially connect mitochondrial defects to necroptosis induction112. Similar to the pathology observed in MS, ALS and AD, the protein levels of RIPK1, RIPK3 and MLKL were increased in the postmortem substantia nigra of individuals with PD compared with controls112. Inhibition of RIPK1 kinase activity by Nec-1 protected PC12 cells [G] from cell death induced by 6-hydroxydopamine, a mitochondrial respiration inhibitor used to induce a model PD113, suggesting that RIPK1 activation might be involved in the loss of dopaminergic neurons in PD. Moreover, leucine-rich repeat kinase 2 (LRRK2), which is encoded by a gene often mutated in PD, was identified in a systematic RNAi screen as promoting the activation of complex I-associated RIPK155. In addition, neural cells differentiated from induced pluripotent stem cells (iPSCs) from PD patients with mutations in the gene encoding optic atrophy 1 (OPA1), a GTPase that regulates mitochondrial fission and fusion, show severe mitochondrial dysfunction and cell death that is inhibited by Nec-1s112. Finally, neurotoxicity in the MPTP-induced PD mouse model is also attenuated by Nec-1s112. These results suggest that mitochondrial dysfunction may sensitize cells to the activation of necroptosis.
PD shares genetic risk factors with Gaucher disease (GD), which is the most common lysosomal storage disease [G] and caused by mutations in the gene encoding glucocerebrosidase (GBA). In two mouse models of GD, the expression of FLIP is increased and correlates with reductions in caspase-8 activity; furthermore, the expression of both RIPK1 and RIPK3 are elevated114. RIPK3 deficiency substantially improved the motor function and other symptoms, and increased survival in a GBA-inhibitor-induced mouse model of GD114. In lysosomal storage diseases, lysosomal stress may promote neuroinflammation, leading to the expression of FLIP, which in turn blocks caspase-8 and allows the induction of necroptosis.
RIPK1 activation and neuroinflammation
In addition to being a key regulator of necroptotic cell death, RIPK1 also promotes neuroinflammation — a parallel deleterious signalling cascade that has emerged as a key driver of neurodegeneration115–117. Risk for neurodegenerative diseases such as AD and ALS is prominently associated with variants of genes in the innate immune system118. Understanding the mechanisms by which RIPK1 interacts with these genetic risk factors to promote cell death and inflammation in the CNS should provide important insights into the pathogenesis of human neurodegenerative diseases.
RIPK1-mediated inflammatory responses in microglia.
The activation of necroptosis robustly induces a pro-inflammatory gene expression cascade through a RIPK1-mediated, cell-autonomous mechanism43,119. In spinal cords from Optn−/− mice, the levels of proinflammatory cytokines, including interleukin-1α (IL-1α), IL-1β, interferon-γ (IFNγ) and TNF were all elevated, and were reduced by RIPK1 inhibition75. The axonal degeneration phenotype of Optn−/− mice can also be mimicked by loss of OPTN specifically from myeloid-cells or microglia75. Thus, the loss of OPTN from microglia, as well as from oligodendrocytes, can also promote the activation of RIPK1 to mediate axonal degeneration. RNA-sequencing analysis of Optn−/− microglia revealed a module of approximately 1300 genes — including those encoding pro-inflammatory microglial markers such as the lipopolysaccharide (LPS) receptor CD14 and CD86 — that are differentially expressed in a RIPK1-dependent manner120. OPTN deficiency may therefore promote the activation of pro-inflammatory microglia, which in turn contribute to the myelination and axonal defects.
The RIPK1-mediated inflammatory response in microglia may also contribute to AD. A hallmark of AD is chronic brain inflammation marked by increases in microglial activation and the levels of pro-inflammatory cytokines121. A GWAS identified three polymorphisms in the promoter of TNF, two of which are linked to increased TNF production, that are associated with late-onset AD122. In addition, individuals with mild cognitive impairment at risk to develop AD show increased cerebral spinal fluid levels of TNF, suggesting that CNS inflammation may occur early in the pathogenesis of AD31. In an AD mouse model, deletion of Tnfrl reduced the production of Aβ, reduced plaque formation and ameliorated the behavioural deficits123.
The levels of RIPK1 in post-mortem samples from individuals with late-onset AD are increased compared with controls and positively correlate with the Braak stage [G]124,125. Of the RIPK1-expressing cells identified in human AD brain samples, a considerable fraction resembled microglia and co-localized with the microglial marker ionized calcium-binding adaptor molecule 1 (IBA1)125. Moreover, in APP/PS1 mice, a mouse model of AD, increased RIPK1 was found in microglia around Aβ plaques96. In the same model, the RIPK1 D138N kinase-dead mutation or the RIPK1 inhibitor Nec-1s reduced the number of plaque-associated microglia and the levels of TNF and IL-1β126. Necroptosis may also directly contribute to neurodegeneration in mice expressing five human familial AD mutations (5xFAD mice)124. Another study suggested that Nec-1 may directly alter Aβ aggregation127. However, the poor in vivo pharmacodynamics of the original Nec-1 126 used in this study make it difficult to explain how the small molecule could reduce Aβ and tau abnormalities in the CNS and improve behavioural symptoms in the APP/PS1 mouse model127.
The RIPK1-mediated inflammatory gene signature may be present in multiple neurological diseases and in ageing. The expression of at least eight RIPK1-regulated genes (included Cst7, Clec7a, Csf1 and Ch25h) was differentially affected in 5xFAD mice, SOD1G93A mice and in ageing microglia75. Interestingly, a common variation near the Ch25h gene (which encodes cholesterol 25-hydroxylase) was identified as a risk factor for late-onset AD in a GW AS study128. Cholesterol 25-hydroxylase — a cell-surface-bound enzyme — mediates the production of 25-hydroxycholesterol. Abberrant production of both CH25H and 25-hydroxycholesterol (25-HC) leading to oligodendrocyte cell death have also been implicated in X-linked adrenoleukodystrophy (X-ALD), a progressive neurodegenerative disorder characterized by cerebral inflammatory demyelination and axonal pathology caused by the loss-of-function mutations in the gene encoding ATP-binding cassette transporter subfamily D member 1 (ABCD1)129. Interestingly, direct administration of 25-HC into the mouse corpus callosum leads to local demyelination129. Thus, the RIPK1-dependent production of this lipid may represent one of the mechanisms mediating deleterious microglial–oligodendrocyte interactions in X-ALD and potentially other neurodegenerative diseases.
Insoluble proteomes in neurodegenerative diseases and necroptosis.
As part of the signalling mechanism of necroptosis induction, activated RIPK1 and RIPK3 form amyloid-like fibrils130. Whereas most RIPK1, RIPK3 and MLKL in control human brains can be extracted using buffers with no detergent or mild detergent, increased levels of insoluble RIPK1, RIPK3 and MLKL are found in post-mortem pathological samples from individuals with MS, ALS or AD and in animal models of MS, ALS and AD75,92,125. Whether this insoluble activated RIPK1 is iuRIPK1 — the form that was recently found to bridge complex I and complex II formation in TNF-induced RIPK1-dependent apoptosis and a subset of necroptosis55 — will be an interesting topic to investigate in future. The formation of insoluble activated RIPK1, RIPK3 and MLKL in human neurodegenerative diseases also raises an intriguing possibility that activation of necroptosis might not only promote necrotic cell death and inflammation, but also might provide a seeding mechanism for promoting pathological protein aggregation by the formation of insoluble activated RIPK1, RIPK3, and phospho-MLKL oligomers during necroptosis that subsequently mediate neurodegeneration.
Protein misfolding can also be induced in the corpus callosum of cuprizone-treated mice. This misfolding is reduced by RIPK1 inhibition or RIPK3 deficiency92, suggesting the activation of necroptosis and the RIPK1-mediated inflammatory response can also promote the accumulation of misfolded proteins. Although MS in humans is considered primarily an autoimmune disease, abnormalities of proteostasis have also been reported in the disorder. A quantitative mass spectrometry analysis of the insoluble proteome in MS lesions from human cortex identified a considerable overlap with the proteins associated with Lewy bodies (LBs) in PD, a classical example of a neurodegenerative disease with inclusion or amyloid pathology92. These data suggest that RIPK1 and RIPK3 might promote protein misfolding and aggregation in many neurological and neurodegenerative diseases. Given the increasingly recognized role of inflammation in driving the pathogenesis of MS131, the contribution of RIPK1-mediated neuroinflammation to protein aggregation and progressive neurodegeneration should be considered.
Ageing and the onset of necroptosis.
Ageing is the most important risk factor for neurodegeneration132. In the adipose tissue of aged rodents, the levels of necroptosis markers are increased and correlate with inflammation133. Molecular mechanisms that mediate the interaction of ageing with genetic risk factors of neurodegenerative diseases that can promote RIPK1 activation are beginning to be elucidated. Mutations causing haploinsufficiency of TBK1 are a major genetic cause of patients with comorbid ALS and frontotemporal dementia (ALS/FTD), accounting for 10.8% of all cases134,135. A recent study showed that TAK1, a key suppressor of RIPK158, is downregulated in ageing human brains 79. Reduced expression of TAK1 in myeloid-lineage cells cooperated with the haploid loss of TBK1, another suppressor of RIPK1, in Tbk1+/− mice to induce the hallmarks of ALS/FTD, including neuroinflammation, aggregation of TAR DNA-binding protein 43 (TDP43), axonal degeneration, neuronal loss and behavioural deficits, all of which were blocked upon inhibition of RIPK1 79. Reduced suppression of RIPK1 kinase activity may represent an important mechanism in ageing human brains to promote neurological diseases.
The natural decline of proteostasis with ageing may contribute to the onset of neurodegeneration, by reducing protein turnover, promoting the accumulation of misfolded proteins and enhancing the activation of RIPK1. This age-dependent decline of proteostasis may involve decreases in the stability of correctly folded proteins, as well as deficits in protein turnover via autophagy [G] and the proteasome. Autophagy is an important intracellular recycling mechanism that through which damaged protein complexes and organelles, including mitochondria, are degraded. Autophagy cargoes are sequestered into autophagosomes, which eventually fuse with lysosomes containing lysosomal proteases that degrade the cargo136. A genome-wide siRNA screen identified that ageing of human brains is associated with increases in the expression of autophagy suppressors and decreases in autophagy mediators 137. The list of genes whose expressions are increased in ageing brains includes those encoding components of the MAP kinase pathway, which inhibits autophagy. In addition, the expression of the key autophagy genes, such as Atg5, Atg7 and beclin 1, were reduced in the aged human brains137,138. In addition to autophagy, proteasomal function also declines during ageing, owing to decreased expression of proteasome subunits, changes in proteasomal function and increased disassembly of proteasomes139. Given that chemical inhibition of the proteasome or lysosome alone is sufficient to promote the activation of RIPK1 in microglia125, proteostatic decline during ageing may promote the activation of RIPK1.
Activation of RIPK1 drives an increase in microglial expression of an endosomal or lysosomal cathepsin inhibitor known as cystatin F (CST7)125. Microglial CST7 expression is strongly induced in several animal models of neurodegenerative diseases, including in SOD1G93A transgenic mice, in a demyelination model, in a prion disease model and with ageing140–142. A single-cell RNA-seq analysis recently revealed Cst7 to be one of the top biomarkers associated with disease-associated microglia in AD mouse model143. In particular, elevated levels of Cst7 were found in microglia around Aβ deposits in the APP/PS1 AD mouse model, whereas dispersed microglia were generally negative for Cst7. Thus, dysfunction of the lysosomal system in microglia, resulting from increases in CST7, might contribute to reduced proteostasis in AD. Interestingly, inhibition of RIPK1 reduces the microglial expression of CST7 in APP/PS1 mice, suggesting the exciting possibility that an age- or disease-associated reduction of lysosomal function in microglial cells may be rescued by blocking the kinase activity of RIPK1125. Indeed, consistent with this notion, inhibition of RIPK1 reduced the Aβ burden in APP/PS1 mice125. Both genetic and pharmacological inhibition of RIPK1 kinase activity enhanced the ability of primary microglia to degrade oligomeric Aβ1–42, consistent with the notion that inflammatory microglia are less proficient at degrading Aβ. These observations suggest that inhibiting RIPK1 activation may promote the ability of microglia to mediate Aβ turnover125.
Conclusion
An emerging theme suggests that key mechanisms of neurodegeneration involve the interaction of cell-autonomous mechanisms mediated by RIPK1 in CNS glial cells to promote the degeneration of axons and neurons in cell-non-autonomous manners (Fig. 4). This deleterious environment is established by activated microglia and astrocytes, as well as infiltrating immune cells such as T cells and macrophages 144,145, through their ability to release disease-associated inflammatory mediators and promote the vicious cycles of neuroinflammation and neurodegeneration. In this environment, RIPK1 not only may act as a key effector of inflammatory cell death, but also may mediate deleterious inflammatory signalling to provide a feedforward mechanism to accelerate neurodegeneration.
Fig. 4 |. Bimodal deleterious RIPK1 activation in neurological disease.
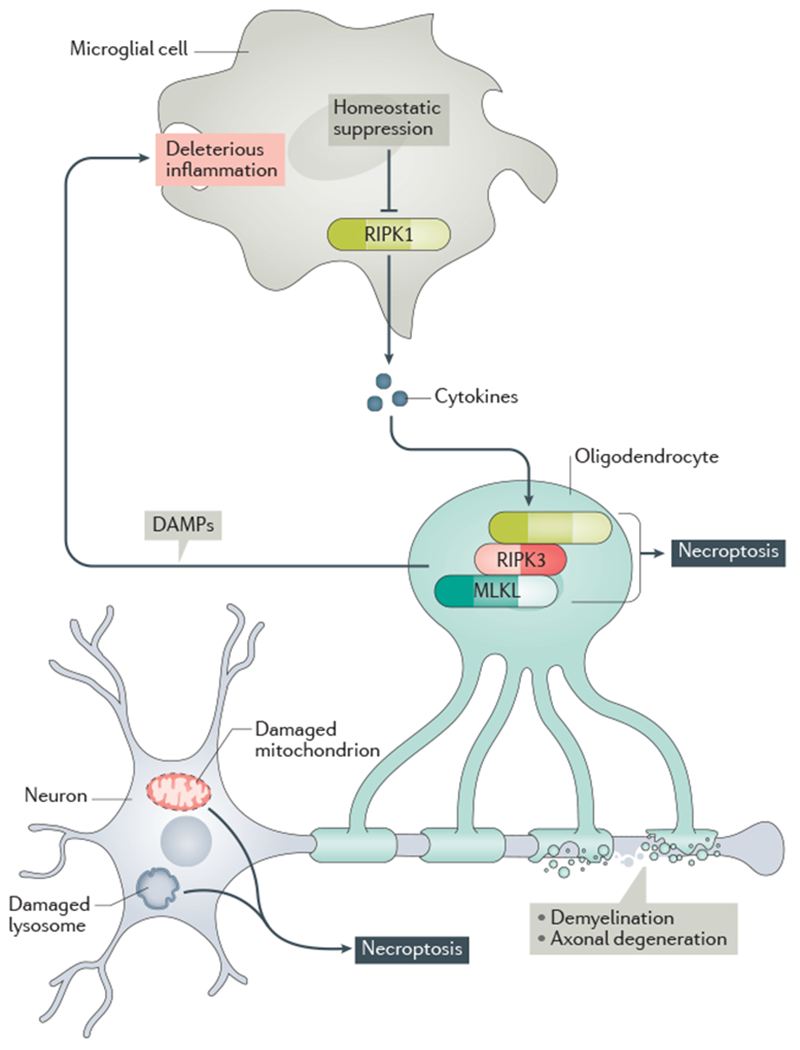
Microglial RIPK1 regulates a degenerative neuroinflammatory milieu in the CNS that can lead to necroptosis of oligodendrocytes and axonal degeneration. In neurons, damaged mitochondria and lysosomes can promote necroptosis. In turn, necroptosis can promote inflammation by driving the cell-autonomous expression of proinflammatory cytokines as well as by releasing of the cellular content from necrotic cells (including damage-associated molecular patterns (DAMPs)) into the CNS. This deleterious axis creates a progressive inflammatory and degenerative environment in the brain to promote the progression of neurodegenerative disease.
Dysregulation of RIPK1 suppression might be important in promoting neuroinflammation and degeneration in the CNS. The activation of RIPK1 is normally suppressed by inhibitory phosphorylation by TAK1 and TAK1-activated kinases TAK1, such as MK2, and TBK1, and by M1 and K48 ubiquitylation (Fig. 2). Furthermore, the activation of NF-κB pathway under inflammatory conditions may sensitize cells to necroptosis by inducing the expression of cFLIPl, which reduces the function of caspase-8146, the enzyme that normally cleaves and inhibits RIPK1147. The decline of these RIPK1 suppression mechanisms in ageing and with injury, stress or infection may have important roles in promoting the onset of multiple neurodegenerative diseases.
In contrast to the critical role of RIPK1 kinase activity in mediating RDA, necroptosis and neuroinflammation, the scaffold function of RIPK1 is essential for cell and animal survival, as Ripk1−/− mice display systemic inflammation and cell death in multiple tissues and die during the postnatal period91,148. Furthermore, rare homozygous loss-of-function RIPK1 mutations in humans lead to immunodeficiency, gut inflammation and progressive polyarthritis149. By contrast, blocking RIPK1 kinase activity, as first demonstrated with the small-molecule inhibitors of necroptosis (necrostatins)2,3 and subsequently confirmed in multiple lines of RIPK1 kinase-dead knock-in mice (Table 2) and human clinical studies150, leads to no deleterious consequences. Inhibition of RIPK1 kinase leads to strong resistance to the TNF-induced systemic inflammatory response151–153. Thus, the kinase activity of RIPK1 may represent an important therapeutic target that can be safely modulated for the treatment of chronic inflammatory CNS diseases by simultaneously inhibiting multiple deleterious mechanisms involved in mediating neurodegeneration.
Acknowledgements
This work was supported by grants from the US National Institute of Neurological Disorders and Stroke (NINDS) (1R01NS082257) and the US National Institute on Aging (NIA) (1R01AG047231; RF1AG055521) (to J.Y.).
Glossary terms
- Death receptors
The receptors for TNF, FasL and TRAILs that contain an intracellular protein-protein interaction domain, known as Death-domain, and can mediate cell death
- Extrinsic cell death
Cell death pathways activated upon stimulation of TNFR1, FAS and certain TRAIL receptors by their cognate ligands to mediate apoptosis and necroptosis
- Intrinsic apoptosis
Intrinsic apoptosis pathway can be activated upon mitochondrial damage to mediate the activation of downstream caspases such as caspase-3
- Middle cerebral artery occlusion (MCAo)
A mouse model of stroke induced by the insertion of a filament into the carotid artery to block cerebral blood flow to induce tissue damage in the brains
- Experimental autoimmune encephalitis
An animal model of multiple sclerosis induced by immunizing with purified myelin, peptides from myelin derived proteins, or passive transfer of T cells reactive to these myelin proteins
- Immune silent
A state that does not induce immune activation or inflammatory response
- Death domain
A protein-protein interaction module composed of a bundle of six alpha-helices
- RIPK1-dependent apoptosis
Stimulation of TNF in cells with TAK1, TBK1 or cIAP1/2 deficiency may promote RIPK1 kinase-dependent activation of caspase-8 and consequently apoptosis which can be inhibited by RIPK1 inhibitor Necstatin-1(Nec-1s)
- Cuprizone model
A rodent model of oligodendrocyte death and reversible demyelination induced by feeding with the copper chelator cuprizone
- Wallerian degeneration
The degeneration of distal axon after a nerve fiber is injuried
- Axonal myelin decompaction
Immature axon myelination pattern with loosely packed myelin sheath
- PC12 cells
A cell line derived from a pheochromocytoma of the rat adrenal medulla
- Lysosomal storage disease
Diseases that result from defects in lysosomal function due to mutations in genes involved lipid metabolism and often leading to neurodegeneration, e.g., Gaucher disease
- Braak stage
A pathology scoring system used for both AD and PD that refers to the extent of distribution of neuropathology in these diseases
- Autophagy
A cellular mechanism involves the formation of double-membrane vesicles, known as autophagosomes, which fuse with lysosomes and then the contents are degraded and recycled
- DFG-out conformation
The inactive conformation of T-loop motif of a kinase, such as receptor-interacting protein kinase 1
- Type II kinase inhibitor
Kinase inhibitors that inactivate the kinase by binding a hydrophobic pocket adjacent to the ATP-binding site. Type II kinase inhibitors are more specific than type I kinase inhibitors, which bind to the ATP-binding pocket itself
- DNA-encoded libraries
Synthetic chemical libraries made by conjugating chemical compounds or building blocks to short DNA fragments that serve as identification bar codes
Footnotes
Competing interests
J.Y. is a consultant of Denali Therapeutics. P.A. declares no competing interests. D.O. is an employee of Sanofi.
Reference:
- 1.Yuan J & Yankner BA Apoptosis in the nervous system. Nature 407, 802–809, doi: 10.1038/35037739 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Degterev A et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1, 112–119 (2005). [DOI] [PubMed] [Google Scholar]; This paper provided the first definition of necroptosis and insights into the functional role of necroptosis in acute neurological injuries.
- 3.Degterev A et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4, 313–321 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrated RIPK1 kinase as the target of necrostatin-1 and the role of RIPK1 as a key mediator of necroptosis.
- 4.Ofengeim D & Yuan J Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol 14, 727–736, doi: 10.1038/nrm3683 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Weinlich R, Oberst A, Beere HM & Green DR Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol 18, 127–136, doi: 10.1038/nrm.2016.149 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Shan B, Pan H, Najafov A & Yuan J Necroptosis in development and diseases. Genes Dev 32, 327–340, doi: 10.1101/gad.312561.118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mullard A Microglia-targeted candidates push the Alzheimer drug envelope. Nat Rev Drug Discov 17, 303–305, doi: 10.1038/nrd.2018.65 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Weisel K et al. Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol Res Perspect 5, doi: 10.1002/prp2.365 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan J & Horvitz HR A first insight into the molecular mechanisms of apoptosis. Cell 116, S53–56, 51 p following S59 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Conradt B, Wu YC & Xue D Programmed Cell Death During Caenorhabditis elegans Development. Genetics 203, 1533–1562, doi: 10.1534/genetics.115.186247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan J, Shaham S, Ledoux S, Ellis HM & Horvitz HR The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell 75, 641–652, doi:0092-8674(93)90485-9 [pii] (1993). [DOI] [PubMed] [Google Scholar]
- 12.Gagliardini V et al. Prevention of vertebrate neuronal death by the crmA gene. Science 263, 826–828 (1994). [DOI] [PubMed] [Google Scholar]
- 13.Degterev A, Boyce M & Yuan J A decade of caspases. Oncogene 22, 8543–8567, doi: 10.1038/sj.onc.1207107 1207107 [pii] (2003). [DOI] [PubMed] [Google Scholar]
- 14.Hyman BT & Yuan J Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat Rev Neurosci 13, 395–406, doi: 10.1038/nrn3228 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Schafer ZT & Kornbluth S The apoptosome: physiological, developmental, and pathological modes of regulation. Dev Cell 10, 549–561, doi: 10.1016/j.devcel.2006.04.008 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Strasser A, Jost PJ & Nagata S The many roles of FAS receptor signaling in the immune system. Immunity 30, 180–192, doi:S1074-7613(09)00069-7 [pii] 10.1016/j.immuni.2009.01.001 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tummers B & Green DR Caspase-8: regulating life and death. Immunol Rev 277, 76–89, doi: 10.1111/imr.12541 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar S, van Raam BJ, Salvesen GS & Cieplak P Caspase cleavage sites in the human proteome: CaspDB, a database of predicted substrates. PLoS One 9, e110539, doi: 10.1371/journal.pone.0110539 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalb R The protean actions of neurotrophins and their receptors on the life and death of neurons. Trends Neurosci 28, 5–11, doi: 10.1016/j.tins.2004.11.003 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Putcha GV, Deshmukh M & Johnson EM Jr. Inhibition of apoptotic signaling cascades causes loss of trophic factor dependence during neuronal maturation. J Cell Biol 149, 1011–1018 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuida K et al. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 384, 368–372, doi: 10.1038/384368a0 (1996). [DOI] [PubMed] [Google Scholar]
- 22.Kuida K et al. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 94, 325–337 (1998). [DOI] [PubMed] [Google Scholar]
- 23.Krajewska M et al. Dynamics of expression of apoptosis-regulatory proteins Bid, Bcl-2, Bcl-X, Bax and Bak during development of murine nervous system. Cell Death Differ 9, 145–157, doi: 10.1038/sj.cdd.4400934 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Troy CM, Akpan N & Jean YY Regulation of caspases in the nervous system implications for functions in health and disease. Prog Mol Biol Transl Sci 99, 265–305, doi: 10.1016/B978-0-12-385504-6.00007-5 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kole AJ, Annis RP & Deshmukh M Mature neurons: equipped for survival. Cell Death Dis 4, e689, doi: 10.1038/cddis.2013.220 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarosiek KA et al. Developmental Regulation of Mitochondrial Apoptosis by c-Myc Governs Age- and Tissue-Specific Sensitivity to Cancer Therapeutics. Cancer Cell 31, 142–156, doi : 10.1016/j.ccell.2016.11.011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kole AJ, Swahari V, Hammond SM & Deshmukh M miR-29b is activated during neuronal maturation and targets BH3-only genes to restrict apoptosis. Genes Dev 25, 125–130, doi : 10.1101/gad.1975411 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wright KM, Smith MI, Farrag L & Deshmukh M Chromatin modification of Apaf-1 restricts the apoptotic pathway in mature neurons. J Cell Biol 179, 825–832, doi: 10.1083/jcb.200708086 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perrelet D et al. Motoneuron resistance to apoptotic cell death in vivo correlates with the ratio between X-linked inhibitor of apoptosis proteins (XIAPs) and its inhibitor, XIAP-associated factor 1. JNeurosci 24, 3777–3785, doi: 10.1523/JNEUROSCI.0413-04.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tarkowski E, Blennow K, Wallin A & Tarkowski A Intracerebral production of tumor necrosis factor-alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J Clin Immunol 19, 223–230 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Tarkowski E, Andreasen N, Tarkowski A & Blennow K Intrathecal inflammation precedes development of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 74, 1200–1205 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu CH et al. Systemic inflammatory response and neuromuscular involvement in amyotrophic lateral sclerosis. NeurolNeuroimmunol Neuroinflamm 3, e244, doi: 10.1212/NXI.0000000000000244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tateishi T et al. CSF chemokine alterations related to the clinical course of amyotrophic lateral sclerosis. J Neuroimmunol 222, 76–81, doi: 10.1016/j.jneuroim.2010.03.004 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Nagatsu T, Mogi M, Ichinose H & Togari A Cytokines in Parkinson’s disease. J Neural Transm Suppl, 143–151 (2000). [PubMed] [Google Scholar]
- 35.Sharief MK & Hentges R Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N Engl J Med 325, 467–472, doi: 10.1056/NEJM199108153250704 (1991). [DOI] [PubMed] [Google Scholar]
- 36.Habbas S et al. Neuroinflammatory TNFalpha Impairs Memory via Astrocyte Signaling. Cell 163, 1730–1741, doi: 10.1016/j.cell.2015.11.023 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Walczak H Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harbor perspectives in biology 5, a008698, doi: 10.1101/cshperspect.a008698 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin-Villalba A et al. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ 8, 679–686 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Hovelmeyer N et al. Apoptosis of oligodendrocytes via Fas and TNF-R1 is a key event in the induction of experimental autoimmune encephalomyelitis. J Immunol 175, 5875–5884 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Nitsch R et al. Human brain-cell death induced by tumour-necrosis-factor-related apoptosis-inducing ligand (TRAIL). Lancet 356, 827–828, doi: 10.1016/S0140-6736(00)02659-3 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Uberti D et al. TRAIL is expressed in the brain cells of Alzheimer’s disease patients. Neuroreport 15, 579–581 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Cannella B, Gaupp S, Omari KM & Raine CS Multiple sclerosis: death receptor expression and oligodendrocyte apoptosis in established lesions. JNeuroimmunol 188, 128–137, doi: 10.1016/j.jneuroim.2007.05.018 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christofferson DE et al. A Novel Role for RIP1 kinase in Mediating TNFα Production. Cell Death Diseases 3, e320 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jouan-Lanhouet S et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ 19, 2003–2014, doi: 10.1038/cdd.2012.90 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siegmund D, Lang I & Wajant H Cell death-independent activities of the death receptors CD95, TRAILR1, and TRAILR2. FEBS J 284, 1131–1159, doi: 10.1111/febs.13968 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Probert L TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 302, 2–22, doi: 10.1016/j.neuroscience.2015.06.038 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Srinivasan K et al. Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nature communications 7, 11295, doi: 10.1038/ncommsll295 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. JNeurosci 34, 11929–11947, doi: 10.1523/JNEUROSCI.1860-14.2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsu H, Huang J, Shu ΗB, Baichwal V & Goeddel DV TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 4, 387–396 (1996). [DOI] [PubMed] [Google Scholar]
- 50.Micheau O & Tschopp J Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Cho YS et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123, doi:S0092-8674(09)00642-4 [pii] 10.1016/j.cell.2009.05.037 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun L et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream ofRIP3 kinase. Cell 148, 213–227, doi:S0092-8674(11)01422-X [pii] 10.1016/j.cell.2011.11.031 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Wang H et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54, 133–146, doi: 10.1016/j.molcel.2014.03.003 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Dondelinger Y et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell reports 7, 971–981, doi: 10.1016/j.celrep.2014.04.026 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Amin P et al. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFalpha-mediated apoptosis. Proc Natl Acad Sci USA 115, E5944–E5953, doi: 10.1073/pnas.1806973115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bertrand MJ et al. cIAPl and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 30, 689–700, doi:S1097-2765(08)00388-2 [pii] 10.1016/j.molcel.2008.05.014 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Chen ZJ Ubiquitination in signaling to and activation of IKK. Immunol Rev 246, 95–106, doi: 10.1111/j.1600-065X.2012.01108.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Geng J et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nature communications 8, 359, doi: 10.1038/s41467-017-00406-w (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dondelinger Y et al. NF-kappaB-Independent Role of IKKalpha/IKKbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol Cell 60, 63–76, doi: 10.1016/j.molcel.2015.07.032 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Jaco I et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol Cell 66, 698–710 e695, doi: 10.1016/j.molcel.2017.05.003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Menon MB et al. p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat Cell Biol 19, 1248–1259, doi: 10.1038/ncb3614 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Elliott PR et al. SPATA2 Links CYLD to LUBAC, Activates CYLD, and Controls LUBAC Signaling. Mol Cell 63, 990–1005, doi: 10.1016/j.molcel.2016.08.001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kupka S et al. SPATA2-Mediated Binding of CYLD to HOIP Enables CYLD Recruitment to Signaling Complexes. Cell reports 16, 2271–2280, doi: 10.1016/j.celrep.2016.07.086 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schlicher L et al. SPATA2 promotes CYLD activity and regulates TNF-induced NF-kappaB signaling and cell death. EMBO reports 17, 1485–1497, doi: 10.15252/embr.201642592 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wei R et al. SPATA2 regulates the activation of RIPK1 by modulating linear ubiquitination. Genes Dev 31, 1162–1176, doi: 10.1101/gad.299776.117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kovalenko A et al. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 424, 801–805, doi: 10.1038/nature01802 nature01802 [pii] (2003). [DOI] [PubMed] [Google Scholar]
- 67.Trompouki E et al. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature 424, 793–796, doi : 10.1038/nature01803 nature01803 [pii] (2003). [DOI] [PubMed] [Google Scholar]
- 68.Draber P et al. LUBAC-Recruited CYLD and A20 Regulate Gene Activation and Cell Death by Exerting Opposing Effects on Linear Ubiquitin in Signaling Complexes. Cell reports 13, 2258–2272, doi: 10.1016/j.celrep.2015.11.009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hitomi J et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135,1311–1323. (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper described a genome-wide siRNA screen for genes that regulate necroptosis.
- 70.Berger SB et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol 192, 5476–5480, doi: 10.4049/jimmunol.1400499 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rahighi S et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 136, 1098–1109, doi: 10.1016/j.cell.2009.03.007 (2009). [DOI] [PubMed] [Google Scholar]
- 72.Hadian K et al. NF-kappaB essential modulator (NEMO) interaction with linear and lys-63 ubiquitin chains contributes to NF-kappaB activation. J Biol Chem 286, 26107–26117, doi: 10.1074/jbc.M111.233163 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nanda SK et al. Polyubiquitin binding to ABIN1 is required to prevent autoimmunity. J Exp Med 208, 1215–1228, doi: 10.1084/jem.20102177 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakazawa S et al. Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nature communications 7, 12547, doi: 10.1038/ncomms12547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ito Y et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353, 603–608, doi: 10.1126/science.aaf6803 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper provided the first genetic connection between ALS and necroptosis.
- 76.Dziedzic SA et al. ABIN-1 regulates RIPK1 activation by linking Met1 ubiquitylation with Lys63 deubiquitylation in TNF-RSC. Nat Cell Biol 20, 58–68, doi: 10.1038/s41556-017-0003-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper described the role of ABIN-1, a ubiquitin-binding protein implicated in ALS and schizophrenia, in inhibiting the activation of RIPK1 and necroptosis.
- 77.Li F et al. Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1-mediated phosphorylation. Autophagy, 1–14, doi: 10.1080/15548627.2017.1391970 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vlantis K et al. NEMO Prevents RIP Kinase 1-Mediated Epithelial Cell Death and Chronic Intestinal Inflammation by NF-kappaB-Dependent and -Independent Functions. Immunity 44, 553–567, doi: 10.1016/j.immuni.2016.02.020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu D et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 174, 1477–1491 e1419, doi: 10.1016/j.cell.2018.07.041 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper revealed the molecular insights by which the reduced levels of TAK1, a key suppressor of RIPK1, in aging of human brains cooperates with the haploidinsufficiency of TBK1 to promote the onset of ALS.
- 80.Harhaj EW & Dixit VM Regulation of NF-kappaB by deubiquitinases. Immunological reviews 246, 107–124, doi: 10.1111/j.1600-065X.2012.01100.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vucic D, Dixit VM & Wertz IE Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol 12, 439–452, doi: 10.1038/nrm3143 (2011). [DOI] [PubMed] [Google Scholar]
- 82.Wertz IE et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430, 694–699, doi: 10.1038/nature02794 nature02794 [pii] (2004). [DOI] [PubMed] [Google Scholar]
- 83.Bosanac I et al. Ubiquitin binding to A20 ZnF4 is required for modulation of NF-kappaB signaling. Mol Cell 40, 548–557, doi: 10.1016/j.molcel.2010.10.009 (2010). [DOI] [PubMed] [Google Scholar]
- 84.Shembade N, Ma A & Harhaj EW Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 327, 1135–1139, doi:327/5969/1135 [pii] 10.1126/science.1182364 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McLaughlin RL et al. Genetic correlation between amyotrophic lateral sclerosis and schizophrenia. Nature communications 8, 14774, doi: 10.1038/ncomms14774 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin Y, Devin A, Rodriguez Y & Liu ZG Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev 13, 2514–2526 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Meng H et al. Death-domain dimerization-mediated activation of RIPK1 controls necroptosis and RIPK1-dependent apoptosis. Proc Natl Acad Sci U S A, doi: 10.1073/pnas.1722013115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Varfolomeev EE et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apol, and DR3 and is lethal prenatally. Immunity 9, 267–276 (1998). [DOI] [PubMed] [Google Scholar]
- 89.Yeh WC et al. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279, 1954–1958 (1998). [DOI] [PubMed] [Google Scholar]
- 90.Dillon CP et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189–1202, doi: 10.1016/j.cell.2014.04.018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rickard JA et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157, 1175–1188, doi: 10.1016/j.cell.2014.04.019 (2014). [DOI] [PubMed] [Google Scholar]
- 92.Ofengeim D et al. Activation of necroptosis in multiple sclerosis. Cell reports 10, 1836–1849, doi: 10.1016/j.celrep.2015.02.051 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper provided the first insight into the involvement and mechanism of RIPK1-mediated necroptosis in a chronic human neurodegenerative disease – multiple sclerosis, and the role of necroptosis in mediating oligodendrocyte cell death.
- 93.Micheau O, Lens S, Gaide O, Alevizopoulos K & Tschopp J NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol 21, 5299–5305, doi: 10.1128/MCB.21.16.5299-5305.2001 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rehker J et al. Caspase-8, association with Alzheimer’s Disease and functional analysis of rare variants. PLoS One 12, e0185777, doi: 10.1371/journal.pone.0185777 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gonzalvez F et al. TRAF2 Sets a threshold for extrinsic apoptosis by tagging caspase-8 with a ubiquitin shutoff timer. Mol Cell 48, 888–899, doi: 10.1016/j.molcel.2012.09.031 (2012). [DOI] [PubMed] [Google Scholar]
- 96.Conforti L, Gilley J & Coleman ΜP Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci 15, 394–409, doi: 10.1038/nrn3680 (2014). [DOI] [PubMed] [Google Scholar]
- 97.Zhou T et al. Implications of white matter damage in amyotrophic lateral sclerosis (Review). Molecular medicine reports 16, 4379–4392, doi: 10.3892/mmr.2017.7186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Adalbert R & Coleman ΜP Review: Axon pathology in age-related neurodegenerative disorders. Neuropathol Appl Neurobiol 39, 90–108, doi: 10.1111/j.1365-2990.2012.01308.x (2013). [DOI] [PubMed] [Google Scholar]
- 99.Raff MC, Whitmore AV & Finn JT Axonal self-destruction and neurodegeneration. Science 296, 868–871, doi: 10.1126/science.1068613 (2002). [DOI] [PubMed] [Google Scholar]
- 100.Wang JT, Medress ZA & Barres BA Axon degeneration: molecular mechanisms of a self-destruction pathway. J Cell Biol 196, 7–18, doi: 10.1083/jcb.201108111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Waller A Experiments on the Section of the Glossopharyngeal and Hypoglossal Nerves of the Frog, and Observations of the Alterations Produced Thereby in the Structure of Their Primitive Fibres. Phil. Trans. R. Soc. Lond. 140, 423–429 (1850). [Google Scholar]
- 102.Vargas ΜE & Barres BA Why is Wallerian degeneration in the CNS so slow? Annu Rev Neurosci 30, 153–179, doi: 10.1146/annurev.neuro.30.051606.094354 (2007). [DOI] [PubMed] [Google Scholar]
- 103.Trapp BD & Nave KA Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci 31, 247–269, doi: 10.1146/annurev.neuro.30.051606.094313 (2008). [DOI] [PubMed] [Google Scholar]
- 104.Butts BD, Houde C & Mehmet H Maturation-dependent sensitivity of oligodendrocyte lineage cells to apoptosis: implications for normal development and disease. Cell Death Differ 15, 1178–1186, doi: 10.1038/cdd.2008.70 (2008). [DOI] [PubMed] [Google Scholar]
- 105.Yoshikawa M et al. Discovery of 7-Oxo-2,4,5,7-tetrahydro-6 H-pyrazolo[3,4-cjpyridine Derivatives as Potent, Orally Available, and Brain-Penetrating Receptor Interacting Protein 1 (RIP 1) Kinase Inhibitors: Analysis of Structure-Kinetic Relationships. JMed Chem, doi: 10.102l/acs.jmedchem.7b01647 (2018). [DOI] [PubMed] [Google Scholar]
- 106.Dadon-Nachum M, Melamed E & Offen D The “dying-back” phenomenon of motor neurons in ALS. J Mol Neurosci 43, 470–477, doi: 10.1007/sl2031-010-9467-1 (2011). [DOI] [PubMed] [Google Scholar]
- 107.Fischer LR & Glass JD Axonal degeneration in motor neuron disease. Neuro-degenerative diseases 4, 431–442, doi: 10.1159/000107704 (2007). [DOI] [PubMed] [Google Scholar]
- 108.Sasaki S & Maruyama S Increase in diameter of the axonal initial segment is an early change in amyotrophic lateral sclerosis. J Neurol Sci 110, 114–120 (1992). [DOI] [PubMed] [Google Scholar]
- 109.Kang SH et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci 16, 571–579, doi: 10.1038/nn.3357 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Re DB et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81, 1001–1008, doi: 10.1016/j.neuron.2014.01.011 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Exner N, Lutz AK, Haass C & Winklhofer KF Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. EMBOJ 31, 3038–3062, doi: 10.1038/emboj.2012.170 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Iannielli A et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell reports 22, 2066–2079, doi: 10.1016/j.celrep.2018.01.089 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wu JR et al. Necrostatin-1 protection of dopaminergic neurons. Neural Regen Res 10, 1120–1124, doi: 10.4103/1673-5374.160108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vitner EB et al. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat Med 20, 204–208, doi: 10.1038/nm.3449 (2014). [DOI] [PubMed] [Google Scholar]; This paper demonstrated the role of RIPK3 and necroptosis in Gaucher’s disease.
- 115.Ransohoff RM How neuroinflammation contributes to neurodegeneration. Science 353, 777–783, doi: 10.1126/science.aag2590 (2016). [DOI] [PubMed] [Google Scholar]
- 116.Chen H, Kankel MW, Su SC, Han SWS & Ofengeim D Exploring the genetics and non-cell autonomous mechanisms underlying ALS/FTLD. Cell Death Differ, doi: 10.1038/s41418-018-0060-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang B et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720, doi: 10.1016/j.cell.2013.03.030 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Efthymiou AG & Goate AM Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener 12, 43, doi: 10.1186/sl3024-017-0184-x (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhu K et al. Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Diseases In press (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kigerl KA et al. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 29, 13435–13444, doi: 10.1523/JNEUROSCI.3257-09.2009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mandrekar-Colucci S & Landreth GE Microglia and inflammation in Alzheimer’s disease. CNS Neurol Disord Drug Targets 9, 156–167 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Collins JS et al. Association of a haplotype for tumor necrosis factor in siblings with late-onset Alzheimer disease: the NIMH Alzheimer Disease Genetics Initiative. Am J Med Genet 96, 823–830 (2000). [DOI] [PubMed] [Google Scholar]
- 123.He P et al. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J Cell Biol 178, 829–841, doi: 10.1083/jcb.200705042 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Caccamo A et al. Necroptosis activation in Alzheimer’s disease. NatNeurosci 20, 1236–1246, doi: 10.1038/nn.4608 (2017). [DOI] [PubMed] [Google Scholar]; This paper provided human pathological evidence for the role of necroptosis in Alzheimer’s disease.
- 125.Ofengeim D et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc Natl Acad Sci U S A 114, E8788–E8797, doi: 10.1073/pnas.1714175114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrated the role of RIPK1 in mediating microglial inflammatory response in AD.
- 126.Degterev A, Maki JL & Yuan J Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ 20, 366, doi: 10.1038/cdd.2012.133 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yang SH et al. Nec-1 alleviates cognitive impairment with reduction of Abeta and tau abnormalities in APP/PS1 mice. EMBO Mol Med 9, 61–77, doi: 10.15252/emmm.201606566 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Papassotiropoulos A et al. Cholesterol 25-hydroxylase on chromosome 10q is a susceptibility gene for sporadic Alzheimer’s disease. Neuro-degenerative diseases 2, 233–241, doi: 10.1159/000090362 (2005). [DOI] [PubMed] [Google Scholar]
- 129.Jang J et al. 25-hydroxycholesterol contributes to cerebral inflammation of X-linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nature communications 7, 13129, doi: 10.1038/ncommsl3129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li J et al. The RIP1/RIP3 Necrosome Forms a Functional Amyloid Signaling Complex Required for Programmed Necrosis. Cell 150, 339–350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper provided the structural evidence for the amyloid conformation of necrosome in necroptosis.
- 131.Abdelhak A, Weber MS & Tumani H Primary Progressive Multiple Sclerosis: Putting Together the Puzzle. Front Neurol 8, 234, doi: 10.3389/fneur.2017.00234 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Winblad B et al. Defeating Alzheimer’s disease and other dementias: a priority for European science and society. Lancet Neurol 15, 455–532, doi: 10.1016/S1474-4422(16)00062-4 (2016). [DOI] [PubMed] [Google Scholar]
- 133.Deepa SS, Unnikrishnan A, Matyi S, Hadad N & Richardson A Necroptosis increases with age and is reduced by dietary restriction. Aging Cell, el2770, doi: 10.1111/acel.12770 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cirulli ET et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441, doi: 10.1126/science.aaa3650 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Freischmidt A, Muller K, Ludolph AC, Weishaupt JH & Andersen PM Association of Mutations in TBK1 With Sporadic and Familial Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. JAMA Neurol 74, 110–113, doi: 10.1001/jamaneurol.2016.3712 (2017). [DOI] [PubMed] [Google Scholar]
- 136.Mizushima N Autophagy in protein and organelle turnover. Cold Spring Harb Symp QuantBiol 76, 397–402, doi: 10.1101/sqb.2011.76.011023 (2011). [DOI] [PubMed] [Google Scholar]
- 137.Lipinski MM et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc Natl Acad Sci U S A 107, 14164–14169, doi:1009485107 [pii] 10.1073/pnas.1009485107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shibata M et al. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem 281, 14474–14485, doi:M600364200 [pii] 10.1074/jbc.M600364200 (2006). [DOI] [PubMed] [Google Scholar]
- 139.Saez I & Vilchez D The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr Genomics 15, 38–51, doi: 10.2174/138920291501140306113344 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chiu IM et al. Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc Natl Acad Sci U S A, doi:0911405106 [pii] 10.1073/pnas.0911405106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nuvolone M et al. Cystatin F is a biomarker of prion pathogenesis in mice. PLoS One 12, e0171923, doi: 10.1371/journal.pone.0171923 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ma J et al. Microglial cystatin F expression is a sensitive indicator for ongoing demyelination with concurrent remyelination. J Neurosci Res 89, 639–649, doi: 10.1002/jnr.22567 (2011). [DOI] [PubMed] [Google Scholar]
- 143.Keren-Shaul H et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290 e1217, doi: 10.1016/j.cell.2017.05.018 (2017). [DOI] [PubMed] [Google Scholar]
- 144.Schetters STT, Gomez-Nicola D, Garcia-Vallejo JJ & Van Kooyk Y Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front Immunol 8, 1905, doi: 10.3389/fimmu.2017.01905 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gate D, Rezai-Zadeh K, Jodry D, Rentsendorj A & Town T Macrophages in Alzheimer’s disease: the blood-borne identity. J Neural Transm (Vienna) 117, 961–970, doi: 10.1007/s00702-010-0422-7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Weinlich R et al. Protective roles for caspase-8 and cFLIP in adult homeostasis. Cell reports 5, 340–348, doi: 10.1016/j.celrep.2013.08.045 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rajput A et al. RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity 34, 340–351, doi: 10.1016/j.immuni.2010.12.018 (2011). [DOI] [PubMed] [Google Scholar]
- 148.Dannappel M et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513, 90–94, doi: 10.1038/nature13608 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cuchet-Lourenco D et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science, doi: 10.1126/science.aar2641 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Harris PA et al. Discovery of Small Molecule RIP1 Kinase Inhibitors for the Treatment of Pathologies Associated with Necroptosis. ACS Medicinal Chemistry Letters 4, 1238–1243, doi: 10.1021/ml400382p (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Polykratis A et al. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol 193, 1539–1543, doi: 10.4049/jimmunol.1400590 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shutinoski B et al. K45A mutation of RIPK1 results in poor necroptosis and cytokine signaling in macrophages, which impacts inflammatory responses in vivo. Cell Death Differ 23, 1628–1637, doi: 10.1038/cdd.2016.51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Duprez L et al. RIP Kinase-Dependent Necrosis Drives Lethal Systemic Inflammatory Response Syndrome. Immunity 35, 908–918, doi:S1074-7613(11)00508-5 [pii] 10.1016/j.immuni.2011.09.020 (2011). [DOI] [PubMed] [Google Scholar]
- 154.Liu Y et al. RIP1 kinase activity-dependent roles in embryonic development of Fadd-deficient mice. Cell Death Differ 24, 1459–1469, doi: 10.1038/cdd.2017.78 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Najjar M et al. Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell reports 10, 1850–1860, doi: 10.1016/j.celrep.2015.02.052 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Xie T et al. Structural Basis of RIP1 Inhibition by Necrostatins. Structure 21, 493–499, doi: 10.1016/j.str.2013.01.016 (2013). [DOI] [PubMed] [Google Scholar]
- 157.Harris PA et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J Med Chem 60, 1247–1261, doi: 10.1021/acs.jmedchem.6b01751 (2017). [DOI] [PubMed] [Google Scholar]
- 158.Harris PA et al. DNA-Encoded Library Screening Identifies Benzo[b][1,4]oxazepin-4-ones as Highly Potent and Monoselective Receptor Interacting Protein 1 Kinase Inhibitors. J Med Chem 59, 2163–2178, doi: 10.1021/acs.jmedchem.5b01898 (2016). [DOI] [PubMed] [Google Scholar]
- 159.Chen Y et al. Necrostatin-1 Improves Long-term Functional Recovery Through Protecting Oligodendrocyte Precursor Cells After Transient Focal Cerebral Ischemia in Mice. Neuroscience 371, 229–241, doi: 10.1016/j.neuroscience.2017.12.007 (2018). [DOI] [PubMed] [Google Scholar]
- 160.Zhang S et al. Necrostatin-1 Attenuates Inflammatory Response and Improves Cognitive Function in Chronic Ischemic Stroke Mice. Medicines (Basel) 3, doi : 10.3390/medicines3030016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.King MD, Whitaker-Lea WA, Campbell JM, Alleyne CH Jr. & Dhandapani KM Necrostatin-1 reduces neurovascular injury after intracerebral hemorrhage. Int J Cell Biol 2014, 495817, doi: 10.1155/2014/495817 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.You Z, Savitz S, Yang J, Degterev A, Yuan J, Cuny GD, Moskowitz MA, Whalen MJ Necrostatin-1 Reduces Histopathology and Improves Functional Outcome After Controlled Cortical Impact In Mice. J Cereb Blood Flow Metab. 28:1564–73. (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wang Y et al. Necroptosis inhibitor necrostatin-1 promotes cell protection and physiological function in traumatic spinal cord injury. Neuroscience 266, 91–101, doi: 10.1016/j.neuroscience.2014.02.007 (2014). [DOI] [PubMed] [Google Scholar]
- 164.Do YJ et al. A novel RIPK1 inhibitor that prevents retinal degeneration in a rat glaucoma model. Exp Cell Res 359, 30–38, doi: 10.1016/j.yexcr.2017.08.012 (2017). [DOI] [PubMed] [Google Scholar]
- 165.Rosenbaum DM et al. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res 88, 1569–1576, doi: 10.1002/jnr.22314 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kim CR, Kim JH, Park HL & Park CK Ischemia Reperfusion Injury Triggers TNFalpha Induced-Necroptosis in Rat Retina. Curr Eye Res 42, 771–779, doi: 10.1080/02713683.2016.1227449 (2017). [DOI] [PubMed] [Google Scholar]
- 167.Dvoriantchikova G, Degterev A & Ivanov D Retinal ganglion cell (RGC) programmed necrosis contributes to ischemia-reperfusion-induced retinal damage. Exp Eye Res 123, 1–7, doi: 10.1016/j.exer.2014.04.009 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dong K et al. Necrostatin-1 protects photoreceptors from cell death and improves functional outcome after experimental retinal detachment. Am J Pathol 181, 1634–1641, doi: 10.1016/j.ajpath.2012.07.029 (2012). [DOI] [PubMed] [Google Scholar]
- 169.Murakami Y et al. Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ 21, 270–277, doi: 10.1038/cdd.2013.109 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cougnoux A et al. Necroptosis in Niemann-Pick disease, type C1: a potential therapeutic target. Cell Death Dis 7, e2147, doi: 10.1038/cddis.2016.16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Newton K et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360, doi: 10.1126/science.1249361 (2014). [DOI] [PubMed] [Google Scholar]
- 172.Lule S et al. Genetic Inhibition of Receptor Interacting Protein Kinase-1 Reduces Cell Death and Improves Functional Outcome After Intracerebral Hemorrhage in Mice. Stroke 48, 2549–2556, doi: 10.1161/STROKEAHA.117.017702 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Newton K et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ 23, 1565–1576, doi: 10.1038/cdd.2016.46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kaiser WJ et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci U S A 111, 7753–7758, doi: 10.1073/pnas.1401857111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Fan H et al. Reactive astrocytes undergo M1 microglia/macrohpages-induced necroptosis in spinal cord injury. Mol Neurodegener 11, 14, doi: 10.1186/s13024-016-0081-8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Liu ZM et al. RIP3 deficiency protects against traumatic brain injury (TBI) through suppressing oxidative stress, inflammation and apoptosis: Dependent on AMPK pathway. Biochem Biophys Res Commun, doi: 10.1016/j.bbrc.2018.02.150 (2018). [DOI] [PubMed] [Google Scholar]
- 177.Trichonas G et al. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proceedings of the National Academy of Sciences of the United States of America 107, 21695–21700, doi: 10.1073/pnas.1009179107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]