

ABSTRACT
Obesity is associated with higher cardio-metabolic risk even in childhood and adolescence; whether this association is mediated by epigenetic mechanisms remains unclear. We examined the extent to which mid-childhood body mass index (BMI) z-score (median age 7.7 years) was associated with cardio-metabolic risk score in early adolescence (median age 12.9 years) via mid-childhood DNA methylation among 265 children in the Project Viva. We measured DNA methylation in leukocytes using the Infinium Human Methylation450K BeadChip. We assessed mediation CpG-by-CpG using epigenome-wide association analyses, high-dimensional mediation analysis, and natural effect models. We observed mediation by mid-childhood DNA methylation at 6 CpGs for the association between mid-childhood BMI z-score and cardio-metabolic risk score in early adolescence in the high-dimensional mediation analysis (accounting for 10% of the total effect) and in the natural effect model (β = 0.04, P = 3.2e-2, accounting for 13% of the total effect). The natural direct effect of BMI z-score on cardio-metabolic risk score was still evident (β = 0.27, P = 1.1e-25). We also observed mediation by mid-childhood DNA methylation at 5 CpGs that was in the opposite direction from the total effect (natural effect model: β = −0.04, P = 2.0e-2). Mediation in different directions implies a complex role of DNA methylation in the association between BMI and cardio-metabolic risk and needs further investigation. Future studies with larger sample size and greater variability in cardio-metabolic risk will further help elucidate the role of DNA methylation for cardio-metabolic risk.
KEYWORDS: Obesity, BMI, cardio-metabolic, DNA methylation, epigenetics, mediation
Introduction
The prevalence of childhood obesity in the United State (U.S.) has increased in the past two decades. Between 2011–2014, 23% of children and adolescents in the U.S. had a body mass index (BMI) at or above the 95th percentile [1]. Childhood obesity has been associated with cardio-metabolic risk factors such as hypertension and insulin resistance, and also with higher risk of cardiovascular disease (CVD) in adulthood [2]. CVD is one of the leading causes of death worldwide and in the U.S. [3,4], and places a significant burden on the healthcare system and economy [5]. Investigation of the mechanisms by which obesity affects cardio-metabolic diseases may provide insights into the prevention of CVD starting early in the life course and may reveal potential molecular targets.
With the accumulation of excess weight, adipose tissue may participate in inducing a pro-inflammatory state by releasing cytokines and adipokines into the circulation [6], which are thought to contribute to higher risk of CVD. However, evidence of mortality benefits from anti-inflammatory drugs in people with coronary artery disease is lacking [7]. As such, other modifiable mechanisms may exist. Epigenetic regulation of gene expression has a central role during development, and epigenetic dysregulation may be a driver of early life programming of diseases that manifest in adult life [8,9]. In preschool children, DNA methylation in multiple genes was found associated with differential body composition measures including BMI [10]. Although the causal direction between obesity and DNA methylation is not totally clear, recent studies suggested elevated BMI could causes changes in DNA methylation [11–13]. For example, DNA methylation at ABCG1 and HIF3A has been identified as a consequence rather than a cause of adiposity using longitudinal analysis, Mendelian randomization and epigenome-wide association studies (EWAS) [11–14]. DNA methylation is also associated with CVD [15]. For example, lower methylation of long-interspersed nucleotide repetitive elements-1 (LINE-1) throughout the genome was associated with higher risk of ischemic heart disease and stroke in a longitudinal study of adult men [16]. Changes in DNA methylation among infants born large-for-gestational-age may affect the expression of certain genes that are related to CVD based on functional enrichment analysis using Gene Ontology [17]. DNA methylation was also found to regulate cardio-metabolic risk factors such as blood pressure [18]. These findings suggest a potential regulatory role of epigenetic variability in the biological processes underlying CVD [9,19]. Obesity undoubtedly increases the risk of CVD [20], but whether it does so via epigenetic mechanisms is unknown. Here, we assessed the extent to which DNA methylation mediates the association between mid-childhood BMI and cardio-metabolic risk in early adolescence, using CpG-by-CpG analysis, high-dimensional mediation analysis, natural effect model, and regional DNA methylation analysis.
Methods
Participants
The Project Viva pre-birth cohort was recruited between 1999 and 2002 in the greater Boston area, Massachusetts (U.S.) [21]. Research staff identified eligible participants attending their initial prenatal visit at Atrius Harvard Vanguard Medical Associates and conducted brief interviews after the visit. We recruited women with a singleton pregnancy, fluent in English, gestational age < 22 weeks, and not planning to move away from the study area during pregnancy. Of the 4,102 eligible participants initially approached, 2,670 (64%) were enrolled in the study [21]. At the time of delivery 2,128 eligible women remained enrolled and had a live birth. Mothers reported maternal characteristics and child sex via interviews and questionnaires. We obtained birth weight and date of delivery from medical records and determined gestational age by subtracting the date of the last menstrual period from the date of delivery. If gestational age according to the 2nd-trimester ultrasound differed from the estimation based on last menstrual period by more than 10 days, we used ultrasound results. Birth weight for gestational age z-score was calculated based on a U.S. national reference [22,23]. We conducted in-person follow-up in mid-pregnancy, at birth, in mid-childhood (age ~ 7 years), and in early adolescence (age ~ 13 years) for phenotype characterization and sample collection.
We measured DNA methylation in whole blood samples from the mid-childhood visit using the Infinium Human Methylation450 BeadChip (Illumina, San Diego, CA). The Infinium Human Methylation450K BeadChip measures DNA methylation at around 485,000 loci, covering 99% of the RefSeq genes, using oligonucleotides that hybridize with corresponding target CpGs, also known as probes [24]. Quality control procedures were performed in 473 whole blood samples in mid-childhood. For sample-level quality control, we excluded samples with low quality (≥ 25% of the probes with a detection p-value ≥ 0.01, n = 6), genotype mismatch (n = 4), or sex mismatch (n = 3), leaving 460 participants with available data on DNA methylation in mid-childhood. For probe-level quality control, we excluded probes in X and Y chromosomes, non-CpG probes, non-specific and cross-reactive probes [25], and single nucleotide polymorphism-associated (SNP-associated) probes with a minor-allele frequency of ≥ 5%. A SNP-associated probe is considered as a probe that has SNPs overlapping the probe, the target CpG, or the single base extension (SBE) of the CpG, and was identified based on the SNP information from Illumina and dbSNP (snp137Common). After quality control, 394,460 CpGs were available for the analysis. Annotation data for the methylation array was based on the Illumina’s v1.2 annotation file or obtained from the UCSC Genome Browser [26]. Both SNP-associated probes and Illumina’s annotation data were obtained using the Bioconductor package IlluminaHumanMethylation450kanno.ilmn12.hg19 [27]. The normal-exponential out-of-band (noob) correction was performed to account for background noise and dye-bias [28]. β-mixture quantile intra-sample normalization procedure (BMIQ) was performed to reduce the potential bias due to type II probes [29]. Batch effects from the sample plate were adjusted for using ComBat [30]. We estimated cell type proportions using the Bioconductor package minfi (CD8T, CD4T, Monocytes, B-cells, NK, and Granulocytes).
Exposure
During the mid-childhood in-person visit (median age 7.7 years), research staff measured standing height with a calibrated stadiometer (Shorr Height Board) and weight with a Tanita Scale. We calculated age- and sex-specific BMI z-score based on the U.S. Centers for Disease Control and Prevention national reference data [31]. Among 8-year-old boys and girls of average height, a 1 unit increase in BMI z-score translates into a 2.2 kg/m2 higher BMI or a 3.6 kg higher weight.
Outcome
The outcome of interest was cardio-metabolic risk score in early adolescence (median age 12.9 years). At the in-person early adolescence visit, trained research assistants measured blood pressure (BP) using biannually calibrated automated oscillometric monitors (The Omron HEM-907XL, Illinois, US) on the child’s upper arm at 1-minute intervals 5 times We used the average of the 5 measures in the data analysis. Research staff measured standing waist circumference with a Lefkin woven measuring tape. Participants provided fasting whole blood samples at the early adolescence visit. We measured plasma fasting glucose enzymatically and fasting insulin using an electrochemiluminescence immunoassay (Roche Diagnostic, Indianapolis, IN), and estimated insulin resistance using the Homeostatic Model of Insulin Resistance (HOMA-IR = glucose × insulin/22.5). We measured triglycerides enzymatically with correction for endogenous glycerol and measured high-density lipoproteins cholesterol (HDL-cholesterol) using a direct enzymatic colorimetric assay.
We then derived the cardio-metabolic risk score using an adapted metabolic syndrome definition, as the mean of sex- and cohort-specific z-scores of 5 components of metabolic risk: 1) systolic blood pressure; 2) waist circumference; 3) log-transformed insulin resistance (HOMA-IR); 4) log-transformed triglycerides; and 5) HDL-cholesterol (scaled inversely). We have used the same method in previous studies in this cohort [32–34].
Mediator
For this study, we assessed the extent to which DNA methylation at each CpG measured in blood collected at the mid-childhood visit (median age 7.7 years) mediated the association of mid-childhood BMI z-score with cardio-metabolic risk score in early adolescence. DNA methylation at each CpG site was defined as the proportion of average methylated fluorescence intensity, also known as β-value (ranged from 0 to 1). We converted β-values into M-values (M = log2[β/(1- β)]) for statistical analysis as M-values are approximately normally distributed [35].
Statistical analysis
Demographic differences for participants included and excluded from this study were assessed using χ2 tests (categorical variables) and t-tests (continuous variables), and effect size analysis [36]. We used least-square linear regression to assess the total effect of BMI z-score on cardio-metabolic risk score, adjusted for maternal education and smoking during pregnancy, and age at the early adolescence visit. We used robust linear regression models (RLM) CpG-by-CpG to separately assess the epigenome-wide associations of (a) BMI z-score with DNA methylation at each site, and (b) DNA methylation at each site with cardio-metabolic risk score (Steps 1 and 2 of Figure 1). To assess the association of BMI z-score with DNA methylation, we adjusted for age at blood draw for DNA methylation, cell type proportion, child sex, maternal education and maternal smoking during pregnancy in EWAS (a). We included the same covariates in EWAS (b) to assess the association of DNA methylation with cardio-metabolic risk score, except that we additionally included age at the early adolescence visit. We considered these models as most appropriate because the covariates adjusted in the models are common causes of BMI and cardio-metabolic risk, i.e., confounders. Also, genomic inflation factors (λ) that were close to 1 (λ = 0.97 for EWAS (a); λ = 1.02 for EWAS (b)) imply systematic bias is unlikely. We controlled the Benjamini-Hochberg false discovery rate (FDR) at 5% to account for multiple comparisons for each of the two EWAS separately.
Figure 1.
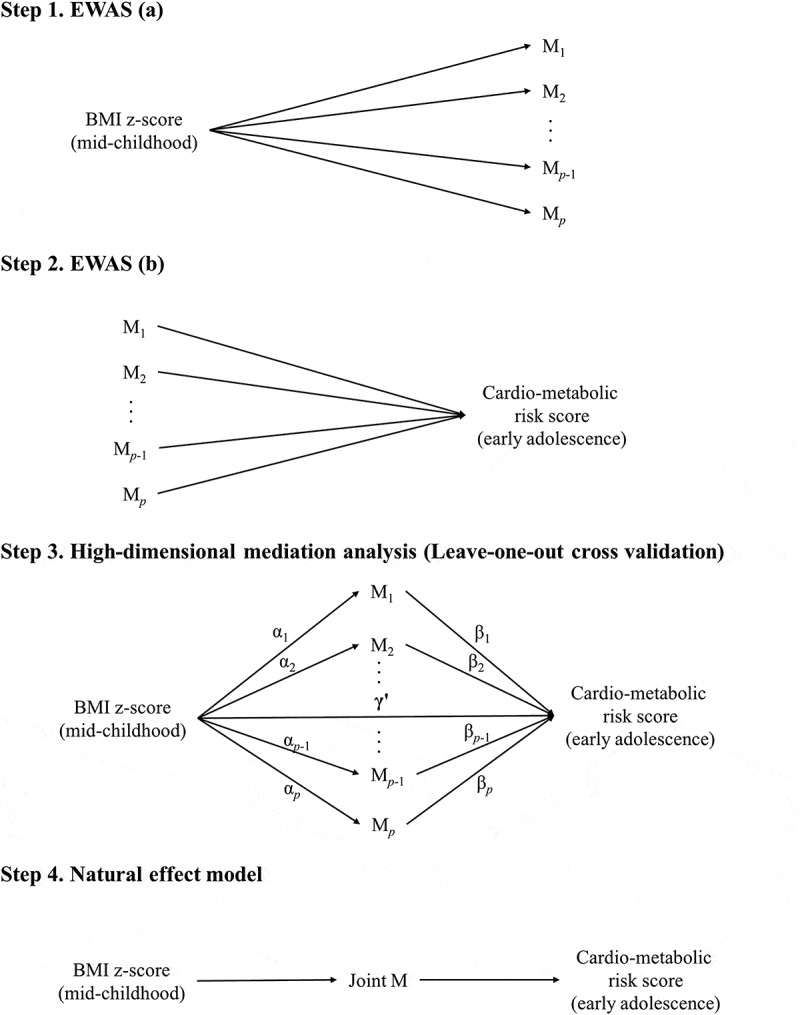
Workflow for assessing the potential mediation effects of DNA methylation in blood in mid-childhood on the associations of BMI z-score in mid-childhood with cardio-metabolic risk score in early adolescence (Mp represents DNA methylation at each CpG. Joint M is the joint natural indirect effect via DNA methylation at the CpGs that were selected in more than 95% of the leave-one-out cross validation tests in the high-dimensional mediation analysis. Illustration for high-dimensional mediation analysis was reproduced according to Zhang et al. 2016 [38] and Boca et al. 2014 [78]).
We performed a high-dimensional mediation analysis (Step 3 of Figure 1) based on the sure independence screening criterion and a variable selection technique (minimax concave penalty), using the HIMA package in R [37,38]. The high-dimensional mediation analysis consists of three steps. First, we identified a subset of CpGs that are among the top n/log(n) largest effects of mediators on the outcome regardless of the p-value, where n is the sample size (i.e., top 48 largest effects given a sample size of 265) [37]. In this study, we used a stricter screening criterion instead of the top 2n/log(n) largest effects in the previous literature to address potential false positive findings. Second, we used minimax concave penalty to evaluate the effects of BMI z-score on the DNA methylation of this subset of CpGs [39]. Third, we examined the FDR of exposure-mediator and mediator-outcome associations based on a joint significance test. To test the robustness of the high-dimensional mediation analysis, we also used the leave-one-out cross-validation approach, in which one participant was left out of each analysis. We reported CpGs selected in more than 95% of the validation tests. We also assessed mediation using a natural effect model (Step 4 of Figure 1) [40,41], using the medflex package in R [42]. The natural effect model circumvents the statistical issues in the traditional mediation analysis, such as confounding and collider bias [43], by decomposing the total causal effect into natural direct and indirect effects [41–43]. Specifically, we assessed the joint natural indirect effect of the potential mediators identified in the leave-one-out cross-validation of the high-dimensional mediation analysis. In a sensitivity analysis, we additionally adjusted for child race, birth weight for gestational age z-score, and maternal pre-pregnancy BMI.
In the regional DNA methylation analysis, differentially methylated regions (DMRs) were identified using the Bioconductor package DMRcate, an approach that is agnostic to genomic annotation and effect direction but not to chromosomal coordinates [44]. Robust estimates of BMI z-score on DNA methylation at each CpG were derived from the Bioconductor package limma [44], and we adjusted for age at blood draw for DNA methylation, child sex, maternal education and smoking during pregnancy in the model. We then assessed the associations of BMI z-score with DMR DNA methylation (exposure-mediator), and of DMR DNA methylation with cardio-metabolic risk score (mediator-outcome) separately using generalized estimating equation (GEE). In GEE, we treated the DNA methylation of each CpG in one DMR as one of the multiple repeated measures. Thus, the results may be more robust than using the median of all available CpGs in one DMR. The FDR was used to account for multiple comparisons of DMRs for each of the two associations separately.
We also performed a secondary analysis to assess mediation by DNA methylation at cg06500161 (ABCG1) selected based on prior literature. DNA methylation at cg06500161 was previously identified to be influenced by adiposity [12,13] by both a Mendelian randomization study and an EWAS. Associations of BMI z-score with DNA methylation at cg06500161, and of DNA methylation at cg06500161 with cardio-metabolic risk score were assessed. We also assessed the natural indirect effect of DNA methylation at cg06500161 for the association of BMI z-score with cardio-metabolic risk score. Table A-1 in Supplementary Material A shows the covariates adjusted for in each analysis.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Results
Among 1,110 children with BMI information in mid-childhood, 10% were overweight (at or above the 85th percentile and below the 95th percentile) and 5% were obese (at or above the 95th percentile). We included 265 participants from the Project Viva who had BMI at the mid-childhood visit, DNA methylation in blood at the mid-childhood visit, cardio-metabolic risk at the early adolescence visit, and all covariates. Table 1 shows that at enrollment, most mothers were college graduate or above (67.9%) and never smoker (70.9%). Participants included in and excluded from this study showed no difference in terms of BMI in mid-childhood but some differences in terms of maternal parity, and age at enrollment, and child birth weight, age at blood draw for DNA methylation, and age at the early adolescence visit. However, the effect sizes of these differences were mostly smaller than 0.2, implying small differences.
Table 1.
Comparisons of covariates between the participants included in and excluded from this study.
| Included (N = 265) |
Excluded (N = 1863) |
|||||
|---|---|---|---|---|---|---|
| Covariates | N | Mean (SD) or N (%) |
N | Mean (SD) or N (%) |
P-valuea | Effect sizeb |
| Maternal characteristics at enrollment | ||||||
| Education | 265 | 1839 | 0.26 | 0.02 | ||
| ≥ College graduate | 180 (67.9) | 1180 (64.2) | ||||
| < College graduate | 85 (32.1) | 659 (35.8) | ||||
| Smoking during pregnancy | 265 | 1842 | 0.34 | 0.03 | ||
| Never | 188 (70.9) | 1255 (68.1) | ||||
| Former | 51 (19.2) | 347 (18.8) | ||||
| During pregnancy | 26 (9.8) | 240 (13.0) | ||||
| Parity (nulliparous) | 265 | 1863 | 2.0e-2 | 0.05 | ||
| No | 156 (58.9) | 955 (51.3) | ||||
| Yes | 109 (41.1) | 908 (48.7) | ||||
| Age (years) | 265 | 32.9 (5.3) | 1863 | 31.7 (5.2) | 2.3e-4 | 0.24 |
| Pre-pregnancy BMI (kg/m2) | 265 | 24.9 (5.2) | 1847 | 24.9 (5.6) | 0.90 | 0.01 |
| Child characteristics | ||||||
| Sex | 265 | 1863 | 0.79 | 0.01 | ||
| Boys | 134 (50.6) | 962 (51.6) | ||||
| Girls | 131 (49.4) | 901 (48.4) | ||||
| Race/ethnicity | 265 | 1844 | 0.15 | 0.06 | ||
| White | 163 (61.5) | 1179 (63.9) | ||||
| Black | 46 (17.4) | 309 (16.8) | ||||
| Hispanic | 16 (6.0) | 112 (6.1) | ||||
| Asian | 7 (2.6) | 88 (4.8) | ||||
| Other | 33 (12.5) | 156 (8.5) | ||||
| Birthweight (grams) | 265 | 3551 (548) | 1862 | 3448 (597) | 0.5e-2 | 0.19 |
| Gestational age (weeks) | 265 | 39.6 (1.6) | 1863 | 39.4 (2.0) | 0.17 | 0.09 |
| Birthweight for gestational age z-score | 265 | 0.31 (1.00) | 1862 | 0.15 (0.97) | 1.3e-2 | 0.16 |
| Age at blood draw for DNAm (years) | 265 | 7.7 (0.7) | 195 | 8.2 (0.9) | 2.0E-7 | 0.50 |
| BMI in mid-childhood (kg/m2) | 265 | 17.4 (3.2) | 845 | 17.2 (2.9) | 0.45 | 0.05 |
| BMIz in mid-childhood | 265 | 0.45 (1.06) | 845 | 0.37 (0.98) | 0.27 | 0.08 |
| Age at the early adolescence visit (years) | 265 | 12.9 (0.6) | 773 | 13.3 (1.0) | 4.4E-12 | 0.50 |
| Cardio-metabolic in early adolescence | 265 | −0.03 (0.63) | 334 | 0.02 (0.62) | 0.30 | 0.09 |
Abbreviation: Standard deviation (SD); Body mass index (BMI); BMI z-score (BMIz); DNA methylation (DNAm).
a χ[2] test was used for categorical variables, t-test was used for continuous variables;
b Cramér’s V was used for the effect size analysis for categorical variables, Cohen’s d was used for the effect size analysis for continuous variables.
Higher BMI z-score in mid-childhood was associated with higher cardio-metabolic risk score in early adolescence (β = 0.31, 95% CI 0.25 to 0.37). Figures 2 and 3 show the QQ-plots for the EWAS (a) and EWAS (b) respectively, and Figures 4 and 5 show the Manhattan plots for the EWAS (a) and EWAS (b) respectively. Figures A-1 and A-2 in Supplementary Material A show that p-values of EWAS (a) were slightly underinflated and null p-values may be expected from EWAS (b). Figures A-3 and A-4 in Supplementary Material A show symmetric patterns with methylation at a few CpGs reached FDR cutoff in the Volcano plots. Table 2 shows the CpG sites with an FDR smaller than 0.05 in each EWAS. BMI z-score in mid-childhood was positively associated with DNA methylation in mid-childhood at cg11412418, cg09571082, and cg17298543 (GNAI1), but negatively associated with DNA methylation in mid-childhood at cg24618739 (PRTFDC1). DNA methylation in mid-childhood at cg17086579 (PITPNM1) was negatively associated with cardio-metabolic risk score in early adolescence. However, the discovered CpG sites in the two EWAS did not overlap.
Figure 2.
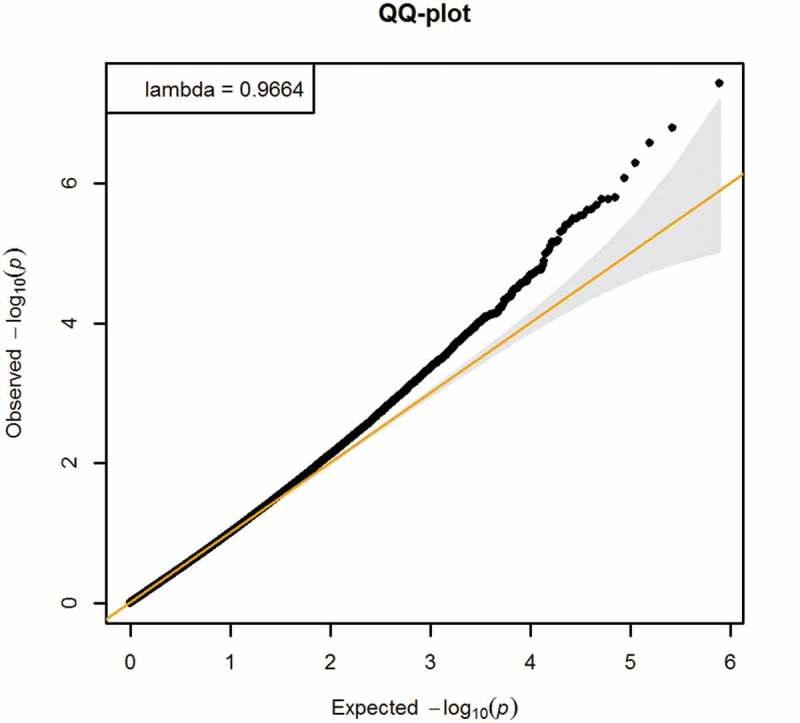
QQ-plot of EWAS of BMI z-score in mid-childhood and DNA methylation in mid-childhood (Adjusted for age at blood draw for DNA methylation, cell type proportion, child sex, maternal education and maternal smoking during pregnancy).
Figure 3.
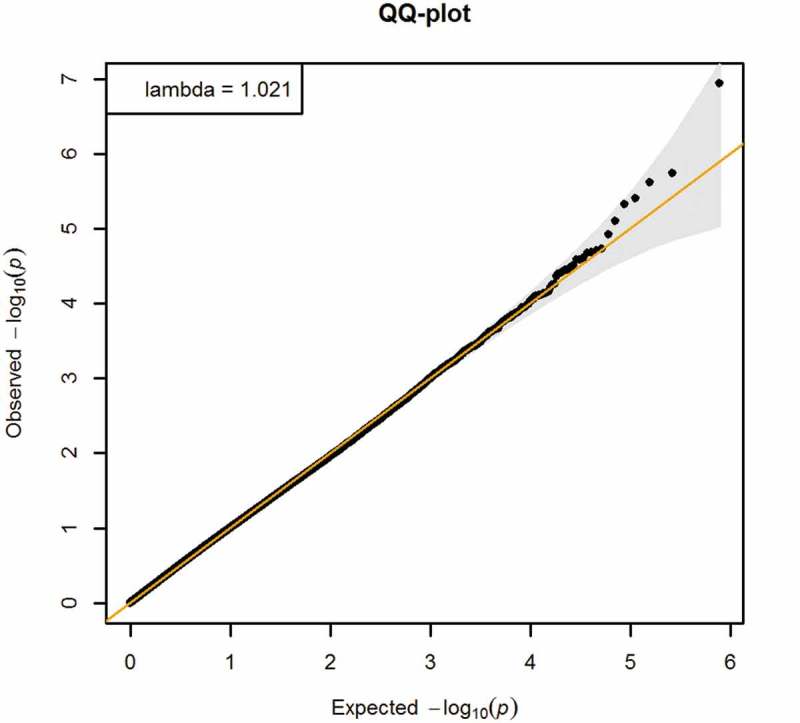
QQ-plot of EWAS of DNA methylation in mid-childhood and cardio-metabolic risk score in early adolescence (Adjusted for age at blood draw for DNA methylation, cell type proportion, child sex, maternal education, maternal smoking during pregnancy, and age at the early adolescence visit).
Figure 4.
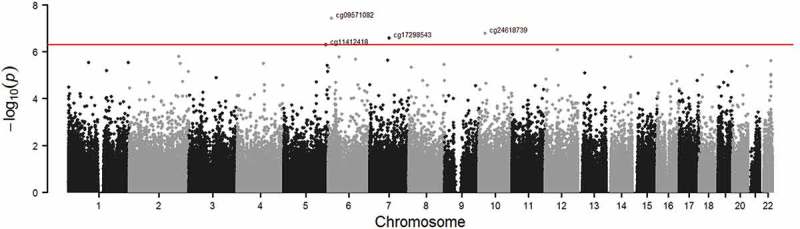
Manhattan plot of EWAS of BMI z-score in mid-childhood and DNA methylation in mid-childhood (Adjusted for age at blood draw for DNA methylation, cell type proportion, child sex, maternal education and maternal smoking during pregnancy; red line: FDR < 0.05).
Figure 5.
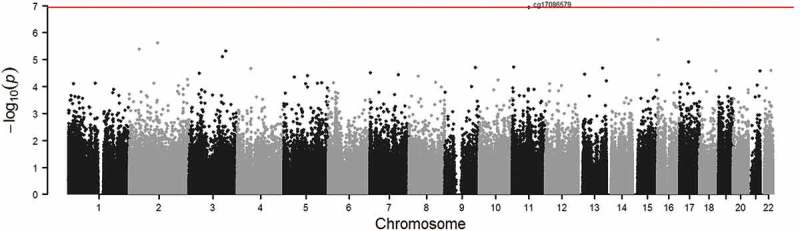
Manhattan plot of EWAS of DNA methylation in mid-childhood and cardio-metabolic risk score in early adolescence (Adjusted for age at blood draw for DNA methylation, cell type proportion, child sex, maternal education, maternal smoking during pregnancy, and age at the early adolescence visit; red line: FDR < 0.05).
Table 2.
Mid-childhood CpG sites associated with BMI z-score in mid-childhood and with cardio-metabolic risk score in early adolescence.
| EWAS | chr | CpG | Change in M-value | FDR | Bonferroni | Gene symbol | Gene region | Relation to Island |
|---|---|---|---|---|---|---|---|---|
| BMIz – DNAma | 5 | cg11412418 | 0.18 | 5.0e-2 | 0.20 | N_Shore | ||
| 6 | cg09571082 | 0.18 | 1.5e-2 | 1.5e-2 | OpenSea | |||
| 7 | cg17298543 | 0.11 | 3.4e-2 | 0.10 | GNAI1 | Body | OpenSea | |
| 10 | cg24618739 | −0.16 | 3.2e-2 | 6.4e-2 | PRTFDC1 | Body | OpenSea | |
| DNAm – cardio-metabolic risk scoreb | 11 | cg17086579 | −0.37 | 4.6e-2 | 4.6e-2 | PITPNM1 | Body | S_Shelf |
Abbreviation: false discovery rate (FDR); Body mass index (BMI); BMI z-score (BMIz); DNA methylation (DNAm).
a Adjusted for child sex, maternal education (college graduate), maternal smoking during pregnancy (never, former, during pregnancy), age at blood draw for DNA methylation, and cell type proportions. Batch effect from plate was adjusted in ComBat. BMIz was included in ComBat model to protect regression variability;
b Adjusted for child sex, maternal education (college graduate), maternal smoking during pregnancy (never, former, during pregnancy), age at the early adolescence visit, age at blood draw for DNA methylation, and cell type proportions. Batch effect from plate was adjusted in ComBat.
Table 3 shows that the variable selection procedure selected 19 CpGs from the top 48 CpGs with the largest effects of DNA methylation in mid-childhood on cardio-metabolic risk score in early adolescence. The mediation by DNA methylation at 12 CpGs were in the same direction as the total effect, i.e., consistent mediation [45], among which 6 CpGs (ATOH8, CDC37/MIR1181, COMT/TXNRD2, DDX10, AS2R40, and TBCD) were selected as potential mediators in more than 95% of the validation tests and jointly explained 10% of the total effect. Although none of them had an FDR smaller than 0.05 in any of the validation tests, the joint natural indirect effect of DNA methylation at these 6 CpGs was evident (NIE: β = 0.04, P = 3.2e-2). This means that one unit higher in BMI z-score (corresponding to 2.2 kg/m2 higher BMI) was indirectly associated with 0.04 higher of cardio-metabolic risk score via DNA methylation at these 6 CpGs. This natural indirect effect accounted for 13% of the total effect. By isolating the joint natural indirect effect of DNA methylation at these 6 CpGs, the remained natural direct effect of BMI z-score on cardio-metabolic risk score was still evident (NIE: β = 0.27, P = 1.1e-25). Joint natural indirect effect of DNA methylation at these 6 CpGs may mediate the associations of BMI z-score with systolic blood pressure (NIE: β = 0.04, P-value = 0.04) and with log-transformed triglycerides (NIE: β = 0.05, P-value = 0.03) but not those with other cardio-metabolic risk components. In a sensitivity analysis, we found the join natural indirect effect of DNA methylation at these 6 CpGs may mediate the associations of subscapular skinfold thickness at mid-childhood with cardio-metabolic risk score (NIE: β = 0.06, P-value = 0.03). This means that one unit higher in subscapular skinfold z-score (corresponding to 5.6 mm thicker subscapular skinfold) was indirectly associated with 0.06 higher of cardio-metabolic risk score via DNA methylation at these CpGs. In a sensitivity analysis adjusted for child race, birth weight for gestational age z-score, and maternal pre-pregnant BMI, 1 out of these 6 potential mediators was also selected in more than 95% of the validation tests (cg22683879 (CDC37/MIR1181)). The mediation estimates by DNA methylation at 7 CpGs were in the opposite direction from the total effect, i.e., inconsistent mediation [45]. Among these 7 CpGs, five (FAIM, PARP6, and SHISA2) were selected as potential mediators in more than 95% of the validation tests. The joint natural indirect effect of DNA methylation at these 5 CpGs was also evident (NIE: β = −0.04, P = 2.0e-2).
Table 3.
High-dimensional mediation analysis for the association of BMI z-score in mid-childhood with cardio-metabolic risk score in early adolescence via DNA methylation in mid-childhood (Results of CpG sites selected by sure independence screening criterion and a variable selection technique (minimax concave penalty)).
| Joint significance testa |
Leave-one-out cross-validationb |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CpG | chr | αc | βd | FDR | % TEe | mean αc | mean βd | Gene symbol | Gene region | Relation to Island |
| cg03205258 | 22 | 0.06 | 0.14 | 0.53 | 2.45% | 0.06 | 0.13 | TXNRD2;COMT | 1stExon;1stExon;5ʹUTR | Island |
| cg01832325 | 11 | 0.04 | 0.15 | 0.53 | 2.11% | NA | NA | HBBP1 | TSS1500 | OpenSea |
| cg25825612 | 17 | −0.04 | −0.17 | 0.53 | 1.95% | −0.04 | −0.14 | TBCD | Body | N_Shelf |
| cg04967734 | 11 | −0.03 | −0.17 | 0.53 | 1.85% | −0.03 | −0.12 | DDX10 | Body | OpenSea |
| cg22683879 | 19 | 0.05 | 0.09 | 0.53 | 1.50% | 0.05 | 0.09 | MIR1181;CDC37 | TSS200;TSS200 | Island |
| cg14732969 | 3 | −0.06 | −0.03 | 0.53 | 0.55% | NA | NA | S_Shore | ||
| cg08033383 | 1 | 0.01 | 0.06 | 0.53 | 0.24% | NA | NA | FAM40A | 1stExon | Island |
| cg08189124 | 11 | 0.03 | 0.25 | 0.54 | 2.17% | NA | NA | N_Shelf | ||
| cg06897686 | 2 | 0.01 | 0.33 | 0.67 | 1.22% | 0.01 | 0.3 | ATOH8 | Body | Island |
| cg24780236 | 7 | −0.02 | −0.2 | 0.67 | 1.14% | −0.02 | −0.19 | TAS2R40 | TSS1500 | OpenSea |
| cg05677579 | 3 | 0.01 | 0.11 | 0.67 | 0.36% | NA | NA | RRP9;PARP3 | TSS1500;TSS200 | Island |
| cg23081542 | 7 | −3.17E-03 | −0.18 | 0.83 | 0.19% | NA | NA | LOC723809 | Body | OpenSea |
| cg01025283 | 3 | −0.05 | 0.15 | 0.23 | −2.25% | −0.05 | 0.16 | FAIM | TSS200;5ʹUTR;1stExon | Island |
| cg26494138 | 2 | −0.05 | 0.24 | 0.23 | −4.18% | −0.05 | 0.20 | S_Shore | ||
| cg23992886 | 7 | 0.02 | −0.14 | 0.54 | −0.89% | NA | NA | MET;MET | Body;Body | OpenSea |
| cg07026904 | 11 | −0.02 | 0.06 | 0.67 | −0.42% | NA | NA | S_Shore | ||
| cg23331961 | 15 | 0.02 | −0.13 | 0.67 | −0.67% | 0.02 | −0.12 | PARP6 | TSS1500 | N_Shore |
| cg11631158 | 13 | 0.01 | −0.09 | 0.76 | −0.43% | 0.01 | −0.09 | SHISA2 | Body | N_Shore |
| cg18647237 | 5 | 4.50E-04 | −0.26 | 0.98 | −0.04% | 4.35E-04 | −0.20 | N_Shore | ||
Abbreviation: false discovery rate (FDR); Body mass index (BMI); BMI z-score (BMIz); DNA methylation (DNAm); the proportion of total effect (% TE)
a In the joint significance test, FDR of the BMIz – DNAm associations (α) and FDR of the DNAm – cardio-metabolic risk score associations (β) were estimated separately, and the larger FDR was reported;
b In the leave-one-out cross-validation, 265 validation tests were performed by excluding one participant from the full dataset. Only the CpGs being selected by the variable selection procedure in more than 95% of the validation tests were reported in this study. Results are the mean coefficients of each CpG in all validation tests;
c Results represent changes of M-value. Adjusted for child sex, maternal education (college graduate), maternal smoking during pregnancy (never, former, during pregnancy), age at blood draw for DNA methylation, and cell type proportions. Batch effect from plate was adjusted in ComBat. BMIz was included in the ComBat model to protect regression variability;
d Results represent changes of M-value. Adjusted for child sex, maternal education (college graduate), maternal smoking during pregnancy (never, former, during pregnancy), age at the early adolescence visit, age at blood draw for DNA methylation, and cell type proportions. Batch effect from plate was adjusted in ComBat. BMIz was included in the ComBat model to protect regression variability;
e ‘% TE’ denotes the proportion of total effect explained by each mediator, calculated as α*β/TE. In this study, higher BMIz in mid-childhood is associated with higher cardio-metabolic risk score in early adolescence.
DMRcate identified 16 DMRs for association with BMI z-score in mid-childhood (Table 4). The number of CpGs in each DMR ranged from 2 to 35 (Table B-1 in Supplementary Material B for the CpGs in each DMR). Figures A-5 to A-6 in the Supplementary Material A show that DNA methylation at CpGs in each DMR were highly correlated. However, no association was identified for these 16 BMI-associated DMR methylation in mid-childhood and cardio-metabolic risk score in early adolescence. BMI z-score was negatively associated with DNA methylation in mid-childhood at 4 DMRs (chr1:248100276–248100614 on OR2L13 (7 CpGs), chr6:28583971–28584464 on ZBED9 (17 CpGs), chr6:28911926–28912166 on LINC01556 (5 CpGs), and chr22:46449430–46450114 on PRR34 and PRR34-AS1 (11 CpGs)).
Table 4.
Regional analysis of the mediation effects of DNA methylation (DNAm) in mid-childhood on the association of body mass index z-score (BMIz) in mid-childhood with cardio-metabolic risk score in early adolescence using generalized estimating equation (GEE).
| BMIz – DNAmb |
DNAm – cardio-metabolic risk scorec |
||||
|---|---|---|---|---|---|
| DMRa | Number of CpGs | α | FDR | β | FDR |
| chr1:248,100,276–248,100,614 | 7 | −0.33 | 2.3e-08 | 2.03e-04 | 0.97 |
| chr1:25,291,385–25,292,215 | 11 | 0.02 | 0.69 | 1.06e-03 | 0.97 |
| chr4:111,397,134–111,397,581 | 7 | −0.08 | 0.13 | −1.44e-03 | 0.97 |
| chr6:28,583,971–28,584,464 | 17 | −0.07 | 1.0e-2 | −2.96e-03 | 0.97 |
| chr6:28,911,926–28,912,166 | 5 | −0.11 | 2.2e-04 | −0.01 | 0.97 |
| chr6:30,459,867–30,460,322 | 2 | −4.45e-03 | 0.86 | −0.03 | 0.97 |
| chr6:31,542,740–31,543,686 | 13 | −0.03 | 0.37 | 1.69e-03 | 0.97 |
| chr6:31,549,929–31,550,090 | 2 | 0.02 | 0.33 | −0.02 | 0.97 |
| chr6:31,690,904–31,692,375 | 35 | −0.06 | 0.13 | 8.15e-04 | 0.97 |
| chr6:32,942,063–32,943,025 | 11 | 0.05 | 0.34 | 6.67e-04 | 0.97 |
| chr8:145,024,774–145,025,610 | 9 | −0.04 | 0.38 | −4.69e-04 | 0.97 |
| chr12:122,711,988–122,712,381 | 9 | 0.03 | 0.23 | −4.09e-03 | 0.97 |
| chr16:54,227,790–54,228,447 | 7 | −0.11 | 0.12 | 2.61e-03 | 0.97 |
| chr19:2,607,726–2,607,903 | 4 | 0.03 | 0.13 | 0.01 | 0.97 |
| chr20:57,582,581–57,583,263 | 15 | 4.44e-04 | 0.98 | 1.68e-03 | 0.97 |
| chr22:46,449,430–46,450,114 | 11 | −0.14 | 3.1e-19 | −0.03 | 0.23 |
Abbreviation: Differentially methylated region (DMR); BMI z-score (BMIz); DNA methylation (DNAm); generalized estimating equation (GEE); false discovery rate (FDR)
a Regions were identified for association with BMI z-score in mid-childhood using DMRcate package, adjusted for child sex, maternal education (college graduate), maternal smoking during pregnancy (never, former, during pregnancy), and age at blood draw for DNA methylation.
b Adjusted for child sex, maternal education (college graduate), maternal smoking during pregnancy (never, former, during pregnancy), age at blood draw for DNA methylation, and cell type proportions. Batch effect from plate was adjusted in ComBat; BMI z-score was included in the ComBat model to protect regression variability;
c Adjusted for child sex, maternal education (college graduate), maternal smoking during pregnancy (never, former, during pregnancy), age at the early adolescence visit, age at blood draw for DNA methylation, and cell type proportions. Batch effect from plate was adjusted in ComBat; BMI z-score was included in the ComBat model to protect regression variability.
In the secondary analysis targeting a region of interest based on prior literature, BMI z-score was positively associated with DNA methylation at cg06500161 (ABCG1) (β = 0.04, P = 1.0e-3). However, higher DNA methylation at this CpG was not associated with cardio-metabolic risk score (β = 0.26, P = 0.17) and no natural indirect effect of DNA methylation at cg06500161 (ABCG1) for the association of BMI z-score with cardio-metabolic risk was evident (NIE: β = 0.01, P = 0.21).
Discussion
In this study, we assessed the association of BMI z-score in mid-childhood with cardio-metabolic risk score in early adolescence and potential epigenetic mechanisms mediating this association. We hypothesized that DNA methylation of leukocytes in mid-childhood partially mediates this association, and we performed a CpG-by-CpG analysis using an EWAS approach, a high-dimensional mediation analysis using a variable selection technique, and we assessed the natural indirect effect using the natural effect model. We also assessed in a regional analysis the association of DNA methylation at DMRs with BMI z-score and with cardio-metabolic risk score. We replicated the previously reported [13] positive association between BMI z-score and DNA methylation for ABCG1 in mid-childhood. We also found that BMI z-score in mid-childhood was negatively associated with DNA methylation in mid-childhood at 4 DMRs using a GEE approach. In the high-dimensional mediation analysis, we identified 6 CpGs that explained 10% of the total effect of BMI z-score in mid-childhood on cardio-metabolic risk score in early adolescence. None of the FDRs was smaller than 0.05. However, the joint natural indirect effect of these 6 CpGs consistently explained 13 % of the total effect. This may imply that the effect of DNA methylation at individual CpGs may not be strong enough to bridge the causal relation between BMI and cardio-metabolic risk.
We also identified potential mediation by 5 CpGs in the opposite direction from the total effect. Considering the small sample size and low variability of cardio-metabolic risk, the hypothesis that DNA methylation mediates the effect of BMI on cardio-metabolic risk needs further examination.
The association of BMI with DNA methylation has been investigated previously, however, whether the change in DNA methylation is the cause or consequence of adiposity remains to be determined. Among 83 BMI-related CpGs that replicated in multiple cohorts, Mendelson et al. used Mendelian randomization to suggest that methylation at cg11024682 (SREBF1) influenced BMI (i.e., methylation as the cause), while BMI affected methylation at 16 other CpGs (i.e., methylation as the consequence) [12]. By investigating 187 CpGs with potential BMI-methylation associations replicated in multiple cohorts, Wahl et al. found that methylation at cg26663590 (NFATC2IP) influenced BMI, while BMI affected DNA methylation at 3 other CpGs [13]. Furthermore, Dick et al. reported a positive association between BMI and DNA methylation at cg22891070, cg27146050, and cg16672562 at HIF3A in adults [14], and Richmond et al. replicated the association of cg27146050 at HIF3A in adolescents [11]. The study by Richmond et al. also supported the direction of BMI causes DNA methylation using Mendelian randomization, although the association was not evident [11]. These findings suggest that the accumulation of excess weight captured as elevated BMI most commonly causes changes in DNA methylation, rather than vice versa.
Both Mendelson et al. and Wahl et al. reported positive associations of BMI with DNA methylation at cg06500161 (ABCG1) [12,13]. In this study, we initially excluded cg06500161 (ABCG1) from our main analysis because it is a SNP-associated probe with a minor allele frequency of 6.5% (rs9982016) in the dbSNP (snp137Common) in all populations. However, the minor allele frequency of rs9982016 is 4.8% among people of European descent according to 1000 Genomes Project Phase 3 (information extracted from Ensembl (http://www.ensembl.org/)) [46]. Furthermore, rs9982016-cg06500161 association is not reported in the mQTL Database of the Avon Longitudinal Study of Parents and Children (ALSPAC) cohort (only SNP-CpG associations with a p-value below 1.0e-7 are reported), and rs9982016 is not associated with DNA methylation level at cg06500161 in a Mexican American cohort (P = 0.19) [47]. Therefore, we performed a secondary analysis assessing mediation by DNA methylation at cg06500161. Consistent with previous studies of DNA methylation in adult blood [12,13], we found that BMI was positively associated with DNA methylation at cg06500161, which is located in ABCG1 regulatory sites [48]. Previous studies also showed that DNA methylation at cg06500161 (ABCG1) in blood was negatively associated with ABCG1 expression [12,13], consistent with the association between weight loss and upregulated ABCG1 expression in an observational study (Table A-2 in Supplementary Material A) [49]. ABCG1 expression was associated with adiposity in a genotype analysis in humans [50], and a decrease in adiposity was found in high-fat diet fed mice with knockout of ABCG1 [50,51]. Taken together, the findings from this study, along with the previous EWAS and Mendelian randomization [12,13], imply a negative effect of BMI on ABCG1 expression via increasing DNA methylation at cg06500161 (ABCG1), which may subsequently inhibit fat mass growth. This body of evidence indicates that the ABCG1 methylation change may be a response to adiposity, and act as a negative feedback to protect the body from the potential harm of excess fat mass. The underlying pathway of elevated ABCG1 expression affecting adiposity may involve the activation of lipoprotein lipase (LPL) and PPARγ [50]. Genetic mutations that lead to LPL deficiency are associated with higher risk of coronary artery disease [52]. PPARγ activation can lower low-density lipoproteins cholesterol (LDL-cholesterol) and improve insulin sensitivity [53,54], which may imply an effect of PPARγ deficiency on higher cardio-metabolic risk. Also, ABCG1 are ATP-binding cassette transporters that modulate cholesterol homeostasis via cholesterol efflux [55,56]. Deficiency of ABCG1 in T cells impairs cholesterol transportation, and may, therefore, inhibit the mammalian target of rapamycin (mTOR) signaling pathway [57], which regulates cardiac metabolism [58]. Furthermore, ATP-binding cassette transporters ABCG5 and ABCG8 are associated with higher LDL-cholesterol [59]. However, whether ABCG1 may exert similar effects is unclear.
To assess mediation by multiple mediators, regressing the outcome on all potential mediators in a single model is suggested [40]. However, traditional regression is not applicable when the number of potential mediators is much larger than the sample size. Therefore, we used a sure independence screening criterion and a variable selection technique to perform a high-dimensional mediation analysis [38]. However, mediation analysis relying on statistical models is vulnerable to confounding and collider bias [43]. Thus, the effect estimates may not be valid. In the high-dimensional mediation analysis, we found mediators in the same direction as the total effect as well as some methylation mediators in the opposite direction. Therefore, we used the natural effect model to assess the joint natural indirect effects of the potential mediators stratified by the direction of mediation [40–42]. The proportion of total effect explained by the indirect effect via DNA methylation at the 6 CpGs in the same direction as the total effect were similar based on the high-dimensional mediation analysis (10%) and the natural effect model (13%). The putative functions of the genes identified in this study (see Tables 2 and 3) were obtained from Genecards and the GWAS Catalog (see Table A-3 in Supplementary Material A). DDX10 is associated with glucose homeostasis traits [60], TAS2R40 is associated with cancer risk [61], FAIM can protect against apoptosis and regulates B-cell signalling and differentiation [62], and PARP6 is associated with cognitive impairment [63]. However, whether these genes play a role in cardio-metabolic risk is unclear given limited evidence from the existing lit erature. Of these CpGs, cg03205258 is on COMT, which regulates catecholamine metabolism and was previously found associated with CVD [64]. In the sensitivity analysis additionally adjusted for additionally adjusted for child race, birth weight for gestational age z-score, and maternal pre-pregnancy BMI, cg22683879 (CDC37/MIR1181) was also selected by the high-dimensional mediation analysis. CDC37 enhances the bioactivity of Hsp90, a molecular chaperone plays a crucial role in the maturation and activation of many steroid receptors including estrogen and androgen [65,66]. Hsp90 also shows cardiac protective effect [67]. Decreased CDC37 gene expression due to elevated DNA methylation may inhibit the activity of Hsp90 and thereby deteriorate cardio-metabolic profile. The observation of inconsistent mediators may imply a complex role of DNA methylation in the association of BMI with cardio-metabolic risk. Mediation in different directions should be interpreted with caution. Maternal pre-pregnancy BMI was previously found negatively associated with cord blood LEP gene methylation, while higher leptin levels may increase risk of obesity by regulating energy metabolism [68]. Therefore, DNA methylation may act as a confounder between BMI and cardio-metabolic risk. However, BMI and DNA methylation were measured at the same time point in this study, it is also possible that unknown confounders affect both BMI and DNA methylation. In addition, other unknown causal mechanisms underlying the association of BMI with cardio-metabolic risk may exist.
We also found associations of BMI z-score with DNA methylation in 4 DMRs that had not previously been identified. One of these, DMR chr1:248100276–248100614, is located on OR2L13, an olfactory receptor gene. Mice experiments have reported an association of impaired olfactory function with weight loss without loss of appetite [69]. However, observational studies reported a negative association of visceral fat and BMI with olfactory sensitivity [70,71]. DMR chr22:46449430–46450114 covers PRR34 (Proline-rich 34) and PRR34-AS1; the latter is an antisense RNA gene which could affect different stages of gene expression [72]. However, little is known about the association of adiposity with proline-rich protein.
This is the first study using epigenome-wide association analyzes, high-dimensional mediation analysis, and natural effect models to investigate the mediation by blood DNA methylation of the association between BMI z-score and cardio-metabolic risk. We identified potential mediators in different directions, which may provide insight into specific targets for intervention. Nevertheless, this study has several limitations. First, the cohort participants all resided in the greater Boston area and had health insurance, and thus results may not be generalizable to other populations, although causal factors usually act consistently. Second, the final sample size was relatively small since we included the participants with complete data on all required variables in the mediation analysis. Thus, the analyzes are underpowered. To address potential false positive findings, we used a stricter screening criterion in the high-dimensional mediation analysis. Third, BMI may not be a strong predictor for total fat mass in children younger than 9 years old [73]; however, our previous study shows that dual x-ray absorptiometry total fat mass was highly correlated with BMI (Spearman r = 0.83) in mid-childhood in this cohort [74]. Also, BMI is likely to be a reliable predictor of subscapular skinfold thickness [75]. Our study showed consistent findings for the associations of subscapular skinfold thickness and BMI with cardio-metabolic risk. In addition, a Mendelian randomization study suggests that higher BMI causes CVD [20]. Therefore, this study may still provide insight into the epigenetic mechanisms underlying the association of adiposity with cardio-metabolic risk. Fourth, the cardio-metabolic risk score was calculated based on five components of metabolic risk including waist circumference, which is correlated with BMI. Therefore, the association of BMI z-score with cardio-metabolic risk score may be overestimated in this study. When assessing the cardio-metabolic risk components separately, only the joint natural indirect effect for the association of BMI z-score with systolic blood pressure and log-transformed triglycerides were evident. This may also imply that components in the global metabolic risk score may involve different biological mechanisms. Fifth, although we identified associations between BMI z-score and DNA methylation in the regional analysis, we could not exclude the possibility of reverse causation. However, a recent Mendelian randomization study and an EWAS support the hypothesis that most commonly BMI influences DNA methylation in adult blood and not vice versa [12,13]. Sixth, we replicated the positive association of BMI z-score with DNA methylation at cg06500161 (ABCG1) in a secondary analysis. However, we cannot exclude the possibility of a SNP effect for this CpG. Seventh, the null findings for the association of DNA methylation with cardio-metabolic risk score may be due to low variability. It may be valuable to replicate this analysis in a cohort with metabolic assessment later in the life course. Eighth, given that DNA methylation at one CpG may affect DNA methylation at another CpG and that unknown mediator-outcome confounders affected by the exposure may exist, this analysis may have violated the assumptions of ‘no confounding between exposure-outcome, mediator-outcome and exposure-mediator relationships and that confounders of the mediator-outcome relationship are not affected by the exposure’ [41]. Future studies may consider using other study designs such as two-step Mendelian randomization to assess the mediation by DNA methylation. Ninth, DNA methylation was measured in whole blood, which may not reflect the DNA methylation profile in tissues relevant to obesity, for instance, adipose tissue [76]. However, a previous study found BMI associates with DNA methylation in both blood and adipose tissue [13], suggesting blood samples could be used as a surrogate – at some selected CpG sites – for adipose tissue for investigating the associations between BMI and DNA methylation. Finally, the Infinium Human Methylation450K BeadChip interrogates only 1.7% of all CpGs in the whole genome [77]. While it covers 99% of the RefSeq genes [24], the design may not lead to the most comprehensive and all-encompassing findings, i.e., associations in other regions of the genome may be missed due to the sparsity of the platform.
Conclusion
Using the high-dimensional mediation analysis and the natural effect model, we identified both consistent and inconsistent mediation by DNA methylation in mid-childhood of the association between BMI z-score in mid-childhood and cardio-metabolic risk score in early adolescence. However, the associations between genes where these potential mediators locate and cardio-metabolic risk are unclear. We need to replicate and expand our findings in future studies with larger sample size and greater variability in cardio-metabolic risk to elucidate the complex role of DNA methylation as in this association.
Funding Statement
This study was supported by the National Institutes of Health [Grants# R01 HL111108, R01 NR013945, R01 HD034568, UG3OD023286].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental Data
Supplemental data for this article can be accessed here
References
- 1.Ogden CL, Carroll MD, Lawman HG, et al. Trends in obesity prevalence among children and adolescents in the United States, 1988-1994 Through 2013-2014. JAMA. 2016;315:2292–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bridger T. Childhood obesity and cardiovascular disease. Paediatrics & Child Health. 2009;14:177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization Top 10 causes of death worldwide. Geneva: World Health Organization; 2017. [Google Scholar]
- 4.Heron M. Deaths: leading causes for 2014. National vital statistics reports. Hyattsville, MD: National Center for Health Statistics; 2016. [Google Scholar]
- 5.Muka T, Imo D, Jaspers L, et al. The global impact of non-communicable diseases on healthcare spending and national income: a systematic review. Eur J Epidemiol. 2015;30:251–277. [DOI] [PubMed] [Google Scholar]
- 6.van Greevenbroek MM, Schalkwijk CG, Stehouwer CD. Dysfunctional adipose tissue and low-grade inflammation in the management of the metabolic syndrome: current practices and future advances. F1000Research 2016, 5(F1000 Faculty Rev):2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harrington RA. Targeting inflammation in coronary artery disease. New Engl J Med. 2017;377:1197–1198. [DOI] [PubMed] [Google Scholar]
- 8.Sun C, Burgner DP, Ponsonby AL, et al. Effects of early-life environment and epigenetics on cardiovascular disease risk in children: highlighting the role of twin studies. Pediatr Res. 2013;73:523–530. [DOI] [PubMed] [Google Scholar]
- 9.Zhong J, Agha G, Baccarelli AA. The role of DNA methylation in cardiovascular risk and disease: methodological aspects, study design, and data analysis for epidemiological studies. Circ Res. 2016;118:119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rzehak P, Covic M, Saffery R, et al. DNA-methylation and body composition in preschool children: epigenome-wide-analysis in the European childhood obesity project (CHOP)-study. Sci Rep. 2017;7:14349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richmond RC, Sharp GC, Ward ME, et al. DNA methylation and BMI: investigating identified methylation sites at HIF3A in a causal framework. Diabetes. 2016;65:1231–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mendelson MM, Marioni RE, Joehanes R, et al. Association of body mass index with DNA methylation and gene expression in blood cells and relations to cardiometabolic disease: a mendelian randomization approach. PLoS Med. 2017;14:e1002215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wahl S, Drong A, Lehne B, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541:81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dick KJ, Nelson CP, Tsaprouni L, et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet. 2014;383:1990–1998. [DOI] [PubMed] [Google Scholar]
- 15.Muka T, Koromani F, Portilla E, et al. The role of epigenetic modifications in cardiovascular disease: A systematic review. Int J Cardiol. 2016;212:174–183. [DOI] [PubMed] [Google Scholar]
- 16.Baccarelli A, Wright R, Bollati V, et al. Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology. 2010;21:819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin XH, Wu DD, Gao L, et al. Altered DNA methylation in neonates born large-for-gestational-age is associated with cardiometabolic risk in children. Oncotarget. 2016;7:86511–86521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richard MA, Huan T, Ligthart S, et al. DNA methylation analysis identifies loci for blood pressure regulation. Am J Hum Genet. 2017;101:888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baccarelli A, Rienstra M, Benjamin EJ. Cardiovascular epigenetics: basic concepts and results from animal and human studies. Circ Cardiovasc Genetics. 2010;3:567–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagg S, Fall T, Ploner A, et al. Adiposity as a cause of cardiovascular disease: a Mendelian randomization study. Int J Epidemiol. 2015;44:578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oken E, Baccarelli AA, Gold DR, et al. Cohort profile: project viva. Int J Epidemiol. 2015;44:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oken E, Huh SY, Taveras EM, et al. Associations of maternal prenatal smoking with child adiposity and blood pressure. Obes Res. 2005;13:2021–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oken E, Kleinman KP, Rich-Edwards J, et al. A nearly continuous measure of birth weight for gestational age using a United States national reference. BMC Pediatr. 2003;3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bibikova M, Barnes B, Tsan C, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. [DOI] [PubMed] [Google Scholar]
- 25.Chen YA, Lemire M, Choufani S, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen KD. IlluminaHumanMethylation450kanno.ilmn12.hg19: annotation for Illumina’s 450k methylation arrays. R package version 0.6.0. 2016. [Google Scholar]
- 28.Triche TJ Jr., Weisenberger DJ, Van Den Berg D, et al. Low-level processing of illumina infinium DNA methylation beadarrays. Nucleic Acids Res. 2013;41:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teschendorff AE, Marabita F, Lechner M, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29:189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. [DOI] [PubMed] [Google Scholar]
- 31.Kuczmarski RJ, Ogden CL, Grummer-Strawn LM, et al. CDC growth charts: united States. Advance Data. 2000;314:1-27. [PubMed] [Google Scholar]
- 32.Haugaard LK, Baker JL, Perng W, et al. Growth in total height and its components and cardiometabolic health in childhood. PloS one. 2016;11:e0163564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cespedes EM, Rifas-Shiman SL, Redline S, et al. Longitudinal associations of sleep curtailment with metabolic risk in mid-childhood. Obesity. 2014;22:2586–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farr OM, Rifas-Shiman SL, Oken E, et al. Current child, but not maternal, snoring is bi-directionally related to adiposity and cardiometabolic risk markers: A cross-sectional and a prospective cohort analysis. Metabolism. 2017;76:70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Du P, Zhang X, Huang CC, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. Bmc Bioinformatics. 2010;11:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fritz CO, Morris PE, Richler JJ. Effect size estimates: current use, calculations, and interpretation. J Exp Psychol Gen. 2012;141:2–18. [DOI] [PubMed] [Google Scholar]
- 37.Fan JQ, Lv JC. Sure independence screening for ultrahigh dimensional feature space. J Roy Stat Soc B. 2008;70:849–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang H, Zheng Y, Zhang Z, et al. Estimating and testing high-dimensional mediation effects in epigenetic studies. Bioinformatics. 2016;32:3150–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang CH. Nearly unbiased variable selection under minimax concave penalty. Ann Stat. 2010;38:894–942. [Google Scholar]
- 40.VanderWeele TJ, Vansteelandt S. Mediation analysis with multiple mediators. Epidemiologic Methods. 2014;2:95–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.VanderWeele TJ. Mediation analysis: a practitioner’s guide. Annu Rev Publ Health. 2016;37:17–32. [DOI] [PubMed] [Google Scholar]
- 42.Steen J, Loeys T, Moerkerke B, et al. medflex: an R package for flexible mediation analysis using natural effect models. J Stat Softw. 2017;76:11. [Google Scholar]
- 43.Richiardi L, Bellocco R, Zugna D. Mediation analysis in epidemiology: methods, interpretation and bias. Int J Epidemiol. 2013;42:1511–1519. [DOI] [PubMed] [Google Scholar]
- 44.Peters TJ, Buckley MJ, Statham AL, et al. De novo identification of differentially methylated regions in the human genome. Epigenet Chromatin. 2015;8:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.MacKinnon DP, Fairchild AJ, Fritz MS. Mediation analysis. Annu Rev Psychol. 2007;58:593–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yates A, Akanni W, Amode MR, et al. Ensembl 2016. Nucleic acids research. 2016;44:D710–D716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mamtani M, Kulkarni H, Dyer TD, et al. Genome- and epigenome-wide association study of hypertriglyceridemic waist in Mexican American families. Clin Epigenetics. 2016;8:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding J, Reynolds LM, Zeller T, et al. Alterations of a cellular cholesterol metabolism network are a molecular feature of obesity-related type 2 diabetes and cardiovascular disease. Diabetes. 2015;64:3464–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johansson LE, Danielsson AP, Parikh H, et al. Differential gene expression in adipose tissue from obese human subjects during weight loss and weight maintenance. Am J Clin Nutr. 2012;96:196–207. [DOI] [PubMed] [Google Scholar]
- 50.Frisdal E, Le Lay S, Hooton H, et al. Adipocyte ATP-binding cassette G1 promotes triglyceride storage, fat mass growth, and human obesity. Diabetes. 2015;64:840–855. [DOI] [PubMed] [Google Scholar]
- 51.Buchmann J, Meyer C, Neschen S, et al. Ablation of the cholesterol transporter adenosine triphosphate-binding cassette transporter G1 reduces adipose cell size and protects against diet-induced obesity. Endocrinology. 2007;148:1561–1573. [DOI] [PubMed] [Google Scholar]
- 52.Khera AV, Won HH, Peloso GM, et al. Association of rare and common variation in the lipoprotein lipase gene with coronary artery disease. JAMA. 2017;317:937–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ivanova EA, Parolari A, Myasoedova V, et al. Peroxisome proliferator-activated receptor (PPAR) gamma in cardiovascular disorders and cardiovascular surgery. J Cardiol. 2015;66:271–278. [DOI] [PubMed] [Google Scholar]
- 54.Leonardini A, Laviola L, Perrini S, et al. Cross-Talk between PPARgamma and insulin signaling and modulation of insulin sensitivity. PPAR Res. 2009;2009:818945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem. 2014;289:24020–24029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yvan-Charvet L, Wang N, Tall AR. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler Thromb Vasc Biol. 2010;30:139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng HY, Gaddis DE, Wu R, et al. Loss of ABCG1 influences regulatory T cell differentiation and atherosclerosis. J Clin Invest. 2016;126:3236–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014;114:549–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.IBC 50K CAD Consortium Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLoS Genet. 2011;7:e1002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palmer ND, Goodarzi MO, Langefeld CD, et al. Genetic variants associated with quantitative glucose homeostasis traits translate to type 2 diabetes in Mexican Americans: the GUARDIAN (Genetics Underlying Diabetes in Hispanics) Consortium. Diabetes. 2015;64:1853–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu W, Jiao X, Thutkawkorapin J, et al. Cancer risk susceptibility loci in a Swedish population. Oncotarget. 2017;8:110300–110310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coccia E, Calleja-Yague I, Planells-Ferrer L, et al. Identification and characterization of new isoforms of human fas apoptotic inhibitory molecule (FAIM). PloS one. 2017;12:e0185327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu X, Pickering EH, Hall SK, et al. Genome-wide association study identifies multiple novel loci associated with disease progression in subjects with mild cognitive impairment. Transl Psychiatry. 2011;1:e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hall KT, Nelson CP, Davis RB, et al. Polymorphisms in catechol-O-methyltransferase modify treatment effects of aspirin on risk of cardiovascular disease. Arterioscler Thromb Vasc Biol. 2014;34:2160–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jackson SE. Hsp90: structure and function. Top Curr Chem. 2013;328:155–240. [DOI] [PubMed] [Google Scholar]
- 66.MacLean M, Picard D. Cdc37 goes beyond Hsp90 and kinases. Cell Stress & Chaperones. 2003;8:114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Latchman DS. Heat shock proteins and cardiac protection. Cardiovasc Res. 2001;51:637–646. [DOI] [PubMed] [Google Scholar]
- 68.Kadakia R, Zheng Y, Zhang Z, et al. Maternal pre-pregnancy BMI downregulates neonatal cord blood LEP methylation. Pediatr Obes. 2017;12(Suppl 1):57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Riera CE, Tsaousidou E, Halloran J, et al. The sense of smell impacts metabolic health and obesity. Cell Metab. 2017;26:198–211 e5. [DOI] [PubMed] [Google Scholar]
- 70.Fernandez-Garcia JC, Alcaide J, Santiago-Fernandez C, et al. An increase in visceral fat is associated with a decrease in the taste and olfactory capacity. PloS one. 2017;12:e0171204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kindleysides S, Beck KL, Walsh DCI, et al. Fat sensation: fatty acid taste and olfaction sensitivity and the link with disinhibited eating behaviour. Nutrients. 2017;9. pii: E87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pelechano V, Steinmetz LM. Gene regulation by antisense transcription. Nat Reviews Genet. 2013;14:880–893. [DOI] [PubMed] [Google Scholar]
- 73.Vanderwall C, Randall Clark R, Eickhoff J, et al. BMI is a poor predictor of adiposity in young overweight and obese children. BMC Pediatr. 2017;17:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boeke CE, Oken E, Kleinman KP, et al. Correlations among adiposity measures in school-aged children. BMC Pediatr. 2013;13:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Freedman DS, Katzmarzyk PT, Dietz WH, et al. Relation of body mass index and skinfold thicknesses to cardiovascular disease risk factors in children: the Bogalusa Heart Study. Am J Clin Nutr. 2009;90:210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang YT, Chu S, Loucks EB, et al. Epigenome-wide profiling of DNA methylation in paired samples of adipose tissue and blood. Epigenetics. 2016;11:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stirzaker C, Taberlay PC, Statham AL, et al. Mining cancer methylomes: prospects and challenges. Trends Genet. 2014;30:75–84. [DOI] [PubMed] [Google Scholar]
- 78.Boca SM, Sinha R, Cross AJ, et al. Testing multiple biological mediators simultaneously. Bioinformatics. 2014;30:214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.