

ABSTRACT
The timely and precise repair of DNA damage, or more specifically DNA double-strand breaks (DSBs) – the most deleterious DNA lesions, is crucial for maintaining genome integrity and cellular homeostasis. An appropriate cellular response to DNA DSBs requires the integration of various factors, including the post-translational modifications (PTMs) of chromatin and chromatin-associated proteins. Notably, the PTMs of histones have been shown to play a fundamental role in initiating and regulating cellular responses to DNA DSBs. Here we review the role of the major histone PTMs, including phosphorylation, ubiquitination, methylation and acetylation, and their interactions during DNA DSB-induced responses.
KEYWORDS: DNA damage, chromatin MODIFICATIONS, Histones
Introduction
Constant exposure to a variety of harmful endogenous and exogenous factors puts eukaryotic cells at continuous risks for thousands of DNA lesions every day [1]. These DNA lesions, if not repaired correctly, can lead to adverse consequences that endanger the fitness and viability of the cell and/or organism [1,2]. A timely and appropriate cellular response to DNA damage is, therefore, crucial and essential, and as such, the cell has evolved a network of signaling pathways to respond to DNA damage, which is collectively termed DNA damage response (DDR) [1–3]. DDRs are diverse in terms of modes of DNA damage repair, including: direct protein-mediated reversal, mismatch repair, base excision repair, nucleotide excision repair, and double-strand break (DSB) repair [1,4]. Different DDRs are utilized depending on the types of DNA lesions and the cell cycle position of the damaged cell [5]. However, DDRs usually follow a general principle: the presence of a lesion in the DNA can cause alterations in DNA structure or replication stalling, which is recognized by sensor proteins. The sensor proteins in turn recruit signaling and downstream effector proteins, which initiate signaling cascades that lead to various cellular processes to resolve the DNA damage or eliminate the cells with irreparable injury [1].
DNA DSBs are among the most deleterious types of DNA damage because they can potentially lead to a loss or alteration of genetic material [6,7]. DSBs are mainly repaired by homologous recombination (HR) and non-homologous end joining (NHEJ), as shown in Figure 1A. HR is the main DSB repair mechanism in the S and G2 phase of the cell cycle when the undamaged homologous chromosome or sister chromatid is available to be used as a template for the repair of the DNA breaks. On the other hand, NHEJ is more dominant in G0 and G1 phases, and unlike HR, NHEJ repairs the DNA DSBs by directly ligating the two broken ends, which might result in small changes to the DNA sequences [8]. Breast cancer 1 (BRCA1) is important for the HR repair pathway, while p53-binding protein 1 (53BP1) plays a crucial role in NHEJ [8,9]. To ensure that the DNA DSBs are repaired with the appropriate repair mechanism, BRCA1 and 53BP1 mutually antagonize each other during the repair process [8].
Figure 1.
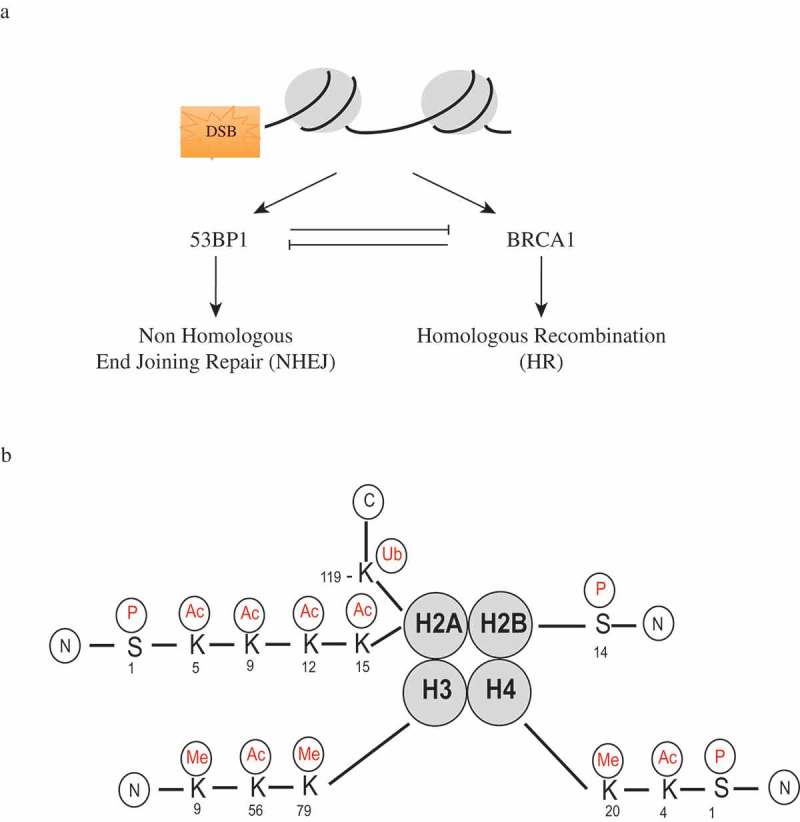
Histone modficiations and DNA double-strand break repair. (a) DNA double-strand breaks can be repaired by non-homologous end joining (NHEJ) or homologous recombination (HR). p53-binding protein 1 (53BP1) plays a crucial role in NHEJ, while breast cancer 1 (BRCA1) is important for HR. 53BP1 and BRCA1 mutually antagonize each other’s actions during the repair process.
The generation and repair of DNA lesions occurs in the context of chromatin, and the chromatin status at sites of DNA damage has been shown to influence the DDR [10]. In eukaryotes, chromatin is comprised of DNA and protein complexes called nucleosomes. Each nucleosome consists of 146 base pairs of DNA wrapped around a core histone octamer comprised of two copies of each histone: H2A, H2B, H3, and H4. The nucleosome is then connected by a linker DNA and histone H1 [4,10]. The chromatin is a highly organized and dynamic structure that regulates DNA accessibility during different biological events. Chromatin generally exists in two forms: a tightly packed heterochromatin and a loosely coiled euchromatin. The highly ordered chromatin structure can be regulated by different processes, one of which is the post-translational modifications (PTMs) of histones [6]. The amino terminus or the “tails” of the histones are protruding from the histone core and open to various PTMs. As a result, the PTMs of histone tails tightly correlate with many biological processes, including DDRs [2,4,10]. As illustrated in Figure 1B, PTMs on the histone tails are diverse, and some of these modifications have been shown to play important roles in DDRs.
In the event of DNA DSBs, histone modifications are critical for the generation and regulation of cellular responses to the damage. Histone PTMs can unravel chromatin structure for DNA accessibility, propagate the initial signaling of the break, facilitate the recruitment of repairing proteins, as well as restore the initial state of chromatin after the DNA breaks are resolved [2–4,6,10]. In this review, we highlight the major histone modifications and their functions in orchestrating the response to DNA DSBs.
Major posttranslational modifications of histone subunits in DNA DSB damage response
Phosphorylation of histone H2A.X
H2A.X is a member of histone H2A family, and it comprises about 2–20% of the H2A proteins in mammalian cells [11,12]. The phosphorylation of serine 139 (S139) on H2A.X (γH2A.X) is one of the prominent DDR-associated histone marks. In general, within seconds after DSBs are induced, H2A.X is rapidly phosphorylated, passing through a half-maximal value at 1 minute [12]. H2A.X molecules in a small region near the DSB site are phosphorylated first, following by the H2A.X molecules at increasing distance from the break site [13]. However, the dynamics of γH2A.X formation are not the same across the genome. γH2A.X foci form more efficiently in euchromatin than in heterochromatin [14,15]. γH2A.X was also shown to spread from DNA DSB sites in a bidirectional but not symmetrical manner, depending on the transcription state of the genes surrounding the DSBs or depending on other repairing and signaling proteins [16,17]. Although there are some exceptions, it is accepted that when a DNA DSB is induced, γH2A.X foci are quickly formed and the γH2A.X levels reflect the number of DSBs in the cell [11].
The phosphatidylinositol 3-kinases (PI-3Ks), a family including DNA-dependent protein kinase (DNA-PK), ataxia telangiectasia mutated (ATM), and ATM and Rad3-related kinase (ATR), are implicated in the phosphorylation of H2A.X in response to DNA DSBs [11]. Although ATM was shown to be the major kinase responsible for modifying H2A.X upon ionizing radiation (IR) [18], under most normal growth conditions, both ATM and DNA-PK can phosphorylate H2A.X in a redundant and overlapping manner [19]. In general, upon the formation of DSBs, PI-3Ks act as DNA damage sensors; they are loaded rapidly onto the break, get activated, and phosphorylate numerous substrates including thousands of H2A.X proteins in the chromatin surrounding the DSBs. The phosphorylation of H2A.X was shown to precede and initiate the accumulation of repair factors and checkpoint proteins, including Mre11-Rad50-Nbs1 (MRN), mediator of DNA damage checkpoint protein 1 (MDC1), breast cancer 1 (BRCA1), p53-binding protein 1 (53BP1), E3 ubiquitin-protein ligase 68 and 168 (RNF68 and RNF168). The phosphorylation of H2A.X is also important for the recruitment of chromatin-remodeling complexes and proteins, such as chromatin-remodeling ATPase complexes INO80 and SWR1 or the histone acetyltransferase TIP60, to DSB sites [3,6,20–22]. However, it was also shown that γH2A.X might have a bigger role in retaining and concentrating these repair-related proteins at sites of DNA damage rather than in recruiting them to the sites of damage [21]. Nevertheless, it is evident that γH2A.X plays an important role in enhancing the repair of DNA lesions by facilitating the opening of the chromatin structure and forming a platform for the accumulation of DNA damage repair factors [11].
Because γH2A.X signaling indicates the sites of DNA DSBs or unresolved DNA damage [11,23], the dephosphorylation of γH2A.X is important to mark the completion of the DNA repair process. There are two proposed mechanisms for γH2A.X to be returned to the original state after the resolution of DNA breaks: γH2A.X might be removed from chromatin by histone exchange or dephosphorylated by a protein phosphatase [24]. There are several phosphatases that have been shown to play a role in dephosphorylating γH2A.X. Protein phosphatase (PP) 4 has been shown to dephosphorylate γH2AX at the sites of DSBs [25], while PP2A directly binds and dephosphorylates γH2A.X at more distal sites. PP2C or wild-type p53-induced phosphatase 1 (WIP1), which dephosphorylates a variety of DNA damage repair proteins, has also been shown to directly reverse γH2A.X [26]. In summary, unlike PI-3Ks that phosphorylate H2A.X to initiate DDRs, phosphatases help restore the un-phosphorylated form of H2A.X to turn off DDRs.
The dephosphorylation of histone H2A.X at tyrosine 142 (Y142) is also essential for the formation of γH2A.X at sites of DSBs. Under non-stressed conditions, Williams–Beuren syndrome transcription factor (WSTF), also known as bromodomain adjacent to zinc finger domain 1B (BAZ1B), phosphorylates Y142 of H2A.X [27]. Under DNA DSB stress, the protein tyrosine phosphatase eyes absent (EYA) dephosphorylates the phosphorylated Y142 of H2A.X. This was shown to be important not only for maintaining a phosphorylation/dephosphorylation cycle of H2A.X C-terminus, ultimately maintaining γH2A.X formation, but also for enhancing the recruitment of other factors to the damage sites [27,28]. The dephosphorylation of Y142 at H2A.X by EYA was also shown to be a determinant of repair/survival versus apoptotic responses to DNA damage by regulating the recruitment of either repair or apoptotic factors to the tail of γH2A.X [28].
Along with histone H2A.X, other histones can also be phosphorylated in response to DNA damage. The serine 14 (S14) on the N-terminal tail of histone H2B is rapidly phosphorylated at the sites of DNA DSBs [29]. Phosphorylated H2B was shown to accumulate into foci after IR-induced damage and work together with γH2AX to establish a heterochromatin-like state around damaged DNA for repair factors to concentrate onto [29]. Interestingly, the phosphorylation of H2B at S14 was also shown to be associated with apoptosis when the DNA damage repair is unsuccessful [30]. Additionally, histone H4 can also be phosphorylated at serine 1, and this phosphorylation event can facilitate the re-joining of DNA breaks in response to methyl methane sulphonate- and phleomycin-induced DSBs in yeast [31]. Briefly, phosphorylation is an important histone modification for the inititiation and the termination of DDRs.
Ubiquitylation of histone H2A
Ubiquitylation has been shown to regulate various cellular processes through constructing a flexible protein-protein communication system with eight structurally and functionally different chains linked through distinct residues [32]. Concordantly, studies have shown that the ubiquitylation of histone tails also plays an important role in DDRs.
One of most well-studied ubiquitylation marks in DDRs is the E3 ubiquitin-protein ligase 68 and 168 (RNF8/RNF168)-mediated histone ubiquitylation. As discussed in the previous section, in response to DNA damage, γH2AX is generated rapidly, which then recruits the mediator protein MDC1 to the chromatin [12,18,33]. MDC1 is then phosphorylated by ATM [34,35] and facilitates the recruitment of a complex of RNF8 and ubiquitin-conjugating enzyme E2 13 (UBC13). The RNF8/UBC13 complex initiates the synthesis of lysine 63 (K63)-linked ubiquitin conjugates at the damaged sites [36–38]. The target of RNF8/UBC13-mediated ubiquitylation was believed to be only histone H2A/H2A.X [37,38], but recently, studies have shown that RNF8/UBC13 might also ubiquitylate non-nucleosomal proteins and linker histone H1 [39,40]. However, despite the diversity of RNF8/UBC13 substrates, the role of K63-linked ubiquitin conjugates that RNF8/UBC13 initiates at DSB sites is to provide an initial binding platform to recruit another E3 ubiquitin ligase RNF168 to the DNA breaks [40,41].
Although RNF8 is the first E3 ubiquitin ligase recruited to the sites of DNA breaks, RNF8 alone is not enough to sustain the ubiquitylation status [42]. Therefore, the presence of RNF168 is required for proper activation of DDRs. RNF168 physically interacts with RNF8-mediated ubiquitylated H2A/H2A.X and accumulates at the break sites [41,42]. RNF168, similar to RNF8, acts in concert with UBC13 to further catalyze the synthesis of K63-linked ubiquitin conjugates to histone H2A/H2A.X on lysine 13–15, amplifying the initial DNA damage-induced ubiquitylation to a threshold required for the physiological function of this compartment [39,41,42]. In turn, RNF8/RNF168-mediated ubiquitylated H2A/H2A.X mediates the recruitment and accumulation of mediator proteins, such as 53BP1 and BRCA1, at the lesions on the chromatin to promote repair mechanisms, cellular responses, and survival following DNA damage stress [41,43]. Recently, RNF168 has also been shown to promote non-canonical lysine 27 (K27)-linked ubiquitin conjugates to histone H2A/H2A.X, which is strictly required for DDRs and can be directly recognized by other DDR mediators, including 53BP1 and BRCA1 [32].
In addition to RNF8 and RNF168, γH2AX and MDC1 also recruit the polycomb group protein B cell-specific moloney murine leukemia virus integration site 1 (BMI1) to the sites of DNA damage [44,45]. At the sites of damage, BMI1 catalyzes the monoubiquitylation of histone H2A/H2A.X at lysine 119 and contributes to efficient DNA repair [44,45]. In the absence of BMI1, the recruitment of 53BP1 and BRCA1 to the DNA damage sites is impaired [45]. In the absence of both BMI1 and RNF8, cells are more sensitive to IR than cells lacking either BMI1 or RNF8 alone, indicating that BMI1 and RNF8 have overlapping roles in the ubiquitylation of histone H2A/H2A.X, and that they cooperatively but independently contribute to a proper and efficient response to DNA DSBs [44,45]
Because the DNA damage-induced ubiquitylation plays an important role in facilitating the cellular response to DNA damage induced stress, histone ubiquitylation cascades need to be fine-tuned. Indeed, studies have shown that histone ubiquitylation is regulated by several mechanisms. The accessibility for the E3 ubiquitin ligases can be limited by the formation of higher order of chromatin structure [46,47]. The size of the RNF168 nuclear pool can be regulated by the E3 ubiquitin-protein ligase thyroid hormone receptor interactor 12 (TRIP12) and the ubiquitin-protein ligase E3 component n-recognin 5 (UBR5) [46,47]. Additionally, the ubiquitylated products at the damage sites can be reversed by deubiquitylating enzymes [46,47].
However, histone H2A/H2A.X is not the only histone that can be ubiquitylated in response to DNA DSBs. Histone H4 is shown to be selectively monoubiquitylated by B-lymphoma and BAL-associated protein (BBAP) at lysine 91, which contributes to the associated DDR [48]. This raises the possibility that histones other than H2A/H2A.X might also be ubiquitylated in response to DNA damage. In summary, in response to DNA DSBs, in a γH2AX- and MDC1-dependent manner, RNF8 and BMI1 are recruited to the sites of DNA breaks. At the sites of DNA damage, RNF8 recruits RNF168 and together, they work with UBC13 to catalyze the formation of ubiquitin conjugates to histone H2A/H2A.X at lysine 13–15. Meanwhile, BMI1 facilitates monoubiquitylation of histone H2A/H2A.X at lysine 119. These ubiquitylation events of histone H2A/H2A.X trigger the ubiquitylation cascade that leads to the recruitment, accumulation, and retention of 53BP1 and BRCA1 to the DNA lesions to promote proper DNA DSB responses [32–45].
Methylation of histone H4
Histone methylation also plays an important role in regulating DDRs. The methylated lysine 20 of histone H4 (H4K20) is one of the histone methylation marks known for their roles in DDRs. The methylation of H4K20 is not induced by DNA damage, but it is pre-existing in the cell [49]. There is no significant change in the levels of H4K20 mono-, di-, or tri-methylation detected in response to DNA damage [50]. However, in non-stressed cell conditions, the methylated H4K20 residues are in the heterochromatin state, and with the introduction of DNA DSBs, an area of open chromatin is generated, exposing the preexisting methylated H4K20 residues [49,50]. When exposed, the methylated H4K20 serves as a docking site required for the recruitment of 53BP1 to the sites of damage [49,50]. Interestingly, although H4K20 methylation levels show no change globally, H4K20 methylation levels increase locally in response to DNA DSBs [50,51]. The localized increase in methylated H4K20 can be mediated by the histone methyltransferase multiple myeloma SET domain (MMSET), which is recruited to DSBs in a γH2AX- and MDC1-dependent manner [51]. The localized increase in methylated H4K20 can also be mediated by the histone methyltransferase suppressor of variegation 4–20 (Suv4-20) [52]. DNA-damage-induced γH2AX recruits proliferating cell nuclear antigen (PCNA), which then facilitates the recruitment of the histone monomethylatransferase PR/SET domain-containing protein 7 (PR-set7) to the DNA damage sites [53]. PR-set7 mono-methylates H4K20 and facilitates subsequent Suv4-20 recruitment and catalysis, required to generate dimethylated H4K20 [52]. The exposure of methylated H4K20 and the localized increased level of methylated H4K20 at DNA damage sites, together, strongly facilitate the recruitment of 53BP1 to DSBs.
Another histone methylation mark that has been shown to be important for the recruitment of 53BP1 to DNA DSB sites is the methylated lysine 79 of histone H3 (H3K79). By screening radiation-sensitive yeast mutants for DNA damage checkpoint defects, disruptor of telomeric silencing 1-like (DOT1L), a conserved H3K79 methyltransferase, was identified [54]. Indeed, when DOT1L is suppressed, the recruitment of 53BP1 to the DSBs is inhibited [54,55]. Similar to H4K20, H3K79 maps to the histone core, which makes the methylated H3K79 inaccessible in normal cell conditions due to the highly-ordered chromatin structure. The disruption of the tightly packed DNA structure by DSBs leads to the exposure of methylated H3K79, resulting in the recognition of 53BP1 [55].
It has been suggested that the simultaneous recognition of γH2AX and methylated H4K20 or methylated H3K79 by two different domains of 53BP1 is needed to provide an affinity to facilitate the recruitment and accumulation of 53BP1 to the DNA DSB sites [50]. However, the initial recruitment of 53BP1 to the sites of DSBs in H2A.X-defficient cells is not impaired, suggesting that the methylated H4K20 or methylated H3K79 are essential for the initial recruitment of 53BP1 while γH2A.X is required for the retention of 53BP1 at DSB sites [55]. Also, the methylated H4K20 and methylated H3K79 work together to ensure the proper recruitment of 53BP1 to the sites of DSBs in the cell. It has been shown that the methylated H3K79 mark, while negligible during S phase, is required for IR-induced 53BP1 foci formation during G1 and G2 phase when the H4K20 methylation level is low [56].
The methylated lysine 9 of histone 3 (H3K9), a heterochromatin-associated mark, also plays an important role in DNA DSB responses. However, unlike the methylated H4K20 and methylated H3K79, methylated H3K9 acts upstream of the signaling cascade. This methylation is mediated by the histone methyltransferase suppressor of variegation 3–9 homolog 1 (Suv39H1) or its homolog Suv39H2 [57–59]. Suv39H1 and Suv39H2 are recruited to DSBs and contribute to the increase of the methylation levels of H3K9 at DSBs [60]. Methylated H3K9 can be directly recognized by the acetyltransferase TIP60; this positively regulates the acetyltransferase activity of TIP60 and leads to the acetylation and activation of the kinase activity of ATM and the initiation of ATM-dependent signaling [61]. Suv39H1/Suv39H2 can also methylate histone H2A.X on lysine 134 at DSBs and facilitate the phosphorylation of H2A.X on serine 139 (γH2A.X) [62]. In short, histone methylation plays an important role in various aspects of DDRs including the unraveling of high-ordered chromatin structure, the initiation of the signaling cascades, the recruitment and retention of proteins at the sites of the DSBs, as well as the facilitation of DNA break repair processes.
Acetylation of histone H3
It has been shown that cells with mutated TIP60 protein that lacks the acetyltransferase activity show defects in DNA DSB repair as well as in apoptotic signaling in response to unrepaired DNA breaks, indicating the crucial role of acetylation in regulating DDRs [63].
The acetylated lysine 56 of histone H3 (H3K56) is one of the acetylation marks shown to play an important role in DDRs. In humans, CREB-binding protein (CBP) and histone acetyltransferase p300 acetylate H3K56, whereas the NAD-dependent deacetylase sirtuin 1 and 2 (SIRT1 and SIRT2) deacetylate the acetylated H3K56. The histone chaperone anti-silencing function 1A (ASF1A) is required for the acetylation of H3K56, while the histone chaperone chromatin assembly factor-1 (CAF-1) is required for the incorporation of histones bearing the acetylated H3K56 into chromatin [64,65]. The acetylation status of histone H3 has been shown to have profound effects on the chromatin structure [66]. H3K56 acetylation is an abundant modification of newly synthesized histone H3 molecules that are incorporated into chromosomes during S phase [66,67]. This acetylation mark largely disappears in G2 phase, and it is maintained at a very low level outside of S phase in normal conditions without DNA damage [66,67]. In the presence of DNA damage, the acetylated levels of H3K56 remain high due to the action of DNA damage checkpoint proteins, and this histone mark co-localizes with other proteins at the sites of DNA DSBs during DDRs. This suggests that the acetylation of H3K56 creates a favorable chromatin environment for DNA repair [64,66–68].
If the acetylation of H3K56 is important for facilitating the DNA repair, the deacetylation of H3K56 is also important for signaling the completion of the DNA repair process. Deacetylation of H3K56 can be carried out by two distinct mechanisms: 1) The rapid recruitment of the histone deacetylases HDAC1 and HDAC2 to the DNA damage sites can promote the hypoacetylation of H3K56 and restore the chromatin status following DDRs [69] and 2) the acetylated core histone can also be degraded by proteosomes following the repair of damaged DNA, and the newly synthesized core histones can be incorporated to form intact nucleosomes [70], marking the completion of the cellular response to DNA damage stress.
Similar to acetylated H3K56, acetylated H4K16 is also indispensible for generating efficient DDRs and influential for DNA DSB repair [71]. In humans, the protein that is responsible for the majority of the acetylation of H4K16 is the male-absent-on-the-first (MOF) protein [72]. In response to DNA damage, MOF has been shown to directly interact with ATM, and this interaction is critical for IR-induced ATM activation [73,74]. Moreover, MOF is important to maintain sufficient acetylation levels of H4K16 to provide a chromatin structure permissive for efficient DNA damage repair [71]. When MOF is depleted in the cell, the acetylation level of H4K16 is reduced, leading to delayed or abrogated IR-induced focus formation of the mediator proteins MDC1, 53BP1 and BRCA1 and their downstream effector proteins at the sites of DNA damage [71,75].
In brief, the acetylation of histone tails is important to create a favorable environment with loosely compacted chromatin structure, that becomes readily accessible to the DNA repair machinery. Once the repair is completed, histones can be deacetylated by histone deacetylases, and the highly compacted chromatin structure is restored.
Interplay of histone modifications during DDRs
Different histone modifications, such as phosphorylation, ubiquitylation, methylation, and acetylation play important roles in different aspects of DDRs (Figure 2). Despite their distinct roles, different histone modifications also work cooperatively to generate proper and efficient cellular responses to DNA damage (Figure 3).
Figure 2.
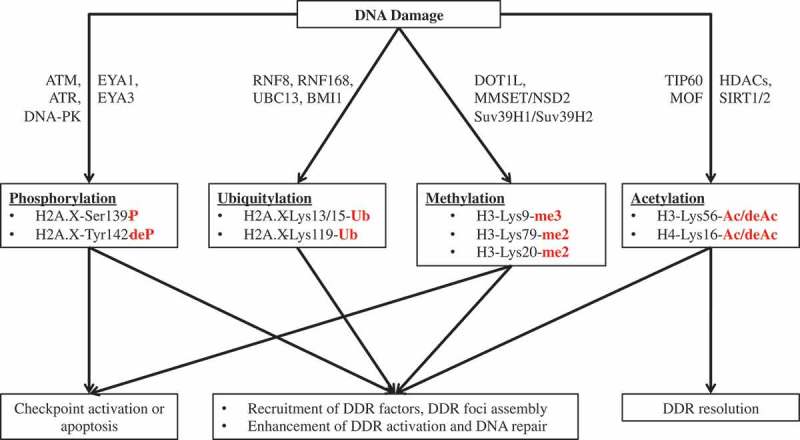
The various functions of different post-translational modifications of histones in DNA damage responses. P: phosphorylation; deP: dephosphorylation; Ub: ubiquitination; Me: methylation; Ac: acetylation; deAc: deacetylation; H: histone; Lys: lysine; Ser: serine; Tyr: tyrosine.
Figure 3.
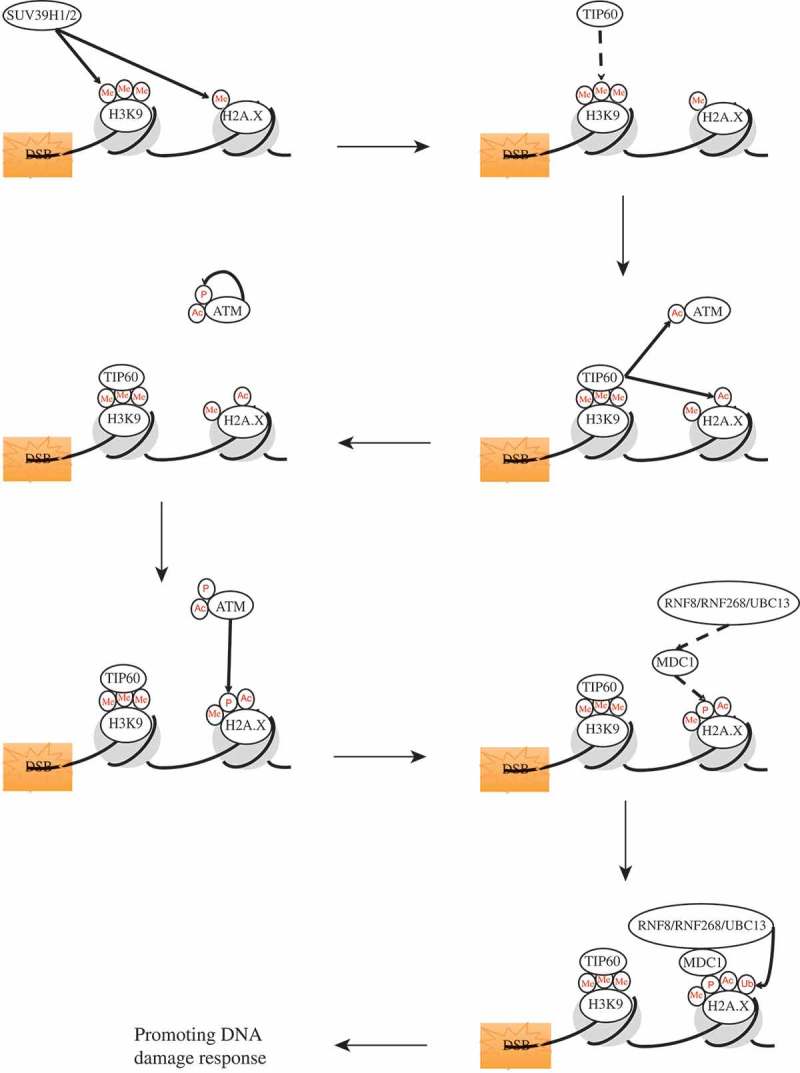
The interplay of different histone modifications in generating and promoting the DNA damage response. Unbroken arrows illustrate the catalysis action of the proteins; broken arrows illustrate the recruitment of the proteins. P: phosphorylation; Ub: ubiquitination; Me: methylation; Ac: acetylation.
When a DNA DSB occurs, the histone methyltransferase Suv39H2 is recruited to the DNA breaks [60]. At the sites of DSB, Suv39H2 methylates H2A.X at lysine 134, facilitating the phosphorylation of H2A.X at serine 139 [62]. Suv39H2, at the break sites, also methylates H3K9, and this methylated H3K9 provides the moiety to recruit TIP60 [60,61]. When TIP60 acetyltransferase directly interacts with methylated H3K9, the acetyltransferase activity of TIP60 is activated, leading to the acetylation and the complete activation of ATM [60,61]. The fully activated TIP60 can also acetylate histone H2A.X, which stimulates the signaling cascade in later steps [76]. Fully activated and acetylated ATM then can interact with Suv39H2-mediated methylated histone H2A.X with high affinity and modify histone H2A.X with the phosphorylation at serine 139 (γH2AX) [11,12,18,62]. γH2AX consequentially recruits the mediator protein MDC1 as well as the E3 ubiquitin ligases RNF8 and RNF168 to the sites of the break [41]. RNF8 and RNF 168, together with UBC13 ubiquitylate H2A.X, which requires its prior acetylation [76]. The sequential acetylation and ubiquitylation of H2A.X facilitate DDRs through enhancing the histone dynamics and promoting the accessibility of the chromatin to the DNA repair machinery [76]. This, however, is only one simple example of how different histone modifications can work together to promote an effectual cellular response to DNA damage stress. Further investigations and studies will most likely uncover other important interactions between different histone modifications in DNA damage responses.
Concluding remarks
DNA damage responses are a mesh of different signaling pathways that activate various cellular processes such as cell cycle checkpoints, DNA break repair and apoptosis to ensure genome integrity in response to DNA damage stress. In response to DNA damage, individual PTMs of histones, such as phosphorylation, ubiquitylation, methylation, and acetylation, have been shown to play important roles both independently and cooperatively in generating and regulating DDRs. Here, we illustrated the roles of these PTMs of histones in the events of DNA DSBs. It is established that the PTMs of histones are important not only in facilitating the reorganization of chromatin structure for the accessibility of DNA repair proteins but also in assisting the recruitment, accumulation, and retention of repair proteins at the sites of DNA DSBs. However, further studies are needed to decipher the relationship between different histone modifications as well as how the dynamic network of histone modifications can be used to determine and to interpret or predict the cellular responses and outcomes in response to DNA DSBs or other types of DNA damage.
Funding Statement
This work was supported by the American Society of Hematology [NA]; Andrew Sabin Family Foundation [NA] and Cancer Prevention Research Institute of Texas [RR150039].
Acknowledgments
We thank Drs. R. Behringer and R. Miller and the members of the Santos Lab for critical reading and suggestions. This work is supported by a Cancer Prevention Research Institute of Texas Recruitment of First-time Tenure-Track Faculty Award (RR150039), an Andrew Sabin Family Foundation Fellow Award and an American Society of Hematology (ASH) Junior Faculty Scholar Award to MAS.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Jackson SP, Bartek J.. The DNA-damage response in human biology and disease. Nature. 2009. October 22;461(7267):1071–1078. PubMed PMID: 19847258; PubMed Central PMCID: PMCPMC2906700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011. March 01;25(5):409–433. PubMed PMID: 21363960; PubMed Central PMCID: PMCPMC3049283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009. May;19(5):207–217. PubMed PMID: 19342239. [DOI] [PubMed] [Google Scholar]
- [4].Cao LL, Shen C, Zhu WG. Histone modifications in DNA damage response. Sci China Life Sci. 2016. March;59(3):257–270. 10.1007/s11427-016-5011-z. PubMed PMID: 26825946. [DOI] [PubMed] [Google Scholar]
- [5].Dasika GK, Lin SC, Zhao S, et al. DNA damage-induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis. Oncogene. 1999. December 20;18(55):7883–7899. PubMed PMID: 10630641. [DOI] [PubMed] [Google Scholar]
- [6].Rossetto D, Truman AW, Kron SJ, et al. Epigenetic modifications in double-strand break DNA damage signaling and repair. Clin Cancer Res. 2010. September 15;16(18):4543–4552. 10.1158/1078-0432.CCR-10-0513. PubMed PMID: 20823147; PubMed Central PMCID: PMCPMC2940951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007. February 23;128(4):707–719. PubMed PMID: 17320508. [DOI] [PubMed] [Google Scholar]
- [8].Daley JM, Sung P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol Cell Biol. 2014. April;34(8):1380–1388. PubMed PMID: 24469398; PubMed Central PMCID: PMCPMC3993578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014. January;15(1):7–18. PubMed PMID: 24326623. [DOI] [PubMed] [Google Scholar]
- [10].Sulli G, Di Micco R, d’Adda Di Fagagna F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer. 2012. October;12(10):709–720. PubMed PMID: 22952011. [DOI] [PubMed] [Google Scholar]
- [11].Bonner WM, Redon CE, Dickey JS, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008. December;8(12):957–967. PubMed PMID: 19005492; PubMed Central PMCID: PMCPMC3094856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rogakou EP, Pilch DR, Orr AH, et al. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998. March 06;273(10):5858–5868. PubMed PMID: 9488723. [DOI] [PubMed] [Google Scholar]
- [13].Rogakou EP, Boon C, Redon C, et al. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999. September 06;146(5):905–916. PubMed PMID: 10477747; PubMed Central PMCID: PMCPMC2169482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kim JA, Kruhlak M, Dotiwala F, et al. Heterochromatin is refractory to gamma-H2AX modification in yeast and mammals. J Cell Biol. 2007. July 16;178(2):209–218. PubMed PMID: 17635934; PubMed Central PMCID: PMCPMC2064441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cowell IG, Sunter NJ, Singh PB, et al. gammaH2AX foci form preferentially in euchromatin after ionising-radiation. PLoS One. 2007. October 24;2(10):e1057 PubMed PMID: 17957241; PubMed Central PMCID: PMCPMC2020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Iacovoni JS, Caron P, Lassadi I, et al. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. Embo J. 2010. April 21;29(8):1446–1457. PubMed PMID: 20360682; PubMed Central PMCID: PMCPMC2868577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Savic V, Yin B, Maas NL, et al. Formation of dynamic gamma-H2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin. Mol Cell. 2009. May 15;34(3):298–310. PubMed PMID: 19450528; PubMed Central PMCID: PMCPMC2744111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Burma S, Chen BP, Murphy M, et al. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001. November 09;276(45):42462–42467. PubMed PMID: 11571274. [DOI] [PubMed] [Google Scholar]
- [19].Stiff T, O’Driscoll M, Rief N, et al. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004. April 01;64(7):2390–2396. PubMed PMID: 15059890. [DOI] [PubMed] [Google Scholar]
- [20].Paull TT, Rogakou EP, Yamazaki V, et al. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10(15):886–895. Jul 27–Aug 10;():. PubMed PMID: 10959836. . [DOI] [PubMed] [Google Scholar]
- [21].Celeste A, Fernandez-Capetillo O, Kruhlak MJ, et al. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. 2003. July;5(7):675–679. PubMed PMID: 12792649. [DOI] [PubMed] [Google Scholar]
- [22].Celeste A, Petersen S, Romanienko PJ, et al. Genomic instability in mice lacking histone H2AX. Science. 2002. May 03;296(5569):922–927. PubMed PMID: 11934988; PubMed Central PMCID: PMCPMC4721576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Martin M, Terradas M, Hernandez L, et al. gammaH2AX foci on apparently intact mitotic chromosomes: not signatures of misrejoining events but signals of unresolved DNA damage. Cell Cycle. 2014;13(19):3026–3036. PubMed PMID: 25486563; PubMed Central PMCID: PMCPMC4614418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chowdhury D, Keogh MC, Ishii H, et al. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell. 2005. December 09;20(5):801–809. PubMed PMID: 16310392. [DOI] [PubMed] [Google Scholar]
- [25].Nakada S, Chen GI, Gingras AC, et al. PP4 is a gamma H2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep. 2008. October;9(10):1019–1026. PubMed PMID: 18758438; PubMed Central PMCID: PMCPMC2527856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cha H, Lowe JM, Li H, et al. Wip1 directly dephosphorylates gamma-H2AX and attenuates the DNA damage response. Cancer Res. 2010. May 15;70(10):4112–4122. PubMed PMID: 20460517; PubMed Central PMCID: PMCPMC2904079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xiao A, Li H, Shechter D, et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature. 2009. January 01;457(7225):57–62. PubMed PMID: 19092802; PubMed Central PMCID: PMCPMC2854499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cook PJ, Ju BG, Telese F, et al. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009. April 02;458(7238):591–596. PubMed PMID: 19234442; PubMed Central PMCID: PMCPMC2692521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fernandez-Capetillo O, Allis CD, Nussenzweig A. Phosphorylation of histone H2B at DNA double-strand breaks. J Exp Med. 2004. June 21;199(12):1671–1677. PubMed PMID: 15197225; PubMed Central PMCID: PMCPMC2212807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cheung WL, Ajiro K, Samejima K, et al. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell. 2003. May 16;113(4):507–517. PubMed PMID: 12757711. [DOI] [PubMed] [Google Scholar]
- [31].Cheung WL, Turner FB, Krishnamoorthy T, et al. Phosphorylation of histone H4 serine 1 during DNA damage requires casein kinase II in S. cerevisiae. Curr Biol. 2005. April 12;15(7):656–660. PubMed PMID: 15823538. [DOI] [PubMed] [Google Scholar]
- [32].Gatti M, Pinato S, Maiolica A, et al. RNF168 promotes noncanonical K27 ubiquitination to signal DNA damage. Cell Rep. 2015. January 13;10(2):226–238. PubMed PMID: 25578731. [DOI] [PubMed] [Google Scholar]
- [33].Lou Z, Minter-Dykhouse K, Franco S, et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell. 2006. January 20;21(2):187–200. PubMed PMID: 16427009. [DOI] [PubMed] [Google Scholar]
- [34].Kolas NK, Chapman JR, Nakada S, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007. December 7;318(5856):1637–1640. PubMed PMID: 18006705; PubMed Central PMCID: PMCPMC2430610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007. May 25;316(5828):1160–1166. PubMed PMID: 17525332. [DOI] [PubMed] [Google Scholar]
- [36].Wang B, Elledge SJ. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci USA. 2007. December 26;104(52):20759–20763. PubMed PMID: 18077395; PubMed Central PMCID: PMCPMC2410075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Huen MS, Grant R, Manke I, et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007. November 30;131(5):901–914. PubMed PMID: 18001825; PubMed Central PMCID: PMCPMC2149842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mailand N, Bekker-Jensen S, Faustrup H, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007. November 30;131(5):887–900. PubMed PMID: 18001824. [DOI] [PubMed] [Google Scholar]
- [39].Mattiroli F, Vissers JH, van Dijk WJ, et al. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell. 2012. September 14;150(6):1182–1195. PubMed PMID: 22980979. [DOI] [PubMed] [Google Scholar]
- [40].Thorslund T, Ripplinger A, Hoffmann S, et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature. 2015. November 19;527(7578):389–393. PubMed PMID: 26503038. [DOI] [PubMed] [Google Scholar]
- [41].Stewart GS, Panier S, Townsend K, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009. February 6;136(3):420–434. PubMed PMID: 19203578. [DOI] [PubMed] [Google Scholar]
- [42].Doil C, Mailand N, Bekker-Jensen S, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009. February 06;136(3):435–446. PubMed PMID: 19203579. [DOI] [PubMed] [Google Scholar]
- [43].Wu J, Huen MS, Lu LY, et al. Histone ubiquitination associates with BRCA1-dependent DNA damage response. Mol Cell Biol. 2009. February;29(3):849–860. PubMed PMID: 19015238; PubMed Central PMCID: PMCPMC2630672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ginjala V, Nacerddine K, Kulkarni A, et al. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol Cell Biol. 2011. May;31(10):1972–1982. PubMed PMID: 21383063; PubMed Central PMCID: PMCPMC3133356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ismail IH, Andrin C, McDonald D, et al. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J Cell Biol. 2010. October 04;191(1):45–60. PubMed PMID: 20921134; PubMed Central PMCID: PMCPMC2953429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gudjonsson T, Altmeyer M, Savic V, et al. TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell. 2012. August 17;150(4):697–709. PubMed PMID: 22884692. [DOI] [PubMed] [Google Scholar]
- [47].Mosbech A, Lukas C, Bekker-Jensen S, et al. The deubiquitylating enzyme USP44 counteracts the DNA double-strand break response mediated by the RNF8 and RNF168 ubiquitin ligases. J Biol Chem. 2013. June 07;288(23):16579–16587. PubMed PMID: 23615962; PubMed Central PMCID: PMCPMC3675593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yan Q, Dutt S, Xu R, et al. BBAP monoubiquitylates histone H4 at lysine 91 and selectively modulates the DNA damage response. Mol Cell. 2009. October 9;36(1):110–120. PubMed PMID: 19818714; PubMed Central PMCID: PMCPMC2913878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sanders SL, Portoso M, Mata J, et al. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004. November 24;119(5):603–614. PubMed PMID: 15550243. [DOI] [PubMed] [Google Scholar]
- [50].Botuyan MV, Lee J, Ward IM, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006. December 29;127(7):1361–1373. PubMed PMID: 17190600; PubMed Central PMCID: PMCPMC1804291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Pei H, Zhang L, Luo K, et al. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011. February 3;470(7332):124–128. PubMed PMID: 21293379; PubMed Central PMCID: PMCPMC3064261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tuzon CT, Spektor T, Kong X, et al. Concerted activities of distinct H4K20 methyltransferases at DNA double-strand breaks regulate 53BP1 nucleation and NHEJ-directed repair. Cell Rep. 2014. July 24;8(2):430–438. PubMed PMID: 25001286; PubMed Central PMCID: PMCPMC4134327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Oda H, Hubner MR, Beck DB, et al. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol Cell. 2010. November 12;40(3):364–376. PubMed PMID: 21035370; PubMed Central PMCID: PMCPMC2999913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wysocki R, Javaheri A, Allard S, et al. Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol Cell Biol. 2005. October;25(19):8430–8443. PubMed PMID: 16166626; PubMed Central PMCID: PMCPMC1265753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Huyen Y, Zgheib O, Ditullio RA Jr.. et al. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004. November 18;432(7015):406–411. PubMed PMID: 15525939. [DOI] [PubMed] [Google Scholar]
- [56].Wakeman TP, Wang Q, Feng J, et al. Bat3 facilitates H3K79 dimethylation by DOT1L and promotes DNA damage-induced 53BP1 foci at G1/G2 cell-cycle phases. Embo J. 2012. May 2;31(9):2169–2181. PubMed PMID: 22373577; PubMed Central PMCID: PMCPMC3343460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Rea S, Eisenhaber F, O’Carroll D, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000. August 10;406(6796):593–599. PubMed PMID: 10949293. [DOI] [PubMed] [Google Scholar]
- [58].O’Carroll D, Scherthan H, Peters AH, et al. Isolation and characterization of Suv39h2, a second histone H3 methyltransferase gene that displays testis-specific expression. Mol Cell Biol. 2000. December;20(24):9423–9433. PubMed PMID: 11094092; PubMed Central PMCID: PMCPMC102198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Krishnan S, Horowitz S, Trievel RC. Structure and function of histone H3 lysine 9 methyltransferases and demethylases. Chembiochem. 2011. January 24;12(2):254–263. PubMed PMID: 21243713. [DOI] [PubMed] [Google Scholar]
- [60].Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, et al. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci USA. 2014. June 24;111(25):9169–9174. PubMed PMID: 24927542; PubMed Central PMCID: PMCPMC4078803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sun Y, Jiang X, Xu Y, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009. November;11(11):1376–1382. PubMed PMID: 19783983; PubMed Central PMCID: PMCPMC2783526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sone K, Piao L, Nakakido M, et al. Critical role of lysine 134 methylation on histone H2AX for gamma-H2AX production and DNA repair. Nat Commun. 2014. December 9;5:5691 PubMed PMID: 25487737; PubMed Central PMCID: PMCPMC4268694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ikura T, Ogryzko VV, Grigoriev M, et al. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000. August 18;102(4):463–473. PubMed PMID: 10966108. [DOI] [PubMed] [Google Scholar]
- [64].Das C, Lucia MS, Hansen KC, et al. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009. May 7;459(7243):113–117. PubMed PMID: 19270680; PubMed Central PMCID: PMCPMC2756583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yuan J, Pu M, Zhang Z, et al. Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle. 2009. June 1;8(11):1747–1753. PubMed PMID: 19411844; PubMed Central PMCID: PMCPMC2776713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Celic I, Masumoto H, Griffith WP, et al. The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr Biol. 2006. July 11;16(13):1280–1289. PubMed PMID: 16815704. [DOI] [PubMed] [Google Scholar]
- [67].Masumoto H, Hawke D, Kobayashi R, et al. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005. July 14;436(7048):294–298. PubMed PMID: 16015338. [DOI] [PubMed] [Google Scholar]
- [68].Vempati RK, Jayani RS, Notani D, et al. p300-mediated acetylation of histone H3 lysine 56 functions in DNA damage response in mammals. J Biol Chem. 2010. September 10;285(37):28553–28564. PubMed PMID: 20587414; PubMed Central PMCID: PMCPMC2937881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Miller KM, Tjeertes JV, Coates J, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010. September;17(9):1144–1151. PubMed PMID: 20802485; PubMed Central PMCID: PMCPMC3018776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Qian MX, Pang Y, Liu CH, et al. Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell. 2013. May 23;153(5):1012–1024. PubMed PMID: 23706739; PubMed Central PMCID: PMCPMC3983474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sharma GG, So S, Gupta A, et al. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol Cell Biol. 2010. July;30(14):3582–3595. PubMed PMID: 20479123; PubMed Central PMCID: PMCPMC2897562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Smith ER, Cayrou C, Huang R, et al. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol Cell Biol. 2005. November;25(21):9175–9188. PubMed PMID: 16227571; PubMed Central PMCID: PMCPMC1265810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Gupta A, Sharma GG, Young CS, et al. Involvement of human MOF in ATM function. Mol Cell Biol. 2005. June;25(12):5292–5305. PubMed PMID: 15923642; PubMed Central PMCID: PMCPMC1140595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Gupta A, Hunt CR, Hegde ML, et al. MOF phosphorylation by ATM regulates 53BP1-mediated double-strand break repair pathway choice. Cell Rep. 2014. July 10;8(1):177–189. PubMed PMID: 24953651; PubMed Central PMCID: PMCPMC4300955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Li X, Corsa CA, Pan PW, et al. MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Mol Cell Biol. 2010. November;30(22):5335–5347. PubMed PMID: 20837706; PubMed Central PMCID: PMCPMC2976376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ikura T, Tashiro S, Kakino A, et al. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol Cell Biol. 2007. October;27(20):7028–7040. PubMed PMID: 17709392; PubMed Central PMCID: PMCPMC2168918. [DOI] [PMC free article] [PubMed] [Google Scholar]