

Abstract
Many filamentous plant pathogens exhibit high levels of genomic variability, yet the impact of this variation on host–pathogen interactions is largely unknown. We have addressed host specialization in the wheat pathogen Zymoseptoria tritici. Our study builds on comparative analyses of infection and gene expression phenotypes of three isolates and reveals the extent to which genomic variation translates into phenotypic variation. The isolates exhibit genetic and genomic variation but are similarly virulent. By combining confocal microscopy, disease monitoring, staining of ROS, and comparative transcriptome analyses, we conducted a detailed comparison of the infection processes of these isolates in a susceptible wheat cultivar. We characterized four core infection stages: establishment, biotrophic growth, lifestyle transition, and necrotrophic growth and asexual reproduction that are shared by the three isolates. However, we demonstrate differentiated temporal and spatial infection development and significant differences in the expression profiles of the three isolates during the infection stages. More than 20% of the genes were differentially expressed and these genes were located significantly closer to transposable elements, suggesting an impact of epigenetic regulation. Further, differentially expressed genes were enriched in effector candidates suggesting that isolate‐specific strategies for manipulating host defenses are present in Z. tritici. We demonstrate that individuals of a host‐specialized pathogen have highly differentiated infection programs characterized by flexible infection development and functional redundancy. This illustrates how high genetic diversity in pathogen populations results in highly differentiated infection phenotypes, which fact needs to be acknowledged to understand host–pathogen interactions and pathogen evolution.
Keywords: confocal microscopy, expression phenotypes, host specialization, infection phenotypes, plant–fungus interactions, Zymoseptoria tritici
1. INTRODUCTION
Plants are colonized by a broad diversity of microbial species. Many plant‐associated microbes are commensals that utilize the plant as a scaffold for their proliferation by feeding on readily available nutrients. Other microbial species engage in more intimate interactions with plants, either as mutualistic symbionts where both partners benefit from the interaction or as antagonistic symbionts where the microbial species exploits the plant for growth and reproduction (Zeilinger et al., 2015). Mutualistic and antagonistic interactions between plants and co‐existing microbial species rely on molecular interactions between plant receptors and signaling pathways and microbial molecules including microbe‐associated molecular patterns (MAMPs) and effectors. The successful establishment of such intimate plant–microbe interactions, in one or the other form, depends on the ability of the microbial partner to suppress and interfere with the immune responses of the plant (Dodds & Rathjen, 2010; Jones & Dangl, 2006; Lo Presti et al., 2015) and to colonize and reproduce associated to plant tissues (Haueisen & Stukenbrock, 2016; van der Does & Rep, 2017). These symbioses are therefore considered to be highly specialized and the result of co‐evolution.
In a small number of model systems, key molecules involved in the antagonistic interaction of plants with their specialized pathogens have been characterized, for example, the Ustilago maydis effector Tin2 that interferes with the anthocyanin biosynthesis pathway in its host maize (Tanaka et al., 2014) or the tyrosine phosphatase HopAO1 of Pseudomonas syringae which inhibits activation of a plant pattern recognition receptor (Macho et al., 2014). While the relevance of these molecular interactions for the studied host–pathogen genotype combinations is undisputed, the consequences at the population level have so far been poorly addressed.
While many plant pathogens are known to be highly specialized to their host, substantial genetic variation is found in many species. Genetic variation can translate into phenotypic variation that can be important for pathogen populations to persist in changing environments (Fisher, Hawkins, Sanglard, & Gurr, 2018; Möller & Stukenbrock, 2017). However, host specialization may require a highly specialized and definite infection program. An intriguing question is which consequences genetic and phenotypic variation entails for the diversity of host interactions in pathogens that are specialized and host‐specific?
In this study, we addressed the extent of phenotypic variation in a fungal plant pathogen characterized by a high level of genomic variability. Our aim was to study phenotypic diversity beyond virulence phenotypes that are limited to the description of quantitative virulence which is based on the quantification of disease symptoms at a particular time of infection. We therefore considered morphological and temporal infection development as well as stage‐specific gene expression to characterize the infection phenotypes of pathogen individuals. We used the wheat pathogen Zymoseptoria tritici (syn. Mycosphaerella graminicola) as a model to investigate how the development of disease symptoms and the transcriptional program induced during infection vary among three field isolates from geographically distinct locations. Zymoseptoria tritici has served as a prominent model in population genetic studies of crop pathogens, and genetic variation has been assessed from local scales in individual lesions on single leaves up to continental scales. The amount of genetic variation in a Z. tritici field population is comparable to the variation found on a continental scale suggesting very high levels of genetic variation in local populations and little subdivision between populations (Linde, Zhan, McDonald, & a., 2002; McDonald et al., 2016; Zhan, Pettway, & McDonald, 2003). Zymoseptoria tritici has been described as hemibiotroph (Ponomarenko, Goodwin, & Kema, 2011) or latent necrotroph (Sánchez‐Vallet, McDonald, Solomon, & McDonald, 2015) with a prolonged epiphytic stage (Fones, Eyles, Kay, Cowper, & Gurr, 2017). Infections are biphasic and characterized by a long asymptomatic, biotrophic phase, followed by necrotrophic growth where the fungus degrades and takes up nutrients from dead host cells (Ponomarenko et al., 2011; Rudd et al., 2015). In spite of the general importance of this pathogen in wheat fields, the life cycle and infection biology of Z. tritici are so far poorly understood.
The haploid genome of Z. tritici comprises a high number of accessory chromosomes ranging from 400 kb to 1 Mb in size in the reference isolate IPO323 (Goodwin et al., 2011; Wittenberg et al., 2009). Recent studies provide evidence for the presence of virulence determinants on the accessory chromosomes; however, the genes responsible for these effects have so far not been identified (Habig, Quade, & Stukenbrock, 2017). Furthermore, several genome‐wide association and quantitative trait loci mapping studies have linked a variety of phenotypic traits to genetic variants and candidate genes (Hartmann, Sánchez‐Vallet, McDonald, & Croll, 2017; Lendenmann, Croll, & McDonald, 2015; Lendenmann, Croll, Stewart, & McDonald, 2014; Mirzadi Gohari et al., 2015; Stewart et al., 2017; Zhong et al., 2017).
Here, we investigated how infection of a susceptible host by genetically and morphologically distinct isolates results in similar quantitative virulence. By combining confocal microscopy, disease monitoring, reactive oxygen species (ROS) localization, and transcriptome analyses, we compiled a detailed characterization of infection phenotypes of three Z. tritici isolates. We hypothesized that high genetic diversity not only increases the evolutionary potential of the pathogen but also results in a variety of host–pathogen interactions that cause a range of different infection phenotypes. Our combined comparative analyses enabled us to characterize infection morphology and gene expression of the three Z. tritici isolates, including a core infection program and isolate‐specific infection phenotypes. We conclude that host specialization of Z. tritici entails a substantial amount of variation in terms of the temporal, spatial, and molecular host–pathogen interactions. We speculate that this phenotypic variation is important for the pathogen to rapidly respond to changing environments, and we underline the need of considering variation at this level in the study of pathogen evolution and the development of disease control strategies.
2. MATERIALS AND METHODS
2.1. Isolates and growth conditions
We used three Zymoseptoria tritici isolates for all experiments: Zt05 (Thygesen, Jørgensen, Jensen, & Munk, 2008), Zt09 (≙ IPO323ΔChr18, a derivate of the reference strain IPO323 (Goodwin et al., 2011) that lost chromosome 18 (Kellner et al., 2014)), and Zt10 (Stukenbrock, Banke, Javan‐Nikkhah, & McDonald, 2007) (Supporting Information Table S1). Fungal cells were inoculated from glycerol stocks onto YMS agar (0.4% [w/v] yeast extract, 0.4% [w/v] malt extract, 0.4% [w/v] sucrose, 2% [w/v] bacto agar) and grown at 18°C for 5 days. Single cells were grown in liquid YMS (200 rpm, 18°C) for 2 days and harvested by centrifugation (3,500 rpm for 10 min).
2.2. Plant infection experiments
We conducted all plant infection experiments in controlled plant growth chambers and inoculated 14‐day‐old seedlings of the winter wheat (Triticum aestivum) cultivar Obelisk (Wiersum Plantbreeding, Winschoten, Netherlands). Fungal inoculum was adjusted to 1 × 108 cells/ml in 0.1% [v/v] Tween 20 (Roth, Karlsruhe, Germany) and brushed onto labeled areas (8 to 12 cm) of the second leaves. The same treatment without fungal cells was applied for mock controls. Plants were incubated for 48 hr at 22°C [day]/20°C [night] and 100% humidity with a 16‐hr light period. Subsequently, humidity was reduced to 70%. Plants were grown for three or four weeks after inoculation, depending on the experiment.
2.3. In planta phenotypic assays
To compare quantitative virulence of Zt05, Zt09, and Zt10 on wheat, we performed three independent, randomized infection experiments with blinded inoculation and evaluation. Inoculated leaf areas of 460 leaves (Zt05: 116, Zt09: 118, Zt10: 118, mock control: 108) were evaluated at 28 days post infection (dpi) by scoring the observed disease symptoms based on the percentage of leaf area covered by necrosis and pycnidia (Poppe, Dorsheimer, Happel, & Stukenbrock, 2015). We differentiated six categories (Supporting Information Figure S1): 0 (no visible symptoms), 1 (1%–20%), 2 (21%–40%), 3 (41%–60%), 4 (61%–80%), and 5 (81%–100%). Statistical differences were evaluated by Mann–Whitney U test considering differences significant if p ≤ 0.01.
To compare the temporal disease development, we manually inspected 40 inoculated leaves per isolate between 9 and 27 dpi and registered the occurrence of first visible symptoms every two days. Individual leaves were documented using a Leica S8APO equipped with a Leica DFC450 camera.
To localize and visualize the reactive oxygen species H2O2 within infected leaf tissue, we conducted 3,3′‐diaminobenzidine (DAB) staining (Thordal‐Christensen, Zhang, Wei, & Collinge, 1997) at 2, 4, 7–11, 14, 16, 18, and 21 dpi and quantified the reddish‐brown precipitate in cleared leaves which indicates an accumulation of H2O2 (Supporting Information Text S1). Leaves were documented prior and post staining.
2.4. Analysis of Z. tritici wheat infection by confocal microscopy
Development of Z. tritici isolates within and on the surface of wheat leaves was analyzed by confocal laser scanning microscopy. We harvested infected wheat leaves at 3–14, 17, 19–21, 24, 25, and 28 dpi and analyzed the interactions between fungal hyphae and wheat tissue. Likewise, we analyzed infected leaves to determine the infection stage of leaf samples used for RNA extraction (see below). In total, we studied 37 infected wheat leaves for Zt05, 34 for Zt09, and 30 for Zt10 (Supporting Information Table S1), analyzed at least 15 infection events per leaf sample by confocal microscopy, and created a total of 113 confocal image z‐stacks. Cleared leaf material was stained with wheat germ agglutinin conjugated to fluorescein isothiocyanate (WGA‐FITC) in combination with propidium iodide (PI) (Supporting Information Text S1 for staining protocol). Microscopy was conducted using a Leica TCS SP5 (Leica Microsystems, Germany) and a Zeiss LSM880 (Carl Zeiss Microscopy, Germany). FITC was excited at 488 nm (argon laser) and detected between 500 and 540 nm. PI was excited at 561 nm (diode‐pumped solid‐state laser) and detected between 600 and 670 nm. Analyses, visualization, and processing of image z‐stacks were performed using Leica Application Suite Advanced Fluorescence (Leica Microsystems, Germany), ZEN black and Zen blue (Carl Zeiss Microscopy, Germany), and AMIRA® (FEITM Visualization Science Group, Germany). Animations of image z‐stacks are .avi format and can be played in VLC media player (available at http://www.videolan.org/vlc/).
2.5. Transcriptome analyses of Z. tritici isolates during wheat infection
Total RNA from Z. tritici‐infected wheat material collected during one infection experiment was isolated using the TRIzol™ reagent (Invitrogen, Karlsruhe, Germany) according to the manufacturer's instructions. One biological sample consists of material from three inoculated second leaves that were pooled and homogenized in liquid nitrogen. One hundred milligram of the resulting powder was used for RNA extraction. Because our analyses revealed isolate‐specific differences in the temporal infection development, we used independent sampling schedules for each isolate to be able to compare their transcriptomes during the same infection stage (Supporting Information Table S2). We collected infected leaf material at up to nine time points per isolate and assigned infection stages by examining central sections (1–2 cm) of each leaf by confocal microscopy (Supporting Information Figure S2). Per isolate and infection stage, the two most representative samples were selected as biological replicates for transcriptome sequencing (Table 1). Preparation of strand‐specific RNA‐seq libraries including polyA enrichment was performed at the Max Planck Genome Center, Cologne, Germany (http://mpgc.mpipz.mpg.de), using the NEBNext UltraTM Directional RNA Library Prep Kit for Illumina according to the manufacturer's protocol (New England Biolabs, Frankfurt/Main, Germany) with an input of 1 µg total RNA. Sequencing, performed using an Illumina HiSeq 2,500 platform, generated strand‐specific, 100‐base, single‐end reads with an average yield of 112 million reads per sample (Supporting Information Table S3).
Table 1.
Summary of the stage‐specific transcriptomes (A–D) of the three Zymoseptoria tritici isolates
| Isolate | Infection stage | Sample | Time point (dpi) | No. of filtered reads | No. of reads mapped to genome | % reads mapped to genome | No. of genes RPKM ≥2a | No. of genes RPKM ≥10a |
|---|---|---|---|---|---|---|---|---|
| Zt05b | A | Zt05_Ta_A_01 | 3 | 107,507,137 | 15,213,307 | 14.15 | 9,302 | 7,455 |
| Zt05_Ta_A_02 | 81,479,903 | 10,509,515 | 12.90 | |||||
| B | Zt05_Ta_B_01 | 8 | 113,732,295 | 15,057,425 | 13.24 | 9,404 | 7,623 | |
| Zt05_Ta_B_02 | 128,271,704 | 14,856,314 | 11.58 | |||||
| C | Zt05_Ta_C_01 | 13 | 91,298,814 | 26,756,807 | 29.31 | 9,538 | 7,982 | |
| Zt05_Ta_C_02 | 135,198,719 | 34,329,884 | 25.39 | |||||
| D | Zt05_Ta_D_01 | 20 | 86,100,462 | 41,871,582 | 48.63 | 9,585 | 7,914 | |
| Zt05_Ta_D_02 | 119,101,106 | 76,123,086 | 63.91 | |||||
| Zt09c | A | Zt09_Ta_A_01 | 4 | 129,342,007 | 5,868,572 | 4.54 | 9,435 | 7,482 |
| Zt09_Ta_A_02 | 92,711,865 | 5,582,561 | 6.02 | |||||
| B | Zt09_Ta_B_01 | 11 | 96,767,482 | 5,034,677 | 5.20 | 9,718 | 7,910 | |
| Zt09_Ta_B_02 | 103,428,015 | 5,964,566 | 5.77 | |||||
| C | Zt09_Ta_C_01 | 13 | 121,529,652 | 31,264,373 | 25.73 | 9,892 | 8,220 | |
| Zt09_Ta_C_02 | 93,253,633 | 27,790,773 | 29.80 | |||||
| D | Zt09_Ta_D_01 | 20 | 110,296,264 | 84,263,562 | 76.40 | 9,867 | 7,949 | |
| Zt09_Ta_D_02 | 101,757,635 | 75,044,880 | 73.75 | |||||
| Zt10d | A | Zt10_Ta_A_01 | 6 | 93,557,587 | 4,895,650 | 5.23 | 8,814 | 7,219 |
| Zt10_Ta_A_02 | 91,111,840 | 4,836,535 | 5.31 | |||||
| B | Zt10_Ta_B_01 | 11 | 94,828,793 | 5,951,869 | 6.28 | 9,068 | 7,407 | |
| Zt10_Ta_B_02 | 110,255,245 | 7,896,227 | 7.16 | |||||
| C | Zt10_Ta_C_01 | 13 | 86,652,710 | 20,804,110 | 24.01 | 9,241 | 7,742 | |
| Zt10_Ta_C_02 | 91,070,127 | 8,620,099 | 9.47 | |||||
| D | Zt10_Ta_D_01 | 24 | 98,493,807 | 29,939,216 | 30.40 | 9,062 | 7,308 | |
| Zt10_Ta_D_02 | 93,690,628 | 34,607,371 | 36.94 |
Overview of RNA‐seq datasets including time point of sampling, number of sequenced reads post filtering, number of mapped reads, percentage of mapped reads, and numbers of transcribed genes.
RPKM values were calculated using Cuffdiff2 and are normalized over all infection stages within the respective isolate (normalization method: geometric, dispersion method: per‐condition).
11,138 genes of IPO323 (94.08%) found by nucleotide blast for Zt05.
11,754 of the 11,839 genes predicted and annotated for IPO323 (Grandaubert et al., 2015); 85 genes located on chromosome 18 were not considered.
10,745 genes of IPO323 (90.76%) found by nucleotide blast for Zt10.
We assessed the quality of the sequencing data with FastQC v0.11.2 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and applied a stringent trimming and filtering protocol using FASTX‐toolkit v0.0.14 (http://hannonlab.cshl.edu/fastx_toolkit/) and Trimmomatic (Bolger, Lohse, & Usadel, 2014) v0.33 (Supporting Information Text S1 for details). The resulting 88‐bp reads were mapped against the genome of the respective isolate with TopHat2 v2.0.9 (Kim et al., 2013). Read alignments were stored in SAM format, and indexing, sorting, and conversion to BAM format were performed using SAMtools v0.1.19 (Li et al., 2009). Relative abundance of transcripts for predicted genes was calculated in RPKM by Cuffdiff2 v2.2.1 (Trapnell et al., 2013). Raw read counts per gene were estimated with HTSeq v0.6.1p1 (Anders, Pyl, & Huber, 2015). Gene coordinates in the Zt05 (Supporting Information Table S4) and Zt10 (Supporting Information Table S5) genomes were obtained by mapping the predicted genes of IPO323 using nucleotide BLAST alignments (e‐value cutoff 1e −3, identity ≥90%, query coverage 90%–110%). Differential gene expression analyses between Z. tritici infection stages and isolates were performed in R (R Core Team, 2017) using the Bioconductor package DESeq2 v1.10.1 (Love, Huber, & Anders, 2014). Significant signals of differential expression for the most strongly changing genes were determined with p adj ≤0.01 and |log2 fold change ≥2| as recommended for RNA‐seq experiments with a low number of replicates (Schurch et al., 2016). The R package topGO (Alexa, Rahnenführer, & Lengauer, 2006) was used to perform Gene Ontology (GO) term enrichment analyses. p Values for each GO term (Grandaubert, Bhattacharyya, & Stukenbrock, 2015) were calculated using Fischer's exact test applying the topGO algorithm “weight01” considering GO term hierarchy. We reported categories significant with p ≤ 0.01 for the ontology “Biological Process.” PFAM domain enrichment analyses were performed using a custom python script, and p values were calculated using chi‐square tests.
To analyze genomic distances between differentially expressed genes and transposable elements (TEs), we annotated TEs as described in Grandaubert et al. (2015) for Zt05 (Supporting Information Table S6) and Zt10 (Supporting Information Table S7) and used the published TE annotation of IPO323 for Zt09 (Grandaubert et al., 2015). Distances between genes of interest and the closest annotated TEs were calculated with bedtools v2.26 (Quinlan & Hall, 2010). Likewise, we used ChIP‐seq peak data (Schotanus et al., 2015) to calculate distances between genes and the closest H3K9me3 and H3K27me3 peaks. Statistical analyses were performed in R. For a detailed overview, see Supporting Information Text S1.
2.6. Pulsed‐field gel electrophoresis
A non‐protoplast protocol (Supporting Information Text S1) was used to produce DNA plugs for separation of small chromosomes (~0.2 to 1.6 Mb) by pulsed‐field gel electrophoresis (PFGE) (Stukenbrock et al., 2010). Chromosomal DNA of Saccharomyces cerevisiae (Bio‐Rad, Munich, Germany) was used as standard size marker. Gels were stained in 1 µg/ml ethidium bromide solution, and chromosome bands were detected with Thyphoon Trio™ (GE Healthcare, Munich, Germany).
2.7. De novo genome assemblies of Z. tritici isolates Zt05 and Zt10 and synteny analyses
DNA of Zt05 and Zt10 was extracted from single cells using the CTAB extraction protocol (Allen, Flores‐Vergara, Krasynanski, Kumar, & Thompson, 2006) and used as input to prepare Pacific Biosciences (PacBio) SMRTbell libraries. Single‐molecule real‐time (SMRT) sequencing was performed on four SMRT cells and run on a PacBio RS II instrument at the Max Planck Genome Center. Genome assemblies of Zt05 and Zt10 based on the generated reads were done as previously described (Plissonneau, Stürchler, & Croll, 2016) using HGAP (Chin et al., 2013) v3.0 included in SMRTanalysis suite v2.3.0. For further details, see Supporting Information Text S1.
Synteny of Z. tritici reference strain IPO323 and the Zt05 and Zt10 unitigs was compared using SyMAP v4.2 (Soderlund, Bomhoff, & Nelson, 2011) applying default settings and considering all unitigs ≥1,000 bp (Zt05) and ≥10,000 bp (Zt10). To estimate the amount of unique DNA in Zt05 and Zt10 in comparison with IPO323 and Zt09, respectively, we generated pairwise genome alignments with Mugsy v1.r2.2 (Angiuoli & Salzberg, 2011) applying default settings. Alignments were analyzed using a custom python script to extract unique DNA blocks with a minimum length of 1 bp.
3. RESULTS
3.1. The three Z. tritici isolates Zt05, Zt09, and Zt10 exhibit high levels of genomic variation
We selected three Z. tritici isolates Zt05, Zt09, and Zt10 that belong to genetically different clades within the species (Grandaubert, Dutheil, & Stukenbrock, 2017) but produce the same amount of disease symptom on the susceptible wheat cultivar Obelisk (see below). These isolates were previously collected in Denmark, the Netherlands, and Iran, respectively (Supporting Information Table S1), and vary in their tolerance to abiotic stressors (Supporting Information Text S1, Figure S3, Table S8). A previous population genomic study using the 39.7 Mb genome of the isolate IPO323 (Goodwin et al., 2011) as reference addressed the extent of genetic variation in these isolates and identified 500,177 single nucleotide polymorphisms (SNPs) in Zt05 and 617,431 SNPs in Zt10, indicating a considerable genetic distance between the three isolates (Grandaubert et al., 2017).
Previous comparative genome analyses have shown that Z. tritici is highly polymorphic at the level of chromosome structure and gene content, including chromosomal rearrangements, genomic orphan regions, and isolate‐specific gene presence/absence patterns (Hartmann et al., 2017; Plissonneau et al., 2016; Plissonneau, Hartmann, & Croll, 2018). To further assess variation in genome structure and content, we performed a karyotype analysis by pulsed‐field gel electrophoresis (PFGE) and generated high‐quality genome assemblies based on long‐read SMRT sequencing of Zt05 and Zt10 (Supporting Information Table S9). The PFGE analyses revealed extensive variation in the karyotypes of the three isolates with no small chromosomes of the same size (Supporting Information Figure S4). The PFGE results suggest that Zt05 and Zt10 possess at least seven and four putative accessory chromosomes, respectively, and show length polymorphisms of the smallest core chromosomes 12 and 13 consistent with variation reported in a previous study (Mehrabi, Taga, & Kema, 2007). In spite of the pronounced differences in genome structure, alignment of the three high‐quality genome assemblies showed a high extent of synteny. By whole‐chromosome synteny analyses using SyMAP, we identified large syntenic DNA blocks for all 21 chromosomes of IPO323 in the Zt05 assembly, while Zt10 lacked homologs of chromosomes 18, 20, and 21 (Supporting Information Figure S5 and Table S9).
To identify the genes that are shared between Zt05, Zt09, and Zt10, we performed nucleotide BLAST analyses using the coding sequences of the 11,839 annotated genes of the reference IPO323 as input (Grandaubert et al., 2015). We identified 11,138 IPO323 genes (94.08%) in Zt05 and 10,745 (90.76%) in Zt10. The gene presence/absence patterns correlate with the absence of large syntenic DNA blocks of chromosomes 18, 20, and 21 in Zt10 (Supporting Information Figure S5B and Table S9). Consequently, 91% of genes on core chromosomes are shared, while only 49% (313 of 643) of the genes located on accessory chromosomes are present in all three isolates (Supporting Information Table S9). Similarly, only 85% (370 of 434) of the previously identified genes encoding candidate secreted effector proteins (CSEPs) (Stukenbrock & Dutheil, 2018) were found in all isolates, indicating that the effector repertoire of Z. tritici is characterized by presence/absence polymorphisms. In total, 10,426 genes were present in the three isolates and considered to be Z. tritici core genes that we included in our further analyses (Supporting Information Table S10). In summary, the genome comparison of Zt05, Zt09, and Zt10 shows a high extent of variation at single nucleotide positions as well as structural variation including differences in the total gene content that is typical in natural isolates of this highly polymorphic pathogen.
3.2. Virulence of the three Z. tritici isolates is very similar but disease develops at different speeds
We compared the virulence phenotypes of Zt05, Zt09, and Zt10 on the highly susceptible wheat cultivar Obelisk and evaluated infections 28 days post inoculation (dpi) by categorizing the percentage of leaf area affected by necrosis (Figure 1a and Supporting Information Figure S6) and covered with pycnidia, the asexual fruiting bodies (Figure 1b and Supporting Information Figure S6). Although we observed different levels of necrosis (two‐sided Mann–Whitney U tests, p ≤ 0.0048), we found no significant differences in the pycnidia levels of the three isolates (two‐sided Mann–Whitney U tests, p ≥ 0.034) (Supporting Information Figure S7). As previously observed (Stewart & McDonald, 2014), quantitative necrosis and pycnidia levels were only weakly linked and coverage of necrotic lesions with pycnidia was highly variable (Supporting Information Figure S1). Hence, for quantitative virulence of Z. tritici, the production of pycnidia is considered the primary measure as it directly reflects reproductive fitness rather than differences in host sensitivity, for example, sensitivity toward pathogen toxins that contribute to necrosis (Stewart & McDonald, 2014). Based on the observed pycnidia levels at 28 dpi, we conclude that the three Z. tritici isolates have similar virulence phenotypes on the wheat cultivar Obelisk.
Figure 1.
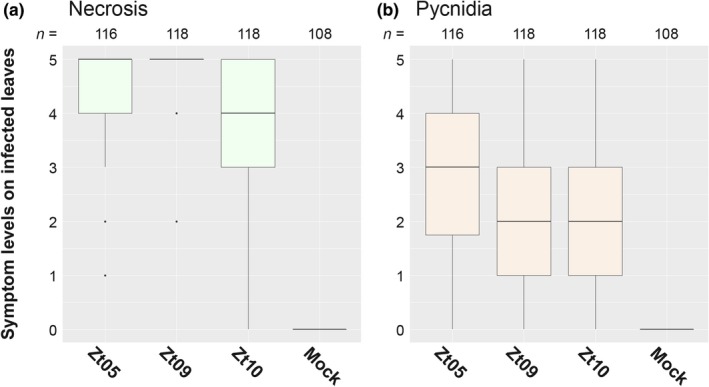
In planta phenotypic assay demonstrates similar pycnidia levels of Zymoseptoria tritici isolates on the susceptible wheat cultivar Obelisk. Quantitative differences in (a) necrosis and (b) pycnidia coverage of inoculated leaf areas were manually assessed at 28 days post inoculation based on six symptom levels: 0 (without visible symptoms), 1 (1% to 20%), 2 (21% to 40%), 3 (41% to 60%), 4 (61% to 80%), and 5 (81% to 100%). The three isolates caused different levels of necrosis (two‐sided Mann–Whitney U tests, p ≤ 0.0048), but pycnidia levels were not different (two‐sided Mann–Whitney U tests, p ≥ 0.034)
Next, we investigated whether disease symptoms develop at a similar pace. We monitored temporal disease progress by screening the leaves every other day for visible necrotic spots and pycnidia. Leaves inoculated with Zt05 and Zt09 showed necrosis and pycnidia significantly earlier than leaves inoculated with Zt10 (one‐sided Mann–Whitney U tests, p ≤ 7.73 × 10−8) (Figure 2a). The median onset of necrosis caused by Zt09 occurred one day after that caused by Zt05 and is significantly later (one‐sided Mann–Whitney U test, p = 0.0089), although the first pycnidia of both isolates developed at the same time (two‐sided Mann–Whitney U test, p = 0.9455). Thus, although the three Z. tritici isolates produced similar quantities of pycnidia in Obelisk, disease develops at different speeds.
Figure 2.
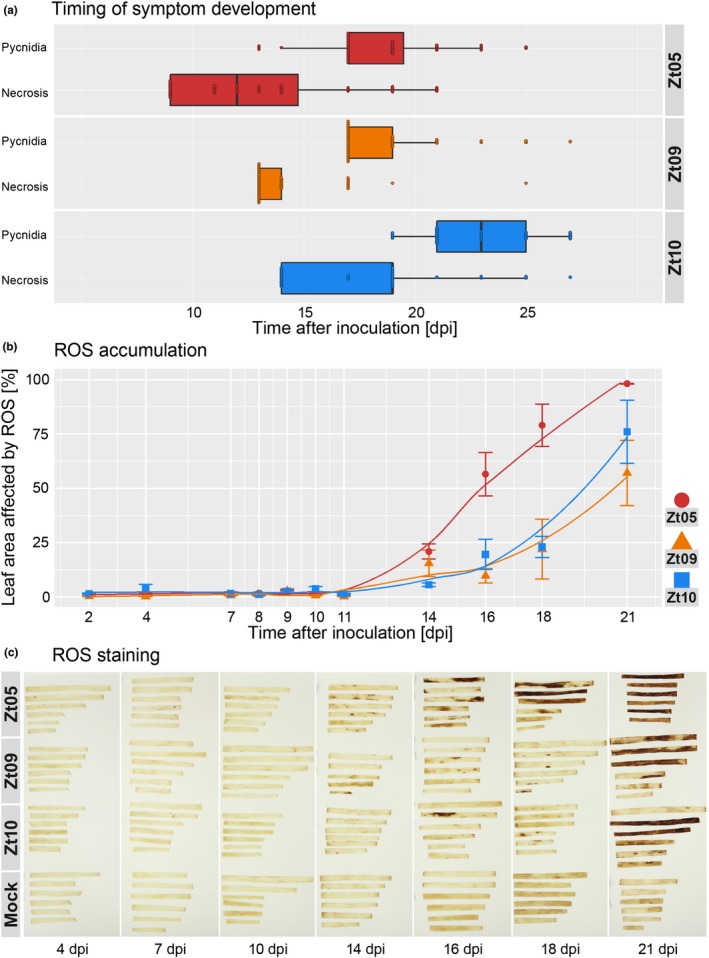
Timing of disease symptom development and H2O2 accumulation varies between wheat leaves infected with different Zymoseptoria tritici isolates. (a) Temporal disease progression for infections with Zt05, Zt09, and Zt10 was measured by manual screening for the first occurrence of necrotic spots and pycnidia. For each isolate, 40 leaves of the wheat cultivar Obelisk were inoculated and tested. No disease developed on seven of the leaves inoculated with Zt10. (b) Reactive oxygen accumulation in infected leaves was visualized by staining with 3,3′‐diaminobenzidine and quantified by analysis of six scanned leaf images per isolate and time point. Mean values of percentage leaf area affected by ROS are plotted. Error bars indicate standard errors of the mean. (c) Dark red‐brown precipitate indicates H2O2 accumulation and appeared first in leaves infected with Zt05 in leaf areas beginning to undergo necrosis
Reactive oxygen species (ROS) play a central role in plant pathogen defense by acting as signaling molecules after pathogen recognition and activating defense responses (Heller & Tudzynski, 2011). We aimed to determine whether the observed differences in temporal disease development of Z. tritici isolates reflect a temporal variation in host response. In some wheat cultivars, a rapid upregulation of defense‐related genes is part of the early responses to Z. tritici infections (Ma, Keller, McDonald, Palma‐Guerrero, & Wicker, 2018; Ray, Anderson, Urmeev, & Goodwin, 2003) while only little transcriptional changes and even the downregulation of such genes were found in the cultivars Riband and Sevin during the asymptomatic phase (Rudd et al., 2015; Yang, Li, & Jørgensen, 2013). Since previous luminol‐based assays for ROS quantification in wheat tissue failed to produce consistent results (Schoonbeek et al., 2015), we visualized the ROS H2O2 by diaminobenzidine (DAB) staining and quantified accumulation by image analysis of the stained leaves. H2O2 is involved in restricting colonization of resistant wheat cultivars but also plays an important role during necrotrophic fungal growth in compatible infections (Shetty et al., 2003, 2007). We observed ROS accumulation coinciding with the onset of necrosis (Figure 2c and Supporting Information Figure S8). However, 11 to 14 days after inoculation, we observed no ROS (Figure 2b,c), possibly because concentrations were low and not detectable. This suggests no or very weak activation of immune responses during this phase of infection in Obelisk. Consistent with the faster disease progress, ROS accumulates earlier in leaves infected with Zt05 than Zt09 and Zt10 (Figure 2b). Thereby, the timing of ROS accumulation in response to the three Z. tritici isolates is consistent with the observed differences in the temporal development of disease symptoms in infected wheat tissue.
3.3. Zymoseptoria tritici infection is characterized by four core developmental stages
Next, we aimed to morphologically characterize host colonization of the three Z. tritici isolates. We conducted detailed confocal microscopy analyses in which we scanned 101 leaves harvested between 3 and 28 days after inoculation (Supporting Information Table S1). Analyses of large z‐stacks of longitudinal optical sections allowed us to infer the spatial and temporal fungal colonization on and in infected leaves. First, we focused on the commonalities in host colonization shared by the isolates. We identified a sequence of four stages that we define as the core infection program of Z. tritici (Figure 3; Supporting Information Text S1).
Figure 3.
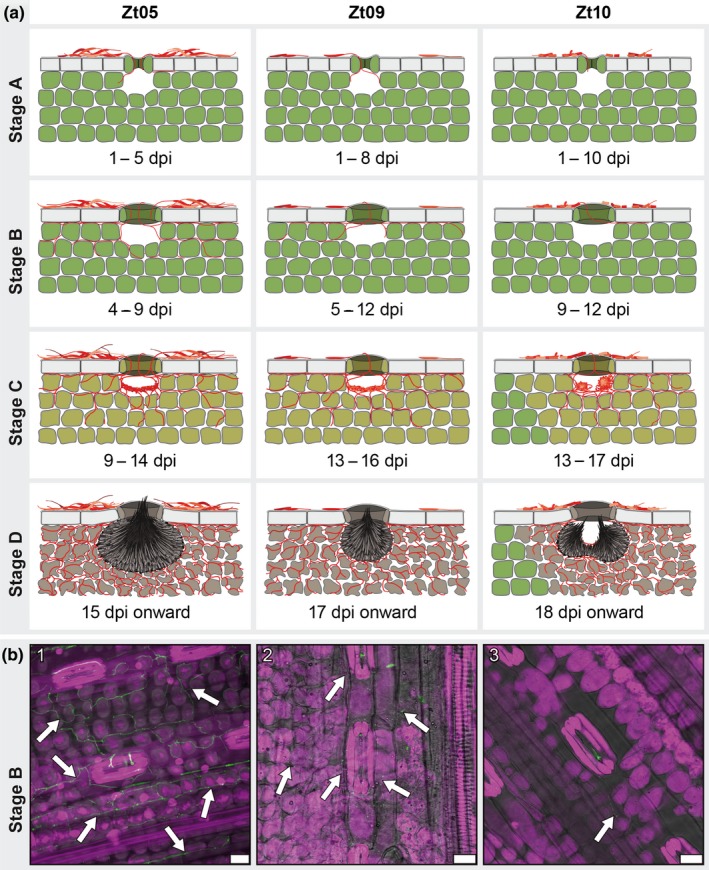
Zymoseptoria tritici wheat infections are characterized by four distinct infection stages and isolate‐specific infection development. (a) Schematic drawings of the key features that characterize the four infection stages of Z. tritici and illustrate the infection phenotypes of isolates Zt05, Zt09, and Zt10 on the wheat cultivar Obelisk. (b) Micrographs showing Z. tritici hyphae (arrows) during biotrophic growth inside wheat leaves. Maximum projections of confocal image z‐stacks. Nuclei and wheat cells are displayed in purple and fungal hyphae or septa in green. The panel shows biotrophic colonization of (1) isolate Zt05 at 7 dpi, (2) Zt09 at 11 dpi, and (3) Zt10 at 9 dpi. Scale bars = 25 µm
Stage A, or infection establishment, involves the penetration of wheat leaf tissue by fungal hyphae. After inoculation, fungal cells germinate on the leaf surface. Germ tubes of Z. tritici develop into infection hyphae, some of which grow in the direction of stomata while others grow epiphytically (Figure 3a stage A; Supporting Information Animations S1 and S2). During stomatal penetration and in the substomatal cavities, infection hyphae grow in tight contact with the guard cells.
Stage B refers to the symptomless, biotrophic intercellular colonization of the wheat mesophyll (Figure 3a stage B; Supporting Information Animations S3 and S4). During this stage, the pathogen explores host tissue without recognition by the host immune system, as indicated by the absence of an accumulation of ROS in infected tissue (Figure 2b,c). Hyphae first grow in the interspace of epidermis and mesophyll, where they spread via the grooves between adjacent epidermal cells before deeper mesophyll cell layers are colonized.
Infection stage C comprises the transition from biotrophic to necrotrophic growth when the first disease symptoms develop (Figure 3a stage C). For asexual reproduction, Z. tritici requires large amounts of nutrients that are released from dead host cells. Fungal hyphae branch extensively and colonize all mesophyll layers, with hyphae growing around individual plant cells as they die off. Primary structures of pycnidia start to develop (Supporting Information Animations S5–S7), and ring‐like scaffolds form in substomatal cavities where hyphae align and build stromata that give rise to conidiogenous cells.
Necrotrophic colonization and asexual reproduction characterize infection stage D (Figure 3a stage D). Hyphae colonize an environment that is nutrient‐rich but also hostile due to highly abundant ROS (Figure 2b). Substomatal cavities are occupied by mature pycnidia that harbor asexual pycnidiospores, which are extruded through stomatal openings. The adjacent mesophyll tissue is heavily colonized by hyphae wrapping around collapsed host cells (Supporting Information Animations S8 and S9), possibly to improve the acquisition of nutrients and protect resources from competing saprotrophs.
Although the four infection stages of Z. tritici can be clearly distinguished, infections by different cells of one isolate are not completely synchronized; different infection stages can be present simultaneously on the same inoculated leaf to some extent.
3.4. Highly differentiated infection phenotypes of the three Z. tritici isolates on Obelisk wheat
While we clearly recognized the four core infection stages for the isolates Zt05, Zt09, and Zt10, we also observed differences (Supporting Information Text S1) that mainly relate to the timing of transitions between the stages and the extent of fungal proliferation. During stage A, infection hyphae of Zt05 enter stomata between 1 and 5 dpi, whereas germ tube formation and stomatal penetration for Zt09 and Zt10 occur later (Figure 3a stage A). We also observed strong epiphyllous growth for Zt05 and, in contrast to the two other isolates, the frequent occurrence of several hyphae entering a single stoma (Figure 3a stage A: Zt05). Stomatal penetrations are evenly distributed for Zt05 and Zt09 in inoculated leaf areas, while Zt10 penetrations are more clustered, leading to patchy infections (Figure 3a stages C, D: Zt10). Zt05 is also the first isolate switching to necrotrophic growth (9 to 14 dpi), followed by Zt09 (13 to 16 dpi) and Zt10 (13 to 17 dpi) (Figure 3a stage C). The isolates enter stage D in the same order. Furthermore, Zt10 forms two pycnidia in one substomatal cavity (Figure 3a stages C and D: Zt10; Supporting Information Animation S7) more often than Zt05 and Zt09.
The most striking difference between the isolates is however the extent of biotrophic colonization during stage B (Figure 3b). Zt05 develops expanded biotrophic hyphal networks with long “runner” hyphae growing longitudinally between epidermis and mesophyll (Figures 3a stage B: Zt05, 3b1; Supporting Information Animation S3). Zt09 produces fewer hyphae that are located mainly between epidermis and mesophyll and in the first mesophyll cell layer (Figures 3a stage B: Zt09, 3b2). Biotrophic growth of Zt10 is strongly reduced and limited to the mesophyll cells adjacent to substomatal cavities (Figures 3a stage B: Zt10, 3b3). Intrafoliar growth by Zt10 beyond that is only found during the lifestyle switch and later. During asymptomatic colonization, Z. tritici depends on successful evasion of host immunity (Jones & Dangl, 2006) and acquires energy initially mainly from stored lipids and only later from host‐derived nutrients (Rudd et al., 2015). Delayed stomatal penetration and reduced internal leaf colonization increase the role of the epiphytic phase where the pathogen is exposed to environmental influences and control measures but could still be transferred to a more favorable host environment (Fones et al., 2017). We speculate that the different extent of fungal growth reflects colonization of different niches associated with the host tissue, deviating strategies to avoid host recognition as well as distinct capabilities to store lipids and exploit the limited nutrient resources during biotrophic colonization.
Taken together, the infection development of the studied Z. tritici isolates is highly divergent, although the final infection outcome is the same (Figure 1). Thereby, instead of one strictly defined infection program, Z. tritici exhibits a variety of host–pathogen interactions that represent equally successful strategies for reproduction in a susceptible wheat cultivar. In particular, we find that infection development of the wheat pathogen can be highly flexible with respect to the timing of the lifestyle transition and the spatial distribution of infecting hyphae inside host tissue.
3.5. Generation of isolate‐ and stage‐specific transcriptomes based on confocal microscopy analyses
Given the morphological and temporal differences in infection development, we next asked how gene expression profiles differ between the Z. tritici isolates Zt05, Zt09, and Zt10 during wheat infection. Previous studies have demonstrated transcriptional reprogramming in Z. tritici during infection (Brunner, Torriani, Croll, Stukenbrock, & McDonald, 2013; Kellner et al., 2014; Keon et al., 2007; Palma‐Guerrero et al., 2016; Rudd et al., 2015; Yang et al., 2013) and shown different transcriptional programs of strains that differ in their virulence phenotypes (Palma‐Guerrero et al., 2017). The transcriptome studies involving different isolates were however limited by the fact that the sequenced samples were collected at predefined time points. Our observations of significant temporal variation in infection development of Z. tritici suggest that predefined sampling time points may be biased and in fact lead to the comparison of transcriptomes of different developmental infection stages.
To account for the temporal variation between Zt05, Zt09, and Zt10, we combined confocal laser scanning microscopy and RNA‐seq and conducted microscopy analyses of tissue from the wheat leaves used for RNA extraction and transcriptome sequencing. Infected leaves were collected at up to nine time points per isolate, and samples for transcriptome sequencing were selected based on their morphological infection stage (Supporting Information Figure S2; Table 1, Supporting Information Table S2). Thereby, we generated stage‐specific RNA‐seq datasets corresponding to the four core infection stages, allowing us to compare the isolate‐specific expression profiles at the same stage of infection development.
We obtained 89.2 to 147.5 million single‐end, strand‐specific reads per replicate (total >2.7 billion reads) that were quality trimmed and filtered. Between 4.54% (early infection) and 76.4% (late infection) of the reads could be mapped to the genome of the respective isolate, reflecting the infection stage‐specific amount of fungal biomass (Table 1, Supporting Information Table S3; Text S1). Across all isolates, transcriptomes of stages A and B, representing biotrophic growth, cluster together and are clearly different from transcriptomes of stages C and D that likewise cluster and represent necrotrophic growth of Z. tritici (Supporting Information Figures S9 and S10). Exploring the transcriptome datasets based on gene read counts shows the greatest variation of biological replicates for Zt10 at stage C (Supporting Information Figure S11), possibly reflecting variability in the infection development of the two biological replicates.
3.6. Core Z. tritici transcriptional program during wheat infection
The mean expression of genes located on accessory chromosomes was 6‐ to 20‐fold lower than the expression levels of genes located on core chromosomes (Supporting Information Table S11). We performed differential gene expression analyses to compare transcription of the 10,426 Z. tritici core genes. We hypothesized that genes involved in stage‐specific infection development and transitions between the four core stages have dynamic, stage‐specific expression profiles that are shared among the three Z. tritici isolates. In total, we identified only 597 genes (5.6%) that fulfilled these two criteria: differential expression between infection stages (DESeq2, p adj ≤0.01, |log2 fold change| ≥2) and shared expression kinetics—and hence represent putative determinants of the Z. tritici core infection stages (Figure 4a). Interestingly, 79 of these genes were differentially expressed between several infection stages, suggesting dynamic, wave‐like expression kinetics (Supporting Information Figure S12).
Figure 4.
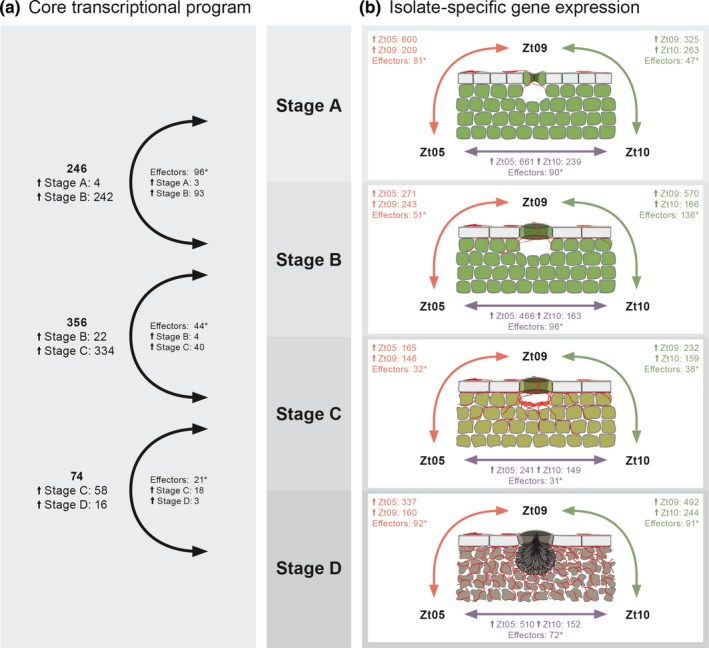
Zymoseptoria tritici core transcriptional program during wheat infection and isolate‐specific expression during the four infection stages. Numbers of significantly differentially expressed genes across all isolates (a) between the four core Z. tritici infection stages and (b) between the isolates within the infection stages (between Zt05 and Zt09: orange arrows, between Zt05 and Zt10: purple arrows, between Zt09 and Zt10: green arrows). Small arrows (↑) with stage or isolate names indicate the number of genes specifically up‐regulated during that stage or in that isolate for the respective comparison. Differential gene expression analyses performed with DESeq2. Genes were considered to be significantly differentially expressed if p adj ≤ 0.01 and |log2 fold change| ≥ 2. *Indicates significant enrichment of effector candidates among differentially expressed genes (Fischer's exact tests, p < 0.001). Effector candidates encode secreted proteins putatively involved in modulating molecular host‐pathogen interactions (Lo Presti et al., 2015)
A total of 246 genes were differentially expressed (Supporting Information Table S12) between stage A and stage B. The vast majority of these (242) were up‐regulated in stage B and are enriched in genes encoding proteins without a functional prediction (146) and with Gene Ontology (GO) groups involved in proteolysis (GO:0006508; 27 genes) and amino acid transmembrane transport (GO:0003333; five genes) (p < 0.01, Fischer's exact test). PFAM domain analysis further shows enrichment of genes encoding cytochrome P450‐like and polyketide synthase‐like proteins that possibly play a role in the production of secondary metabolites (p < 0.001, χ 2 test).
Comparing biotrophic growth and the transition to necrotrophic growth, only 22 genes are up‐regulated in the same manner in the three isolates in stage B while 334 genes are significantly induced during stage C (Supporting Information Table S13). Genes up‐regulated during the necrotrophic stage are enriched with GO groups involved in metabolic processes (GO:0008152; 97 genes), in particular L‐arabinose metabolic processes (GO:0046373; four genes), and transmembrane transport (GO:0055085; 25 genes). Similarly, the PFAM analysis shows an enrichment of genes encoding transporters; CAZymes including different groups of glycosyl hydrolases, serine hydrolases, alpha‐L‐arabinofuranosidases, and cutinases that play important roles as plant tissue and cell wall‐degrading enzymes (Kubicek, Starr, & Glass, 2014); polyketide synthases; and cytochrome P450s. These transcriptional changes reflect the previously described metabolic reprogramming of Z. tritici during the transition from biotrophic to necrotrophic growth (Rudd et al., 2015). Instead of feeding from intracellular lipids, the fungus switches to utilize plant‐derived nutrients (Rudd et al., 2015) and rapidly develops large hyphal networks and the primal structures of the asexual pycnidia in the infected wheat mesophyll tissue.
Between stages C and D, only 74 genes were differentially expressed (Supporting Information Table S14) indicating overall similar transcription profiles during the two necrotrophic stages. Among the 58 genes up‐regulated in stage C, we identified GO groups involved in arabinan metabolic processes (GO:0031221; one gene) and an enrichment of PFAM domains related to cytochrome P450s, polyketide synthases, hydrophobic surface‐binding protein A, tyrosinases, and to beta‐ketoacyl‐ACP synthases, which are known to be involved in fatty acid production and the generation of new cell membrane. Only 16 genes were significantly up‐regulated in all three isolates during the transition from stage C to stage D, which is when the pycnidia mature. These genes are predicted to encode proteins similar to CAZymes, transporters, and proteins containing RNA‐binding domains. Upregulation of a secreted catalase‐like protein‐encoding gene shows the importance of ROS detoxification, which is highly abundant in necrotic leaf tissue as shown by DAB staining (Figure 2c).
In summary, we identified a small number of genes that show the same expression dynamics in the three isolates suggesting that the transitions between the four stages of the core infection program of Z. tritici are facilitated by a comparably small set of core genes. These genes may be involved in determining the stage‐specific infection development and include candidates for core virulence determinants (see below).
3.7. Core biotrophic and necrotrophic effector candidates with shared expression profiles in Z. tritici isolates
Given their importance in plant–pathogen interactions, we particularly focused our analyses on genes encoding candidate secreted effector proteins (CSEPs) (Lo Presti et al., 2015). Zymoseptoria tritici CSEP‐encoding genes were previously predicted (Stukenbrock & Dutheil, 2018) using the machine learning approach EffectorP (Sperschneider et al., 2015) and are significantly enriched among the core differentially expressed genes (p ≤ 2.7 × 10−13, Fischer's exact tests) (Figure 4a), indicating highly dynamic transcription of core effectors during all stages of wheat infection. We filtered the differentially expressed CSEP genes according to their expression profiles (Supporting Information Figures S13 and S14) to identify putative key genes facilitating biotrophic and necrotrophic growth in wheat (Tables 2 and 3).
Table 2.
Zymoseptoria tritici core biotrophic effector candidate genes
| Criteria | Significantly up‐regulated at stage B | |||
| Low/no expression during stage C and D | ||||
| RPKM stage B > RPKM stage C in Zt05 and Zt09 | ||||
| Expression profiles |
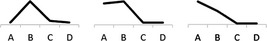
|
|||
| Putative functions | Bypass host recognition during biotrophic colonization | |||
| Candidate genes | 25 (Supporting Information Figure S13) | |||
| Candidates encoding hypothetical proteins | 23 (Supporting Information Table S15) | |||
| Candidates with predicted function | Gene | Function/Functional prediction | PFAM domains | GO terms associated |
| MgNLP (Motteram et al., 2009) Zt09_chr_13_00229 | Necrosis and ethylene‐inducing peptide 1‐like protein MgNLP (Kettles, Bayon, Canning, Rudd, & Kanyuka, 2017; Motteram et al., 2009) | PF05630 | GO:0008150; GO:0003674; GO:0005575 | |
| Zt09_chr_2_00129 | Hypothetical protein, secreted phospholipase A2 precursor | PF06951 | GO:0004623; GO:0005509; GO:0005576; GO:0016042 | |
Summary of core Z. tritici biotrophic effector candidate genes that were identified based on their specific expression profiles within the Z. tritici core transcriptional program during wheat infection. Functional annotation, PFAM, and GO term information from Grandaubert et al. (2015).
Table 3.
Zymoseptoria tritici core necrotrophic effector candidate genes
| Criteria | Significantly up‐regulated at stage C | |||
| Low/no expression during stage A and B | ||||
| Expression profiles |
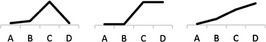
|
|||
| Putative functions | Facilitate transition from biotrophy to necrotrophy | |||
| Induction of necrosis | ||||
| Candidate genes | 35 (Supporting Information Figure S14) | |||
| Candidates encoding hypothetical proteins | 24 (Supporting Information Table S16) | |||
| Candidates with predicted function | Gene | Functional prediction | PFAM domains | GO terms associated |
| Zt09_chr_3_00584 | PCWDE, similar to alpha‐1, Glycosyl transferases group 1 | PF00128; PF00534; PF08323 | GO:0003824; GO:0043169; GO:0005975; GO:0009058 | |
| Zt09_chr_9_00308 | PCWDE, similar to carbohydrate‐binding module family 63 protein, expansin‐like | — | GO:0008150; GO:0003674; GO:0005575 | |
| Zt09_chr_3_01063 | PCWDE, similar to pectate lyase | PF00544 | GO:0008150; GO:0003674; GO:0005575 | |
| Zt09_chr_12_00112 | cutinase‐like protein | PF01083 | GO:0050525; GO:0016787; GO:0005576; GO:0008152 | |
| Zt09_chr_2_00663 | similar to Chain A, cutinase‐like protein | PF01083 | GO:0050525; GO:0016787; GO:0005576; GO:0008152 | |
| Zt09_chr_2_01151 | PCWDE, similar to acetyl xylan esterase, | PF01083 | GO:0016787; GO:0008152 | |
| Zt09_chr_6_00446 | PCWDE,similar to acetyl xylan esterase | PF01083 | GO:0016787; GO:0008152 | |
| Zt09_chr_10_00107 | PCWDE, similar to glycoside hydrolase family 12 protein | PF01670 | GO:0008810; GO:0004553; GO:0000272 | |
| Zt09_chr_2_01205 | PCWDE, similar to putative extracellular cellulase CelA/allergen Asp F7‐like protein | PF03330 | GO:0008150; GO:0003674; GO:0005575 | |
| Zt09_chr_6_00626 | similar to metalloprotease | PF05572 | GO:0008237 | |
| Zt09_chr_9_00038 | similar to hydrophobin | PF06766 | GO:0005576 | |
PCWDE: putative plant cell wall‐degrading enzyme.
Summary of core Z. tritici necrotrophic effector candidate genes that were identified based on their specific expression profiles within the Z. tritici core transcriptional program during wheat infection. Functional annotation, PFAM, and GO term information from Grandaubert et al. (2015).
During symptomless stage B, 78 CSEP genes are specifically up‐regulated, highlighting that a large suite of Z. tritici effectors is induced after stomatal penetration and is required for biotrophic colonization of wheat mesophyll. By filtering the up‐regulated genes for lower expression during stage C (RKPM stage B > RPKM stage C in Zt05 and Zt09), we narrowed down these genes to 25 core biotrophic CSEP genes (Table 2, Supporting Information Table [Link], [Link]; Figure S13) that mostly encode hypothetical proteins. In comparison with Zt05 and Zt09, expression of the biotrophic effectors in Zt10 is in general lower, possibly reflecting the strongly limited biotrophic colonization of this isolate (Figure 3b).
Thirty five CSEP genes are specifically up‐regulated at stage C (Supporting Information Figure S14) and represent candidates for necrotrophic core effectors (Table 3, Supporting Information Table S16). These genes may be involved in the transition from biotrophic to necrotrophic growth and the induction of necrosis. Nine CSEP genes encode putative plant cell wall‐degrading enzymes and cutinase‐like proteins, demonstrating that the lifestyle switch to necrotrophy involves intensified degradation of plant tissue and cell wall components. The gene Zt09_chr_9_00038 encodes a putative hydrophobin; hydrophobins are small fungal‐specific proteins with various functions (Aimanianda et al., 2009; Wösten, 2001), i.a. as toxins in plant–pathogen interactions (Takai, 1974). Likewise strongly induced during the transition to necrotrophy is the gene Zt09_chr_7_00263 that encodes a putative secreted metalloprotease, which are known fungal virulence factors in animal and plant pathogens (Karimi Jashni et al., 2015; Naumann, Wicklow, & Price, 2011; O'Connell et al., 2012; Vu et al., 2014).
In summary, our comparative approach allowed us to identify a set of core Z. tritici effector candidates, which are consistently expressed during infection of the cultivar Obelisk in the different genetic background of Zt05, Zt09, and Zt10.
3.8. Isolate‐specific transcriptional changes during wheat infection
The 597 genes that we identified as differentially expressed between the stages show the same expression profile in each of the three isolates and we consider them as part of the core Z. tritici transcriptional infection program. However, we observed that transcript levels of many genes strongly deviate between the isolates during the specific infection stages.
To further study how the infection phenotypes of the Z. tritici isolates relate to differences in gene expression, we compared expression profiles during the infection stages (Figure 4b). In total, 2,377 (~22.8%) of the 10,426 shared genes are differentially expressed between the Z. tritici isolates during wheat infection (Table 4, Supporting Information Tables S17, S19) reflecting the extent of spatial, temporal, and quantitative differences in development that we observe by confocal microscopy (Figure 3a). A similar genome portion is differentially expressed in Swiss isolates during infection of the cultivar Drifter (Palma‐Guerrero et al., 2017) indicating a high extent of functional redundancy and flexibility in the infection program of Z. tritici.
Table 4.
Genes with isolate‐specific expression profiles during wheat infection
| Genes differentially expressed between isolatesa | 2,377 genes in total | ||
| 22.8% of all 10,426 core genes | |||
| Comparison (all four stages) | DE genes b | Regulation | |
| Zt05‐Zt09 | 1,311 | ↑Zt05: 917 ↑Zt09: 436 | |
| Zt05‐Zt10 | 1,482 | ↑Zt05: 1086 ↑Zt10: 412 | |
| Zt09‐Zt10 | 1,514 | ↑Zt09: 1062 ↑Zt10: 541 | |
| GO groups enrichedc | GO term | DE genes in GO group | GO ID |
| Transmembrane transport | 159 (of 491) | GO:0055085 | |
| Carbohydrate metabolic process | 82 (of 242) | GO:0005975 | |
| Proteolysis | 64 (of 237) | GO:0006508 | |
| Amino acid transmembrane transport | 17 (of 36) | GO:0003333 | |
| Oxidation–reduction process | 159 (of 627) | GO:0055114 | |
| Lipid catabolic process | 6 (of 15) | GO:0016042 | |
| Candidate secreted effector genes | 245 of 370 effector candidate genes | ||
| Comparison (all four stages) | DE effector genes d | Regulation | |
| Zt05‐Zt09 | 161 | ↑Zt05: 120 ↑Zt09: 50 | |
| Zt05‐Zt10 | 167 | ↑Zt05: 141 ↑Zt10: 31 | |
| Zt09‐Zt10 | 193 | ↑Zt09: 167 ↑Zt10: 60 | |
DE: differentially expressed. ↑: significantly up‐regulated in the isolate.
Summary of genes that are differentially expressed between the three Z. tritici isolates Zt05, Zt09, and Zt10 during the four infection stages.
Differentially expressed genes identified by DESeq2, p adj ≤0.01, |log2 fold change| ≥2.
774 genes are differentially expressed in at least two isolate comparisons.
Gene Ontology (GO) group enrichment analyses by topGO for ontology “Biological Process,” p ≤ 0.01.
198 effector candidate genes are differentially expressed in at least two isolate comparisons.
For all isolate comparisons, the identified differentially expressed genes are significantly enriched in CSEP genes (p ≤ 1.18 × 10−7, Fischer's exact tests) (Figure 4b) indicating isolate‐specific effector expression (Supporting Information Table S19). Figure 5 exemplifies the isolate‐specific expression kinetics of five CSEP genes. These five genes encode one hypothetical effector (Zt09_chr_12_00427) and secreted proteins with various functions: a hydrophobin (Zt09_chr_9_00020) also shown previously to be differentially expressed between Swiss field isolates (Palma‐Guerrero et al., 2017), a DNase (Zt09_chr_2_01162), and the ribonuclease Zt6 (Zt09_chr_3_00610), which possesses ribotoxin‐like activity and is cytotoxic against plants and various microbes (Kettles, Bayon, Sparks, et al., 2017). The gene Zt09_chr_4_00039 encodes a protein with homology to the phytotoxin cerato‐platanin of Ceratocystis fimbriata which was shown to induce necrosis and defense responses in plane trees (Pazzagli et al., 1999). During all infection stages, Zt09_chr_4_00039 is significantly higher expressed in Zt09 and might contribute to the higher necrosis levels caused by Zt09 (Figure 1a).
Figure 5.
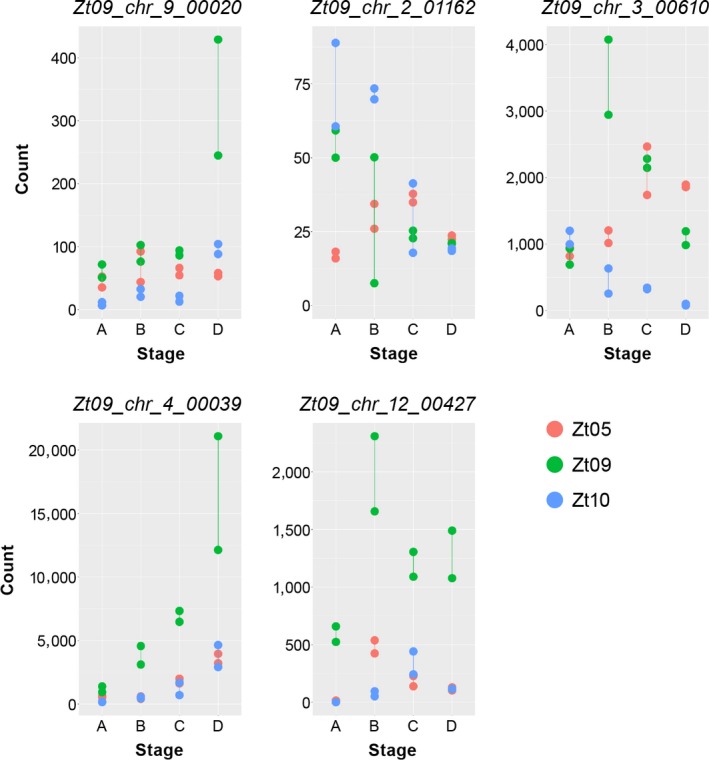
Five effector candidates with highly differentiated expression profiles in the three isolates during wheat infection. The plots display normalized read counts for five effector candidate genes for the twelve RNA‐seq datasets. Read counts were normalized across the four core infection stages (A to D) and the three Z. tritici isolates Zt05, Zt09, and Zt10 by applying the regularized log transformation (rlog) function of DESeq2 (Love et al., 2014) and represent a measure of relative gene expression between the infection stages and between the isolates
In addition to the differences in the expression of CSEP genes, we also noted isolate‐specific expression patterns for genes located on accessory chromosomes. For example, three neighboring genes located on chromosome 19 in Zt09 (Zt09_chr_19_00071 to Zt09_chr_19_00073) are significantly higher expressed in Zt10 during all four infection stages (Supporting Information Figure S15; Table S18), and other genes located ~70 kb downstream are likewise specifically up‐regulated in Zt10 compared to one or both of the other isolates. We consider the possibility that the genomic environment, including the presence of transposable elements, may influence the observed variation in transcription activity of an entire region. To address this hypothesis, we investigated the specific region in the genome alignment of the three isolates. In Zt10, these genes are located on unitig 16, which is syntenic to the accessory chromosome 19 of Zt09. Transposable elements (RLG elements) are located downstream of Zt09_chr_19_00072 and upstream of Zt09_chr_19_00073 in Zt09, but are not present within the vicinity of these genes on unitig 16 in Zt10. Instead, the right arm of unitig 16 of Zt10 (~100 Mb) is inverted compared to Zt09 and Zt05, and the inversion starts upstream of the gene Zt09_chr_19_00073, reflecting a significant change in sequence structure that might influence transcriptional regulation on this accessory chromosome.
3.9. Transposable elements are associated with the differentially expressed genes
Detailed analyses of the Z. tritici transcriptomes revealed considerable variation in the transcriptional landscapes among isolates. The extent of genetic differentiation between Zt05, Zt09, and Zt10 likely accounts for much of the transcriptional variation in the form of SNPs in regulatory sequences. However, other layers of gene regulation may also contribute to the heterogeneous transcriptional landscapes. We hypothesized that epigenetic transcriptional regulation, such as co‐regulation of sequences associated with transposable elements, could impact gene expression variation. In a previous study, we showed that transposable elements and the accessory chromosomes of Z. tritici are enriched with the histone modifications H3K9me3 and H3K27me3, which are associated with repressive regions of chromatin (Schotanus et al., 2015). In Fusarium graminearum, the histone modification H3K27me3 is associated with gene clusters encoding secondary metabolites and pathogenicity‐related traits (Connolly, Smith, & Freitag, 2013). It is possible that variation in the distribution of histone modifications like H3K27me3 across the genome sequences of Zt05, Zt09, and Zt10 contributes to the dramatic variation in expression phenotypes.
To test this, we assessed the distances of all genes to the closest annotated transposable element. In the genomes of all three isolates, we found that isolate‐specific differentially expressed genes are located significantly closer to transposable elements than genes that were not differentially expressed (Mann–Whitney U tests, p < 2.2 × 10−16). Although more than 50% of these differentially expressed genes are located downstream of the closest transposable element, we did not observe an overall enrichment of TEs in their putative promotor regions 2 kb upstream (Supporting Information Table S20). Among the differentially expressed genes, isolate‐specific up‐regulated genes (genes that are significantly up‐regulated in one isolate in contrast to the others) are significantly enriched within a distance of 2 kb to transposable elements in Zt05 and Zt09 (Fisher's exact tests, p ≤ 0.0094), but not in Zt10. We note, however, that the transposable element annotation in Zt10 is not as complete as in Zt05 and Zt09 as the annotation was based on the Illumina short read assembly which is 6.7 Mb smaller than the de novo genome assembly of SMRT Sequencing reads. We also analyzed the in vitro histone 3 methylation data for Zt09 (Schotanus et al., 2015) and found that differentially expressed genes are indeed significantly closer to H3K9me3 and H3K27me3 peaks (Mann–Whitney U tests, p < 2.2 × 10−16). Further, we observed significant enrichment of genes up‐regulated in Zt09 in comparison with Zt05 and Zt10 within a distance of 2 kb to H3K9me3 and H3K27me3 peaks (Fisher's exact tests, p ≤ 1.55 × 10−15), but down‐regulated genes were only enriched in the vicinity of H3K27me3 peaks (distance ≤2 kb, Fisher's exact test, p = 1.35 × 10−15). Poor transcription of genes located on the accessory chromosomes was explained by enrichment of H3K27me3 covering the entire chromosomes and H3K9me3, which is mostly associated with repetitive DNA (Schotanus et al., 2015). Our findings indicate that during host infection, chromatin state of repeat‐rich genome compartments is highly dynamic and changes between “active” euchromatin and “repressive” heterochromatin, as suggested in Leptosphaeria maculans (Soyer et al., 2014). Further, our observation suggests that the fine‐scale distribution of epigenetic marks likely differs between the genomes of Z. tritici isolates and contributes to the isolate‐specific gene expression phenotypes that we observed. To further visualize the transcriptional landscape across the three Z. tritici genomes, we calculated expression values (RPKM) of 1‐kb windows (Supporting Information Table S11) and plotted them in heatmaps along the chromosomes and unitigs (Supporting Information Figures S16–S18). This approach allows visualization of the transcriptional landscapes at a high resolution. Thereby, we identified heterogeneous gene expression patterns across chromosomes, such as on chromosome 19 in Zt10 (Supporting Information Figure S18) and demonstrated conservation of previously identified patterns, such as transcriptional silencing of the right arm of chromosome 7 (Kellner et al., 2014; Rudd et al., 2015) in Zt05, Zt09, and Zt10 (Supporting Information Figure S17). This chromosomal segment has characteristics of an accessory chromosome, as it is significantly enriched with H3K27me3 that mediates transcriptional silencing (Schotanus et al., 2015). While syntenic chromosomal regions generally have a similar composition of transcribed and silenced loci, the fine‐scale distribution of transcriptional cold and hot spots is clearly different between the genomes of the three isolates studied here.
4. DISCUSSION
Many plant pathogens have a narrow host range and are specialized to colonize and reproduce in a small number of host plant species or host genotypes (Haueisen & Stukenbrock, 2016; van der Does & Rep, 2017). By combining advanced microscopy and comparative transcriptome analyses, we addressed the consequences of host specialization in the specialized wheat pathogen Z. tritici. We specifically asked whether one particular virulence phenotype results from one specific infection program. Our infection experiments and transcriptome data analyses reveal an unexpectedly high extent of plasticity in the infection program. The three studied Z. tritici isolates differ significantly in their genomic composition, and we show that the genetic variation and isolate‐specific transcriptional reprogramming during the different stages of host infection translate into highly distinct phenotypes that deviate temporally, spatially, and morphologically. Gene expression profiles associated with host colonization show a high degree of variability: More than 20% of the core genes are differentially expressed between these three Z. tritici isolates during the four infection stages indicating different feeding and molecular host interaction strategies. Candidates for effectors and putatively phytotoxic and antimicrobial molecules are enriched among the differentially expressed genes, suggesting that the three isolates employ different molecular strategies to interact with wheat and its associated microbiome. Effectors with cytotoxic activity have been proposed to shape the host environment by manipulating host and the associated microbiome in favor of the pathogen (Snelders, Kettles, Rudd, & Thomma, 2018). The three isolates show different abilities to grow epi‐ and endophytically during the symptomless stages of infection and consequently are differentially exposed to biotic and abiotic influences, such as host responses, microbial competitors, or pathogen control treatments. Strikingly, the highly variable infection programs result in the same level of virulence, showing that “host specialization” in Z. tritici involves a very flexible strategy to exploit wheat tissue for growth and reproduction. This flexibility may be facilitated by the fact that pathogenesis does not involve the formation of complex penetration or feeding structures that require fine‐tuned developmental programs. In powdery mildew which need to form appressoria as well as haustoria to establish host infections, comparative transcriptomics during early pathogenesis identified a comparatively low number of isolate‐specifically expressed genes in two barley powdery mildew strains (Hacquard et al., 2013) and showed that transcriptional programs are similar even during incompatible infections and interactions with nonhost species (Hacquard et al., 2013; Hu et al., 2018).
As necrotic lesions are usually composed of several distinct Z. tritici genotypes (Linde et al., 2002), it would be highly relevant to investigate whether strains in one lesion have similar or different infection phenotypes. Isolates with different infection strategies colonizing the same leaf could complement each other or have antagonistic effects. Future multi‐isolate studies must however consider possible developmental asynchrony during infection and should focus on isolates showing similar temporal infection development. Our findings likewise underline the importance to consider not only quantitative infection outcomes but also qualitative aspects, like the timing of disease progress over an appropriate period of time, to be able to compare the infections of different pathogen isolates and detect meaningful differences in their infection phenotypes (Habig et al., 2017; Meile et al., 2018).
An intriguing question that emerges from our analyses is which factors cause deviation in gene expression phenotypes in Z. tritici. Genetic variants associated with transcriptional regulation likely contribute to differences in gene regulation. However, we hypothesize that variation in epigenetic traits promotes different transcriptional programs. Genome‐wide patterns of transcriptional activity (Supporting Information Figures S16–S18) indeed suggest some variation in the physical distribution of transcriptionally active and silent regions, which may result from distinct epigenetic landscapes related to histone modifications or DNA methylation.
The capacity of Z. tritici populations to harbor and maintain a broad diversity of infection programs might facilitate flexibility and dynamic responses to changes in their host environment, for example, fungicide treatments during the season or the introduction of new host genotypes. Furthermore, the significant variation in expression of virulence factors such as effectors, secondary metabolites, and plant cell wall‐degrading enzymes suggests that different individuals of Z. tritici employ different molecular strategies to interact with the same host environment. We hypothesize that highly diverging infection phenotypes are not exclusive among isolates of Z. tritici and are likely found in populations of other pathogens that retain high levels of genetic diversity. Variation in infection and expression profiles contributes another layer of polymorphism in pathogen populations that has so far been neglected in most systems. Many studies identifying virulence factors focus on the molecular interactions between reference genotypes—one pathogen isolate and one host genotype. Our work challenges the relevance of these specific interactions in the field on population scales and emphasizes the importance to consider the diversity of pathogen infection programs to improve our understanding of pathogen evolution and the ecology of plant pathogens.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTION
Conceptualization: JH and EHS. Wheat infection experiments and in vitro stress assay: JH. In planta ROS assay: JH and HS. Karyotyping and synteny analyses: JH and MM. Confocal microscopy analyses and data visualization: JH and HA. Genome sequencing and de novo genome assemblies: MM and CJH. Transcriptome sequencing, data curation, and comparative transcriptome analyses: JH. Gene and TE annotations, software for enrichment analyses: JG. Preparation and writing of manuscript: JH and EHS. Editing of original manuscript: JH, EHS, MM, and HS.
DATA ACCESSIBILITY
All generated RNA‐seq datasets have been deposited at the NCBI Gene Expression Omnibus and are accessible with the accession number GSE106136. De novo genome assemblies of isolates Zt05 and Zt10 are available under accession numbers PEBP00000000 and PEBO00000000. The genome sequence of the reference isolate IPO323 used for transcriptome analysis of Zt09 is available at: http://genome.jgi.doe.gov/Mycgr3/Mycgr3.home.html. Genome assemblies based on whole‐genome shotgun sequencing (Illumina) were also used; the assembly for Zt10 is available at GenBank (Zt10 = STIR04_A26b) GCA_000223645.2. Sequencing data and assembly for Zt05 (Zt05 = MgDk09_U34) are available through NCBI BioProject PRJNA312067 (Grandaubert et al., 2017).
Supporting information
ACKNOWLEDGMENTS
We thank Ronny Kellner for insightful comments to a previous version of this manuscript, Julien Y. Dutheil for support with comparative transcriptome analyses, and Petra Happel for help with the in vitro stress assays. This work was supported by intramural funding of the Max Planck Society and a personal grant from the State of Schleswig‐Holstein to Eva H. Stukenbrock. The funders had no role in study design, data collection and analyses, decision to publish, or preparation of the manuscript.
Haueisen J, Möller M, Eschenbrenner CJ, et al. Highly flexible infection programs in a specialized wheat pathogen. Ecol Evol. 2019;9:275–294. 10.1002/ece3.4724
REFERENCES
- Aimanianda, V. , Bayry, J. , Bozza, S. , Kniemeyer, O. , Perruccio, K. , Elluru, S. R. , … Latgé, J. P. (2009). Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature, 460, 1117–1121. [DOI] [PubMed] [Google Scholar]
- Alexa, A. , Rahnenführer, J. , & Lengauer, T. (2006). Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics, 22, 1600–1607. 10.1093/bioinformatics/btl140 [DOI] [PubMed] [Google Scholar]
- Allen, G. C. , Flores‐Vergara, M. A. , Krasynanski, S. , Kumar, S. , & Thompson, W. F. (2006). A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. NatureProtocols, 1, 2320–2325. 10.1038/nprot.2006.384 [DOI] [PubMed] [Google Scholar]
- Anders, S. , Pyl, P. T. , & Huber, W. (2015). HTSeq—A Python framework to work with high‐throughput sequencing data. Bioinformatics, 31, 166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angiuoli, S. V. , & Salzberg, S. L. (2011). Mugsy: Fast multiple alignment of closely related whole genomes. Bioinformatics, 27, 334–342. 10.1093/bioinformatics/btq665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner, P. C. , Torriani, S. F. F. , Croll, D. , Stukenbrock, E. H. , & McDonald, B. A. (2013). Coevolution and life cycle specialization of plant cell wall degrading enzymes in a hemibiotrophic pathogen. Molecular Biology and Evolution, 30, 1337–1347. 10.1093/molbev/mst041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, C.‐S. , Alexander, D. H. , Marks, P. , Klammer, A. A. , Drake, J. , Heiner, C. , … Korlach, J. (2013). Nonhybrid, finished microbial genome assemblies from long‐read SMRT sequencing data. Nature Methods, 10, 563–569. [DOI] [PubMed] [Google Scholar]
- Connolly, L. R. , Smith, K. M. , & Freitag, M. (2013). The Fusarium graminearum Histone H3 K27 methyltransferase KMT6 regulates development and expression of secondary metabolite gene clusters. PLoS Genetics, 9(10), e1003916 10.1371/journal.pgen.1003916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds, P. N. , & Rathjen, J. P. (2010). Plant immunity: Towards an integrated view of plant–pathogen interactions. Nature Reviews Genetics, 11, 539–548. 10.1038/nrg2812 [DOI] [PubMed] [Google Scholar]
- Fisher, M. C. , Hawkins, N. J. , Sanglard, D. , & Gurr, S. J. (2018). Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science, 742, 739–742. 10.1126/science.aap7999 [DOI] [PubMed] [Google Scholar]
- Fones, H. N. , Eyles, C. J. , Kay, W. , Cowper, J. , & Gurr, S. J. (2017). A role for random, humidity‐dependent epiphytic growth prior to invasion of wheat by Zymoseptoria tritici . Fungal Genetics and Biology, 106, 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin, S. B. , Ben M'Barek, S. , Dhillon, B. , Wittenberg, A. H. J. , Crane, C. F. , Hane, J. K. , … Kema, G. H. J. (2011). Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genetics, 7, e1002070 10.1371/journal.pgen.1002070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandaubert, J. , Bhattacharyya, A. , & Stukenbrock, E. H. (2015). RNA‐seq based gene annotation and comparative genomics of four fungal grass pathogens in the genus zymoseptoria identify novel orphan genes and species‐specific invasions of transposable elements. G3 (Bethesda, Md.), 5, 1323–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandaubert, J. , Dutheil, J. Y. , & Stukenbrock, E. H. (2017). The genomic determinants of adaptive evolution in a fungal pathogen. bioRxiv. 10.1101/176727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habig, M. , Quade, J. , & Stukenbrock, E. H. (2017). Forward genetics approach reveals host‐genotype dependent importance of accessory chromosomes in the fungal wheat pathogen Zymoseptoria tritici . Mbio, 8, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacquard, S. , Kracher, B. , Maekawa, T. , Vernaldi, S. , Schulze‐Lefert, P. , Loren, V. , & van Themaat, E. (2013). Mosaic genome structure of the barley powdery mildew pathogen and conservation of transcriptional programs in divergent hosts. Proceedings of the National Academy of Sciences of the United States of America, 110, E2219–E2228. 10.1073/pnas.1306807110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann, F. E. , Sánchez‐Vallet, A. , McDonald, B. A. , & Croll, D. (2017). A fungal wheat pathogen evolved host specialization by extensive chromosomal rearrangements. The ISME Journal, 1189–1204. 10.1038/ismej.2016.196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haueisen, J. , & Stukenbrock, E. H. (2016). Life cycle specialization of filamentous pathogens ‐ colonization and reproduction in plant tissues. Current Opinion in Microbiology, 32, 31–37. 10.1016/j.mib.2016.04.015 [DOI] [PubMed] [Google Scholar]
- Heller, J. , & Tudzynski, P. (2011). Reactive oxygen species in phytopathogenic fungi: Signaling, development, and disease. Annual Review of Phytopathology, 49(49), 369–390. [DOI] [PubMed] [Google Scholar]
- Hu, Y. , Liang, Y. , Zhang, M. , Tan, F. , Zhong, S. , Li, X. , … Luo, P. (2018). Comparative transcriptome profiling of Blumeria graminis f. sp. tritici during compatible and incompatible interactions with sister wheat lines carrying and lacking Pm40. PLoS ONE, 13, 1–19. 10.1371/journal.pone.0198891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, J. D. G. , & Dangl, J. L. (2006). The plant immune system. Nature, 444, 323–329. 10.1038/nature05286 [DOI] [PubMed] [Google Scholar]
- Karimi Jashni, M. , Dols, I. H. M. , Iida, Y. , Boeren, S. , Beenen, H. G. , Mehabi, R. , … De Wit, P. J. G. M. (2015). Synergistic action of serine‐ and metallo‐proteases from Fusarium oxysporum f. sp. lycopersici cleaves chitin‐binding tomato chitinases, reduces their antifungal activity and enhances fungal virulence. Molecular Plant‐Microbe Interactions, 28, 996–1008. [DOI] [PubMed] [Google Scholar]
- Kellner, R. , Bhattacharyya, A. , Poppe, S. , Hsu, T. Y. , Brem, R. B. , & Stukenbrock, E. H. (2014). Expression profiling of the wheat pathogen Zymoseptoria tritici reveals genomic patterns of transcription and host‐specific regulatory programs. GenomeBiology and Evolution, 6, 1353–1365. 10.1093/gbe/evu101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keon, J. , Antoniw, J. , Carzaniga, R. , Deller, S. , Ward, J. L. , Baker, J. M. , … Rudd, J. J. (2007). Transcriptional adaptation of Mycosphaerella graminicola to programmed cell death (PCD) of its susceptible wheat host. Molecular Plant‐Microbe Interactions, 20, 178–193. [DOI] [PubMed] [Google Scholar]
- Kettles, G. J. , Bayon, C. , Canning, G. , Rudd, J. J. , & Kanyuka, K. (2017). Apoplastic recognition of multiple candidate effectors from the wheat pathogen Zymoseptoria tritici in the nonhost plant Nicotiana benthamiana. New Phytologist, 213, 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettles, G. , Bayon, C. , Sparks, C. A. , Canning, G. , Kanyuka, K. , & Rudd, J. J. (2017). Characterisation of an antimicrobial and phytotoxic ribonuclease secreted by the fungal wheat pathogen Zymoseptoria tritici . New Phytologist, 44, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Pertea, G. , Trapnell, C. , Pimentel, H. , Kelley, R. , & Salzberg, S. L. (2013). TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. GenomeBiology, 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicek, C. P. , Starr, T. L. , & Glass, N. L. (2014). Plant cell wall‐degrading enzymes and their secretion in plant‐pathogenic fungi. Annual Review of Phytopathology, 52, 427–451. 10.1146/annurev-phyto-102313-045831 [DOI] [PubMed] [Google Scholar]
- Lendenmann, M. H. , Croll, D. , & McDonald, B. A. (2015). QTL mapping of fungicide sensitivity reveals novel genes and pleiotropy with melanization in the pathogen Zymoseptoria tritici . Fungal Genetics and Biology, 80, 53–67. 10.1016/j.fgb.2015.05.001 [DOI] [PubMed] [Google Scholar]
- Lendenmann, M. H. , Croll, D. , Stewart, E. L. , & McDonald, B. A. (2014). Quantitative trait locus mapping of melanization in the plant pathogenic fungus Zymoseptoria tritici . G3 (Bethesda, Md.), 4, 2519–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , … Durbin, R. (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linde, C. C. , Zhan, J. , & McDonald, B. A. (2002). Population structure of Mycosphaerella graminicola: From lesions to continents. Phytopathology, 92, 946–955. [DOI] [PubMed] [Google Scholar]
- Lo Presti, L. , Lanver, D. , Schweizer, G. , Tanaka, S. , Liang, L. , Tollot, M. , … Kahmann, R. (2015). Fungal effectors and plant susceptibility. Annual Review of Plant Biology, 66, 513–545. 10.1146/annurev-arplant-043014-114623 [DOI] [PubMed] [Google Scholar]
- Love, M. I. , Huber, W. , & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, X. , Keller, B. , McDonald, B. A. , Palma‐Guerrero, J. , & Wicker, T. (2018). Comparative transcriptomics reveals how wheat responds to infection by Zymoseptoria tritici . Molecular Plant‐Microbe Interactions, 31, 420–431. [DOI] [PubMed] [Google Scholar]
- Macho, A. P. , Schwessinger, B. , Ntoukakis, V. , Brutus, A. , Segonzac, C. , Roy, S. , … Zipfel, C. (2014). A bacterial tyrosine phosphatase inhibits plant pattern recognition receptor activation. Science, 343, 1509–1512. 10.1126/science.1248849 [DOI] [PubMed] [Google Scholar]
- McDonald, M. C. , McGinness, L. , Hane, J. K. , Williams, A. H. , Milgate, A. , & Solomon, P. S. (2016). Utilizing gene tree variation to identify candidate effector genes in Zymoseptoria tritici . G3 (Bethesda, Md.), 6: 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrabi, R. , Taga, M. , & Kema, G. H. J. (2007). Electrophoretic and cytological karyotyping of the foliar wheat pathogen Mycosphaerella graminicola reveals many chromosomes with a large size range. Mycologia, 99, 868–876. [DOI] [PubMed] [Google Scholar]
- Meile, L. , Croll, D. , Brunner, P. C. , Plissonneau, C. , Hartmann, F. E. , Mcdonald, B. A. , & Sanchez‐Vallet, A. (2018). A fungal avirulence factor encoded in a highly plastic genomic region triggers partial resistance to septoria tritici blotch. New Phytologist, 219(3), 1048–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadi Gohari, A. , Ware, S. B. , Wittenberg, A. H. J. , Mehrabi, R. , Ben M'Barek, S. , Verstappen, E. C. P. , … Kema, G. H. (2015). Effector discovery in the fungal wheat pathogen Zymoseptoria tritici . Molecular Plant Pathology, 16, 931–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller, M. , & Stukenbrock, E. H. (2017). Evolution and genome architecture in fungal plant pathogens. Nature Reviews Microbiology, 15, 756–771. 10.1038/nrmicro.2017.76 [DOI] [PubMed] [Google Scholar]
- Motteram, J. , Küfner, I. , Deller, S. , Brunner, F. , Hammond‐Kosack, K. E. , Nürnberger, T. , & Rudd, J. J. (2009). Molecular characterization and functional analysis of MgNLP, the sole NPP1 domain‐containing protein, from the fungal wheat leaf pathogen Mycosphaerella graminicola . Molecular Plant‐Microbe Interactions, 22, 790–799. [DOI] [PubMed] [Google Scholar]
- Naumann, T. A. , Wicklow, D. T. , & Price, N. P. J. (2011). Identification of a chitinase‐modifying protein from Fusarium verticillioides: Truncation of a host resistance protein by a fungalysin metalloprotease. Journal of Biological Chemistry, 286, 35358–35366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell, R. J. , Thon, M. R. , Hacquard, S. , Amyotte, S. G. , Kleemann, J. , Torres, M. F. , … Vaillancourt, L. J. (2012). Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nature Genetics, 44, 1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma‐Guerrero, J. , Ma, X. , Torriani, S. F. F. , Zala, M. , Francisco, C. S. , Hartmann, F. E. , … McDonald, B. A. (2017). Comparative transcriptome analyses in Zymoseptoria tritici reveal significant differences in gene expression among strains during plant infection. Molecular Plant‐Microbe Interactions, 30, 231–244. [DOI] [PubMed] [Google Scholar]
- Palma‐Guerrero, J. , Torriani, S. F. F. , Zala, M. , Carter, D. , Courbot, M. , Rudd, J. J. , … Croll, D. (2016). Comparative transcriptomic analyses of Zymoseptoria tritici strains show complex lifestyle transitions and intraspecific variability in transcription profiles. Molecular Plant Pathology, 17, 845–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazzagli, L. , Cappugi, G. , Manao, G. , Camici, G. , Santini, A. , & Scala, A. (1999). Purification, characterization, and amino acid sequence of cerato‐ platanin, a new phytotoxic protein from Ceratocystis fimbriata f. sp. platani. Journal of Biological Chemistry, 274, 24959–24964. [DOI] [PubMed] [Google Scholar]
- Plissonneau, C. , Hartmann, F. E. , & Croll, D. (2018). Pangenome analyses of the wheat pathogen Zymoseptoria tritici reveal the structural basis of a highly plastic eukaryotic genome. BMC Biology, 16, 1–16. 10.1186/s12915-017-0457-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plissonneau, C. , Stürchler, A. , & Croll, D. (2016). The evolution of orphan regions in genomes of a fungal pathogen of wheat. Mbio, 7(5), e01231‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarenko, A. , Goodwin, S. B. , & Kema, G. H. J. (2011). Septoria tritici blotch (STB) of wheat. Plant Health Instructor, 1–7. [Google Scholar]
- Poppe, S. , Dorsheimer, L. , Happel, P. , & Stukenbrock, E. H. (2015). rapidly evolving genes are key players in host specialization and virulence of the fungal wheat pathogen Zymoseptoria tritici (Mycosphaerella graminicola). PLOS Pathogens, 11, e1005055 10.1371/journal.ppat.1005055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, A. R. , & Hall, I. M. (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2017). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Ray, S. , Anderson, J. M. , Urmeev, F. I. , & Goodwin, S. B. (2003). Rapid induction of a protein disulfide isomerase and defense‐related genes in wheat in response to the hemibiotrophic fungal pathogen Mycosphaerella graminicola . Plant Molecular Biology, 53, 701–714. 10.1023/B:PLAN.0000019120.74610.52 [DOI] [PubMed] [Google Scholar]
- Rudd, J. J. , Kanyuka, K. , Hassani‐Pak, K. , Derbyshire, M. , Andongabo, A. , Devonshire, J. , … Courbot, M. (2015). Transcriptome and metabolite profiling of the infection cycle of Zymoseptoria tritici on wheat reveals a biphasic interaction with plant immunity involving differential pathogen chromosomal contributions and a variation on the hemibiotrophic lifestyle definition. Plant Physiology, 167, 1158–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez‐Vallet, A. , McDonald, M. C. , Solomon, P. S. , & McDonald, B. A. (2015). Is Zymoseptoria tritici a hemibiotroph? Fungal Genetics and Biology, 79, 29–32. 10.1016/j.fgb.2015.04.001 [DOI] [PubMed] [Google Scholar]
- Schoonbeek, H. , Wang, H. , Stefanato, F. L. , Craze, M. , Bowden, S. , Wallington, E. , … Ridout, C. J. (2015). Arabidopsis EF‐Tu receptor enhances bacterial disease resistance in transgenic wheat. New Phytologist, 206, 606–613. 10.1111/nph.13356 [DOI] [PubMed] [Google Scholar]
- Schotanus, K. , Soyer, J. L. , Connolly, L. R. , Grandaubert, J. , Happel, P. , Smith, K. M. , … Stukenbrock, E. H. (2015). Histone modifications rather than the novel regional centromeres of Zymoseptoria tritici distinguish core and accessory chromosomes. Epigenetics & Chromatin, 8, 41 10.1186/s13072-015-0033-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurch, N. J. , Schofield, P. , Gierliński, M. , Cole, C. , Sherstnev, A. , Singh, V. , … Barton, G. J. (2016). How many biological replicates are needed in an RNA‐seq experiment and which differential expression tool should you use? RNA, 22, 839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty, N. P. , Kristensen, B. K. , Newmana, M. A. , Møller, K. , Gregersen, P. L. , & Jørgensen, H. J. L. (2003). Association of hydrogen peroxide with restriction of Septoria tritici in resistant wheat. Physiological and Molecular Plant Pathology, 62, 333–346. 10.1016/S0885-5765(03)00079-1 [DOI] [Google Scholar]
- Shetty, N. P. , Mehrabi, R. , Lütken, H. , Haldrup, A. , Kema, G. H. J. , Collinge, D. B. , & Jørgensen, H. J. L. (2007). Role of hydrogen peroxide during the interaction between the hemibiotrophic fungal pathogen Septoria tritici and wheat. New Phytologist, 174, 637–647. 10.1111/j.1469-8137.2007.02026.x [DOI] [PubMed] [Google Scholar]
- Snelders, N. C. , Kettles, G. J. , Rudd, J. J. , & Thomma, B. P. H. J. (2018). Plant pathogen effector proteins as manipulators of host microbiomes? Molecular Plant Pathology, 19, 257–259. 10.1111/mpp.12628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderlund, C. , Bomhoff, M. , & Nelson, W. M. (2011). SyMAP v3. 4: A turnkey synteny system with application to plant genomes. Nucleic Acids Research, 39 10.1093/nar/gkr123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyer, J. L. , El Ghalid, M. , Glaser, N. , Ollivier, B. , Linglin, J. , Grandaubert, J. , … Fudal, I. (2014). Epigenetic control of effector gene expression in the plant pathogenic fungus Leptosphaeria maculans . PLoS Genetics, 10, e1004227 10.1371/journal.pgen.1004227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperschneider, J. , Gardiner, D. M. , Dodds, P. N. , Tini, F. , Covarelli, L. , Singh, K. B. , … Taylor, J. M. (2015). EffectorP: Predicting fungal effector proteins from secretomes using machine learning. New Phytologist, 210, 743–761. [DOI] [PubMed] [Google Scholar]
- Stewart, E. L. , & McDonald, B. A. (2014). Measuring quantitative virulence in the wheat pathogen Zymoseptoria tritici using high‐throughput automated image analysis. Phytopathology, 104, 985–992. [DOI] [PubMed] [Google Scholar]
- Stewart, E. L. , Croll, D. , Lendenmann, M. H. , Sanchez‐Vallet, A. , Hartmann, F. E. , Palma‐Guerrero, J. , … McDonald, B. A. (2017). QTL mapping reveals complex genetic architecture of quantitative virulence in the wheat pathogen Zymoseptoria tritici . Molecular Plant Pathology, 19, 201–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukenbrock, E. H. , Banke, S. , Javan‐Nikkhah, M. , & McDonald, B. A. (2007). Origin and domestication of the fungal wheat pathogen Mycosphaerella graminicola via sympatric speciation. Molecular Biology and Evolution, 24, 398–411. [DOI] [PubMed] [Google Scholar]
- Stukenbrock, E. H. , & Dutheil, J. Y. (2018). Fine‐scale recombination maps of fungal plant pathogens reveal dynamic recombination landscapes and intragenic hotspots. Genetics, 208, 1209–1229. 10.1534/genetics.117.300502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukenbrock, E. H. , Jørgensen, F. G. , Zala, M. , Hansen, T. T. , McDonald, B. A. , & Schierup, M. H. (2010). Whole‐genome and chromosome evolution associated with host adaptation and speciation of the wheat pathogen Mycosphaerella graminicola. PLoS Genetics, 6, e1001189 10.1371/journal.pgen.1001189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai, S. (1974). Pathogenicity and cerato‐ulmin production in Ceratocystis ulmi . Nature, 252, 124–126. 10.1038/252124a0 [DOI] [PubMed] [Google Scholar]
- Tanaka, S. , Brefort, T. , Neidig, N. , Djamei, A. , Kahnt, J. , Vermerris, W. , … Kahmann, R. (2014). A secreted Ustilago maydis effector promotes virulence by targeting anthocyanin biosynthesis in maize. Elife, 3, 1–27. 10.7554/eLife.01355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thordal‐Christensen, H. , Zhang, Z. , Wei, Y. , & Collinge, D. B. (1997). Subcellular localization of H2O2 in plants. H2O2 accumulation in papillae and hypersensitive response during the barley‐powdery mildew interaction. Plant Journal, 11, 1187–1194. 10.1046/j.1365-313X.1997.11061187.x [DOI] [Google Scholar]
- Thygesen, K. , Jørgensen, L. N. , Jensen, K. S. , & Munk, L. (2008). Spatial and temporal impact of fungicide spray strategies on fungicide sensitivity of Mycosphaerella graminicola in winter wheat. European Journal of Plant Pathology, 123, 435–447. 10.1007/s10658-008-9381-3 [DOI] [Google Scholar]
- Trapnell, C. , Hendrickson, D. G. , Sauvageau, M. , Goff, L. , Rinn, J. L. , & Pachter, L. (2013). Differential analysis of gene regulation at transcript resolution with RNA‐seq. Nature Biotechnology, 31, 46–53. 10.1038/nbt.2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Does, H. C. , & Rep, M. (2017). Adaptation to the host environment by plant‐pathogenic fungi. Annual Review of Phytopathology, 55, 427–450. 10.1146/annurev-phyto-080516-035551 [DOI] [PubMed] [Google Scholar]
- Vu, K. , Tham, R. , Uhrig, J. P. , Thompson, G. R. , Pombejra, S. N. , Jamklang, M. , … Gelli, A. (2014). Invasion of the central nervous system by cryptococcus neoformans requires a secreted fungal Metalloprotease. Mbio, 5, 1–13. 10.1128/mBio.01101-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenberg, A. H. J. , van der Lee, T. A. J. , Ben M'Barek, S. , Ware, S. B. , Goodwin, S. B. , Kilian, A. , … Schouten, H. J. (2009). Meiosis drives extraordinary genome plasticity in the haploid fungal plant pathogen Mycosphaerella graminicola. PLoS ONE, 4, e5863 10.1371/journal.pone.0005863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wösten, H. A. (2001). Hydrophobins: Multipurpose proteins. Annual Review of Microbiology, 55, 625–646. 10.1146/annurev.micro.55.1.625 [DOI] [PubMed] [Google Scholar]
- Yang, F. , Li, W. , & Jørgensen, H. J. L. (2013). Transcriptional reprogramming of wheat and the hemibiotrophic pathogen septoria tritici during two phases of the compatible interaction (Y‐H Lee, Ed.). PLoS ONE, 8, e81606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilinger, S. , Gupta, V. K. , Dahms, T. E. S. , Silva, R. N. , Singh, H. B. , Upadhyay, R. S. , … Nayak, S. C. (2015). Friends or foes? Emerging insights from fungal interactions with plants. FEMS Microbiology Reviews, 40(2), 182–207. 10.1093/femsre/fuv045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan, J. , Pettway, R. E. , & McDonald, B. A. (2003). The global genetic structure of the wheat pathogen Mycosphaerella graminicola is characterized by high nuclear diversity, low mitochondrial diversity, regular recombination, and gene flow. Fungal Genetics and Biology, 38, 286–297. 10.1016/S1087-1845(02)00538-8 [DOI] [PubMed] [Google Scholar]
- Zhong, Z. , Marcel, T. C. , Hartmann, F. E. , Ma, X. , Plissonneau, C. , Zala, M. , … Palma‐Guerrero, J. (2017). A small secreted protein in Zymoseptoria tritici is responsible for avirulence on wheat cultivars carrying the Stb6 resistance gene. New Phytologist, 214, 619–631. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All generated RNA‐seq datasets have been deposited at the NCBI Gene Expression Omnibus and are accessible with the accession number GSE106136. De novo genome assemblies of isolates Zt05 and Zt10 are available under accession numbers PEBP00000000 and PEBO00000000. The genome sequence of the reference isolate IPO323 used for transcriptome analysis of Zt09 is available at: http://genome.jgi.doe.gov/Mycgr3/Mycgr3.home.html. Genome assemblies based on whole‐genome shotgun sequencing (Illumina) were also used; the assembly for Zt10 is available at GenBank (Zt10 = STIR04_A26b) GCA_000223645.2. Sequencing data and assembly for Zt05 (Zt05 = MgDk09_U34) are available through NCBI BioProject PRJNA312067 (Grandaubert et al., 2017).