

Abstract
Lead discovery in osteosarcoma has been hampered by the lack of new agents, limited representative clinical samples and paucity of accurate preclinical models. We developed orthotopic patient-derived xenografts (PDXs) that recapitulated the molecular, cellular and histologic features of primary tumors, and screened PDX-expanded short-term cultures and commercial cell lines of osteosarcoma against focused drug libraries. Osteosarcoma cells were most sensitive to HDAC, proteasome, and combination PI3K/MEK and PI3K/mTOR inhibitors, and least sensitive to PARP, RAF, ERK and MEK inhibitors. Correspondingly, PI3K signaling pathway genes were up-regulated in metastatic tumors compared to primary tumors. In combinatorial screens, as a class, HDAC inhibitors showed additive effects when combined with standard-of-care agents gemcitabine and doxorubicin. This lead discovery strategy afforded a means to perform high-throughput drug screens of tumor cells that accurately recapitulated those from original human tumors, and identified classes of novel and repurposed drugs with activity against osteosarcoma.
1. Introduction
Osteosarcoma is the most common bony malignancy among children and adolescents. Survival outcomes remain poor for patients with metastatic disease at diagnosis1, with 5-year survival rates of only 30%, compared to 70% for those with localized disease2,3. Approximately 30–40% of patients with localized disease at diagnosis will develop local or distant recurrences4 – 90% of which occur in the lung. Thus, improved treatment options are needed, especially for the metastatic form of this disease.
Chemotherapeutic agents used in current standard-of-care protocols have remained largely unchanged in the last 20–30 years, with methotrexate, doxorubicin, and cisplatin commonly used upfront5, and gemcitabine and docetaxel for relapse or refractory disease. Patients with greater than 90% chemotherapy-induced necrosis of the primary tumor at resection have improved outcomes compared to those with less necrosis6,7. These findings have prompted the intensification of adjuvant chemotherapy regimens in an attempt to increase survival outcomes, but without clear improvements in response thus far3,8,9.
Genomic analyses have identified molecular markers with prognostic significance, such as p53 deficiency, which occurs in virtually all osteosarcomas10,11. However these have not been successfully translated into effective therapeutic strategies12. For example, increased levels of HER2 expression predicts a poor response to chemotherapy and decreased overall survival, but a Children’s Oncology Group (COG) trial of trastuzumab with standard chemotherapy failed to show a benefit in children with metastatic osteosarcoma13. Similarly, IGF-1 has been implicated in the tumorigenic and metastatic potential of osteosarcoma14,15, but the use of a somatostatin analog to antagonize growth hormone signaling also failed to show a significant clinical benefit16, thus leading to interest in other isoforms of the IGF-1 receptor instead17. Future efforts to develop molecular targeted therapy for osteosarcoma may benefit from basic and translational research to provide scientific justification and translational relevance before moving into clinical trials18.
Major barriers to providing data in support of new trials are paucity of fresh patient tumor material for live-cell analysis, and preclinical models that faithfully recapitulate the tumors at primary and metastatic sites and at recurrence. We developed a method for the generation of orthotopic patient derived xenograft (PDX) models of osteosarcoma. We demonstrate how the models recapitulate the molecular, cellular and histologic characterization of primary tumors, but are limited in modeling the metastatic phenotype in-vivo. We thus used the PDX platform instead to expand tumor cell populations for combinatorial drug screening, as a means to identify novel therapeutic combinations.
2. Materials and methods
2.1. Tumor samples
Osteosarcoma samples were obtained from diagnostic biopsies or therapeutic resections of human patients at St Jude Children’s Research Hospital. The xenografts SJOS013, SJOS001107_D, SJOS001107_M, and SJOS001105 were gifts from Dr. Andrew Davidoff (St Jude Children’s Research Hospital, Memphis, TN). Xenograft tumors were harvested and disaggregated by mechanical and chemical means. Tumor pieces were suspended in 100 mL Dulbecco’s phosphate buffered saline (without calcium and magnesium, DPBS) with 2 mg/ml Type 2 collagenase (Worthington cat LS004176) and Trypsin 0.06–0.012X, and agitated with a bead stirrer at 200r.p.m. for 90 minutes at 37°C. Trypsinization was stopped using soybean trypsin inhibitor, and DNAse, and 1M magnesium chloride were added. The resulting slurry was strained, centrifuged and red blood cells were lysed. The final cell pellet was resuspended in DPBS with 10% fetal bovine serum (FBS; Biomet cat. S01520).
2.2. Cell lines
Three human osteosarcoma cell lines were used: U2OS (ATCC HTB-96), and SaOS2 (ATCC HTB-85), and LM7, which was a gift from Dr. Eugenie Kleinerman (Children’s Cancer Hospital, the University of Texas MD Anderson Cancer Center, Houston, TX). The LM7 cell line was created by passaging 106 SaOS2 cells through lungs of athymic mice 7 times19,20. U2OS was grown on McCoy’s5A (ATCC cat. 30–2007) with 10% FBS and 1% penicillin-streptomycin (Gibco cat. 10378–016), at 37°C in 5% CO2. SaOS2 was grown in similar media and growth conditions except with 15% FBS. LM7 was grown on DMEM (Lonza cat. 12–614F), with 10% FBS, 1mM non-essential amino acids (Gibco cat. 11140–050), 1mM sodium pyruvate (Gibco cat. 11360–070), 2mM penicillin-streptomycin with glutamine (Gibco cat. 10378–016), and 2X MEM vitamin solution (Gibco cat. 11120–052).
2.3. Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee at St Jude Children’s Research Hospital (IACUC protocols 393 and 455). All animals were fed Purina Rodent Lab Diet #5013 (LabDiet, St. Louis, MO) ad libitum and housed in a 12–12 light cycle in accordance with approved IACUC protocols. CD-1 immunodeficient nude mice homozygous for the Foxn1nu mutation (stock number 002019), and in NOD scid gamma mice (NSG, stock number 005557) were purchased from Jackson Laboratories.
2.4. Drug screening
Tumors were dissociated into single cell suspensions (Day 0). Cells were plated in 384 well dishes in DMEM and drugs added in triplicate dose response (10 concentrations) using a pin tool 24 hours later (Day 1). 72 hours later, CellTiter-Glo was added and ATP levels quantitated on a plate reader (Day 4). Positive and negative control compounds were included in each experiment for data normalization. Validated data was entered into a central database and curve fitting was performed to generate an EC50 for each drug.
Primary single agent cytoxicity screens were performed on curated drug libraries from the Chemical Biology and Therapeutics department of St Jude Children’s Research Hospital (http://edrugs.stjude.org), containing 373 drugs in 432 formulations. Compounds from all libraries were dissolved in DMSO as stock solutions, except cisplatin which was dissolved in aqueous buffer and plated fresh with each assay. Using a 384 pintool, 70nL of compound was transferred into each 25uL well of the 384-well assay plate. Relative light units (RLUs) were determined 72 hours after drug delivery. Secondary curveshift drug screening was performed by combining standard-of-care agents with drug classes shortlisted from the primary drug screen, in order to look for additive and more-than-additive effects. Differences in the areas under dose-response curves of test agents with and without addition of standard-of-care agents were compared and ranked. Dose-response curves were derived by comparing the relative activity to the negative control agent as reference. Combinations that produced positive increases in the AUC and an EC50 of <10uM were defined as ‘hits’ in the curveshift experiments. Combinations were deemed additive or synergistic when the net ΔAUC was equal to or greater than the sum of individual AUCs, respectively.
Tertiary synergy drug screening was performed by testing ‘hit’ combinations with graduated concentrations of each drug applied in X- and Y- axes of 384-well assay plates. A response surface method, the Bivariate Response to Additive Interacting Doses (BRAID) analysis, was used to model the magnitude of combined action between two drugs, as previously described21.
3. Results
3.1. Orthotopic osteosarcoma PDXs recapitulate gross and microscopic features of primary limb tumors
Through the Childhood Solid Tumor Network (CSTN)22, primary and metastatic osteosarcoma tumors (from pulmonary and extra-pulmonary sites) were obtained from patients at diagnosis and recurrence (Table S1). Tumor pieces (5mm3 or 0.1mg) were initially implanted into the subcutaneous space on the flanks of NSG mice. Resultant tumors were processed into single cell suspensions, resuspended in Matrigel™ and injected into the bone marrow to establish orthotopic tumor models (Fig. 1A). Immediately following injection, tumor cells remained suspended within the medullary cavity as a localized bolus (Fig. 1B, C). Within 2–3 months, gross tumors formed with both soft tissue and bony components similar to patient tumors and to those found in an osteosarcoma genetically engineered mouse model (GEMM) induced by inactivation of Rb and p53 in the osteoblastic lineage23.The orthotopic tumors displayed characteristic gross and radiographic features including sclerosis, cortical disruption and soft tissue extension, mirroring the phenotype of primary human and GEMM tumors (Fig. 1D–L).
FIGURE 1: Orthotopic osteosarcoma tumors recapitulate radiographic and gross disease phenotype.
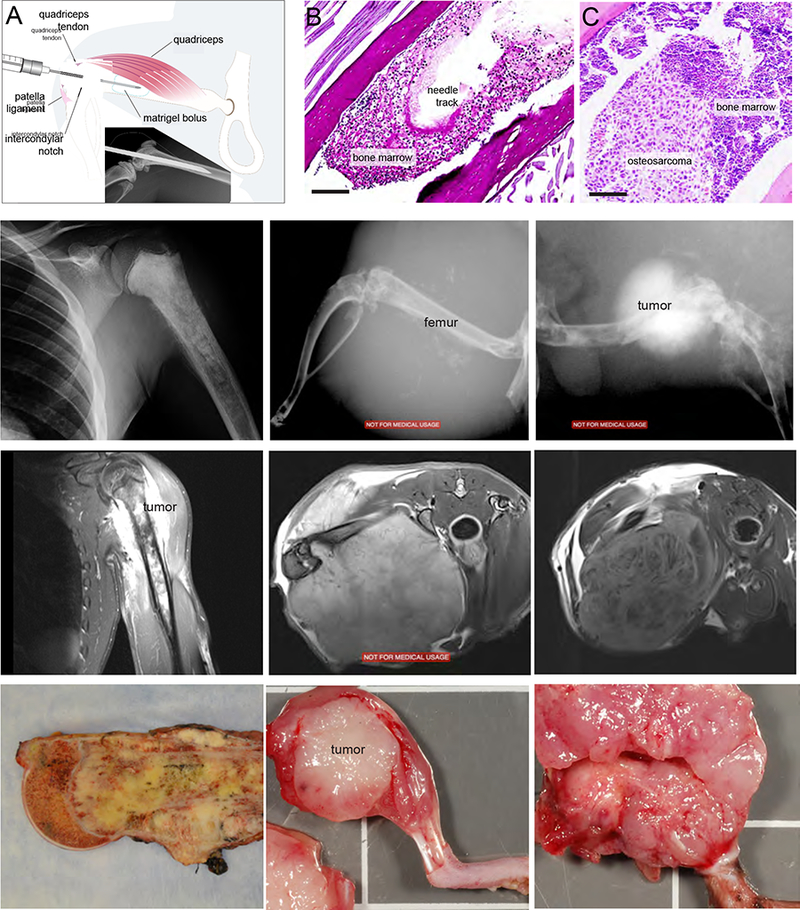
(A) Method of injection of Matrigel-tumor cell suspension into bone marrow cavity and radiograph of mouse femur at the time of injection (inset). (B-C) Photomicrographs of Matrigel bolus in bone marrow cavity at 1 minute post-injection (Periodic Acid-Schiff (PAS), 20X), and the resulting tumor (PAS, 4X). Representative X-rays (D-F), MRIs (G-I) and gross images (J-L) of primary tumors from patient, the corresponding xenograft, and a genetic mouse model of osteosarcoma, respectively.
Tumors also displayed characteristic histologic features, including deposition of osteoid matrix, malignant cartilage and hyalinized collagen, cellular pleomorphism, multinucleated osteoclast-like giant cells, and multipolar mitotic figures (Fig. 2A). We also scored 11 ultracellular features in a blinded manner for osteosarcoma tumors from patients, PDXs and GEMMs (Table S2). All PDX tumors displayed dilated rough endoplasmic reticulum and villous cell surfaces – unique characteristics involved in facilitation of extracellular matrix secretion (Fig. 2B, Fig. S1). To compare the gene-expression profiles, we performed RNA sequencing on 9 patient-PDX tumor pairs and calculated correlation coefficients from the log2-transformed values of fragments per kilobase of transcript per million mapped reads (FPKM) for each pair. Patient-PDX tumor pairs showed strong correlation with median coefficients of 0.92 (range: 0.79–0.96) (Fig. 2C).
FIGURE 2: Osteosarcoma xenograft tumors recapitulate the histological and molecular features of patient tumors.
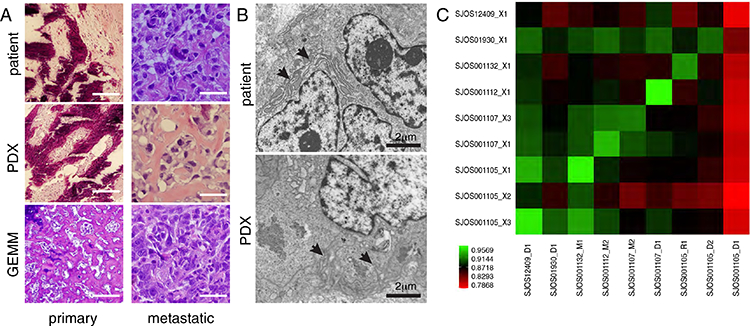
(A) Representative H&E sections from a primary and a metastatic patient tumor and corresponding xenografts (SJOS001112 and SJOS001121, respectively), and primary and metastatic tumors from a GEMM of osteosarcoma. (B) Representative transmission electron micrographs of a PDX from a metastatic patient tumor and corresponding xenograft (SJOS001112 and SJOS001112_X1, respectively) showing abundant dilated rough endoplasmic reticulum (arrows), cytoplasmic collagen, scalloped cell surface, and large and poorly developed golgi apparatus (5000X). (C) Correlational matrix of RNA-seq data for 9 patient tumor and xenograft pairs (Spearman coefficient, Fragments per kilobase of transcript per million mapped reads).
3.2. Osteosarcoma PDXs demonstrate moderate capacity for lung metastases
We studied the metastatic phenotype of our orthotopic PDX model, particularly since pulmonary metastasis is the primary cause of death in patients with osteosarcoma. As the bone marrow is a highly vascular compartment, we hypothesized that bone marrow injection could also lead to seeding of the lung. To test this, we euthanized mice within 1–2 minutes after a bone marrow injection of osteosarcoma cells suspended in Matrigel™ and performed histopathologic analysis of the lungs, and compared them with animals with tail vein injections. Emboli containing tumor cells in matrigel were readily detected in the lungs of mice receiving bone marrow or tail vein injections (Fig. 3A–D, Fig. S2). To determine if those lung emboli could go on to form lung nodules with histopathologic features of patient metastases, 1×106 cells each from a metastatic tumor PDX (SJOS001107), primary tumor PDX (SJOS001121), GEMM tumor and a commercial cell line (SaOS2) were resuspended in Matrigel™ and injected into the bone marrow of a pair of CD-1 nude mice, and tail veins of another pair of mice. When the primary tumor reached 20% of the body weight of the mice with bone marrow injections, both groups were euthanized and lungs were harvested for histopathological examination and quantification. At median 81 days after injection, pulmonary nodules formed in 7 of 8 (87.5%) mice following bone marrow injection (Fig. 3E) and 4 of 8 mice (50%) following tail vein injections (Fig. 3F). However, in the PDX and cell line xenografts, the appearance of pulmonary metastases on conventional X-ray imaging, CT-scan and histology were less robust compared to spontaneous metastases seen in patients and GEMMs (Fig. S3). The majority of mice in all cohorts had evidence of lung metastases on histopathological examination (11/16), but only half of these could be detected using X-ray or CT imaging (5/16). As time for observation for lung metastases was limited by the growth of the primary tumor, we surgically removed the tumor-bearing limb in a further pair of PDX and observed the animals up to 6 months further after surgery (Fig. S4), but did not observe a significant increase in number of nodules after this additional time. Taken together the data suggested that the orthtopic PDXs also recapitulated the latent pulmonary metastatic disease phenotype.
FIGURE 3: Orthotopic bone marrow injection of osteosarcoma in vivo recapitulates metastatic human disease phenotype.
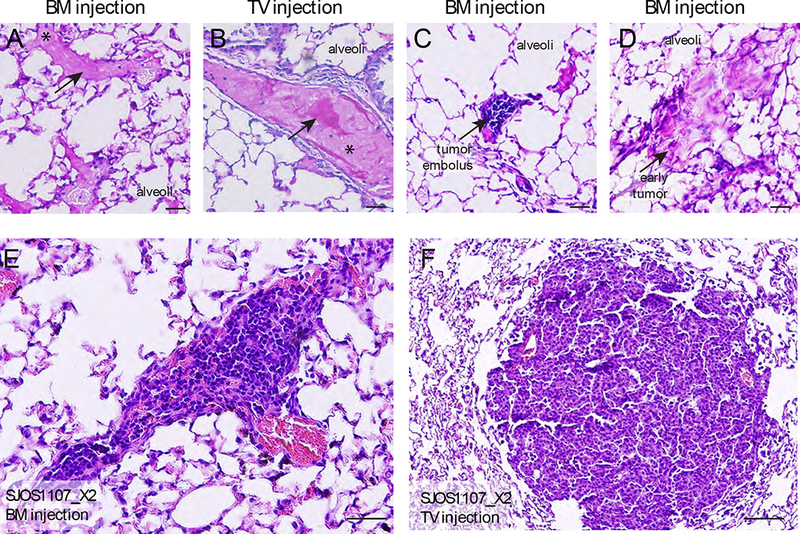
(A-B) Pulmonary dissemination of matrigel following bone marrow and tail vein injection at 1 minute post-injection (PAS, 20X). (C) Histologic section of lung tissue immediately following bone marrow injection, demonstrating embolus of osteosarcoma tumor cells suspended in Matrigel, with surrounding platelets (PAS, 20X). (D) Small pulmonary foci of osteosarcoma at 2 months following bone marrow injection (PAS, 20X). (E-J) Representative gross photographs and H&E sections of lungs with pulmonary metastases in nude mice injected with osteosarcoma cells via the bone marrow cavity, and tail vein.
3.3. High throughput screening of osteosarcoma PDX cells
The orthotopic PDX model also afforded a means to rapidly expand the original cell population within its original micro-environment. In order to identify new drug combinations for treatment of metastatic or recurrent osteosarcoma, we tested 372 drugs in 432 formulations on 4 short-term cultures of PDX-expanded osteosarcoma cells and 3 commerical cell lines using a high throughput drug screen in-vitro (Fig. 4A, Table S3). Validated data was entered into a central database and curve fitting was performed to generate an EC50 for each drug (Fig. 4B, C). Overall, commercial cell lines showed more exaggerated responses to chemotherapy compared to PDX cells, with U2OS and SaOS2 clustering separately with lower log EC50 (uM) values compared to LM7 and PDXs. Single-agent drug screen data is available through the Childhood Solid Tumor Network (https://www.stjude.org/research/resources-data/childhood-solid-tumor-network/available-resources/data.html).
FIGURE 4: High-throughput drug screen identifies classes with and without efficacy against osteosarcoma.
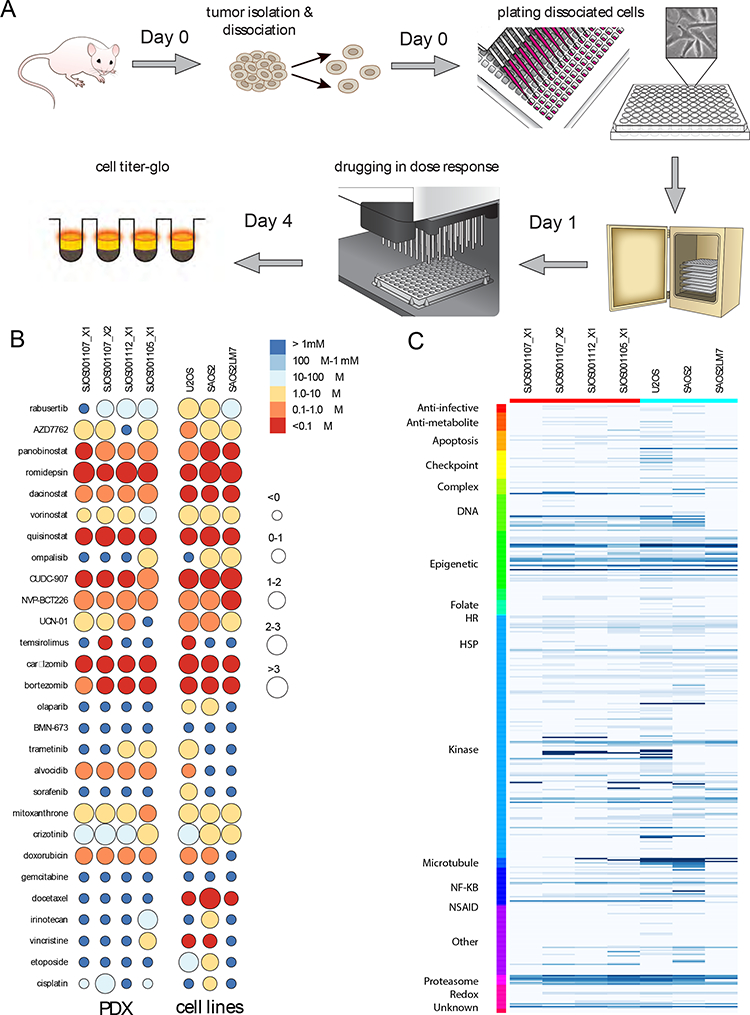
(A) Schematic overview of the high-throughput in-vitro screening assay using early phase cultures of xenograft cells and cell lines. (B) 21 test compounds that yielded the greatest and least cytotoxic effects in the single agent drug screen, and 7 standard-of-care agents (color scale: EC50 values, circle size: magnitude of activity from 0 – >3 log10 units). (C) Single agent drug screen of 373 compounds in 432 formulations sorted by mechanism of action, with dark blue showing low EC50 and light blue showing high EC50 values for compounds that fit a dose-response curve and had at least 1 data point above 1 log10 unit. .
Drug classes that showed the greatest efficacy against osteosarcoma cells were microtubule inhibitors, topoisomerase inhibitors, HDAC inhibitors, proteasome inhibitors, PI3K and mTOR inhibitors (Fig. 4B, Table S3). Other compounds of interest included rabusertib, a CHK1 inhibitor, and CUDC-907, a combination HDAC-PI3K inhibitor (Fig. 4B, Table S3). Notably, DNA alkylators, PARP inhibitors, TGFβ inhibitors, gamma secretase inhibitors, farnesyltransferase inhibitors, retinoids and compounds perturbing HMG-CoA metabolism had very little activity in our screen. BRAF, VEGF, JAK/STAT and BCR-ABL inhibitors as well as antimetabolites showed mixed results (Fig. 4B and Table S3).
Next, we performed screening of these drugs in combination with standard-of-care agents doxorubicin and gemcitabine (Fig. 5A–C),which were selected because of prior evidence of potentiation with HDAC and PI3K inhibitors24,25. These were added to the media at fixed concentrations of 460nM and 52.5 μM, respectively, equivalent to the corresponding mean EC50 from our single-agent drug screening assays, and also equivalent to human plasma Cmax, as determined from pediatric phase I/II studies. Increase in median AUC was seen with 5 drug classes in combination with doxorubicin and gemcitabine: MEK/ERK inhibitors, ALK inhibitors, HDAC inhibitors, combination PI3K/mTOR with MEK/ERK inhibitors, and combination PI3K/mTOR with HDAC inhibitors (Fig. 5D). Mostly PI3K/mTOR inhibitors and HDAC inhibitors showed positive ΔAUC at EC50 below 10 uM (Fig. 5E). Selected PI3K inhibitors showed additional cytotoxicity, notably GSK-2126458 with gemcitabine. As a class, HDAC inhibitors showed additive cytotoxicity with doxorubicin and gemcitabine. PI3K and HDAC inhibitors, and PI3K and MEK inhibitors were additive especially with doxorubicin. Patterns of additivity with doxorubicin and gemcitabine were mirrored when these experiments were repeated with commercial cell lines, except that the extent of potentiation was increased (Fig. S5).
FIGURE 5: Candidate drug classes demonstrate potentiation with standard-of-care agents.
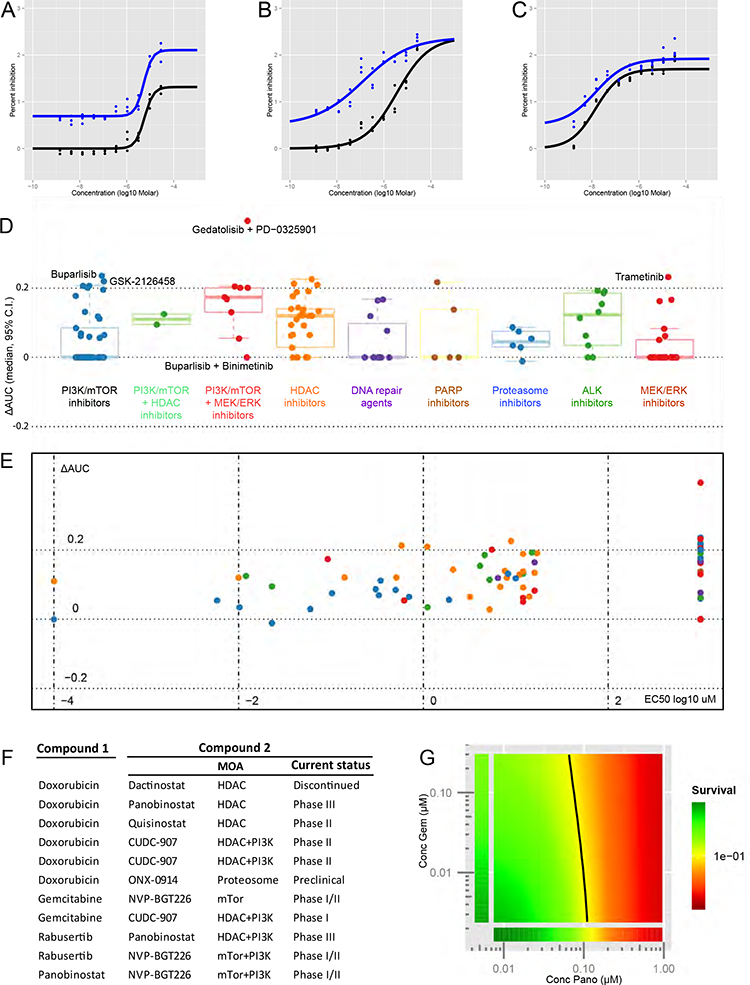
(A-C) Representative dose-response curves of percent inhibition against concentration (log10 uM) with and without addition of single concentration of doxorubicin, against early phase culture of SJOS001112, demonstrating additive (Vorinostat, A), and synergistic (Bortezomib, B; and Rabusertib, C) effects with varying magnitudes of change in AUC. (D-E) Curveshift screens of 135 test agents from 9 selected candidate drug classes in combination with fixed doses of doxorubicin and gemcitabine, illustrated as (D) boxplots depicting change in ΔAUC (median, 95% C.I.), and (E) corresponding scatterplots of ΔAUC (y-axis) against EC50 log10 uM (x-axis)(median data for all xenograft-expanded cell lines tested). (F) Shortlisted ‘hit’ combinations of doxorubicin, gemcitabine, GSK2126458, panobinostat, and rabusertib (compound 1), and test agents (compound 2) that resulted in positive increase in AUC with an EC50 of <10 uM for the latter. (G) Representative response surface model fit for a selected ‘hit’ combination of gemcitabine and panobinostat, each of which were combined across a range of physiologically relevant concentrations. ‘Survival’ is the average logarithm of the reduction in CellTiter-Glo signal relative to the negative control for each dose-pair (as a color scale ranging from 1e+0 (green) to 1e-2 (red)) and is plotted as a best-fit response surface.
To explore the possible interaction of PI3K and HDAC inhibition in osteosarcoma, as evidenced by the effects seen with CUDC-907, PI3K inhibitors were combined with Panobinostat and HDAC inhibitors were combined with GSK2126458. Also, to explore the possible role of CHK1 inhibition, rabusertib was combined with HDAC inhibitors and other tyrosine kinase inhibitors. Combinations that produced a positive increase in the area under the concentration-time curve (AUC) and an EC50 of <10 uM were defined as ‘hits’. ‘Hit’ combinations common to all 3 cell lines, and all 3 xenografts were compared (Fig. 5F). Combinations of PI3K and HDAC inhibitors consistently showed additive effects when applied together. HDAC inhibitors panobinostat and quisinostat and the mTOR inhibitor NVP-BGT226 showed additive effects with standard-of-care agents doxorubicin and gemcitabine.
To verify that these additive effects were achievable across a range of concentrations that could be achieved in human plasma, ‘hit’ combinations were tested in graduated doses on X- and Y-axes of assay plates seeded with xenograft cells (Fig. 5G).
3.4. Drug class susceptibility identified through high-throughput screen correlates with pathway dysregulation in osteosarcoma tumors
To verify the results of the drug screen, we explored differences in the molecular signature of osteosarcoma tumor types to see if the identified drug classes correlated with dysregulated signaling pathways. We analyzed differentially expressed genes in primary and metastatic human osteosarcoma samples using RNA-seq data from the Pediatric Cancer Genome Project. We selected for the top 200 differentially up- and down-regulated genes between primary and metastatic tumors, and identified the signaling pathways represented by these dysregulated genes. KEGG network analysis showed that PI3K signaling and inositol phosphate metabolism were the 2 signaling networks represented by the top 200 differentially up-regulated genes, and the Wnt signaling pathway was represented by the 200 most differentially down-regulated genes (Fig. S6).
4. Discussion
Current barriers to the advancement of therapeutic strategies for osteosarcoma include a lack of preclinical models of osteosarcoma that accurately and reliably represent the phenotype and biology of human disease and a lack of novel and repurposed drug classes that have been explored as therapeutic leads. Genomic analyses of osteosarcoma have revealed a highly complex genomic landscape although with few consistently recurrent somatic mutations. As such, few actionable genetic aberrations have been identified and translated successfully from preclinical studies into clinical use. We developed a patient-derived orthotopic xenograft model of osteosarcoma that recapitulated the primary phenotype of the disease. Using it to expand the limited patient-derived cell population, we screened the cells with a high-throughput in-vitro assay to identify novel and repurposed drug classes with activity against osteosarcoma.
Intra-femoral injection of patient-derived osteosarcoma cells resulted in development of extremity tumors that resembled the histologic, ultrastructural, and radiographic features of the primary tumor. Passive dissemination of tumor cell emboli to the lungs led to development of nodules reminiscent of pulmonary metastases in humans and similar to that seen in other spontaneous metastatic models derived through intra-medullary injection of human osteosarcoma cell lines26,27. While this modeled the possible hematogenous mode of dissemination to the lungs, in humans, metachronous pulmonary metastases often occur after a latent period of 2–3 years following treatment28,29, thus the experimental timelines required to model spontaneous sustained pulmonary disease may be correspondingly longer. Indeed after observing the animals for up to 6 months post-injection, subclinical pulmonary metastases were observed in mice that had tumor cells injected via both intravenous and intra-osseous routes.
The PDX platform also allowed us to sustain the biological makeup of metastatic tumor cells, and through short-term in-vitro culture, permit the interrogation of their drug-response phenotype. Our high-throughput in-vitro cytotoxicity assay showed that microtubule inhibitors, topoisomerase inhibitors, HDAC inhibitors, and proteasome inhibitors had particular effect against osteosarcoma. Multistep inhibition of the PI3K/Akt/mTOR pathway, especially with concurrent inhibition of MEK, showed greater efficacy than inhibition of single nodes of the pathway alone. In synergy experiments, the HDAC inhibitors panobinostat and quisinostat, and the mTOR inhibitor NVP-BGT226 showed additive effects when combined with standard-of-care agents doxorubicin and gemcitabine across dose ranges that could be achieved in human plasma. Other drugs with prominent single-agent activity at in-vitro concentrations equivalent to human dose ranges included rabusertib, a CHK1 inhibitor, and CUDC-907, a combination HDAC and PI3K inhibitor. These are feasible therapeutic leads that can be explored further for osteosarcoma treatment.
4.1. Preclinical models of human osteosarcoma
Human-derived cell lines such as U2OS, SaOS2 and HOS have been well established and characterized, but have been unable to produce in vivo metastases until transformed with the Ki-ras oncogene, as evidenced by the KRIB and 143B models30. Oncogenic transformation alters molecular signaling pathways and confounds the genetic profile of these lines and potentially also in vivo responses to therapy31. The LM7 cell line was derived from serial culture of lung nodules resulting from tail vein injections of SaOS2 cells, without oncogenic transformation, and yielded more florid pulmonary metastases. However similar upregulation of Wnt pathway signaling was noted in this cell line compared to the transformed HOS/143B line. Our phenotypic screen of the LM7 cell line also showed its drug susceptibility pattern to be more similar to its parent SaOS2 cell line than xenograft-derived tumor cells.
The greatest technical hurdle to high-throughput patient-derived orthotopic xenograft models of bone tumors is the delivery of human tumor cells into the orthotopic site of the bony medullary cavity. Using dissociated single-cell suspensions of source tumors, our non-invasive intra- femoral injection technique avoids the trauma of cortical disruption and fracture seen in other implantation models32. The use of dissociated cell suspensions allows for high-throughput propagation of these tumor models, and quantification of the starting tumor burden in the models, which allows for a more uniform enrolment of xenograft models onto preclinical in-vivo studies33. Prolonged cell cultures derived from xenotransplanted models may be limited by the infiltration of murine fibroblasts, which potentially can overgrow the human cell population34. This is overcome in our model by limiting the use of these cells to short-term culture for in-vitro drug screening.
4.2. Therapeutic leads for osteosarcoma
Our findings confirm recent observations about the PI3K pathway as a potential vulnerability in osteosarcoma35,36. However, targeting the PI3K/Akt pathway is limited by concurrent cross-activation of other survival and growth-related pathways, which has been evidenced by poor single-agent activity in cancers. Potential strategies to overcome this limitation include concurrent inhibition of PI3K and other signaling pathways. Combination PI3K and MEK inhibition has been shown to be efficacious in solid tumor xenografts carrying KRAS mutations, HER2 amplifications or PTEN deletions and mutations, and clinical studies combining the MEK inhibitor GDC-0973 and the PI3K inhibitor GDC-0941 are underway37–39. Synergistic effects of HDAC and PI3K inhibitors have also been observed in multiple hematogenous malignancies and solid tumors40. Both of these combinations were observed to be effective against metastatic osteosarcoma xenograft cells in our in-vitro system, therefore PI3K inhibition should be considered as a therapeutic strategy for osteosarcoma in future.
HDAC inhibitors have been shown to induce differentiation, growth arrest, senescence, apoptosis, and produce anti-angiogenic effects in multiple tumor types, including osteosarcoma41–45. In the normal development of bone, HDAC enzymes regulate the development of osteoblasts and osteoclasts46,47, and HDAC inhibitors have been shown to induce accelerated maturation and differentiation of bone through the inhibition of transcription factor Runx2 and regulation of the Wnt/β-catenin pathway48. In addition, oxidative phosphorylation, cytoskeleton modeling, cell cycle and ubiquitin-proteasome pathways are most perturbed in osteosarcoma following HDAC inhibition49. Chromatin decondensation from HDAC inhibition has also been shown to contribute to increased topoisomerase I and II inhibition in osteosarcoma50–52. Preclinical data with cell line-derived xenografts of osteosarcoma showed that HDAC inhibition enhanced NK cell killing through upregulation of activating receptor NKG2D and its ligands. However, at present, only the HDAC inhibitors belinostat and entinostat are employed in clinical trials admitting patients with osteosarcoma. Taken together with our findings, HDAC inhibitors as a class should be further studied to identify specific therapeutic leads for osteosarcoma therapy. Based on our findings, panobinostat and romidepsin are potential HDAC inhibitors that can be combined with the nucleoside analog gemcitabine in protocols for relapse or refractory disease at dose ranges that are achievable in pediatric human plasma. Incorporation of HDAC inhibitors into upfront regimens utilizing doxorubicin should also be explored, given the preclinical data supporting synergistic effects of HDAC and topoisomerase inhibition.
Supplementary Material
HIGHLIGHTS.
Few new agents are identified for osteosarcoma due to the paucity of actionable somatic mutations
Patient-derived orthotopic xenografts can expand tumor cells as short term early-phase cultures
High-throughput screens identify novel and repurposed drugs with activity against osteosarcoma
HDAC inhibitors have particular efficacy and are potentiated in combination with anthracyclines
Acknowledgements
Funding sources: This work was supported by Cancer Center Support [grant CA21765], American Lebanese Syrian Associated Charities, National Medical Research Council Singapore [grant 008–185] (A. Loh), St. Baldrick’s Foundation and the National Pediatric Cancer Foundation (E. Stewart), National Institutes of Health [grants EY014867, EY018599, and CA168875], the Howard Hughes Medical Institute, Alex Lemonade Stand, Tully Family and Peterson Foundations (M.A. Dyer).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statements
All authors have participated in conception and design, or analysis and interpretation of the data; drafting the article or revising it critically for important intellectual content; and approval of the final version.
This manuscript has not been submitted to, nor is under review at, another journal or other publishing venue.
The authors have no affiliation with any organization with a direct or indirect financial interest in the subject matter discussed in the manuscript
All research work involving the use of human subjects was carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki), with prior approval from the Institutional Review Board of St Jude Children’s Research Hospital.
All animal experiments complied with ARRIVE guidelines, and were performed with prior approval from the Institutional Animal Care and Use Committee (IACUC) of St Jude Children’s Research Hospital.
References
- 1.Kaste SC, Pratt CB, Cain AM, Jones-Wallace DJ & Rao BN Metastases detected at the time of diagnosis of primary pediatric extremity osteosarcoma at diagnosis: imaging features. Cancer 86, 1602–1608 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Kager L et al. Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 21, 2011–2018, doi: 10.1200/JCO.2003.08.132 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Luetke A, Meyers PA, Lewis I & Juergens H Osteosarcoma treatment - where do we stand? A state of the art review. Cancer treatment reviews 40, 523–532, doi: 10.1016/j.ctrv.2013.11.006 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Kempf-Bielack B et al. Osteosarcoma relapse after combined modality therapy: an analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS). Journal of clinical oncology : official journal of the American Society of Clinical Oncology 23, 559–568, doi: 10.1200/JCO.2005.04.063 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Ferrari S et al. Neoadjuvant chemotherapy with high-dose Ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the Italian and Scandinavian Sarcoma Groups. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 23, 8845–8852, doi: 10.1200/JCO.2004.00.5785 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Bielack SS et al. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 20, 776–790 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Kim MS, Cho WH, Song WS, Lee SY & Jeon DG time dependency of prognostic factors in patients with stage II osteosarcomas. Clin Orthop Relat Res 463, 157–165, doi: 10.1097/BLO.0b013e318142b27d (2007). [DOI] [PubMed] [Google Scholar]
- 8.Berend KR et al. Adjuvant chemotherapy for osteosarcoma may not increase survival after neoadjuvant chemotherapy and surgical resection. Journal of surgical oncology 78, 162–170 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Souhami RL et al. Randomised trial of two regimens of chemotherapy in operable osteosarcoma: a study of the European Osteosarcoma Intergroup. Lancet 350, 911–917, doi: 10.1016/S0140-6736(97)02307-6 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Ueda Y et al. Analysis of mutant P53 protein in osteosarcomas and other malignant and benign lesions of bone. Journal of cancer research and clinical oncology 119, 172–178 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toguchida J et al. Prevalence and spectrum of germline mutations of the p53 gene among patients with sarcoma. The New England journal of medicine 326, 1301–1308, doi: 10.1056/NEJM199205143262001 (1992). [DOI] [PubMed] [Google Scholar]
- 12.Bishop MW, Janeway KA & Gorlick R Future directions in the treatment of osteosarcoma. Current opinion in pediatrics 28, 26–33, doi: 10.1097/MOP.0000000000000298 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ebb D et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: a report from the children’s oncology group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 30, 2545–2551, doi: 10.1200/JCO.2011.37.4546 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burrow S, Andrulis IL, Pollak M & Bell RS Expression of insulin-like growth factor receptor, IGF-1, and IGF-2 in primary and metastatic osteosarcoma. Journal of surgical oncology 69, 21–27 (1998). [DOI] [PubMed] [Google Scholar]
- 15.MacEwen EG et al. IGF-1 receptor contributes to the malignant phenotype in human and canine osteosarcoma. Journal of cellular biochemistry 92, 77–91, doi: 10.1002/jcb.20046 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Mansky PJ et al. Treatment of metastatic osteosarcoma with the somatostatin analog OncoLar: significant reduction of insulin-like growth factor-1 serum levels. J Pediatr Hematol Oncol 24, 440–446 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Avnet S et al. Insulin receptor isoform A and insulin-like growth factor II as additional treatment targets in human osteosarcoma. Cancer research 69, 2443–2452, doi: 10.1158/0008-5472.CAN-08-2645 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Khanna C et al. Toward a drug development path that targets metastatic progression in osteosarcoma. Clinical cancer research : an official journal of the American Association for Cancer Research 20, 4200–4209, doi: 10.1158/1078-0432.CCR-13-2574 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jia SF, Worth LL & Kleinerman ES A nude mouse model of human osteosarcoma lung metastases for evaluating new therapeutic strategies. Clinical & experimental metastasis 17, 501–506 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Jia SF, Worth LL, Turan M, Duan Xp XP & Kleinerman ES Eradication of osteosarcoma lung metastasis using intranasal gemcitabine. Anticancer Drugs 13, 155–161 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Twarog NR, Stewart E, Hammill CV & Shelat AA BRAID: A Unifying Paradigm for the Analysis of Combined Drug Action. Sci Rep 6, 25523, doi: 10.1038/srep25523 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart E et al. The childhood solid tumor network: A new resource for the developmental biology and oncology research Communities. Dev Biol, doi: 10.1016/j.ydbio.2015.02.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walkley CR et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev 22, 1662–1676, doi:22/12/1662[pii]10.1101/gad.1656808 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao AM, Ke ZP, Shi F, Sun GC & Chen H Chrysin enhances sensitivity of BEL-7402/ADM cells to doxorubicin by suppressing PI3K/Akt/Nrf2 and ERK/Nrf2 pathway. Chemico-biological interactions 206, 100–108, doi: 10.1016/j.cbi.2013.08.008 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Bezler M, Hengstler JG & Ullrich A Inhibition of doxorubicin-induced HER3-PI3K-AKT signalling enhances apoptosis of ovarian cancer cells. Molecular oncology 6, 516–529, doi: 10.1016/j.molonc.2012.07.001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berlin O et al. Development of a novel spontaneous metastasis model of human osteosarcoma transplanted orthotopically into bone of athymic mice. Cancer research 53, 4890–4895 (1993). [PubMed] [Google Scholar]
- 27.Chaffee BK & Allen MJ A clinically relevant mouse model of canine osteosarcoma with spontaneous metastasis. In Vivo 27, 599–603 (2013). [PMC free article] [PubMed] [Google Scholar]
- 28.Bacci G et al. High grade osteosarcoma of the extremities with lung metastases at presentation: treatment with neoadjuvant chemotherapy and simultaneous resection of primary and metastatic lesions. Journal of surgical oncology 98, 415–420, doi: 10.1002/jso.21140 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Ferrari S et al. Postrelapse survival in osteosarcoma of the extremities: prognostic factors for long-term survival. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 21, 710–715 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Dass CR, Ek ET & Choong PF Human xenograft osteosarcoma models with spontaneous metastasis in mice: clinical relevance and applicability for drug testing. Journal of cancer research and clinical oncology 133, 193–198, doi: 10.1007/s00432-006-0157-x (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ek ET, Dass CR & Choong PF Commonly used mouse models of osteosarcoma. Critical reviews in oncology/hematology 60, 1–8, doi: 10.1016/j.critrevonc.2006.03.006 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Crnalic S, Hakansson I, Boquist L, Lofvenberg R & Brostrom LA A novel spontaneous metastasis model of human osteosarcoma developed using orthotopic transplantation of intact tumor tissue into tibia of nude mice. Clinical & experimental metastasis 15, 164–172 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Stewart E et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell reports 9, 829–841, doi: 10.1016/j.celrep.2014.09.028 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohseny AB, Hogendoorn PC & Cleton-Jansen AM Osteosarcoma models: from cell lines to zebrafish. Sarcoma 2012, 417271, doi: 10.1155/2012/417271 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupte A BE, Wan SS, Stewart E, Loh A, Shelat AA, Gould CM, Chalk AM,Taylor S, Lackovic K, Karlström Å, Mutsaers AJ, Desai J, Madhamshettiwar PB,Zannettino AC, Burns C, Huang DC, Dyer MA, Simpson KJ, Walkley CR. Systematic Screening Identifies Dual PI3K and mTOR Inhibition as a Conserved Therapeutic Vulnerability in Osteosarcoma. Clinical cancer research : an official journal of the American Association for Cancer Research 21, 3216–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perry JA, K. A, Tonzi P, Van Allen EM, Carter SL, Baca SC, Cowley GS,Bhatt AS, Rheinbay E, Pedamallu CS, Helman E, Taylor-Weiner A, McKenna A, DeLuca DS, Lawrence MS, Ambrogio L, Sougnez C, Sivachenko A, Walensky LD, Wagle N, Mora J, de Torres C, Lavarino C, Dos Santos Aguiar S, Yunes JA, Brandalise SR, Mercado-Celis GE, Melendez-Zajgla J, Cárdenas-Cardós R, Velasco-Hidalgo L,Roberts CW, Garraway LA, Rodriguez-Galindo C, Gabriel SB, Lander ES, Golub TR, Orkin SH, Getz G, Janeway KA. . Complementary genomic approaches highlight thePI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci USA, E5564–5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johanna Bendell PL, Kwak Eunice, Pandya Susan, Musib Luna, Jones Cheryl, De Crespigny Alex, Belvin Marcia, McKenzie Meghan, Gates Mary R., Chan Iris and Shapiro Geoffrey. in Proceedings of the 102nd Annual Meeting of the American Association for Cancer Research Vol. 71 (AACR, Orlando, FL, 2011. Apr 2–6;). [Google Scholar]
- 38.Wee S et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer research 69, 4286–4293, doi: 10.1158/0008-5472.CAN-08-4765 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Engelman JA et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nature medicine 14, 1351–1356, doi: 10.1038/nm.1890 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qian C et al. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clinical cancer research : an official journal of the American Association for Cancer Research 18, 4104–4113, doi: 10.1158/1078-0432.CCR-12-0055 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Rikimaru T et al. Clinical significance of histone deacetylase 1 expression in patients with hepatocellular carcinoma. Oncology 72, 69–74, doi: 10.1159/000111106 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Miyake K et al. Expression of hypoxia-inducible factor-1alpha, histone deacetylase 1, and metastasis-associated protein 1 in pancreatic carcinoma: correlation with poor prognosis with possible regulation. Pancreas 36, e1–9, doi: 10.1097/MPA.0b013e31815f2c2a (2008). [DOI] [PubMed] [Google Scholar]
- 43.Weichert W HDAC expression and clinical prognosis in human malignancies. Cancer letters 280, 168–176, doi: 10.1016/j.canlet.2008.10.047 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Hirose T et al. p53-independent induction of Gadd45 by histone deacetylase inhibitor: coordinate regulation by transcription factors Oct-1 and NF-Y. Oncogene 22, 7762–7773, doi: 10.1038/sj.onc.1207091 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Watanabe K, Okamoto K & Yonehara S Sensitization of osteosarcoma cells to death receptor-mediated apoptosis by HDAC inhibitors through downregulation of cellular FLIP. Cell death and differentiation 12, 10–18, doi: 10.1038/sj.cdd.4401507 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Schroeder TM & Westendorf JJ Histone deacetylase inhibitors promote osteoblast maturation. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 20, 2254–2263, doi: 10.1359/JBMR.050813 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Westendorf JJ Histone deacetylases in control of skeletogenesis. Journal of cellular biochemistry 102, 332–340, doi: 10.1002/jcb.21486 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Schroeder TM, Nair AK, Staggs R, Lamblin AF & Westendorf JJ Gene profile analysis of osteoblast genes differentially regulated by histone deacetylase inhibitors. BMC genomics 8, 362, doi: 10.1186/1471-2164-8-362 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wittenburg LA, Ptitsyn AA & Thamm DH A systems biology approach to identify molecular pathways altered by HDAC inhibition in osteosarcoma. Journal of cellular biochemistry 113, 773–783, doi: 10.1002/jcb.23403 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marchion DC et al. Sequence-specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. Journal of cellular biochemistry 92, 223–237, doi: 10.1002/jcb.20045 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Wittenburg LA, Bisson L, Rose BJ, Korch C & Thamm DH The histone deacetylase inhibitor valproic acid sensitizes human and canine osteosarcoma to doxorubicin. Cancer chemotherapy and pharmacology 67, 83–92, doi: 10.1007/s00280-010-1287-z (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Munster P et al. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: a clinical and translational study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 25, 1979–1985, doi: 10.1200/JCO.2006.08.6165 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.