

Abstract
Evocalcet is a novel calcimimetic agent for the treatment of secondary hyperparathyroidism (SHPT). This study evaluated the effects of evocalcet on inhibition and induction of cytochrome P450 (CYP) isozymes. Although drug interactions arising from reversible inhibition of CYP isozymes by evocalcet were considered unlikely based on the results of in vitro studies and static model analyses, the potential for evocalcet to cause time‐dependent inhibition of CYP3A or induction of several CYP isozymes could not be ruled out. Therefore, a clinical drug‐drug interaction (DDI) study to evaluate the effects of evocalcet on the pharmacokinetics (PKs) of probe substrates for CYP isozymes (CYP1A2, CYP2B6, CYP2C8, CYP2C9, and CYP3A) was conducted in healthy male volunteers using a novel cocktail combination. Evocalcet did not significantly affect the PKs of the probe substrates, confirming that CYP‐mediated interactions were unlikely.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Evocalcet, a novel calcimimetic agent, has no substantial inhibitory effects on CYP isozymes in vitro.
what question did this study address?
This study assessed the effects of evocalcet on time‐dependent inhibition and induction of CYP isozymes in vitro and the correlations between in vitro data and clinical evaluation of potential DDIs.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Evocalcet has no marked effects on the PKs of drugs that are substrates for CYP isozymes (CYP1A2, CYP2B6, CYP2C8, CYP2C9, and CYP3A) in vivo.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Evocalcet can be used without considering the risk of CYP‐mediated DDIs.
Secondary hyperparathyroidism (SHPT), characterized by elevation of serum parathyroid hormone (PTH) levels, is a common disorder in patients with chronic kidney disease.1 As chronic kidney disease progresses, the increased serum PTH levels result in high‐turnover bone disease and increased levels of serum calcium and phosphate. This abnormal mineral metabolism often leads to cardiovascular calcification, fractures, and increased risks of all‐cause and cardiovascular mortality.2, 3, 4, 5
Cinacalcet hydrochloride (cinacalcet) is an oral calcimimetic agent that acts allosterically on calcium receptors on parathyroid cells to suppress PTH secretion.6, 7 Cinacalcet has been widely used for management of PTH, calcium, and phosphorus levels in patients undergoing dialysis with SHPT.8, 9, 10, 11, 12, 13 As a result, cinacalcet has helped to dramatically reduce the number of parathyroidectomy surgeries.14
However, cinacalcet treatment is occasionally associated with gastrointestinal (GI) symptoms.15 GI intolerance typically results in decreased patient compliance, and some patients with SHPT cannot receive a therapeutic dose because of their poor tolerance.16, 17 Cinacalcet is also a potent inhibitor of cytochrome P450 (CYP) 2D6, which has raised concerns about interactions with a number of drugs.18, 19 Therefore, there is an unmet need for better tolerated new calcimimetic agents without potential for drug‐drug interactions (DDIs).
Evocalcet (MT‐4580/KHK7580) is a new calcimimetic agent with a dose‐dependent PTH‐lowering effect that only marginally affected gastric emptying in a nonclinical study.20 Noninferiority of evocalcet to cinacalcet for suppression of intact PTH, with fewer GI‐related adverse events (AEs), was demonstrated in a randomized clinical trial of patients with SHPT undergoing hemodialysis.21 Therefore, evocalcet may be an alternative to existing calcimimetics for management of SHPT.
The US Food and Drug Administration, European Medicines Agency, and Pharmaceuticals and Medical Devices Agency guidelines recommend that the potential for DDIs in new drugs should be assessed in vitro and in vivo.22, 23, 24, 25 Evocalcet did not cause reversible inhibition of eight CYP isozymes (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2E1, and CYP3A) within the concentration range of 0.15–50 μmol/L in vitro.20 Evocalcet was shown to competitively inhibit CYP2D6‐specific metabolic activity, with 50.7% residual activity at the maximum tested evocalcet concentration of 50 μmol/L. In the present study, static model analyses for the reversible inhibition were conducted and the potential for evocalcet to cause time‐dependent inhibition and induction of CYP isozymes were evaluated in accordance with the guidelines. Although drug interactions arising from reversible inhibition were considered unlikely, the potential to cause time‐dependent inhibition of CYP3A or induction of several CYP isozymes could not be ruled out due to the inability to calculate inhibition and induction parameters. Because DDIs could not be ruled out by in vitro studies, a clinical study was conducted in healthy male volunteers to evaluate the potential for evocalcet to cause DDIs using a cocktail approach.
Methods
In vitro CYP competitive inhibition
The in vitro method used to characterize the direct inhibitory effects of evocalcet on the specific activities of nine CYP isozymes (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4) was described previously.20
The Ki value for evocalcet for each CYP isozyme was estimated by dividing the half‐maximal inhibitory concentration (IC50) by 2. The maximum concentration (Cmax) of evocalcet at steady state ([I]) was obtained by a population pharmacokinetic (PopPK) analysis using clinical pharmacokinetic (PK) data from several trials in patients with SHPT. A total of 1,855 concentration‐time points from 128 subjects were used in the analysis. The PopPK analysis was conducted using a nonlinear mixed‐effects model; a two‐compartment model with first‐order absorption was selected as the PopPK model and [I] was predicted using predicted PopPK parameters. The maximum evocalcet concentration in the GI tract ([I g]) was obtained by dividing the maximum therapeutic dose (12 mg) by 250 mL. The R value and alternative R value (predicted ratio of area under the plasma concentration time curve (AUC)s for the substrate drug in the presence and absence of evocalcet) were calculated as shown below.22, 24
| (1) |
| (2) |
In vitro CYP time‐dependent inhibition
The time‐dependent inhibition of each CYP isozyme by evocalcet was assessed by a traditional IC50 shift approach.26 Briefly, each CYP isoenzyme was pre‐incubated with evocalcet for 30 minutes in the presence of an nicotinamide adenine dinucleotide phosphate‐generating system in human liver microsomes, and the residual activity was compared with the activity without pre‐incubation.
In vitro CYP induction
The potential for evocalcet to induce CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP3A4 was assessed in vitro using primary cultures of human hepatocytes freshly isolated from three white donors. Human hepatocytes were freshly isolated at Quotient Bioresearch (Rushden) using a two‐step collagenase perfusion procedure.27, 28, 29 Liver tissues were obtained from the University Hospital of Leicester under ethical consent (REC reference 14/EM/0019 approved by the NRES committee region‐East Midlands). Donor 1 was a 76‐year‐old woman, and donors 2 and 3 were 71‐year‐old men. The viability of the hepatocytes from donors 1, 2, and 3 was 83%, 90%, and 73%, respectively, determined using trypan blue exclusion. The isolated hepatocytes were diluted in hepatocyte maintenance medium (Lonza) containing 5% fetal bovine serum, seeded into collagen‐coated 24‐well plates and placed in a humidified incubator at 37°C and 5% CO2 overnight to allow for cell attachment. Once the hepatocytes had been assessed for attachment and deemed acceptable, the medium was gently removed and replaced with fresh warmed hepatocyte maintenance medium and returned to the incubator. The hepatocytes were then maintained in the medium for ~2 days before initiation of experiments.
The cells were incubated with evocalcet (0.2–20 μmol/L), omeprazole (2–50 μmol/L; positive control for CYP1A2), phenobarbital (0.02–1 mmol/L; positive control for CYP2B6), or rifampicin (0.1–25 μmol/L; positive control for CYP2C8, CYP2C9, CYP2C19, and CYP3A4) for 72 hours, followed by mRNA assessment. Total RNA was extracted from the hepatocytes with a PureLink RNA Mini Kit (Life Technologies, Carlsbad, CA) and reverse‐transcribed to cDNA using an iScript cDNA synthesis kit (Bio‐Rad Laboratories, Hercules, CA) according to the manufacturer's instructions. Quantitative polymerase chain reaction was performed with a CFX96 Touch real‐time polymerase chain reaction instrument (Bio‐Rad Laboratories). A 96‐well optical plate and Sso Advanced SYBR Green Supermix (Bio‐Rad Laboratories) were used according to the manufacturer's instructions. The cDNA concentrations were normalized to those of β‐actin (Δct). The relative mRNA expression was determined by a 2−ΔΔct calculation.
Clinical DDI study
Prior to the start of the study, the study protocol was approved by the Hakata Clinic Institutional Review Board. The study was conducted in compliance with the International Ethical Guidelines for Biomedical Research Involving Human Subjects, Good Clinical Practice Guidelines, The Declaration of Helsinki, and local laws. All subjects provided written informed consent.
In this open‐label clinical pharmacology study in Japanese male healthy volunteers, the effects of multiple oral doses of 6 mg evocalcet administered for 14 days on the activity of various CYP isozymes were evaluated, using the PKs of substrates for the CYP isozymes as indicators. Theophylline (100 mg, Theodur; Mitsubishi Tanabe Pharma Corporation, Osaka, Japan), efavirenz (200 mg, Stocrin; MSD K.K., Tokyo, Japan), repaglinide (0.25 mg, Surepost; Sumitomo Dainippon Pharma, Osaka, Japan), diclofenac sodium (25 mg, Voltaren; Novartis Pharma K.K., Tokyo, Japan), and tadalafil (5 mg, Zalutia; Eli Lilly Japan K.K., Hyogo, Japan) were used as the probe drugs for CYP1A2, CYP2B6, CYP2C8, CYP2C9, and CYP3A, respectively.
Eligible subjects were admitted to the clinical site on the day before (day −1) the day of substrate drug administration (day 1). The subjects received a cocktail of the probe drugs administered orally on day 1, followed by multiple oral doses of 6 mg evocalcet administered once daily on days 4–20, and a cocktail of the probe drugs administered orally on day 18. The PKs of the probe drugs were evaluated on days 1–4 and 18–21. Subjects who completed the scheduled observations and examinations on day 22 were discharged if no clinically significant abnormalities requiring hospitalized observation were noted, and if the investigator or subinvestigator judged that their discharge was appropriate. Subsequently, the subjects returned to the clinical site on day 27 to undergo required observations and examinations.
The safety profile (i.e., AEs, vital signs, clinical laboratory measurements, and 12‐lead electrocardiography) was characterized during the study.
Plasma concentrations of probe substrates
Blood samples for analysis of plasma concentrations of probe substrates were collected before and at 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 12, 24, 36, 48, and 72 hours after administration of the cocktail on days 1 and 18 using blood collection tubes containing K2EDTA. The tubes were immediately mixed by inversion and centrifuged to obtain plasma.
The plasma concentrations of the probe substrates were measured using validated methods. The analytes were extracted from the plasma samples using an OASIS μElution plate (Waters, Milford, MA) and their concentrations were determined by high‐performance liquid chromatography (HPLC)‐tandem mass spectrometry. The lower limit of quantification (LLOQ) was 50 ng/mL for theophylline, 1 ng/mL for efavirenz, 5 pg/mL for repaglinide, 0.4 ng/mL for diclofenac, and 1 ng/mL for tadalafil.
Plasma concentrations of evocalcet
Blood samples for analysis of plasma evocalcet concentrations were collected before the first administration of evocalcet (day 4), before administration of evocalcet on day 12, before administration of evocalcet and the cocktail on day 18, and at 72 hours after administration of the cocktail on day 18 (day 21). The blood samples were collected using blood collection tubes containing K2EDTA. The tubes were immediately mixed by inversion and centrifuged to obtain plasma.
The plasma concentrations of evocalcet were measured using validated methods. Evocalcet was extracted from the plasma samples with stable isotope‐labeled evocalcet as an internal standard using an OASIS μElution plate and their concentrations were determined by HPLC‐tandem mass spectrometry. Chromatographic separation was accomplished using an LC‐20A or UFLC system (Shimadzu, Kyoto, Japan) with a mobile phase comprising water/formic acid (1,000:1, v/v) and acetonitrile/formic acid (1,000:1, v/v). The HPLC system was coupled to a tandem mass spectrometer (API 5000; AB Sciex Pte., Concord, Ontario, Canada), with an electrospray ionization source. The mass spectrometer was operated in the selected reaction‐monitoring mode. Mass transitions (Q1/Q3) of m/z 375/155 and 381/155 were used for evocalcet and the internal standard in the positive‐ion mode, respectively. LLOQ was 0.134 nmol/L (0.05 ng/mL), with a linear calibration range of 0.134–267 nmol/L (0.05–100 ng/mL).
PK analysis
The PK parameters for the probe substrates were calculated for each subject by a noncompartmental analysis using Phoenix WinNonlin 6.1 software (Pharsight, Mountain View, CA). The plasma Cmax and time to reach Cmax (tmax) were obtained directly from experimental observations. The AUC from time zero to the last measureable time point (AUC0–t) was calculated by a linear trapezoidal method using the actual elapsed time. The AUC from time zero to infinity (AUC∞) was calculated by the following equation: AUC∞ = AUC0–t+Ct/k el, where Ct is the plasma concentration of the probe substrate at the last measureable time point and k el is the elimination rate constant.
Statistical methods
The sample size was not based on a statistical power calculation. The coefficient variations (CVs) for the AUCs of the probe substrates ranged from 15.9–32.6% in their package inserts. Forty subjects provided a probability of more than 85% and 99% such that the upper boundary of the 90% confidence interval (CI) of the AUC ratio (with evocalcet/without evocalcet) would not exceed 1, assuming a subject discontinuation rate of about 10%, CV for the AUC of the probe substrates of 35%, and true AUC ratio (with evocalcet/without evocalcet) of 0.8 and 0.67, respectively.
The PK parameters (Cmax, AUC0–t, and AUC∞) were log‐transformed and analyzed for each probe substrate using a linear mixed‐effects model. The point estimates and corresponding 90% CIs for the ratios of geometric means (i.e., the ratios of the geometric mean with evocalcet/without evocalcet) were calculated. All statistical analyses were performed using SAS 9.2 software (SAS Institute, Cary, NC).
Results
In vitro CYP competitive inhibition
The residual activities of the nine CYP isozymes (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A) in the competitive inhibition assays using human liver microsomes were all > 50% at the maximum evocalcet concentration (50 μmol/L) in duplicate experiments (Table S1).20 Although IC50 values could not be determined, it was clear that the values were > 50 μmol/L.
The potential for competitive inhibition of the nine CYP isozymes was evaluated by a basic static model using the predicted steady state Cmax after administration of a 12 mg oral dose ([I]: 2.25 μmol/L, Figure S1), maximum concentration in the GI tract ([I g]: 12 mg/250 mL = 128 μmol/L), and unbound fraction in plasma (f u: 0.018) assuming an IC50 value of 50 μmol/L.22, 24 The R values (1 + f u×[I]/K i) were < 1.002 and the alternative R values (1 + [I g]/K i) were < 6.13 for the CYP isozymes evaluated. Because all R values were below the cutoff value for competitive interactions,22, 24 clinical studies to confirm the lack of competitive inhibition of CYP isozymes were determined to be unnecessary.
In vitro CYP time‐dependent inhibition
Evocalcet did not show time‐dependent inhibition of CYP isozyme‐specific metabolic activities except for that of CYP3A (Table S1). Evocalcet inhibited CYP3A‐specific metabolic activity in a time‐dependent manner, but it was not possible to determine the IC50 value because the residual metabolic activity ranged from 65.4–66.2% at the maximum concentration of 50 μmol/L (Table S1). The time‐dependent inhibition of CYP3A activity by evocalcet was evaluated by calculating the inactivation parameters (K i, k inact) for CYP3A activity by a dilution method using human liver microsomes. Although time‐dependent inhibition of CYP3A activity by evocalcet was observed, there was no trend for saturation of the kobs values even at the maximum evocalcet concentration of 50 μmol/L. Therefore, inactivation parameters could not be calculated (data not shown).
In vitro CYP induction
The potential of evocalcet to induce CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP3A4 was assessed using primary cultures of human hepatocytes isolated from three donors. The mRNA expression levels of CYP1A2, CYP2B6, CYP2C8, CYP2C9, and CYP3A4 increased with increasing evocalcet concentration (Figures [Link], [Link], [Link], [Link], [Link] – S7). All induction ratios were less than those for the positive controls (Table 1). The potential for evocalcet to induce CYP2C19 could not be evaluated because the mRNA induction ratio for the CYP2C19 positive control was low.
Table 1.
Induction of CYPs mRNA in human hepatocytes
| CYP isozymes | Test compound | Concentration (μmol/L) | Maximum fold induction | ||
|---|---|---|---|---|---|
| Donor 1 | Donor 2 | Donor 3 | |||
| CYP1A2 | Evocalcet | 0.2–20 | 4.26 | 1.12 | 29.1 |
| Omeprazole | 2–50 | 99.0 | 88.9 | 1,080 | |
| CYP2B6 | Evocalcet | 0.2–20 | 8.67 | 3.40 | 4.75 |
| Phenobarbital | 20–1000 | 11.6 | 27.1 | 23.7 | |
| CYP2C8 | Evocalcet | 0.2–20 | 9.47 | 6.97 | 3.40 |
| Rifampicin | 0.1–25 | 17.3 | 19.0 | 7.10 | |
| CYP2C9 | Evocalcet | 0.2–20 | 2.84 | 2.31 | 1.60 |
| Rifampicin | 0.1–25 | 5.20 | 4.75 | 3.20 | |
| CYP2C19 | Evocalcet | 0.2–20 | 1.53 | 0.900 | 1.02 |
| Rifampicin | 0.1–25 | 1.60 | 2.43 | 1.03 | |
| CYP3A4 | Evocalcet | 0.2–20 | 4.46 | 3.24 | 13.8 |
| Rifampicin | 0.1–25 | 14.1 | 15.2 | 18.2 | |
CYP, cytochrome P450.
Clinical DDI study
Forty healthy male Japanese volunteers were enrolled. All subjects completed the study and were included in the analyses. The mean age (range) was 26.0 years (20–39 years), mean body weight (range) was 62.9 kg (52.7–76.6 kg), and mean body mass index (range) was 21.0 kg/m2 (18.5–24.7 kg/m2).
The mean plasma evocalcet concentration before the first administration of evocalcet on day 4 was below the LLOQ (<0.134 nmol/L). The mean (SD) plasma evocalcet trough concentrations were 200 (119), 272 (198), and 246 (185) nmol/L on days 12, 18, and 21, respectively, confirming that evocalcet had attained steady state by day 18. The trough concentrations were comparable to those in another multiple‐dose clinical study on healthy volunteers (unpublished data).
The time courses of the plasma concentrations and PK parameters for the CYP probe drugs are presented in Figure 1 and Table 2, respectively. There were no marked differences between the plasma concentration‐time curves for any of the CYP probe drugs with and without evocalcet. The 90% CIs for the geometric mean ratios for AUC0–t and AUC∞ (with evocalcet/without evocalcet) were within the range of 0.8–1.25 for all probe drugs, except for theophylline (90% CIs of 1.192–1.326 and 1.190–1.326, respectively). The 90% CIs for the geometric mean ratios for Cmax were within the range of 0.8–1.25 for all probe drugs, except for diclofenac (90% CI of 0.752–1.099).
Figure 1.
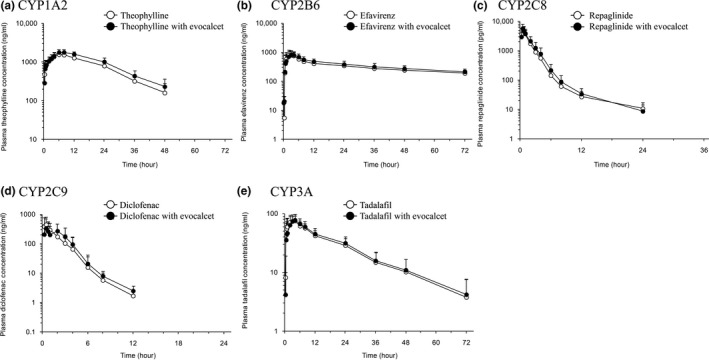
Mean plasma concentration‐time profiles of (a) theophylline, (b) efavirenz, (c) repaglinide, (d) diclofenac, and (e) tadalafil with and without evocalcet. Data are presented as arithmetic mean + SD. CYP, cytochrome P450.
Table 2.
Pharmacokinetic parameters of probe substrates with or without evocalcet
| Geometric mean (CV%) | Geometric mean ratio (90% CI) | ||
|---|---|---|---|
| Probe drug alone | Probe drug with evocalcet | With/without evocalcet | |
| Theophylline | |||
| AUC0–t ng*h/mL, (same later in this table) | 36,450 (29.8) | 45,830 (22.9) | 1.257 (1.192–1.326) |
| AUC∞ (ng*h/mL) | 38,440 (29.3) | 48,280 (22.9) | 1.256 (1.190–1.326) |
| Cmax (ng/mL) | 1,622 (19.5) | 1,856 (16.3) | 1.145 (1.096–1.196) |
| Efavirenz | |||
| AUC0–t (ng*h/mL) | 22,440 (23.8) | 24,880 (23.8) | 1.111 (1.069–1.154) |
| AUC∞ (ng*h/mL) | 37,560 (40.9) | 43,940 (36.1) | 1.168 (1.097–1.244) |
| Cmax (ng/mL) | 1027 (30.3) | 988.4 (24.9) | 0.962 (0.888–1.043) |
| Repaglinide | |||
| AUC0–t (ng*h/mL) | 9.786 (32.7) | 10.87 (34.6) | 1.111 (1.042–1.184) |
| AUC∞ (ng*h/mL) | 10.22 (33.2) | 10.98 (35.1) | 1.104 (1.021–1.195) |
| Cmax (ng/mL) | 5.731 (37.0) | 5.579 (37.9) | 0.973 (0.864–1.097) |
| Diclofenac | |||
| AUC0–t (ng*h/mL) | 893.9 (21.8) | 974. 7 (23.3) | 1.090 (1.020–1.166) |
| AUC∞ (ng*h/mL) | 905.8 (21.3) | 1,000 (21.4) | 1.111 (1.050–1.175) |
| Cmax (ng/mL) | 576.1 (46.1) | 523.5 (49.7) | 0.909 (0.752–1.099) |
| Tadalafil | |||
| AUC0–t (ng*h/mL) | 1621 (28.7) | 1709 (27.5) | 1.054 (1.008–1.103) |
| AUC∞ (ng*h/mL) | 1,719 (34.2) | 1,815 (30.8) | 1.056 (1.008–1.106) |
| Cmax (ng/mL) | 83.22 (17.7) | 82.16 (22.0) | 0.987 (0.930–1.048) |
AUC0‐t, area under the plasma concentration time curve from time zero to the last measureable time point; AUC∞, area under the plasma concentration time curve from zero to infinity; CI, confidence interval; Cmax, peak plasma concentration; CV, coefficient variation.
No AEs occurred before exposure to evocalcet (days 1–3). Hepatic steatosis and alanine aminotransferase increase were each reported in 1 of 40 subjects (3%) after evocalcet administration (on and after day 4). The increase in alanine aminotransferase was considered to be drug related. No AEs resulting in death or other significant or serious AEs occurred during this study.
Discussion
Calcimimetic treatment is an emerging therapy for SHPT in patients undergoing dialysis. Cinacalcet, an oral calcimimetic agent, is used for SHPT in patients on maintenance dialysis. However, cinacalcet potently inhibits CYP2D6, and, therefore, has potential for DDIs with CYP2D6 substrates.18, 19 Furthermore, noninferiority of evocalcet to cinacalcet for suppression of intact PTH, with fewer GI‐related AEs, was demonstrated in a randomized clinical trial in patients with SHPT receiving hemodialysis.21
An in vitro competitive inhibition study involving CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A suggested that interactions in vivo from reversible inhibition of CYP isozymes, including CYP2D6, by evocalcet were unlikely because the R values and alternative R values were below the cutoff values (R value: 1.02; alternative R value: 11).30, 31, 32 Evocalcet caused time‐dependent inhibition of CYP3A in human liver microsomes, but k inact and Ki values could not be calculated. Evocalcet also caused concentration‐related and > 100% increases in CYP mRNA levels for CYP1A2, CYP2B6, CYP2C8, CYP2C9, and CYP3A4 in primary cultured human hepatocytes, but it was not possible to calculate the induction parameters (maximum effect (Emax) and EC50) for the individual CYP isozymes due to the inability to attain sufficiently high evocalcet concentrations because of its limited solubility and a small increase in mRNA, meaning that reasonable fits to the data could not be obtained, although a nonlinear regression analysis with a four‐parameter sigmoidal equation was conducted. As a result, the potential for DDIs associated with the induction of CYP1A2, CYP2B6, CYP2C8, CYP2C9, and CYP3A and time‐dependent inhibition of CYP3A could not be determined in vitro. Thus, a clinical DDI study was conducted.
In healthy adult male subjects given a single oral dose of evocalcet between 1 and 20 mg, Cmax and AUC∞ increased in a dose‐dependent manner (unpublished data). Although the approved dose in Japan for patients with SHPT receiving maintenance dialysis is between 1 and 12 mg/day, a dose of 6 mg/day was chosen for the clinical DDI study. The dose was reduced from 12 mg/day to 6 mg/day because one of six healthy volunteers who received daily doses of 12 mg experienced an AE (tetany; unpublished data). Potential interactions at doses exceeding 6 mg were not evaluated in this clinical study. Evocalcet was dosed for 14 days before administration of the probe drugs to allow adequate time for attainment of steady state (7 days of daily administration) and for CYP induction to occur. CYP3A induction by rifampicin occurs within 1 week after multiple daily doses,33 suggesting the potential for evocalcet to induce CYP3A can be evaluated after 14 days of daily administration (7 days after attainment of steady state).
The effects of evocalcet were evaluated in a clinical DDI study on the CYP isozymes examined in vitro. Although the necessity for a clinical DDI study on CYP2C19 could not be determined from the in vitro results, a potential effect of evocalcet on CYP2C19 induction could be presumed from its effects on CYP2C8, CYP2C9, and CYP3A because CYP2C and CYP3A are co‐induced by pregnane X receptors.25
A cocktail approach, involving simultaneous administration of substrates for multiple CYP isozymes to subjects, is effective for evaluation of whether a test drug has the potential for clinically significant DDIs with multiple enzymes. Many cocktail combinations have been reported and their usefulness for DDI evaluation has been proven.34, 35, 36, 37, 38, 39, 40 However, the use of repaglinide in a cocktail as a substrate for CYP2C8 and the combination of substrates used in this study for several CYP isozymes (CYP1A2, CYP2B6, CYP2C8, CYP2C9, and CYP3A) has not been reported. In this study, theophylline, efavirenz, repaglinide, diclofenac sodium, and tadalafil were chosen as substrates for CYP1A2, CYP2B6, CYP2C8, CYP2C9, and CYP3A, respectively, because all of these agents were approved for oral administration in Japan to support a clinical trial. In addition, all five substrates are listed as examples of probe drugs in the guidelines published by regulatory agencies.23, 24, 25 Hence, the use of these drugs as substrates for individual CYP isozymes was considered appropriate. All drugs were administered as a cocktail. It was judged there was little risk to safety because of previous experience with each substrate having been administered to healthy volunteers at higher doses. Moreover, it was judged that a PK interaction among the substrates was unlikely because DDIs among the substrates selected for the cocktail have not been reported. Although efavirenz has the potential to induce CYP2B6 and CYP3A,41 this was unlikely to occur after a single administration. These findings suggest that the cocktail approach utilized in this study should be appropriate. As the safety of the selected combination of five substrates in humans as a cocktail has not been previously reported, only 5 of the 40 subjects were initially dosed with the cocktail to evaluate its safety. Thereafter, the other 35 subjects were enrolled. No AEs considered to be safety‐related were observed. These findings demonstrated the safety of a single dose of the cocktail administered in combination with multiple 6 mg daily doses of evocalcet for 14 days in healthy subjects.
The 90% CIs for the geometric mean ratios of AUC0–t, AUC∞, and Cmax (with evocalcet/without evocalcet) were within the range of 0.8–1.25 for efavirenz, repaglinide, and tadalafil, respectively. Therefore, evocalcet was judged not to have PK interactions with substrates of CYP2B6, CYP2C8, and CYP3A. The 90% CIs for the geometric mean ratios for the AUC0–t (1.192–1.326) and AUC∞ (1.190–1.326) for theophylline and that for Cmax (0.752–1.099) for diclofenac exceeded the lower or upper range of 0.8–1.25, but the deviations from this range were slight and not considered to be clinically relevant. The 90% CI of the Cmax for theophylline and those of the AUC0–t and AUC∞ for diclofenac were within the range of 0.8–1.25. These results indicate that evocalcet does not have a marked effect on the PKs of drugs that are substrates of CYP1A2 or CYP2C9. Given that the inducibility of CYP2C can be generally ranked as CYP2C8 ≥ CYP2C9 > CYP2C19,42 based on the results for repaglinide and diclofenac as substrates for CYP2C8 and CYP2C9, respectively, induction of CYP2C19 by evocalcet is unlikely. Although the potential for DDIs at an evocalcet dose of 12 mg was not studied, induction of CYPs at the higher dose is unlikely. The predicted unbound steady‐state Cmax after administration of a 12 mg oral dose is 40.5 nmol/L; significantly less than the concentration tested in the in vitro induction study (i.e., 200 nmol/L) where induction was not found for any CYP isozymes tested.
In conclusion, drug interactions resulting from reversible inhibition of CYP isozymes by evocalcet are unlikely, based on in vitro studies using a basic static model. Although the potential for time‐dependent inhibition of CYP3A and induction of CYP isozymes could not be determined from the in vitro studies, it was confirmed in a clinical study utilizing a cocktail approach that evocalcet had no marked effect on the PKs of drugs that are substrates for CYP1A2, CYP2B6, CYP2C8, CYP2C9, and CYP3A. These findings support that evocalcet is unlikely to result in DDIs when co‐administered with drugs that are CYP substrates.
Funding
This study was sponsored by Kyowa Hakko Kirin and Mitsubishi Tanabe Pharma Corporation under a collaboration agreement.
Conflict of Interest
K.N., H.M., Y.E., S.O., H.N., Y.N., T.U., and R.S. are employees of Kyowa Hakko Kirin. A.K. is an employee of Mitsubishi Tanabe Pharma Corporation. M.S. reports no conflict of interest relevant to this study. M.F. has received consulting fees from Kyowa Hakko Kirin and Ono Pharmaceutical, lecture fees from Kyowa Hakko Kirin, Bayer Yakuhin, Torii Pharmaceutical, and Ono Pharmaceutical, and grants from Kyowa Hakko Kirin and Bayer Yakuhin. T.A. has received consulting fees from Kyowa Hakko Kirin, Astellas Pharma, Bayer Yakuhin, Fuso Pharmaceutical Industries, Japan Tobacco, Ono Pharmaceutical, and NIPRO Industry, and lecture fees from Kyowa Hakko Kirin, Chugai Pharmaceutical, Bayer Yakuhin, Kissei Pharmaceutical, Torii Pharmaceutical, and Ono Pharmaceutical.
Author Contributions
K.N. wrote the manuscript; H.M., M.S., Y.E., S.O., H.N., Y.N., T.U., A.K., R.S., M.F., and T.A. designed the research; M.S. performed the research; K.N., H.M., and A.K. analyzed the data.
Supporting information
Table S1. Inhibition of cytochrome P450 (CYP) activities in human liver microsomes.
Figure S1. Predicted plasma evocalcet concentration‐time profiles after multiple oral administration of 12 mg evocalcet once a day.
Figure S2. Potential for evocalcet to induce human cytochrome P450 (CYP)1A2 mRNA levels in donors 1, 2, and 3.
Figure S3. Potential for evocalcet to induce human cytochrome P450 (CYP)2B6 mRNA levels in donors 1, 2, and 3.
Figure S4. Potential for evocalcet to induce human cytochrome P450 (CYP)2C8 mRNA levels in donors 1, 2, and 3.
Figure S5. Potential for evocalcet to induce human cytochrome P450 (CYP)2C9 mRNA levels in donors 1, 2, and 3.
Figure S6. Potential for evocalcet to induce human cytochrome P450 (CYP)2C19 mRNA levels in donors 1, 2, and 3.
Figure S7. Potential for evocalcet to induce human cytochrome P450 (CYP)3A4 mRNA levels in donors 1, 2, and 3.
Acknowledgments
The authors thank Kyowa Hakko Kirin and Mitsubishi Tanabe Pharma Corporation who sponsored this work under a collaboration agreement.
References
- 1. Cunningham, J. , Locatelli, F. & Rodriguez, M. Secondary hyperparathyroidism: pathogenesis, disease progression, and therapeutic options. Clin. J. Am. Soc. Nephrol. 6, 913–921 (2011). [DOI] [PubMed] [Google Scholar]
- 2. Tentori, F. et al Mortality risk for dialysis patients with different levels of serum calcium, phosphorus, and PTH: the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am. J. Kidney Dis. 52, 519–530 (2008). [DOI] [PubMed] [Google Scholar]
- 3. Taniguchi, M. et al Serum phosphate and calcium should be primarily and consistently controlled in prevalent hemodialysis patients. Ther. Apher. Dial. 17, 221–228 (2013). [DOI] [PubMed] [Google Scholar]
- 4. Raggi, P. et al Cardiac calcification in adult hemodialysis patients. A link between end‐stage renal disease and cardiovascular disease? J. Am. Coll. Cardiol. 39, 695–701 (2002). [DOI] [PubMed] [Google Scholar]
- 5. Tentori, F. et al High rates of death and hospitalization follow bone fracture among hemodialysis patients. Kidney Int. 85, 166–173 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nemeth, E.F. et al Pharmacodynamics of the type II calcimimetic compound cinacalcet HCl. J. Pharmacol. Exp. Ther. 308, 627–635 (2004). [DOI] [PubMed] [Google Scholar]
- 7. Kawata, T. et al Direct in vitro evidence of the suppressive effect of cinacalcet HCl on parathyroid hormone secretion in human parathyroid cells with pathologically reduced calcium‐sensing receptor levels. J. Bone Miner. Metab. 24, 300–306 (2006). [DOI] [PubMed] [Google Scholar]
- 8. Nagano, N. Pharmacological and clinical properties of calcimimetics: calcium receptor activators that afford an innovative approach to controlling hyperparathyroidism. Pharmacol. Ther. 109, 339–365 (2006). [DOI] [PubMed] [Google Scholar]
- 9. Fukagawa, M. et al Cinacalcet (KRN1493) effectively decreases the serum intact PTH level with favorable control of the serum phosphorus and calcium levels in Japanese dialysis patients. Nephrol. Dial. Transplant. 23, 328–335 (2008). [DOI] [PubMed] [Google Scholar]
- 10. Wetmore, J.B. et al A randomized trial of cinacalcet versus vitamin D analogs as monotherapy in secondary hyperparathyroidism (PARADIGM). Clin. J. Am. Soc. Nephrol. 10, 1031–1040 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Akizawa, T. et al Decreases in PTH in Japanese hemodialysis patients with secondary hyperparathyroidism: associations with changing practice patterns. Clin. J. Am. Soc. Nephrol. 6, 2280–2288 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fukagawa, M. et al Prescription patterns and mineral metabolism abnormalities in the cinacalcet era: results from the MBD‐5D study. Clin. J. Am. Soc. Nephrol. 7, 1473–1480 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fukuma, S. , Kurita, N. , Fukagawa, M. , Akizawa, T. & Fukuhara, S. Impact of cinacalcet introduction on MBD management: the MBD‐5D study in Japan. Kidney Int. Suppl. 3, 436–441 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tominaga, Y. , Kakuta, T. , Yasunaga, C. , Nakamura, M. , Kadokura, Y. & Tahara, H. Evaluation of parathyroidectomy for secondary and tertiary hyperparathyroidism by the Parathyroid Surgeons’ Society of Japan. Ther. Apher. Dial. 20, 6–11 (2016). [DOI] [PubMed] [Google Scholar]
- 15. Palmer, S.C. et al Cinacalcet in patients with chronic kidney disease: a cumulative meta‐analysis of randomized controlled trials. PLoS Med. 10, e1001436 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Block, G.A. et al Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N. Engl. J. Med. 350, 1516–1525 (2004). [DOI] [PubMed] [Google Scholar]
- 17. Gincherman, Y. , Moloney, K. , McKee, C. & Coyne, D.W. Assessment of adherence to cinacalcet by prescription refill rates in hemodialysis patients. Hemodial. Int. 14, 68–72 (2010). [DOI] [PubMed] [Google Scholar]
- 18. Harris, R.Z. , Salfi, M. , Sullivan, J.T. & Padhi, D. Pharmacokinetics of cinacalcet hydrochloride when administered with ketoconazole. Clin. Pharmacokinet. 46, 495–501 (2007). [DOI] [PubMed] [Google Scholar]
- 19. Nakashima, D. et al Effect of cinacalcet hydrochloride, a new calcimimetic agent, on the pharmacokinetics of dextromethorphan: in vitro and clinical studies. J. Clin. Pharmacol. 47, 1311–1319 (2007). [DOI] [PubMed] [Google Scholar]
- 20. Kawata, T. et al A novel calcimimetic agent, evocalcet (MT‐4580/KHK7580), suppresses the parathyroid cell function with little effect on the gastrointestinal tract or CYP isozymes in vivo and in vitro. PLoS One 13, e0195316 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fukagawa, M. , Shimazaki, R. Akizawa, T. & Evocalcet Study Group . Head‐to‐head comparison of the new calcimimetic agent evocalcet with cinacalcet in Japanese hemodialysis patients with secondary hyperparathyroidism. Kidney Int. 94, 818–825 (2018). [DOI] [PubMed] [Google Scholar]
- 22. Food and Drug Administration . Draft Guidance/Guidance for Industry. In vitro metabolism‐ and transporter‐mediated drug‐drug interaction studies.<https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM581965.pdf> (2017). Accessed April 25, 2018.
- 23. Food and Drug Administration . Draft Guidance/Guidance for Industry. Clinical drug interaction studies ‐ study design, data analysis, and clinical implications<https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf> (2017). Accessed April 25, 2018.
- 24. European Medicines Agency . Guideline on the investigation of drug interactions<http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf> (2012). Accessed April 25, 2018.
- 25. Ministry of Health, Labour and Welfare, Japan . Guideline of drug interaction studies for drug development and appropriate provision of information <https://www.pmda.go.jp/files/000206158.pdf>(2014). Accessed April 25, 2018.
- 26. Obach, R.S. , Walsky, R.L. & Venkatakrishnan, K. Mechanism‐based inactivation of human cytochrome p450 enzymes and the prediction of drug‐drug interactions. Drug Metab. Dispos. 35, 246–255 (2007). [DOI] [PubMed] [Google Scholar]
- 27. Richert, L. et al Tissue collection, transport and isolation procedures required to optimize human hepatocyte isolation from waste liver surgical resections. A multilaboratory study. Liver Int. 24, 371–378 (2004). [DOI] [PubMed] [Google Scholar]
- 28. David, P. et al Metabolic capacities in cultured human hepatocytes obtained by a new isolating procedure from non‐wedge small liver biopsies. Hum. Exp. Toxicol. 17, 544–553 (1998). [DOI] [PubMed] [Google Scholar]
- 29. Pichard, L. , Raulet, E. , Fabre, G. , Ferrini, J.B. , Ourlin, J.‐C. & Maurel, P. Human hepatocyte culture. Methods Mol. Biol. 320, 283–293 (2006). [DOI] [PubMed] [Google Scholar]
- 30. Vieira, M. et al Evaluation of various static in vitro‐in vivo extrapolation models for risk assessment of CYP3A inhibition potential of an investigational drug. Clin. Pharmacol. Ther. 95, 189–198 (2014). [DOI] [PubMed] [Google Scholar]
- 31. Huang, S.‐M. , Temple, R. , Throckmorton, D.D. & Lesko, L.J. Drug interaction studies: study design, data analysis, and implications for dosing and labeling. Clin. Pharmacol. Ther. 81, 298–304 (2007). [DOI] [PubMed] [Google Scholar]
- 32. Tachibana, T. , Kato, M. , Watanabe, T. , Mitsui, T. & Sugiyama, Y. Method for predicting the risk of drug‐drug interactions involving inhibition of intestinal CYP3A4 and P‐glycoprotein. Xenobiotica 39, 430–443 (2009). [DOI] [PubMed] [Google Scholar]
- 33. Fromm, M.F. , Busse, D. , Kroemer, H.K. & Eichelbaum, M. Differential induction of prehepatic and hepatic metabolism of verapamil by rifampicin. Hepatology 24, 796–801 (1996). [DOI] [PubMed] [Google Scholar]
- 34. Streetman, D.S. et al Combined phenotypic assessment of CYP1A2, CYP2C19, CYP2D6, CYP3A, N‐acetyltransferase‐2, and xanthine oxidase with the “Cooperstown cocktail”. Clin. Pharmacol. Ther. 68, 375–383 (2000). [DOI] [PubMed] [Google Scholar]
- 35. Chainuvati, S. et al Combined phenotypic assessment of cytochrome p450 1A2, 2C9, 2C19, 2D6, and 3A, N‐acetyltransferase‐2, and xanthine oxidase activities with the “Cooperstown 5 + 1 cocktail”. Clin. Pharmacol. Ther. 74, 437–447 (2003). [DOI] [PubMed] [Google Scholar]
- 36. Blakey, G.E. et al Pharmacokinetic and pharmacodynamic assessment of a five‐probe metabolic cocktail for CYPs 1A2, 3A4, 2C9, 2D6 and 2E1. Br. J. Clin. Pharmacol. 57, 162–169 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ryu, J.Y. et al Development of the “Inje cocktail” for high‐throughput evaluation of five human cytochrome P450 isoforms in vivo. Clin. Pharmacol. Ther. 82, 531–540 (2007). [DOI] [PubMed] [Google Scholar]
- 38. Nordmark, A. , Andersson, A. , Baranczewski, P. , Wanag, E. & Ståhle, L. Assessment of interaction potential of AZD2066 using in vitro metabolism tools, physiologically based pharmacokinetic modelling and in vivo cocktail data. Eur. J. Clin. Pharmacol. 70, 167–178 (2014). [DOI] [PubMed] [Google Scholar]
- 39. Williams, D. et al Use of a cocktail probe to assess potential drug interactions with cytochrome P450 after administration of belatacept, a costimulatory immunomodulator. Br. J. Clin. Pharmacol. 83, 370–380 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Armani, S. et al Drug interaction potential of osilodrostat (LCI699) based on its effect on the pharmacokinetics of probe drugs of cytochrome P450 enzymes in healthy adults. Clin. Drug Investig. 37, 465–472 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Efavirenz [package insert]. Morgantown, WV: Mylan Pharmaceuticals Inc. (2015).
- 42. Chen, Y. & Goldstein, J.A. The transcriptional regulation of the human CYP2C genes. Curr. Drug Metab. 10, 567–578 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Inhibition of cytochrome P450 (CYP) activities in human liver microsomes.
Figure S1. Predicted plasma evocalcet concentration‐time profiles after multiple oral administration of 12 mg evocalcet once a day.
Figure S2. Potential for evocalcet to induce human cytochrome P450 (CYP)1A2 mRNA levels in donors 1, 2, and 3.
Figure S3. Potential for evocalcet to induce human cytochrome P450 (CYP)2B6 mRNA levels in donors 1, 2, and 3.
Figure S4. Potential for evocalcet to induce human cytochrome P450 (CYP)2C8 mRNA levels in donors 1, 2, and 3.
Figure S5. Potential for evocalcet to induce human cytochrome P450 (CYP)2C9 mRNA levels in donors 1, 2, and 3.
Figure S6. Potential for evocalcet to induce human cytochrome P450 (CYP)2C19 mRNA levels in donors 1, 2, and 3.
Figure S7. Potential for evocalcet to induce human cytochrome P450 (CYP)3A4 mRNA levels in donors 1, 2, and 3.