

Abstract
The purpose of this study was to measure the electrocardiographic (ECG) effects of WCK 2349 (the L‐alanine ester prodrug of levonadifloxacin) at a supratherapeutic oral dose of 2,600 mg. A total of 48 healthy volunteers were randomized to treatment with placebo, WCK 2349, or oral moxifloxacin, 400 mg, in a crossover‐designed thorough QT study. A supratherapeutic mean maximum levonadifloxacin concentration (C max) of 43.3 μg/mL was achieved at 3.1 hours. A therapeutic dose of 1,000 mg b.i.d. in a previous study in patients resulted in a C max of 17.8 μg/mL. WCK 2349 exerted no significant effect on baseline‐ and placebo‐corrected QTcF (QT interval corrected for heart rate (HR) by the Fridericia formula), QRS, or PR interval. HR was transiently accelerated by a maximum of 14.4 (95% confidence interval, 11.80–16.92) beats per minute (bpm) at 3 hours. Concentration–effect modeling predicted a mean increase of 8.0 bpm at C max at the standard therapeutic dose. A therapeutic dose of 1,000 mg b.i.d. of WCK 2349 is not expected to cause clinically significant ECG effects, except for a possible transient increase in HR, which seems to be clinically insignificant.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Because WCK 2349 is a new antibiotic, its electrocardiographic (ECG) effects in humans have not been previously studied.
what question did this study address?
The primary purpose of this study was to fulfill the regulatory requirement to rigorously define the effects of the drug on cardiac repolarization. This requirement was successfully addressed, and the effect of WCK 2349 on other ECG variables was defined as well.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The study demonstrates that WCK 2349 has no cardiac safety liabilities and can be used without predosing or postdosing ECG. In addition, a modest increase in heart rate may occur.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The findings contribute our clinical approach to use of this new benzoquinolizine fluoroquinolone and offer an alternative to QT‐prolonging antibiotics.
WCK 2349 (I: alalevonadifloxacin) is a novel L‐alanine ester prodrug of levonadifloxacin, a benzoquinolizine fluoroquinolone being developed as an oral antibacterial agent that displays excellent coverage for methicillin‐resistant Staphylococcus aureus. This agent is being developed for the treatment of acute bacterial skin and skin structure infections and hospital‐acquired bacterial pneumonia caused by methicillin‐resistant S. aureus. Levonadifloxacin bears an unusual acidic side chain, 4‐hydroxy piperidine. Most of the marketed quinolones have basic side chains, usually terminating with an NH2 group. Attachment of the acidic side chain to the C‐8 position of the benzoquinolizine core enhances levonadifloxacin potency against Gram‐positive bacteria and overcomes resistance associated with quinolones. It also helps to enhance levonadifloxacin bactericidal activity against both Gram‐positive and Gram‐negative pathogens in an acidic environment. On the basis of the novelty in chemical structure of levonadifloxacin, worldwide patents have been granted.
The therapeutic dose of WCK 2349 is 1,000 mg b.i.d. In a previous study in 59 patients, this dose resulted in a mean plasma maximum concentration (C max) at steady state of 17.8 ± 3.67 μg/mL.1 The purpose of this investigation was to assess electrocardiographic (ECG) effects of WCK 2349 at supratherapeutic plasma concentrations, primarily to exclude a significant increase in QTcF (QT interval corrected for heart rate (HR) by the Fridericia2 formula), in accordance with the ICH‐E‐14 Guidance.3
Fluoroquinolones, like sitafloxacin, grepafloxacin, and moxifloxacin, have been associated with QT prolongation risk, primarily because of inhibition of potassium channels encoded by the human ether‐à‐go‐go‐related gene. Levonadifloxacin did not cause inhibition of the human ether‐à‐go‐go‐related gene potassium channel at the tested supratherapeutic concentration (≈27‐fold human free therapeutic C max). An ECG telemetry study in monkeys at ≈4.7 fold the therapeutic C max revealed no ECG effects of levonadifloxacin.4 The supratherapeutic dose of 2,600 mg used in this study was identified from a preliminary dose escalation that demonstrated safety and tolerance and achieved a mean C max of more than twofold above the mean C max during clinical use.
Patients and Methods
Study design
This study and the consent form, which was read and signed by each participant, were approved by Chesapeake Research Review (Columbia, MD), a commercial investigational review board. In a three‐armed, crossover study, 48 normal volunteers were randomly allocated to receive single oral doses of WCK 2349, 2,600 mg; placebo; or moxifloxacin, 400 mg orally. Subjects checked in to the clinical pharmacology unit (Spaulding Clinical Research, West Bend, WI) on day ‐1. Baseline ECG and other data were recorded, and treatment was administered on days 1, 6, and 11, allowing for a 5‐day washout between treatments. Subjects remain confined during the entire protocol. Subjects were discharged on day 12 and followed up by telephone 5 days later.
Subjects
Inclusion criteria for normal volunteers recruited to this study included an age range of 18–55 years, a body mass index between 18 and 33 kg/m2, and normal medical history, physical examination, 12‐lead ECG, and laboratory test results. Subjects were excluded for a resting pulse rate <45 beats per minute (bpm) or >100 bpm, a systolic blood pressure <90 or >150 mmHg, a diastolic blood pressure <50 or >95 mm Hg, QTcF >430 milliseconds (males) or >450 milliseconds (females), and use of medications within five elimination half‐lives of entry into the study.
Electrocardiography
Continuous 12‐lead ECG data were collected with a Mortara Surveyor telemetry system (Mortara Instrument, Milwaukee, WI). Three 10‐second ECGs separated by 1 minute from one another were extracted from the telemetry data stream at each timepoint (hours 0, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 16, 24, and 36). ECG intervals were measured automatically by the Mortara Veritas algorithm and then were overread in a high‐resolution computer workstation (Mortara Escribe) by board‐certified cardiologists blinded to treatment and chronology at the Spaulding Clinical Core ECG Laboratory. All recordings obtained in an individual subject were read with the same cardiologist blinded.
Pharmacokinetic sampling
Blood samples were collected in K2 EDTA vacutainers immediately after each ECG extraction timepoint in the three groups and centrifuged to obtain plasma, which was stored at −20°C. Moxifloxacin plasma samples were stored but not assayed, because ECG changes in the moxifloxacin arm behaved as expected in this study. Levonadifloxacin plasma concentrations were determined using a validated liquid chromatography–tandem mass spectrometry method.5
Pharmacokinetic parameters were determined by noncompartmental analysis using Phoenix WinNonlin (Certara, Mountain View, CA), Version 6.4.
Statistical analysis
Assuming a one‐sided 0.05 significance level, an average SD of at most 9 milliseconds (observed from previous studies), up to 12 postdose assessment timepoints, one baseline for each regimen, and three replicate ECGs at each ECG assessment timepoint, a total of 48 subjects were enrolled. At least 42 completers were anticipated to achieve at least 90% power to exclude a prolongation of ≥10 milliseconds for all postdose placebo‐ and baseline‐adjusted QTcF intervals (ddQTcF).6 We assumed that the prolongation from placebo has at most a 3‐milliseconds baseline‐adjusted mean. Use of the union–intersection test permits simultaneous testing at all postdose timepoints.
The primary end point was placebo‐adjusted change in QTcF from baseline, determined by a linear mixed‐effects model in which differences between WCK 2349 and placebo, and between moxifloxacin and placebo, were determined. Subject was included as a random effect, and treatment, hour, treatment×hour, and treatment sequence were included as fixed effects. The two‐sided upper 90% confidence interval (CI; equivalent to a one‐sided upper 95% CI) for the difference in QTcF changes from baseline between each treatment and placebo was calculated individually at each timepoint. A similar linear mixed‐effects model was used to estimate the slope of the plasma concentration relative to the placebo‐ and baseline‐adjusted QTcF.
Algebraic changes in ddHR, ddPR, and ddQRS were also examined. Because of a significant increase in ddHR, an ad hoc concentration–ddHR analysis, using a similar mixed‐effects model, was performed.
Assay sensitivity was assessed by the same mixed‐effects model limited to three timepoints (hours 1, 2, and 3). Adequate sensitivity was defined as a lower two‐side 90% CI >5 milliseconds at one or more of the three timepoints after adjustment for multiplicity.
Results
Enrollment and completion
A total of 48 healthy volunteers (mean age, 30.0 years) were enrolled. Sixteen were female (33%). All subjects completed treatment, except for one, who dropped out after receiving WCK 2349 and did not receive the other two treatments.
Drug exposure and pharmacokinetics
The desired supratherapeutic exposure of levonadifloxacin was achieved at a dose of 2,600 mg. The observed mean plasma C max was 43.3 μg/mL at 3.1 hours. The area under the curve (AUC) at 0–24 hours was 412.29 μg×h/mL. T max (time from dosing to maximum plasma concentration) was 3.1 hours. The plasma concentration–time plot is shown in Figure 1.
Figure 1.
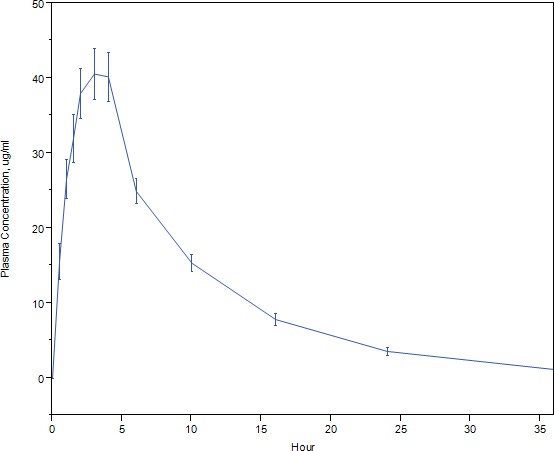
The plasma levonadifloxacin concentration–time profile. The mean levonadifloxacin plasma concentration and its 95% confidence interval at each timepoint are displayed against time in hours.
Moxifloxacin was administered as an active control to determine if the trial design and conduct were able to detect a QT effect. Samples for measurement of moxifloxacin plasma concentrations were drawn and stored, but not analyzed, because the expected effects of moxifloxacin were observed.
ECG effects
Figure 2 shows unadjusted ddQTcF for WCK 2349 and moxifloxacin at each timepoint. Because the cardiovascular changes are associated with C max, the ddQTcF of WCK 2349 at the C max timepoint of 3 hours was 2.3 milliseconds and ranged from 0.4 to 3.6 milliseconds near its vicinity timepoints between 0.5 and 10 hours. WCK 2349 demonstrated ddQTcF of ≈5 milliseconds at later timepoints (16, 24, and 36 hours), which could not be attributed to the parent drug because the plasma concentrations of levonadifloxacin were declining. Moxifloxacin at the 3‐hour timepoint showed a ddQTcF of 9.5 milliseconds, which ranged from 2.7 to 9.6 milliseconds between the 0.5‐ and 10‐hour time period.
Figure 2.
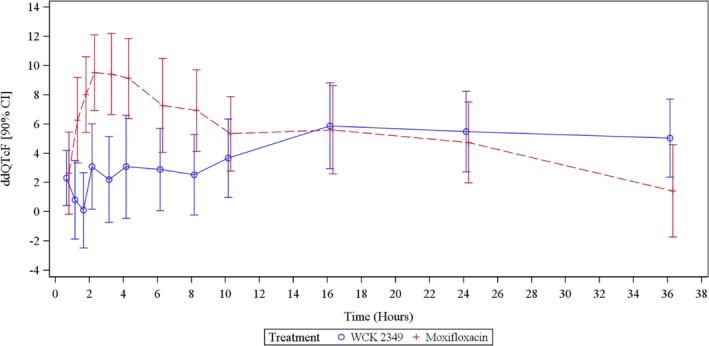
ddQTcF in the WCK 2349 and moxifloxacin arms. The error bars represent the 90% two‐sided confidence intervals. The mean hourly change in ddQTcF was modest and well below the level of regulatory concern (10‐millisecond upper confidence interval) in the WCK 2349 group, and the expected increase above this level was observed in the moxifloxacin group.
The modest increase in ddQTcF at 16–36 hours is not explained by late‐appearing metabolites. Levonadifloxacin sulfate is the major metabolite identified in the circulation, accounting for ≈40–50% of the levonadifloxacin plasma concentrations in humans. This metabolite was measured in this study and found to account for 30% of levonadifloxacin exposure (AUC). In telemetry studies in monkeys, in which the sulfate is the major metabolite, as in humans, no significant change was observed in cardiovascular parameters, including systolic and diastolic blood pressure, HR, and cardiac conduction times (PR, QRS, and QTc interval durations). Two glucuronide‐conjugated metabolites along with three oxidative metabolites have been detected in urine in trace amounts. Among these metabolites, only one oxidative metabolite was observed in trace amounts in the circulation.
Table 1 shows the mixed‐effects model ddQTcF and 90% CI at each timepoint. Mean ddQTcF for WCK 2349 was maximal at hour 16 (6.0 milliseconds). The upper bound of the 90% two‐sided CI remained <10 milliseconds throughout. In the case of moxifloxacin, the mean ddQTcF approached 10 milliseconds at several timepoints, and the lower boundary of the 90% CI exceeded 5 milliseconds at multiple timepoints.
Table 1.
ddQTcF (mixed‐effects model) by treatment and timepoint
| Timepoint (h) | WCK 2349 (N = 47) | Moxifloxacin (N = 47) | ||
|---|---|---|---|---|
| Mean | 90% CI | Mean | 90% CI | |
| 0.5 | 2.7 | 0.2 to 5.2 | 2.7 | 0.2 to 5.3 |
| 1 | 1.2 | −1.3 to 3.7 | 6.3 | 3.8 to 8.9 |
| 1.5 | 0.4 | −2.2 to 2.9 | 8.1 | 5.6 to 10.6 |
| 2 | 3.4 | 0.8 to 5.9 | 9.6 | 7.1 to 12.1 |
| 3 | 2.3 | −0.2 to 4.8 | 9.5 | 7.0 to 12.0 |
| 4 | 3.2 | 0.7 to 5.7 | 9.2 | 6.7 to 11.7 |
| 6 | 3.2 | 0.7 to 5.8 | 7.4 | 4.8 to 9.9 |
| 8 | 2.8 | 0.3 to 5.3 | 7.0 | 4.5 to 9.5 |
| 10 | 3.6 | 1.1 to 6.1 | 5.4 | 2.9 to 8.0 |
| 16 | 6.0 | 3.5 to 8.5 | 5.7 | 3.1 to 8.2 |
| 24 | 5.8 | 3.3 to 8.3 | 4.8 | 2.3 to 7.4 |
| 36 | 5.2 | 2.7 to 7.8 | 1.5 | −1.0 to 4.1 |
Data are given in milliseconds. CI = confidence interval; ddQTcF = placebo‐and baseline‐adjusted QT interval corrected for heart rate by the Fridericia formula.
Categorical change in QTc was a secondary end point in this study. Categorical changes in HR, PR, and QRS were also tabulated. QTcF exceeded 450 milliseconds on at least one ECG at no timepoints in the placebo group, in one subject at two timepoints in the WCK 2349 arm, and in one subject at one timepoint and two at another timepoint in the moxifloxacin group. QTcF did not exceed 480 or 500 milliseconds at any timepoint during any treatment. Change of QTcF over baseline did not exceed 30 or 60 milliseconds in any cases. Categorical changes in the other intervals were rare, with no suggestion of a treatment effect.
Assay sensitivity was assessed, with control for multiplicity by limiting the comparisons to four timepoints (hours 1, 2, 3, and 4) and using the Tukey–Kramer procedure. Two of the four timepoints excluded 5 milliseconds from the CI, confirming assay sensitivity. QTcF did not exceed 480 milliseconds in any subject at any timepoint, and ddQTcF did not exceed 30 milliseconds at any timepoint in the WCK 2349 treatment arm. As shown in Figure 3, the plasma concentration–ddQTcF slope of −0.017 msec/(μg×mL−1) was modestly negative. Assay sensitivity was confirmed by a lower one‐sided 95% CI for ddQTcF of >5 milliseconds at hours 4 and 6 after dosing of moxifloxacin.
Figure 3.
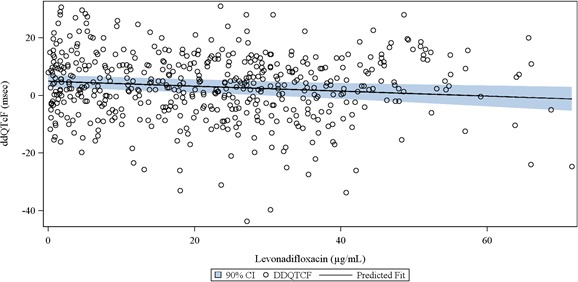
The plasma levonadifloxacin concentration–ddQTcF relationship. The linear mixed‐effects model showed a modest negative gradient (black fit line). The confidence band (blue shading) remained <10 milliseconds throughout.
The HR was the only ECG variable that changed to a clinically important extent in the WCK 2349 arm. Table 2 shows unadjusted ddHR, and the latter is shown graphically in Figure 4 . The maximum mean hourly change in ddHR in the WCK 2349 arm was 14.4 bpm (95% CI, 11.8–16.9 bpm) at 3.1 hours. The maximum observed HR in the WCK 2349 arm was 109 bpm, and HR exceeded 100 bpm on at least one ECG in three subjects, all at 6 hours, whereas in one of these subjects, it was also seen at 8 hours postdose. However, none of the subjects exceeded 120 bpm, a criterion for clinical significance. Mixed‐effects concentration–HR modeling was added as an ad hoc analysis because of the significant increase in HR. The plasma concentration–ddHR relationship was positive (Figure 5 ), with an intercept of 0.9456 bpm and a slope of 0.3937 bpm/(μg×mL−1).
Table 2.
ddHR (unadjusted) by treatment and timepoint
| Timepoint (h) | WCK 2349 (N = 47) | Moxifloxacin (N = 47) | ||
|---|---|---|---|---|
| Mean | 95% CI | Mean | 95% CI | |
| 0.5 | 4.1 | 2.43 to 5.81 | 1.9 | 0.10 to 3.60 |
| 1 | 7.8 | 5.41 to 10.09 | 2.3 | −0.31 to 4.82 |
| 1.5 | 10.3 | 8.04 to 12.50 | 2.7 | 0.39 to 4.95 |
| 2 | 13.4 | 10.75 to 16.12 | 2.5 | 0.38 to 4.66 |
| 3 | 14.4 | 11.80 to 16.92 | 2.1 | −0.06 to 4.33 |
| 4 | 13.7 | 11.17 to 16.20 | 1.9 | −0.09 to 3.92 |
| 6 | 11.4 | 8.99 to 13.80 | 1.7 | −0.73 to 4.07 |
| 8 | 11.2 | 8.98 to 13.50 | 3.5 | 1.45 to 5.52 |
| 10 | 7.8 | 5.18 to 10.48 | 1.3 | −1.25 to 3.86 |
| 16 | 4.2 | 1.74 to 6.76 | 2.6 | 0.38 to 4.73 |
| 24 | 2.7 | 0.46 to 5.03 | 1.3 | −1.03 to 3.68 |
| 36 | 1.3 | −1.39 to 3.90 | −0.9 | −3.42 to 1.54 |
Data are given as beats per minute. CI = confidence interval; ddHR = placebo‐ and baseline‐adjusted heart rate.
Figure 4.
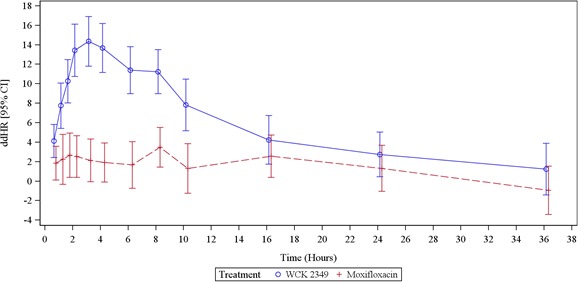
ddHR in the WCK 2349 arm. The mean hourly change in ddHR was consistently positive in the WCK 2349 arm. The mean change reached 14.4 bpm at 3 hours. ddHR changes in the moxifloxacin arm were also generally positive but to a lesser extent.
Figure 5.
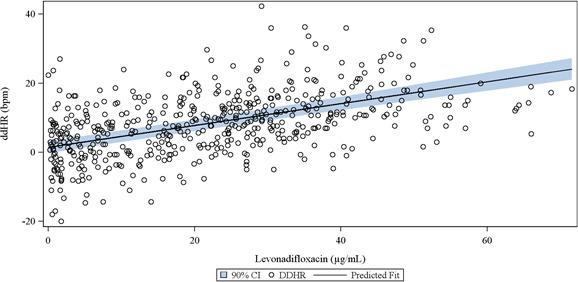
The plasma levonadifloxacin concentration–ddHR relationship. The linear mixed‐effects model showed a positive gradient; 95% confidence bounds are displayed.
PR and QRS interval durations were not clinically significantly affected by WCK 2349. PR change from baseline, corrected for the placebo change, was consistently negative. The largest change at any timepoint was −6.3 milliseconds at hour 6. QRS interval change was variably positive and negative, with a maximum change of −2.4 milliseconds at hour 4.
Overall safety
Overall, WCK 2349, administered orally at a dose of 2,600 mg, was well tolerated. Of 48 subjects, 26 reported at least one treatment‐emergent adverse effect (TEAE) during the study. All TEAEs were mild in severity, except for a single moderate TEAE of prolonged QT interval in a subject receiving WCK 2349 at dose of 2,600 mg that was considered possibly related to study drug. This subject was discontinued from the study because of this TEAE. There were no deaths or serious adverse events during the study. No clinically significant trends were observed in clinical laboratory results, vital sign measurements, safety 12‐lead ECG results, or physical examination findings.
Discussion
An insignificant effect of WCK 2349 on QTcF at an oral dose of 2,600 mg, on the basis of both the mixed‐effects model of mean ddQTcF at each timepoint and the ddQTcF–plasma concentration model, was the primary finding in this study. The therapeutic mean plasma C max of levonadifloxacin is 25.2 μg/mL (range, 15.7–36.76 μg/mL), and the mean AUC at 0–12 hours is 153.3 μg×h/mL. At the single supratherapeutic dose used in this study (2,600 mg), the plasma levonadifloxacin mean C max was 43.3 μg/mL (range, 27.09–71.56 μg/mL) and the AUC at 0–24 hours was 412.3 μg×h/mL, resulting in C max and AUC values of 1.7‐ and 2.7‐fold of therapeutic exposures, respectively. Because WCK 2349 is administered intravenously in the hospital setting, overdosing will be rare. There are no known metabolic inhibitors, and renal and hepatic impairment do not cause a significant increase in plasma concentration. Thus, the supratherapeutic dose of 2,600 mg used in this study was adequate to cover all real‐use scenarios and clinically recommended doses of ≤1,000 mg are not anticipated to affect cardiac repolarization. Several fluoroquinolones, macrolides, and other antibiotic classes prolong QT and require cautious use in patients with preexisting or a risk of QT prolongation.7 Patients receiving antibiotics are often unusually vulnerable to QT prolongation and ventricular arrhythmia because of metabolic inhibitor interactions, electrolyte abnormalities, or renal, cardiac, and hepatic comorbidities.8, 9, 10
WCK 2349 caused a substantial, transient, asymptomatic increase in HR. The maximum change was 14.4 bpm at the supratherapeutic dose of 2,600 mg. Modeling predicts a maximum increase at steady state of 8.0 bpm in patients receiving the clinical dose of 1,000 mg b.i.d., although previous studies with less robust ECG did not show an increase in HR. At least six other macrolide and fluoroquinolone antibiotics are known to increase HR.11 Although increase in HR was observed with these antibiotics at supratherapeutic levels, none of them manifested any serious adverse events when administered to infected patients at their therapeutic doses. The mechanism responsible for HR elevation is not known for WCK 2349 or the others. Given that the HR increase was accompanied by a decrease in the PR interval, an autonomic mechanism (sympathetic activation, vagal withdrawal, or both) is suggested. Other possibilities would include If (the hyperpolarization‐activated pacemaker current) agonism, because this current regulates both HR and AV nodal conduction velocity.12, 13
It is not known if WCK 2349 could cause HR acceleration in the clinical setting in which infected patients are usually febrile and already tachycardic, although tachycardia after WCK 2349 has not been observed in patients to date. It is possible that further autonomic adjustments resulting in additional rate increase would be minimal or absent in the presence of infection‐related tachycardia.
We conclude that WCK 2349 has no significant ECG effects at the supratherapeutic dose, except for its HR effect. However, in the various phase I and phase II studies performed with oral WCK 2349 and intravenous WCK 771 (L‐ arginine salt of levonadifloxacin), no adverse events related to HR increase have been reported so far. WCK 2349 provides a new antibiotic choice for patients infected with methicillin‐susceptible and methicillin‐resistant strains of S. aureus and other organisms. The 1,000 mg b.i.d. therapeutic dose of WCK 2349 is not anticipated to affect cardiac conduction or repolarization. Thus, WCK 2349 can be administered in patients who are at risk of QT prolongation.
Ethics and Consent
The study was approved by an Institutional Review Board (Chesapeake Research Review, Columbia, MD), and each subject signed a written consent form for participation in the study.
Funding
No funding was received for this work.
Conflict of Interest
J.W.M. is a consultant to Spaulding Clinical Research and to Wockhardt. R.C., A.P., R.G., and A.B. are or were employees of Wockhardt.
Authors Contributions
J.W.M., R.C., and A.B. wrote the manuscript; J.W.M., R.C., A.P., R.G., and A.B. designed the research; J.W.M. performed the research; J.W.M. analyzed the data; R.C. contributed new reagents/analytical tools.
ClinicalTrials.gov Identifier: NCT02217930, registered August 15, 2014; URL: https://clinicaltrials.gov/ct2/results?cond=%26term=NCT02217930%26cntry=%26state=%26city=%26dist=
References
- 1. Wockhardt Bio AG . WCK 2349 Investigator's Brochure. Version Number 70/WCK 2349. 2016:62. Wockhardt, Mumbai, India. [Google Scholar]
- 2. Fridericia, L.S. Die systolendauer im elektrokardiogramm bei normalen menschen und bei herzkranken. Acta Med. Scand. 53, 489 (1920). [Google Scholar]
- 3. US Food and Drug Administration . Guidance for industry: E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs <www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073153.pdf.> (2005). [PubMed]
- 4. Wockhardt Bio AG . WCK 771 Investigator's Brochure. Version Number V 60/WCK 771. 2016:93. Wockhardt, Mumbai, India. [Google Scholar]
- 5. Yeole, R.D. et al Simple liquid chromatography‐tandem mass spectrometry method for determination of novel anti‐methicillin‐resistant Staphylococcus aureus fluoroquinolone WCK 771 in human serum. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 846, 306–312 (2007). [DOI] [PubMed] [Google Scholar]
- 6. Zhang, J. & Machado, S.G. Statistical issues including design and sample size calculation in thorough QT/QTc studies. J. Biopharm. Stat. 18, 451–467 (2008). [DOI] [PubMed] [Google Scholar]
- 7. Mason, J.W. Antimicrobials and QT prolongation. J. Antimicrob. Chemother. 72, 1272–1274 (2017). [DOI] [PubMed] [Google Scholar]
- 8. Mortensen, E.M. et al Association of azithromycin with mortality and cardiovascular events among older patients hospitalized with pneumonia. JAMA 311, 2199–2208 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Owens, R.C. Jr. QT prolongation with antimicrobial agents: understanding the significance. Drugs 64, 1091–1124 (2004). [DOI] [PubMed] [Google Scholar]
- 10. Simko, J. , Csilek, A. , Karaszi, J. & Lorincz, I. Proarrhythmic potential of antimicrobial agents. Infection 36, 194–206 (2008). [DOI] [PubMed] [Google Scholar]
- 11. Mason, J.W. & Moon, T.E. Moxifloxacin increases heart rate in humans. Antibiotics (Basel). 6, E5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Herrmann, S. , Layh, B. & Ludwig, A. Novel insights into the distribution of cardiac HCN channels: an expression study in the mouse heart. J. Mol. Cell. Cardiol. 51, 997–1006 (2011). [DOI] [PubMed] [Google Scholar]
- 13. Verrier, R.L. et al If inhibition in the atrioventricular node by ivabradine causes rate‐dependent slowing of conduction and reduces ventricular rate during atrial fibrillation. Heart Rhythm 11, 2288–2296 (2014). [DOI] [PubMed] [Google Scholar]