

Abstract
Orally dispersible tablet (ODT) formulations of levo praziquantel (L‐PZQ) and racemic PZQ (rac‐PZQ) are being developed to treat schistosomiasis in preschool‐aged children. Two crossover studies (N = 32 and 36, respectively) assessed the relative bioavailability of these ODTs vs. Cysticide in adults. Bioavailability for L‐PZQ of ODT rac‐PZQ and Cysticide at 40 mg/kg was comparable (L‐PZQ area under the concentration‐time curve from zero to infinity (AUC 0–∞) test/reference ratio (90% confidence interval (CI)): 96% (84–111%)), whereas relative bioavailability of ODT L‐PZQ 20 mg/kg was ~40% that of Cysticide 40 mg/kg (test/reference: 40% (35–46%)). AUC 0‐∞ and peak plasma concentration (Cmax) were highly variable in both studies. For both ODTs, L‐PZQ AUC 0–∞ showed greater than dose‐proportional increase over the ranges tested and a significant food effect. Safety was comparable among formulations. The lower bioavailability of ODT L‐PZQ, as well as the high variability and nondose‐proportionality of pharmacokinetic (PK) parameters, highlighted the need for a dedicated pediatric dose‐finding study for the selection of the most appropriate formulation and dose (L‐PZQ ODT or rac‐PZQ ODT).
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
ODT formulations of L‐PZQ and rac‐PZQ are being developed to treat schistosomiasis in children < 4 years old. No PK data were available for these formulations.
what question did this study address?
Relative bioavailabilities (in adults) of racemate and of L‐PZQ ODTs were compared with that of a reference (racemic) PZQ formulation.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
For ODT rac‐PZQ, relative bioavailability was comparable to that of reference PZQ. For ODT L‐PZQ, it was ~40% that of the reference. With both ODTs, PK profiles were erratic and exposure increased nondose‐proportionally.
Results suggest a possible PK interaction between L‐PZQ and D‐PZQ when given as racemate. There were no new safety signals with either formulation.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
It may not be possible to construct appropriate models for pediatric extrapolation for products like PZQ with variable and erratic PK profiles. Limits in the understanding of dose‐exposure relationships for PZQ and its enantiomers have also become apparent.
Schistosomiasis is one of the most prevalent parasitic diseases in Africa and is very important in terms of its public health burden and economic impact. It is caused by parasitic flatworms.1 Globally, it affects at least 206.5 million people, mainly in Africa;2, 3 ~50% of cases involve children. More than 700 million people are at risk of infection via contaminated water. Schistosomiasis accounts for some 20,000 deaths yearly, and it is the second most important tropical disease (after malaria) in terms of morbidity. Disease control relies on preventive chemotherapy, targeting the entire at‐risk population with single oral doses of praziquantel (PZQ) 40 mg/kg annually. PZQ (2‐(cyclohexalcarbanoyl)‐1,2,3,6,7,11b‐hexahydro‐4H‐pyrazino(2,1‐alpha)isoquinolin‐4‐one), developed in the 1970s, is on the World Health Organization (WHO) Model List of Essential Medicines for treatment of schistosomiasis in adults and school‐aged children and is currently the only WHO‐recommended drug for schistosomiasis in humans.4
Preschool‐aged children (PSAC) account for 10–20 million cases of schistosomiasis annually but are presently not included in control programs because the absence of an appropriate pediatric formulation could make their inclusion disruptive and unsafe. In 2012, the WHO formally acknowledged that infants and PSAC are at significant risk of schistosomiasis and qualify for PZQ treatment. The WHO presently recommends that PSAC are treated for schistosomiasis within child health services by weight monitoring: they should be immunized, dewormed, and given micronutrient supplements.4
The extent and severity of schistosomiasis in PSAC are poorly understood.6, 7 However, studies in Nigeria,8, 9, 10 Ghana,11 and Uganda12 identified schistosome infection in very early childhood. In a study in Niger, prevalence among children < 5 years old exceeded the threshold at which large‐scale PZQ administration is recommended.13
Although PZQ is registered for use in children ≥ 4 years old, clinical data on younger children are very limited. In particular, the effective dose in younger children is unknown, as pharmacokinetics (PKs) and dose‐finding studies are scarce.14 Moreover, PZQ treatment poses particular challenges in this population due to the size of existing tablets and the drug's extreme bitterness. Crushed tablets have been used in pediatric trials,15, 16 but dosing is imprecise and the effect of crushing on bioavailability is unknown. Crushing may also increase perception of bitterness. A formulation permitting accurate dosing and enhanced compliance in young children would address a clear medical need.
The Pediatric Praziquantel Consortium (PPC), an international not‐for‐profit partnership, was established in 2012. The PPC aims to reduce the global disease burden of schistosomiasis by addressing the medical needs of infected PSAC. Its mission is to develop, register, and provide access to a pediatric PZQ formulation suitable for this age group.
The PPC is investigating orally dispersible/disintegrating tablet (ODT) formulations of 150 mg PZQ that can be administered dispersed in water, permitting more precise dosing in young children than the existing 500–600 mg tablets.
PZQ is a racemic mixture of levo‐PZQ (L or R‐ (‐)‐PZQ, responsible for anthelminthic activity) and dextro‐PZQ (D or S‐ (+)‐PZQ, thought to be responsible for bitterness) in equal proportions. It was believed that a formulation containing only the active enantiomer Levo‐PZQ (L‐PZQ) would be more palatable than racemic PZQ (rac‐PZQ) and could be efficacious at lower doses, potentially leading to improved tolerability and benefit/risk balance.
Accordingly, two ODT formulations were developed: one containing equal proportions of L‐PZQ and D‐PZQ (ODT rac‐PZQ) and the other containing L‐PZQ alone (ODT L‐PZQ). Both contain 150 mg of active ingredient (either racemic or L‐PZQ) with mannitol and sweeteners for taste masking. The clinical development program of the ODTs initially considered the pediatric PK/pharmacodynamic extrapolation strategy described by the International Conference of Harmonisation guideline on clinical investigation of medicinal products in the pediatric population.17 The purpose was to identify the targeted drug exposure of each of the new formulations in PSAC by using modeling and simulation tools on adult exposure data, in order to predict the effective dose in this population. To this end, two open‐label, randomized phase I studies were conducted in healthy adults to assess relative bioavailability of the ODT formulations compared with a reference formulation (Cysticide 500 mg oral tablets; Merck KGaA, Darmstadt, Germany). EMR200585‐001 was a single‐dose, four‐period crossover study comparing ODT rac‐PZQ with Cysticide, and EMR200661‐001 was a single‐dose, five‐period crossover study comparing ODT L‐PZQ with Cysticide (Figure 1). Both were performed at PAREXEL International's Early Phase Clinical Unit Bloemfontein (University of the Free State, Bloemfontein, South Africa).
Figure 1.
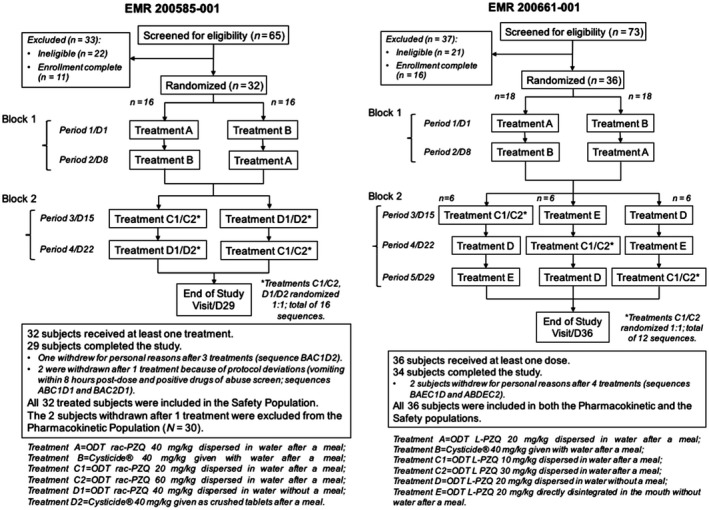
Study design and subject disposition. ODT, orally dispersible tablet.
The primary objective was to assess relative bioavailability of L‐PZQ in the ODTs dispersed in water compared with Cysticide after single oral administration of 40 mg/kg (for ODT rac‐PZQ and Cysticide) or 20 mg/kg (for ODT L‐PZQ) under fed conditions. Secondary objectives included investigation of dose proportionality, food effect on L‐PZQ bioavailability, and the safety, tolerability, and palatability of the new formulations. EMR200661‐001 also investigated the effect of disintegrating the ODT directly in the mouth (without water) on L‐PZQ bioavailability. EMR200585‐001 assessed relative bioavailability of crushed vs. noncrushed Cysticide tablets.
MATERIALS AND METHODS
Both protocols and related consent documents were approved by the University of the Free State Ethics Committee and by the South African Medicines Control Council. Both studies were conducted in accordance with the Declaration of Helsinki, the International Council for Harmonization Note for Guidance on Good Clinical Practice, and applicable regulations.
Selection and description of participants
Both studies enrolled healthy men 18–55 years old, weighing ≥ 55.0 to < 95.0 kg. Eligibility criteria were the same for both trials. This ensured good comparability of the demographic characteristics across both PK studies and allowed high‐density PK sampling, which would not be feasible in pediatric populations.
Overall study design
The study designs for EMR200585‐001 and EMR200661‐001 differed only in the number of periods, and the treatments to be compared (Figure 1). Volunteers were screened after providing written informed consent. Eligible subjects were admitted on day −1 and randomized to a treatment sequence. They received the first treatment on the morning of day 1, followed by a 24‐hour in‐house period for observation and PK sampling. Safety assessments occurred predose and 4 and 24 hours postdose. PK samples were collected predose and 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 5.5, 6, 8, 12, 16, and 24 hours postdose (± 5% in minutes). Subsequent treatment periods followed the same schedule, with a washout of ≥ 7 days between periods. An end‐of‐trial visit took place 3–10 days after the last treatment.
Fasting subjects fasted overnight before treatment administration. Fed subjects consumed a standardized breakfast between 30 and 10 minutes before dosing, consisting of 100 g All‐Bran flakes, 40 g bread, 250 g low‐fat milk, 5 g Marmite (yeast extract), 10 g white sugar; 16.38% protein, 9.92% fat, 73.72% carbohydrates; 658.6 calories. All subjects fasted for 4 hours postdose and received a standardized lunch after 4 hours. They were instructed not to consume alcohol, caffeine, or xanthine‐containing products from 48 hours before until 24 hours after each administration and to minimize physical activity while hospitalized.
Treatments
Three PZQ formulations were investigated: ODT rac‐PZQ (Farmaguinhos/Fiocruz, Brazil; 150 mg ODTs), ODT L‐PZQ (Merck KGaA; 150 mg ODTs), and a reference racemic PZQ formulation (Cysticide; Merck KGaA; 500 mg tablets).
Randomization
In both studies, treatment periods were divided into two blocks to minimize impact of dropouts: sequences were arranged to complete assessment of relative bioavailability in the first two periods (block 1), with investigation of different doses and modes of administration in subsequent periods (block 2). Randomization was separate for the two blocks, resulting in a total of 16 sequences for EMR200585‐001 and 12 for EMR200661‐001 (Figure 1).
Outcomes
The primary end point of both studies was the time zero to infinity area under the concentration‐time curve (AUC0–∞) of L‐PZQ. Secondary end points included peak plasma concentration (Cmax), time of maximum plasma concentration (Tmax), apparent terminal half‐life (t½), AUC0–t, and relative bioavailability for L‐PZQ, D‐PZQ, and racemate PZQ, as well as AUC0–∞ of D‐PZQ, racemate PZQ, and (L) trans‐OH‐PZQ.
Safety was assessed through the recording of adverse events, vital signs, 12‐lead electrocardiograms (ECGs), and laboratory investigations (chemistry, hematology, and urinalysis).
Subjects assessed palatability using modified 100‐mm visual analogue scale (VAS) scales incorporating facial hedonic scales. Flavor, smell, sweetness, and overall liking of the medicine were rated immediately after dosing, and taste in the mouth and acceptability to swallow 2–5 minutes later.
Analytical methods
L‐PZQ, D‐PZQ, and (L) trans‐OH‐PZQ were measured using validated enantioselective assays. Racemate PZQ levels were calculated by addition of L‐PZQ and D‐PZQ levels. The lower limit of quantification for all analytes was 5 ng/mL.
Pharmacokinetic analysis
PK parameters were calculated by Merck KGaA's Department of Quantitative Pharmacology (Darmstadt, Germany) using Phoenix/WinNonlin (version 6.3 and were evaluated using standard noncompartmental methods).
Dose‐dependent parameters (AUC and Cmax) were adjusted to the planned dose through multiplication by the planned dose divided by the actual dose. This was necessary because the different formulation strengths of the tablets resulted in slight dose differences between test and reference treatments and deviations from the nominal dose. Only whole tablets were given because division of the reference 500 mg tablets would not have been sufficiently precise.
AUCs were calculated using the mixed log‐linear trapezoidal method. Actual sampling time was used for PK evaluation; where it was missing, scheduled time was used. No other missing data were imputed. Predose samples were considered to be taken at zero hours. Concentrations below lower limit of quantification occurring before the last quantifiable data point were taken as zero when calculating the AUC of single‐dose profiles; those occurring thereafter were not considered in the calculation of the terminal elimination rate.
Statistics
No formal hypotheses were defined. Sample size was based on the expected precision of the treatment ratios to be estimated. A precision of 20% or better was considered reasonable. Assuming within‐subject coefficients of variation of 51% for Cmax (data on file) and 26.5% for AUC18 and taking the width of the 90% confidence intervals (CIs) for the ratio of test/reference geometric means for Cmax and AUC as the measure of precision, the 90% CIs would be within the intervals (0.88*R, 1.13*R) for AUC and (0.80*R, 1.25*R) for Cmax with 26 evaluable subjects, where R is the estimated treatment ratio. EMR200585‐001 assumed a dropout rate of ~20%, requiring randomization of 32 subjects. EMR200661‐001 assumed a dropout rate of ~27%; to ensure balance among sequences, either 30 or 36 subjects had to be randomized.
Statistical analysis was performed using SAS for Windows (release 9.1.2 or later; SAS Institute, Cary, NC) according to analysis plans finalized before database lock. All data were evaluated as observed, with no imputation of missing values.
The PK populations included all treated subjects who had PK data available for the first two treatment periods and no deviations likely to affect comparability of PK results. The safety populations included all subjects who received at least one treatment and had follow‐up safety data.
Statistical analyses (including summary statistics) were performed on the adjusted parameter values for dose‐dependent parameters.
For the analysis of relative bioavailability, a mixed model was applied to log‐transformed AUC0–∞, with treatment and sequence as fixed effects and subject within sequence as a random effect. Based on the residual (within‐subject) variation, 90% CIs for the ratios of geometric means were calculated. AUC0–t and Cmax for all analytes and AUC0–∞ for D‐PZQ and racemate PZQ were analyzed in the same way. For Tmax, the Hodges‐Lehmann shift estimate was given for treatment difference together with the 90% CI according to Tukey. This methodology was used for all relevant estimates of treatment differences. Secondary PK parameters and plasma concentrations were summarized descriptively.
Safety and palatability were analyzed descriptively. In EMR200661001, palatability scores were compared between treatments using SAS PROC MIXED with sequence, number of tablets administered, and treatment arm in the model.
RESULTS
EMR200585‐001 (ODT rac‐PZQ vs. Cysticide)
Subject population
Sixty‐five male volunteers were screened between January 21, 2015 and February 11, 2015, and 32 were randomized (2 per sequence). All 32 received at least one treatment (separated by 7‐day washouts) and were included in the safety population. Twenty‐nine subjects completed the study. One subject withdrew after three treatments for personal reasons, and two were withdrawn after one treatment due to protocol deviations, and were excluded from the PK population (Figure 1).
Nineteen randomized subjects (59.4%) were black, 11 (34.4%) were white, and 2 (6.3%) were of “other” ethnicities. Mean (SD) age, weight, and body mass index were 29.0 (6.4) years, 73.46 (10.91) kg, and 24.13 (2.72) kg/m2, respectively.
PK results
After administration of either ODT rac‐PZQ (treatment A) or Cysticide (treatment B), mean plasma L‐PZQ concentrations were approximately a quarter of those of D‐PZQ despite the 1:1 ratios of the enantiomers in the tablets (Figure 2). Plasma concentrations of the metabolite L‐trans‐OHPZQ were > 70times higher than those of the parent but were comparable between formulations (data not shown).
Figure 2.
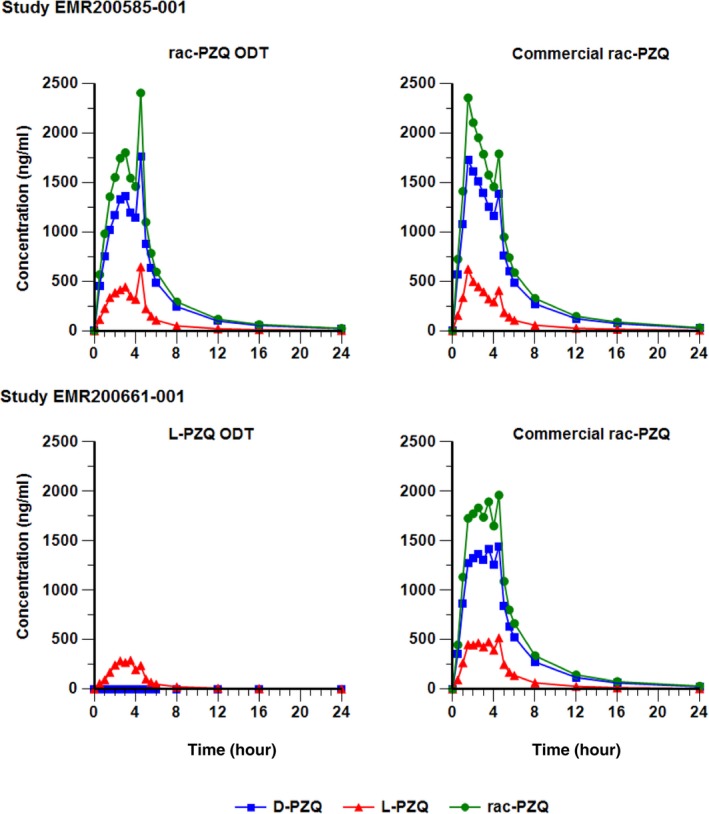
Mean concentration‐time profiles of Levo‐praziquantel (L‐PZQ), dextro‐PZQ (D‐PZQ), and total racemate PZQ after administration of orally dispersible tablet (ODT) racemic PZQ (rac‐PZQ; EMR200585 001), ODT L‐PZQ (EMR200661 001), or Cysticide (both studies; pharmacokinetic populations, linear scale).
The shapes of the mean L‐PZQ profiles differed somewhat between formulations, with a Tmax of 3.0 hours after administration of ODT rac‐PZQ and 1.5 hours after Cysticide (ranges were 1–4.5 hours for both formulations; Table 1). This difference was not statistically significant. Interestingly, additional small peaks at 4.5 hours postdose were frequent with both formulations, possibly due to the meal provided at 4 hours. The shapes of individual concentration/time profiles were erratic and varied considerably between subjects (Figure 3).
Table 1.
Summary of L‐PZQ pharmacokinetic parameters (EMR 200585‐001 and EMR 200661‐001; PK populations)
| Study EMR 200585‐001 | Study EMR 200661‐001 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L‐PZQ | A | B | C1 | C2 | D1 | D2 | A | B | C1 | C2 | D | E |
| Dose mg/kg | 40 | 40 | 20 | 60 | 40 | 40 | 20 | 40 | 10 | 30 | 40 | 40 |
| IMP | Rac‐ODT | Cys | Rac‐ODT | Rac‐ODT | Rac‐ODT | Cys | L‐ODT | Cys | L‐ODT | L‐ODT | L‐ODT | L‐ODT |
| Condition | Fed | Fed | Fed | Fed | Fasted | Crushed,fed | Fed | Fed | Fed | Fed | Fasted | Dispersed in mouth, fed |
| N | 30 | 30 | 14/15 | 15 | 14 | 14/15 | 36 | 36 | 17/18 | 17 | 35 | 36 |
| Cmax (ng/mL) | 881.3 | 762.6 | 154.6 | 1,548 | 189.3 | 446.3 | 378.8 | 727.3 | 89.9 | 1,051.8 | 131.1 | 471.2 |
| (57.2) | (59.5) | (80.4) | (32.6) | (104.0) | (87.6) | (114.1) | (63.3) | (92.2) | (83.6) | (110.6) | (99.5) | |
| 245–2,860 | 218–2,010 | 41.6–542 | 1,020–2,560 | 50.4–1,030 | 65.8–1,150 | 68.4–2,038.1 | 222.4–2,249.4 | 28.8–388.3 | 307.4–3,869.1 | 14.9–648.6 | 71.3–1,694.0 | |
| Tmax (h) | 3.00 | 1.50 | 4.00 | 3.00 | 4.25 | 2.50 | 2.50 | 2.50 | 2.25 | 3.00 | 2.00 | 4.00 |
| 1.00–4.50 | 1.00–4.50 | 1.50–5.00 | 0.50–4.50 | 0.50–4.50 | 0.50–8.00 | 0.50–4.50 | 1.00–4.50 | 1.00–4.50 | 1.00–4.50 | 0.50–4.50 | 0.50–5.00 | |
| AUC0–∞ (h*ng/mL) | 1,969.6 | 2,047.9 | 345.4 | 4,871.3 | 924.9 | 1,537.7 | 825.2 | 2,066.0 | 216.7 | 2,324.9 | 506.2 | 954.5 |
| (47.2) | (60.2) | (69.5) | (42.2) | (67.7) | (70.6) | (101.0) | (64.7) | (102.8) | (76.4) | (100.0) | (96.4) | |
| 561–3,710 | 488–4795 | 129–1,017 | 1,994–8,639 | 326–2,790 | 678–5,294 | 222–4,015 | 665–6,466 | 73–1,147 | 880–10,054 | 59–2,049 | 152–4,532 | |
| t1/2 (h) | 3.305 | 3.830 | 1.916 | 4.202 | 4.168 | 3.072 | 2.777 | 3.827 | 1.604 | 3.361 | 2.837 | 2.625 |
| (49.1) | (46.8) | (72.2) | (37.8) | (34.9) | (43.5) | (53.9) | (46.3) | (88.6) | (31.9) | (62.3) | (65.2) | |
| 1.41–10.62 | 1.68–14.56 | 0.83–7.23 | 2.27–8.17 | 2.25–7.51 | 1.69–7.25 | 1.01–6.49 | 1.44–10.1 | 0.623–7.58 | 1.82–5.46 | 0.869–24.9 | 0.452–6.65 | |
| CL/f (L/h) | 716.8 | 691.3 | 2,028.4 | 438.5 | 1,466.9 | 971.1 | 1665 | 667.3 | 3,091 | 923.5 | 2,729 | 1,440 |
| (45.0) | (57.9) | (60) | (45.8) | (60.6) | (69.5) | (94.3) | (60.1) | (92.9) | (71.4) | (95.4) | (89.3) | |
| 332–2,673 | 248–2,244 | 685–4,727 | 213–1,195 | 590–3,374 | 261–2,263 | 419–7,100 | 194–1,900 | 770–8,940 | 254–2,760 | 826–29,200 | 255–8,530 | |
AUC0–∞, area under the plasma concentration‐time curve from zero to infinity; CL/f, apparent clearance; Cmax, maximum plasma concentration; Cys, Cysticide; EMR, electronic medical record; L‐PZQ, levo‐praziquantel; N, number of subjects included in the analysis; ODT, orally dispersible tablet; PK, pharmacokinetic; rac‐PZQ, racemate praziquantel; t1/2, terminal half‐life; Tmax, time to maximum concentration.
The table presents L‐PZQ PK parameters by geometric mean, GeoCV%, and range (minimum‐maximum); for Tmax median and range are presented. Dose‐dependent parameters are adjusted to the planned dose.
For study EMR 200585–001: A = ODT rac‐PZQ 40 mg/kg dispersed in water, fed; B = Cysticide 40 mg/kg given with water, fed; C1 = ODT rac‐PZQ 20 mg/kg dispersed in water, fed; C2 = ODT rac‐PZQ 60 mg/kg dispersed in water, fed; D1 = ODT rac‐PZQ 40 mg/kg dispersed in water, fasted; D2 = Cysticide 40 mg/kg as crushed tablets, fed.
For study EMR 200661001: A = ODT LPZQ 20 mg/kg dispersed in water, fed; B = Cysticide® 40 mg/kg given with water, fed; C1 = ODT L‐PZQ 10 mg/kg dispersed in water, fed; C2 = ODT L‐PZQ 30 mg/kg dispersed in water, fed; D = ODT L‐PZQ 20 mg/kg dispersed in water, fasted; E = ODT L‐PZQ 20 mg/kg directly disintegrated in the mouth without water, fed.
Figure 3.
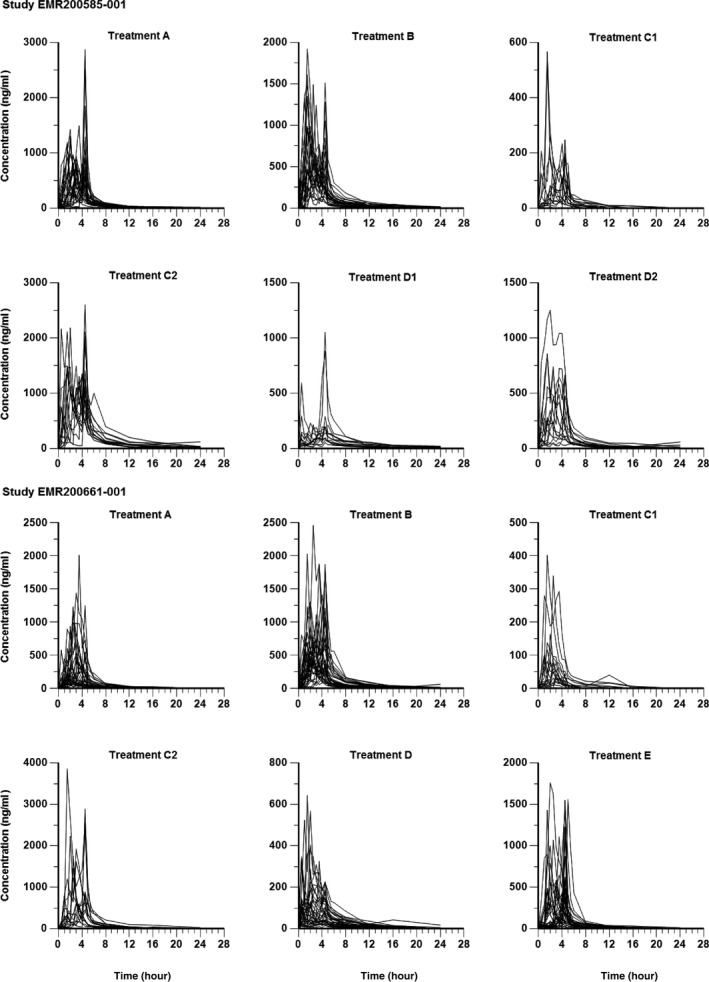
Plasma concentration‐time profiles for levo‐praziquantel by treatment, linear scale (EMR 200585 001 and EMR 200661‐001, pharmacokinetic populations).
ODT rac‐PZQ dispersed in water (treatment A) and Cysticide (treatment B) showed comparable relative bioavailability after single oral administration of 40 mg/kg under fed conditions. The test/reference ratio for the primary end point, the AUC0–∞ of the active enantiomer L‐PZQ, was 96% (90% CI: 84–111%; Table 2). For D‐PZQ and rac‐PZQ, the corresponding test/reference ratios were 91% (90% CI: 84–99%) and 92% (90% CI: 84–101%), respectively.
Table 2.
Statistical analysis of L‐PZQ AUC 0‐∞ (EMR 200585‐001 and EMR 200661‐001; PK populations)
| Parameter (h*ng/mL) | Treatment | N | Geo‐LS mean | Test/reference ratio (%) | 90% CI of ratio | CV(%) |
|---|---|---|---|---|---|---|
| EMR 200585001 | ||||||
| AUC0‐∞ | A (ODT rac‐PZQ 40 mg/kg dispersed in water; fed) | 30 | 2,017.268 | 96.2 | 83.9–110.2 | 32.5 |
| B (Cysticide 40 mg/kg given with water; fed) | 30 | 2,047.876 | ||||
| AUC0‐∞ | C1 (ODT rac‐PZQ 20 mg/kg dispersed in water; fed) | 14 | 371.0268 | 18.4 | 15.3–22.0 | 32.5 |
| A (ODT rac‐PZQ 40 mg/kg dispersed in water; fed) | 30 | 2,017.268 | ||||
| AUC0‐∞ | C2 (ODT rac‐PZQ 60 mg/kg dispersed in water; fed) | 15 | 4,492.878 | 222.7 | 186.8–265.6 | 32.5 |
| A (ODT rac‐PZQ 40 mg/kg dispersed in water; fed) | 30 | 2,017.268 | ||||
| AUC0‐∞ | A (ODT rac‐PZQ 40 mg/kg dispersed in water; fed) | 30 | 2,017.268 | 238.1 | 198.7–285.3 | 32.5 |
| D1 (ODT rac‐PZQ 40 mg/kg dispersed in water; fasted) | 14 | 847.2601 | ||||
| AUC0‐∞ | D2 (Cysticide 40 mg/kg as crushed tablets; fed) | 14 | 1,721.742 | 82.1 | 68.5–98.3 | 32.5 |
| B (Cysticide 40 mg/kg given with water; fed) | 30 | 2,097.487 | ||||
| EMR 200661001 | ||||||
| AUC0‐∞ | A (ODT L‐PZQ 20 mg/kg dispersed in water; fed) | 36 | 825.2068 | 39.9 | 34.7–46.0 | 36.8 |
| B (Cysticide 40 mg/kg given with water; fed) | 36 | 2,066.002 | ||||
| AUC0–∞ | C1 (ODT L‐PZQ 10 mg/kg dispersed in water; fed) | 17 | 225.5903 | 27.3 | 22.5–33.2 | 39.2 |
| A (ODT L‐PZQ 20 mg/kg dispersed in water; fed) | 36 | 825.2068 | ||||
| AUC0‐∞ | C2 (ODT L‐PZQ 30 mg/kg dispersed in water; fed) | 17 | 2,147.473 | 260.2 | 214.0–316.4 | 39.2 |
| A (ODT L‐PZQ 20 mg/kg dispersed in water; fed) | 36 | 825.2068 | ||||
| AUC0‐∞ | A (ODT L‐PZQ 20 mg/kg dispersed in water; fed) | 36 | 825.2068 | 167.1 | 143.9–194.0 | 39.2 |
| D (ODT L‐PZQ 20 mg/kg dispersed in water; fasted) | 35 | 493.9041 | ||||
| AUC0–∞ | E (ODT L‐PZQ 20 mg/kg directly disintegrated in the mouth without water; fed) | 36 | 954.5461 | 115.7 | 99.8–134.1 | 39.2 |
| A (ODT L‐PZQ 20 mg/kg dispersed in water; fed) | 36 | 825.2068 | ||||
AUC0–∞, area under the plasma concentration‐time curve from zero to infinity (adjusted to planned dose); CI, confidence interval; CV%, coefficient of variability percentage; EMR, electronic medical record; Geo‐LSMean, geometric least squares mean; L‐PZQ, levo‐praziquantel; N, number of subjects included in the analysis; ODT, orally dispersible tablet; PK, pharmacokinetic; rac‐PZQ, racemate praziquantel.
Variability was high with both formulations: the coefficient of variation for geometric mean (GeoCV%) for L‐PZQ AUC0–∞ was 47% for ODT rac‐PZQ and 60% for Cysticide (Table 1). Corresponding figures for rac‐PZQ were 28% and 40.7%, and those for L‐trans‐OH‐PZQ were 25% and 26% (data not shown).
Results for the secondary parameter AUC from time zero to time of last measurable concentration (AUC0‐t) were comparable with the primary end point results. For the Cmax, the test/reference ratios for L‐PZQ, D‐PZQ, and rac‐PZQ were 116%, 107%, and 109%, respectively.
Dose proportionality
Increase in exposure was greater than dose‐proportional with ODT rac‐PZQ over the dose range of 20, 40, and 60 mg/kg (treatments C1, A, and C2). The 2‐fold decrease in dose from 40 to 20 mg/kg resulted in a > 5‐fold (ratio 18.4%) decrease in L‐PZQ AUC0–∞, and the 1.5‐fold increase from 40 to 60 mg/kg resulted in a > 2.2‐fold (ratio 222.7%) increase in AUC0–∞ (Table 2). Similar trends were observed for D‐PZQ and rac‐PZQ dose‐proportionality (data not shown).
Food effect
A significant food effect was observed for ODT rac‐PZQ. Administration of 40 mg/kg dispersed in water after a meal resulted in higher exposure than the same treatment under fasting conditions (treatments A and D1). The ratio of fed to fasted for L‐PZQ AUC0–∞ was 238% (90% CI: 198.7–285.3%; Table 2). Results were similar for D‐PZQ and racemate PZQ (fed/fasted ratios 169% and 181%, respectively; data not shown). Administration of ODT rac‐PZQ under fasting conditions slightly (nonsignificantly) delayed peak concentration compared with administration after a meal (median L‐PZQ Tmax 4.3 vs. 3.0 hours; Table 1).
Tablets crushed vs. noncrushed
Administration of crushed Cysticide tablets resulted in lower exposure than the same product uncrushed, both under fed conditions (treatments D2 and B, respectively). For L‐PZQ AUC0–∞, the ratio of crushed to uncrushed was 82% (90% CI: 68.5–98.3%; Table 2). For rac‐PZQ AUC0–∞, the corresponding ratio was 86% (90% CI: 75.4–98.4%; data not shown).
Safety results
There were no deaths, serious adverse events (SAEs), or withdrawals due to treatment‐emergent adverse events (TEAEs) Table 3. Most of the total of 24 TEAEs reported in 12 subjects was mild, only 2 TEAEs (both were headache) were graded as moderate (during treatments A and C2). All TEAEs had resolved by the end of the trial (Table 3).
Table 3.
Summary of treatment‐emergent adverse events (EMR 200585‐001 and EMR 200661‐001; safety populations)
| EMR 200585‐001 | Treatment A (N = 31) | Treatment B (N = 32) | Treatment C1 (N = 15) | Treatment C2 (N = 15) | Treatment D1 (N = 14) | Treatment D2 (N = 15) |
|---|---|---|---|---|---|---|
| No. of TEAEs | E | E | E | E | E | E |
| Any TEAEs | 7 | 4 | 2 | 8 | 0 | 3 |
| Serious TEAEs | 0 | 0 | 0 | 0 | 0 | 0 |
| TEAEs resulting in discontinuation | 0 | 0 | 0 | 0 | 0 | 0 |
| TEAEs of severe intensity | 0 | 0 | 0 | 0 | 0 | 0 |
| IMP‐related TEAEs | 5 | 3 | 0 | 8 | 0 | 1 |
| No. of subjects experiencing TEAEs | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) |
| Any TEAEs | 5 (16.1) | 4 (12.5) | 1 (6.7) | 6 (40.0) | 0 (0.0) | 3 (20.0) |
| Serious TEAEs | 0 | 0 | 0 | 0 | 0 | 0 |
| TEAEs resulting in discontinuation | 0 | 0 | 0 | 0 | 0 | 0 |
| TEAEs of severe intensity | 0 | 0 | 0 | 0 | 0 | 0 |
| IMP‐related TEAEs | 4 (12.9) | 3 (9.4) | 0 (0.0) | 6 (40.0) | 0 (0.0) | 1 (6.7) |
|
Notes (EMR 200585‐001) EMR, electronic medical record; IMP, Investigational Medicinal Product; N, number of subjects; n, number of AEs; ODT, orally dispersible tablet; PZQ, praziquantel; TEAEs, treatment‐emergent adverse events. A = New ODT‐PZQ formulation at 40 mg/kg dispersed in water after a meal. B = Current PZQ formulation at 40 mg/kg given with water after a meal. C = New ODT‐PZQ formulation at 20 mg/kg dispersed in water after a meal (C1) or new ODT‐PZQ formulation at 60 mg/kg dispersed in water after a meal (C2). D = New ODT‐PZQ formulation at 40 mg/kg dispersed in water without a meal (D1) or current PZQ formulation at 40 mg/kg given as crushed tablets with water after a meal (D2). | ||||||
| EMR200661001 | Treatment A (N = 36) | Treatment B (N = 36) | Treatment C1 (N = 18) | Treatment C2 (N = 17) | Treatment D (N = 35) | Treatment E (N = 36) |
| No. of TEAEs | E | E | E | E | E | E |
| Any TEAEs | 12 | 23 | 5 | 7 | 7 | 1 |
| Serious TEAEs | 0 | 0 | 0 | 0 | 0 | 0 |
| TEAEs resulting in discontinuation | 0 | 0 | 0 | 0 | 0 | 0 |
| TEAEs of severe intensity | 0 | 0 | 0 | 0 | 0 | 0 |
| IMP‐related TEAEs | 9 | 23 | 2 | 4 | 4 | 1 |
| No. of subjects experiencing TEAEs | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) |
| Any TEAEs | 8 (22.2%) | 17 (47.2%) | 4 (22.2%) | 4 (23.5%) | 5 (14.3%) | 1 (2.8%) |
| Serious TEAEs | 0 | 0 | 0 | 0 | 0 | 0 |
| TEAEs resulting in discontinuation | 0 | 0 | 0 | 0 | 0 | 0 |
| TEAEs of severe intensity | 0 | 0 | 0 | 0 | 0 | 0 |
| IMP‐related TEAEs | 7 (19.4%) | 17 (47.2%) | 1 (5.6%) | 3 (17.6%) | 4 (11.4%) | 1 (2.8%) |
|
Notes (EMR200661‐001)EMR, electronic medical record; IMP, Investigational Medicinal Product; N, number of subjects; n, number of AEs; ODT, orally dispersible tablet; PZQ, praziquantel; TEAEs, treatment‐emergent adverse events. A = MSC2499550A formulation at 20 mg/kg dispersed in water, after a meal. B = Current PZQ formulation (Cysticide) at 40 mg/kg given with water after a meal. C = MSC2499550A formulation at 10 (C1) or 30 (C2) mg/kg (randomized 1 to 1) given dispersed in water, after a meal. D = MSC2499550A formulation at 20 mg/kg given dispersed in water without a meal. E = MSC2499550A formulation at 20 mg/kg directly disintegrated in the mouth without water after a meal. | ||||||
The most frequent TEAEs were nausea and headache (seven events each). Seventeen TEAEs in 10 subjects were considered treatment‐related; these consisted of nausea, headache, dizziness, rash, and vomiting. Although numbers were small, frequencies of both total and treatment‐related TEAEs seemed to increase with dose.
No laboratory, vital signs, or ECG parameters showed clinically significant changes after treatment for any subject.
Palatability
Palatability was generally comparable across treatments; however, the crushed Cysticide tablets were consistently rated much worse than any other treatment (100 mm VAS; data not shown).
EMR200661‐001 (ODT L‐PZQ vs. Cysticide)
Subject population
Seventy‐three male volunteers were screened between October 14, 2014 and October 22, 2014, and 36 were randomized (3 per sequence). All 36 received at least one treatment (separated by 7‐day washouts); 34 completed the study, and 2 withdrew after four treatments for personal reasons (Figure 1). All randomized subjects were included in both the PK and the safety populations.
Twenty‐seven randomized subjects (75.0%) were black, six (16.7%) were white, and three (8.3%) were of “other” ethnicities. Mean (SD) age, weight, and body mass index were 26.3 (7.0) years, 70.67 (9.88) kg, and 24.20 (2.87) kg/m2, respectively.
PK results
Mean plasma concentration‐time profiles for L‐PZQ differed between ODT L‐PZQ and Cysticide, with mean L‐PZQ levels ≈40% lower after administration of ODT L‐PZQ, despite the comparable amounts of L‐PZQ in the tablets (Figure 4). There was no conversion of L‐PZQ to D‐PZQ, as D‐PZQ levels after ODT L‐PZQ administration were zero (Figure 2). Again, small additional peaks occurred at 4.5 hours postdose with both formulations, possibly related to the meal provided at 4 hours. As in EMR200585‐001, individual concentration‐time profiles were erratic and varied considerably between subjects (Figure 3).
Figure 4.
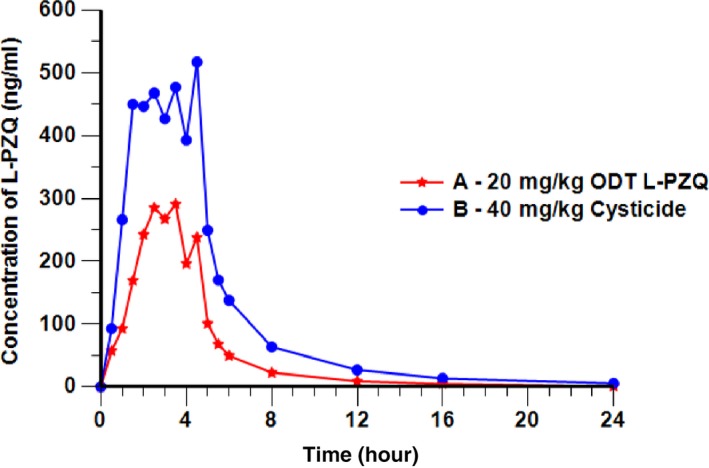
Mean concentration‐time profile of Levo‐praziquantel (L‐PZQ) after administration of orally dispersible tablet (ODT) L‐PZQ or Cysticide (EMR 200661‐001, pharmacokinetic population; linear scale).
As in EMR200585‐001, L‐PZQ concentrations were approximately a quarter of those of D‐PZQ after administration of Cysticide (Figure 2), and levels of L‐trans‐OH‐PZQ were > 70 times higher than those of the parent but comparable between formulations (data not shown), suggesting that the pathway leading to this metabolite might not explain the observed difference in L‐PZQ levels.
As measured by AUC0‐∞, the relative bioavailability of L‐PZQ when given as a single enantiomer (ODT L‐PZQ 20 mg/kg dispersed in water; treatment A) was around 40% that of Cysticide (treatment B) after single oral administration under fed conditions (Figure 4). For the primary end point of L‐PZQ AUC0‐∞, the test/reference ratio was 40% (90% CI: 35–46%; Table 2). Similarly, for the secondary parameters L‐PZQ AUC0‐t, and Cmax, the test/reference ratios were 39% (90% CI: 34–46%) and 52% (90% CI: 42–65%; data not shown).
Overall, variability of the PK parameters was high. The GeoCV% for L‐PZQ AUC0‐∞ was 101% for ODT L‐PZQ and 65% for Cysticide.
Dose proportionality
Increase in exposure was greater than dose‐proportional with ODT L‐PZQ over the dose range of 10, 20, and 30 mg/kg (treatments C1, A, and C2). The 2‐fold decrease in dose from 20 to 10 mg/kg resulted in a > 3‐fold (ratio 27.3%) decrease in L‐PZQ AUC0‐∞, and the 1.5‐fold increase from 20 to 30 mg/kg resulted in a > 2‐fold (ratio 260.2) increase in AUC0‐∞ (Table 2).
Food effect
A food effect was observed for ODT L‐PZQ. Administration of 20 mg/kg after a meal (treatment A) resulted in higher exposure than the same dose given under fasting conditions (treatment D). Fed/fasted ratios were 167.1% for L‐PZQ AUC0‐∞ (Table 2) and 295.6% for Cmax. Median Tmax was comparable when treatment was administered without food (2.0 hours vs. 2.5 hours; Table 1).
Method of administration
Disintegrating ODT L‐PZQ tablets directly in the mouth (treatment E) resulted in comparable exposure to administration dispersed in water (treatment A), with no indication of buccal uptake. The ratio of disintegrated to dispersed for L‐PZQ AUC0‐∞ was 115.7% (90% CI: 100–134%; Table 2).
Safety results
There were no deaths, SAEs, or treatment discontinuations due to TEAEs Table 3. In total, 55 TEAEs in 23 subjects were reported, most of which were mild, and 4 TEAEs were graded as moderate (treatment B: headache and upper abdominal pain; treatment C2: two events (both headache)). All TEAEs had resolved by the end of the trial.
The most frequent TEAEs were headache and nausea (27 and 7 events, respectively). Forty‐three TEAEs in 19 subjects were considered treatment related; these consisted of headache, nausea, dyspepsia, abdominal discomfort, upper abdominal pain, fatigue, diarrhea, dizziness, dysgeusia, and vomiting.
No laboratory, vital signs, or ECG parameters showed clinically significant mean or median changes after treatment, and no individual changes were considered clinically significant.
Palatability
VAS palatability scores favored administration of ODT L‐PZQ dispersed in water over direct disintegration in the mouth without water and favored administration of ODT L‐PZQ dispersed in water after a meal over administration without a meal. Palatability was significantly better for ODT L‐PZQ than for Cysticide in terms of flavor, sweetness, overall liking of medicine, and acceptability to swallow (data not shown).
DISCUSSION
In the phase I studies, the relative bioavailability of ODT rac‐PZQ was comparable with that of the reference PZQ formulation (Cysticide) when equivalent doses were administered under fed conditions. However, the relative bioavailability of L‐PZQ given as a single enantiomer (ODT L‐PZQ) was only about 40% that of Cysticide.
Plasma L‐PZQ concentrations were approximately a quarter of those of D‐PZQ after administration of either ODT rac‐PZQ or Cysticide, despite the 1:1 ratio of the enantiomers in the tablets. This finding has been reported previously19; the reason for the difference is unknown.
Both ODT formulations showed greater than dose‐proportional increases in exposure over the ranges tested, and both showed increased exposure under fed conditions. PK parameters were quite variable in both studies.
Safety results for both ODT formulations were consistent with previous experience with PZQ. TEAEs were comparable in nature and frequency between equivalent doses of ODT rac‐PZQ and Cysticide, although the numbers of subjects and events were small. At similar L‐PZQ doses, fewer TEAEs were reported with ODT L‐PZQ than with the racemic reference formulation. Lower ODT rac‐PZQ doses seemed to be better tolerated than higher doses. PZQ has known gastrointestinal and minor neurological side effects; the TEAEs considered to be related to ODT PZQ were generally consistent with this profile, and most were expected. No new types of TEAEs were observed, and no SAEs were reported. All TEAEs were mild to moderate in intensity and all had resolved spontaneously by the end of the studies. It is expected that these favorable safety profiles will be maintained in the pediatric population once appropriate dosing is determined, as the safety profiles of existing formulations have been similar between adults and children.20, 21, 22
The finding that L‐PZQ exposure was lower with ODT L‐PZQ in comparison to the reference PZQ formulation was unexpected and the explanation is unknown. There was no evidence of conversion of L‐PZQ to D‐PZQ, and levels of L‐trans‐OH‐PZQ were comparable after administration of ODT L‐PZQ compared with rac‐PZQ, suggesting that the pathway leading to this metabolite might not explain the difference in systemic exposure. PZQ metabolism is not well understood. It is hypothesized that the D‐PZQ in the racemic mixture might protect the L‐PZQ from metabolic degradation but the exact mechanism remains to be understood. It is unlikely that the difference in exposure is related to the ODT formulation, as there are no major differences in excipient composition compared with the reference tablets, and exposure with the ODT rac‐PZQ was comparable with that with the standard formulation.
The shapes of the individual concentration/time profiles were highly variable with early and late Cmax, broad and sharp peaks, and multiple peaks. Overall between‐subject variability was very high, which hindered the construction of reliable population PK models that would permit extrapolation of adult PK data to children.23 The lower bioavailability of L‐PZQ ODT, the high variability and the nondose linearity of the PK parameters indicated the need for a proper pediatric dose‐finding study. Therefore, both rac‐PZQ ODT and L‐PZQ ODT are being investigated in a phase II dose‐finding study in PSAC (MS200661‐0005; NCT02806232) in order to identify the ODT formulation and dose with the optimal benefit risk profile that can then be used in a pediatric phase III trial.
Trial registration
ClinicalTrials.gov identifiers NCT02352713 (study EMR200585001) and NCT02271984 (study EMR200661001).
Funding
These studies were conducted and their results analyzed by the Pediatric Praziquantel Consortium (PPC), an international not‐for‐profit partnership established in 2012 by Merck KGaA, Darmstadt, Germany (Merck KGaA). Apart from in‐kind contributions by all partners, the Consortium received grant support from the Bill & Melinda Gates Foundation (BMGF; Grant No. OPP1063223; www.gatesfoundation.org) and from the Global Health Innovative Technology fund (GHIT, Tokyo, Japan etc, Tokyo, Japan; Grant Nos. 2013212 and 2014206; www.ghitfund.org). Neither the BMGF nor the GHIT had any role in the decision to publish or in preparation of the manuscript.
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
W.B., D.B., E.H.‐M., E.K.‐L., P.W., and O.Y. wrote the manuscript. W.B., E.K.‐L., and P.W. designed the research. D.B. performed the research. W.B., E.H.‐M., P.W., and O.Y. analyzed the data.
Acknowledgments
The authors thank Annette Dubois for her medical writing support (funded by a grant from the Global Health Technology Fund in accordance with Good Publication Practice guidelines) and Christian Lüpfert (Translational Quantitative Pharmacology, Merck KGaA) for his scientific contributions and his critical review of the manuscript.
These studies were conducted and their results analyzed by the Pediatric Praziquantel Consortium (PPC), an international not‐for‐profit partnership established in 2012 by Merck KGaA, Astellas Pharma (Astellas), the Swiss Tropical and Public Health Institute (Swiss TPH), and Lygature (formerly TI Pharma). Farmaguinhos and Simcyp (a Certara company) joined the Consortium as full partners in 2014; Simcyp left the Consortium in 2017. Imperial College London's Schistosomiasis Control Initiative (SCI) joined in 2016. All partners have agreed to the publication of the manuscript.
References
- 1. Wu, G.Y. & Halim, M.H. Schistosomiasis: progress and problems. World J. Gasteroenterol. 6, 12–19 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. World Health Organization [Internet] . World Health Organization, Geneva, c2016. Fact Sheet No. 115: Schistosomiasis. [about 6 screens]. <http://www.who.int/mediacentre/factsheets/fs115/en/index.html> (2016). Accessed 6 December 2016.
- 3. Colley, D.G. , Bustinduy, A.L. , Secor, W.E. , King, C.H. Human schistosomiasis. Lancet 383, 2253–2264 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. World Health Organization [Internet] . World Health Organization, Geneva, c2016. Schistosomiasis: Strategy. <http://www.who.int/schistosomiasis/strategy/en/> (2017) Accessed 17 February 2017.
- 5. World Health Organization . Report of a meeting to review the results of studies on the treatment of schistosomiasis in preschool‐age children. 32 (World Health Organization, Geneva, 2011). <apps.who.int/iris/bitstream/10665/44639/1/9789241501880_eng.pdf>. [Google Scholar]
- 6. Allen, H.E. , Crompton, D.W. , de Silva, N. , LoVerde, P.T. & Olds, G.R. New policies for using anthelmintics in high risk groups. Trends Parasitol. 18, 381–382 (2002). [DOI] [PubMed] [Google Scholar]
- 7. Geary, T.G. et al Unresolved issues in anthelmintic pharmacology for helminthiases of humans. Int. J. Parasitol. 40, 1–13 (2010). [DOI] [PubMed] [Google Scholar]
- 8. Mafiana, C.F. , Ekpo, U.F. & Ojo, D.A. Urinary schistosomiasis in preschool children in settlements around Oyan Reservoir in Ogun State, Nigeria: implications for control. Trop. Med. Int. Health 8, 78–82 (2003). [DOI] [PubMed] [Google Scholar]
- 9. Opara, K.N. , Udoidung, N.I. & Ukpong, I.G. Genitourinary schistosomiasis among pre‐primary schoolchildren in a rural community within the Cross River Basin, Nigeria. J. Helminthol. 81, 393–397 (2007). [DOI] [PubMed] [Google Scholar]
- 10. Ekpo, U.F. , Laja‐Deile, A. , Oluwole, A.S. , Sam‐Wobo, S.O. & Mafiana, C.F. Urinary schistosomiasis among preschool children in a rural community near Abeokuta, Nigeria. Parasit. Vectors 3, 58 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bosompem, K.M. et al Infant schistosomiasis in Ghana: A survey in an irrigation community. Trop. Med. Int. Health 9, 917–922 (2004). [DOI] [PubMed] [Google Scholar]
- 12. Odogwu, S.E. et al Schistosoma mansoni in infants (aged <3 years) along the Ugandan shoreline of Lake Victoria. Ann. Trop. Med. Parasitol. 100, 315–326 (2006). [DOI] [PubMed] [Google Scholar]
- 13. World Health Organization . Prevention and control of schistosomiasis and soil‐transmitted helminthiasis: report of a WHO expert committee. (WHO Technical Report Series No. 912) p. 63 (World Health Organization, Geneva, Switzerland, 2002). <whqlibdoc.who.int/trs/WHO_TRS_912.pdf>. [PubMed] [Google Scholar]
- 14. Coulibaly, J.T. , Panic, G. , Silué, K.D. , Kovacˇ, J. , Hattendorf, J. & Keiser, J. Efficacy and safety of praziquantel in preschool‐aged and school‐aged children infected with Schistosoma mansoni: a randomised controlled, parallel‐group, dose‐ranging, phase 2 trial. Lancet Glob. Health. 5, e688–e698 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Betson, M. , Sousa‐Figueiredo, J.C. , Rowell, C. , Kabatereine, N.B. & Stothard, J.R. Intestinal schistosomiasis in mothers and young children in Uganda: investigation of field‐applicable markers of bowel morbidity. Am. J. Trop. Med. Hyg. 83, 1048–1055 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garba, A. et al Schistosomiasis in infants and preschool‐aged children: infection in a single Schistosoma haematobium and a mixed S. haematobium S. mansoni foci of Niger. Acta Trop. 115, 212–219 (2010). [DOI] [PubMed] [Google Scholar]
- 17. European Medicines Agency . ICH E11(R1) step 5 guideline on clinical investigation of medicinal products in the pediatric population. EMA/CPMP/ICH/2711/1999. (EMA, London, 2017) <https://www.ema.europa.eu/ich-e11r1-step-5-guideline-clinical-investigation-medicinal-products-pediatric-population>. [Google Scholar]
- 18. Jung, H. , Medina, R. , Castro, N. , Corona, T. & Sotelo, J. Pharmacokinetic study of praziquantel administered alone and in combination with cimetidine in a single day therapeutic regimen. Antimicrob. Agents Chemother. 41, 1256–1259 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Westhoff, F. & Blaschke, G. High‐performance liquid chromatographic determination of the stereoselective biotransformation of the chiral drug praziquantel. J. Chromatogr. 578, 265–271 (1992). [DOI] [PubMed] [Google Scholar]
- 20. Zwang, J. & Olliaro, P.L. Clinical efficacy and tolerability of praziquantel for intestinal and urinary schistosomiasis – a meta‐analysis of comparative and non‐comparative clinical trials. PLoS Negl. Trop. Dis. 8, e3286 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Coulibaly, J.T. , N'Gbesso, Y.K. , Knopp, S. , Keiser, J. , N'Goran, E.K. & Utzinger, J. Efficacy and safety of praziquantel in preschool‐aged children in an area co‐endemic for Schistosoma mansoni and S. haematobium. PLoS Negl. Trop. Dis. 6, e1917 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Navaratnam, A.M. , Sousa‐Figueiredo, J.C. , Stothard, J.R. , Kabatereine, N.B. , Fenwick, A. & Mutumba‐Nakalembe, M.J. Efficacy of praziquantel syrup versus crushed praziquantel tablets in the treatment of intestinal schistosomiasis in Ugandan preschool children, with observation on compliance and safety. Trans. R. Soc. Trop. Med. Hyg. 106, 400–407 (2012). [DOI] [PubMed] [Google Scholar]
- 23. Bonate, P.L. et al Extrapolation of praziquantel pharmacokinetics to a pediatric population: a cautionary tale. J. Pharmacokinet. Pharmacodyn. 45, 747–762 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]