

Abstract
A milestone step in translational science to transform basic scientific discoveries into therapeutic applications is the advancement of a drug candidate from preclinical studies to initial human testing. First‐in‐human (FIH) trials serve as the link to advance new promising drug candidates and are conducted primarily to determine the safe dose range for further clinical development. Cross‐functional collaboration is essential to ensure efficient and successful FIH trials. The aim of this publication is to serve as a tutorial for conducting FIH trials for both small molecule and biological drug candidates with topics covering regulatory requirements, preclinical safety testing, study design considerations, safety monitoring, biomarker assessment, and global considerations. An emphasis is placed on FIH trial design considerations, including starting dose selection, study size and population, dose escalation scheme, and implementation of adaptive designs. In light of the recent revision of the European Medicines Agency (EMA) guideline on FIH trials to promote safety and mitigate risk, we also discuss new measures introduced in the guideline that impact FIH trial design.
Successful execution of first‐in‐human (FIH) trials requires sponsors to approach the task with thorough consideration and planning across many disciplines, which need to be seamlessly integrated. From a pharmacology perspective, both in vitro and in vivo animal data help provide information on the drug candidate's potency and pharmacologic profile to confirm relevance and potential for the proposed clinical indication. Preclinical pharmacokinetic (PK) data provide construction of exposure‐response curves needed to provide an estimation of the therapeutically relevant dose range for the FIH studies to be efficient and informative. In vitro metabolism and drug‐drug interaction (DDI) studies inform the need to assess DDI risks early on in a development program. Toxicological assessment of the drug candidates is not only needed to meet regulatory requirements, but also ideally leads to understanding of on‐target and off‐target pharmacology so translational risk to humans can be minimized.
Clinical pharmacologists who are usually the “owners” of the FIH studies should ensure the study design takes into consideration all preclinical learnings on the drug candidates. Moreover, FIH studies also need to be recognized as “bridges” to future clinical development, therefore, study design and conduct are best served by working cross‐functionally to engage key stakeholders. Taking into account unmet medical needs and the competitive landscape, clinical development scientists in relevant therapeutic areas should provide input on study design elements, including the specific disease considerations and any potential exploration of efficacy signals. Formulation scientists provide input to strike a balance between pilot and later stage development/commercialization dosage forms to obtain early relative bioavailability information. Safety physicians should meticulously lay out plans for safety monitoring, which is a critical component of any FIH trial. Clinical operations colleagues are critical partners to help with study site selection, startup activities, institutional review board (IRB)/ethics committee (EC) approvals, contract research organization (CRO) collaboration, and study oversight to ensure smooth execution and quality of the studies. Key stakeholders and their main responsibilities are depicted in Figure 1.
Figure 1.
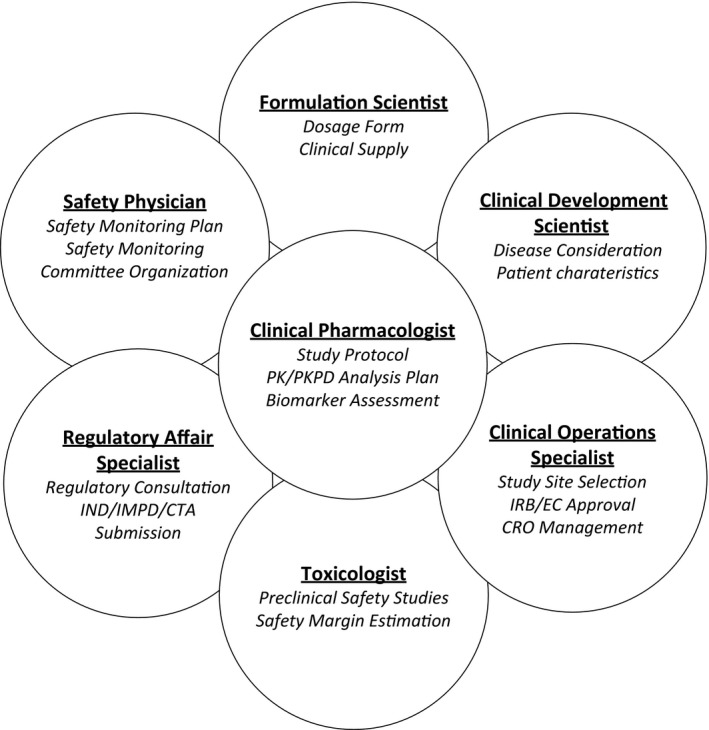
Key stakeholders and their main responsibilities in planning a first‐in‐human study. CTA, clinical trial application; CRO, contract research organization; EC, ethics committee; IMPD, Investigational Medicinal Product Dossier; IND, investigational new drug; IRB, institutional review board; PK, pharmacokinetic; PKPD, pharmacokinetic pharmacodynamic.
FIH studies are always preceded by submission of regulatory filings to health authorities (e.g., investigational new drug (IND) applications in the United States, Investigational Medicinal Product Dossier in Europe, etc.) in the countries where studies are conducted. Although both the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) have provided helpful guidance documents on how to approach FIH studies,1, 2 it is not possible to find answers to every question that exists for complex drug development programs, especially if the questions are unique to the therapeutic indication. In some cases, sponsors may benefit from regulatory interactions prior to submission filing to incorporate learnings into the FIH study design, not only to minimize risks of clinical hold due to safety concerns but also to ensure return on investment when early exploration of efficacy or biomarkers are pursued.
Recognizing the complexity and multidisciplinary nature of conducting initial trials for potential therapeutic agents in humans, this tutorial aims to provide some insights on key elements mentioned above to help sponsors be more successful in design and execution of FIH trials. Given the significant differences in characterization and regulatory expectation for cell and gene therapies as well as vaccines, FIH trials for these modalities are beyond the scope of this tutorial.
Preclinical safety testing
There are numerous references, as well as guidance documents for developing preclinical safety programs to support an FIH trial.2, 3, 4, 5 In particular, the International Conference on Harmonization (ICH) M3(R2) Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals provides recommendations of what ICH regulatory authorities expect for a safety evaluation program to enable an FIH trial.6 Additional information on requirements specific for biotechnology‐derived pharmaceuticals (biologics) are provided in ICH S6(R1)7 and guidance related to nonclinical evaluation for anticancer pharmaceuticals are provided in ICH S9.8 This background of regulatory expectations is helpful to keep in mind in developing a preclinical safety testing strategy tailored for the particular needs of a drug development program (i.e., modality and indication).
There are several main goals of preclinical safety testing: i) identification of organ toxicity, ii) the relationship to drug exposure, iii) determination of on‐target and off‐target effects, iv) potential relevance to humans, and v) identification/qualification of safety biomarkers to monitor in the clinic. The approach to achieve these goals will be dependent on a few factors. First, the type of therapeutic (e.g., small molecule or biologic) will dictate what set of studies are essential, as well as those expected by regulatory authorities, to enable an FIH clinical trial (Table 1). Second, the therapeutic indication (e.g., cardiovascular, central nervous system (CNS), oncology, and rare/orphan disease) will dictate whether additional assessments and considerations are necessary to better understand dose‐related efficacy and toxicity in target organs to estimate a safety margin. Finally, the scope and design of the FIH trial and subsequent studies (e.g., treatment duration) must be considered in order to design appropriate pivotal preclinical studies that will both enable the planned clinical studies and adequately inform of potential safety risks for the intended patient population or healthy volunteers.
Table 1.
ICH recommended preclinical studies enabling FIH trials
| Study type | Small molecules | Large moleculesa | GLP compliance Requirement |
|---|---|---|---|
| Pharmacodynamics | No | ||
| In vitro (MOA) | X | X | |
| In vivo (MOA and therapeutic effect) | X | X | |
| Safety pharmacology (ICH S7A62 and S7B63) | Yes | ||
| In vitro (concentration‐effect relationship) | X | X | |
| In vivo (dose‐response for CNS, CV, respiratory effects) | X | X | |
| Pharmacokinetics (ICH M3(R2)6) | |||
| In vitro metabolism (across species microsomal metabolism) | X | NA | No |
| In vitro plasma protein binding | X | NA | No |
| Toxicokinetics from repeat dose GLP toxicity studies (ICH S3A64) | X | X | Yes |
| Genotoxicity battery (ICH S2(R1)7) | Yes | ||
| In vitro Ames test | X | *a | |
| In vitro and/or in vivo mammalian cell chromosomal damage evaluation | X | *a | |
| Single‐dose / dose range finding | No and Yesb | ||
| Rodent single‐dose (could be MTD study) | X | NA | |
| Nonrodent single‐dose (could be MTD study) | Xd | Xe | |
| Repeat dose toxicityc (ICH M3(R2)6) | Yes | ||
| Rodent multidose | X | Optionale | |
| Nonrodent multidose | Xd | Xe | |
| Other studies | No | ||
| Immunotoxicity (ICH S865) | X | X | |
| Photosafety (ICH S1010) | X | X | |
| Abuse liabilityf | X | X | |
CNS, central nervous system; CV, cardiovascular; FIH, first‐in‐human; GLP, good laboratory practice; ICH, International Conference on Harmonization; MOA, mechanism of action; MTD, maximum tolerated dose; NA, not applicable.
Refer to ICH S6 (R1).7
Not typically required.
If single‐dose study is pivotal (i.e., used to support a single‐dose FIH trial), it should be GLP compliant, which is more typical for large molecules.
Duration and dosing route dependent on clinical trial design (Table 1 in ref. 6).
Species selection dependent on similarity in metabolism to humans.
Often nonhuman primate or minipig; dependent on presence of target and relative potency of the drug candidate against the target.
Tissue cross‐reactivity dictates which species should be studied. If the biologic is cross‐reactive in both rodents/nonrodents, then both species should be studied. If the biologic is cross‐reactive in only one species (most often nonhuman primate), then only that species is studied. If the biologic is not cross‐reactive to any species, then consider a transgenic or surrogate biologic.
For drugs with abuse potential based on MOA/similarity to known drugs of abuse.
For small molecules, refer to ICH guidance S2(R1) for recommendations on genotoxicity testing, which does not apply to biologics.9 Initially, a gene mutation assay (e.g., in vitro Ames test) will support a single‐dose trial; the standard test battery includes studies in mammalian systems to detect chromosomal damage, which is recommended to support repeat‐dosing clinically.
For safety pharmacology, ICH guidances S7A and S7B provide further details to the core battery of studies described in ICH M3(R2) recommended for FIH trials for the assessment of cardiovascular (CV), CNS, and respiratory systems. Initially, an in vitro CV assessment to determine the potential for QT prolongation, a predictor of cardiac arrhythmia, through assessment of compound inhibition of the human ether‐a‐go‐go channel is conducted. Other channels that play a role in QT prolongation, including sodium channel (Nav1.5) and calcium channel (Cav1.2) are also usually assessed in vitro. In addition, to support the FIH trial, an in vivo QT evaluation is recommended, which is typically initiated after dose‐ranging toxicity studies. Dose levels should be selected that exceed anticipated human exposure but be limited by the expected tolerability in the animal species used. This study can be conducted standalone or incorporated in the nonrodent pivotal toxicology study. In section 2.9 of the ICH S7A, conditions under which studies may not be necessary (e.g., locally applied products) and considerations for biologics are described.
The results of the in vitro studies (i.e., genotoxicity and CV safety pharmacology) will provide starting information to determine the relative risk based on activity levels for these safety end points compared with pharmacodynamic activity levels. In parallel to these in vitro assessments, initial in vivo toxicity studies are conducted to determine the maximum tolerated dose (MTD) or up to the maximum feasible dose in animal test species, starting with rodents. Nonrodent species should be chosen that are most pharmacologically (e.g., target sequence homology and relative binding affinity) and, for small molecules, metabolically relevant to humans. Typically, canine and nonhuman primates are used for small and large molecule studies, respectively.
Pivotal toxicology studies to support at least the dosing regimen and duration for the FIH trial are expected to be conducted in compliance with good laboratory practice (GLP). The dose range should be broad to establish a no observed adverse effect level (NOAEL) with a sufficient margin of exposure over the maximum exposure intended in the FIH trial. For small molecules, this is routinely done in rodents and nonrodent species, whereas for large molecules, a nonrodent species, typically nonhuman primates, alone would be sufficient.
Finally, an initial assessment of the phototoxic potential should be conducted based on photochemical properties of the drug and information on other members in the class, which is described in ICH S10 Photosafety Evaluation of Pharmaceuticals.10
Sponsors interested in earlier access to human data before generating a full package of preclinical safety data as listed above may consider conducing exploratory clinical trials, which, per ICH guidance,6 involve limited human exposure, have no therapeutic intent, and are not intended to examine clinical tolerability. These FIH studies typically examine PK, pharmacodynamic (PD), and/or biomarkers. Five different approaches with corresponding preclinical testing programs are provided in the guidance document, which vary in extent to support microdosing up to 14‐day dosing into the therapeutic range.6
Starting dose selection
Determining the dose range that should be studied is an important objective prior to taking a new drug candidate into an FIH trial. Typically, the dose is determined by utilizing all relevant IND‐enabling preclinical results from pharmacology, toxicology, and PK studies and any available human experience with other therapies that share the mechanism of action (MOA). Most importantly, the starting dose must mitigate the risk of toxicity while balancing the need to elicit pharmacologic activity, especially when dosing patients with more grievous conditions, such as cancer. It is also important to determine the dose range to be studied, including escalation steps, as the data collected will inform the doses to be studied later in development when efficacy is of primary interest. Extraneous dose cohorts due to either starting the dose too low and/or escalating too cautiously will increase overall study size and duration and, therefore, should be minimized.
The EMA and the FDA have published guidance documents for investigators to follow when determining an appropriate starting dose. The EMA guidance assists in the transition from preclinical to early clinical development and covers many of the risks inherent to FIH trials and discusses mitigation strategies to manage these risks.2 The FDA guidance aims at avoiding toxicity at initial doses by using the generally accepted benchmark for safety, the NOAEL obtained from the most sensitive toxicology test species as a starting point for determining a reasonably safe starting dose.1
There is no singular approach to selecting a proper FIH starting dose due to the uniqueness of each new drug candidate and the many different assumptions that have to be made for translation across species.1, 2, 11, 12, 13 New challenges exist each time for extrapolation of doses from animals to humans and what may have worked well for one drug candidate may not be appropriate the next time around. For these reasons it is difficult to establish a standard uniform approach. However, the regulatory guidance documents do provide a framework to follow.
Methods for calculating the starting dose are either empirical or mechanistic (Table 2). As mentioned, the most widely used empirical method follows the FDA guidance1 for estimating a maximum safe starting dose by converting the NOAEL to a human equivalent dose with the use of allometric scaling. However, this method has its disadvantages, including the use of a somewhat arbitrary safety factor to ensure safety of the starting dose, the dose is based on minimal risk of toxicity rather than based on pharmacologic activity, and it does not address dose escalation or the maximum allowable dose. The EMA guidance2 outlines a more mechanistic approach based on state‐of‐the‐art modeling incorporating all relevant preclinical pharmacology data including ex vivo and in vitro studies in human tissues. This guidance highlights selection of a minimal anticipated biological effect level, as this is becoming increasingly important as more targeted therapies are being developed that are very specific and potent with predicted pharmacologically active doses well below a dose that is thought to be safe based on the NOAEL.11
Table 2.
Methods for estimating a starting dose in FIH clinical trials
| Method | Advantages | Disadvantages |
|---|---|---|
| MRSD approach (dose‐by‐factor) | Good safety record, easy to calculate | Empirical approach based only on dose, arbitrary safety factor applied, neglects pharmacological activity, and dose escalation |
| Similar MOA | Easy to use; minimal data required | Only applicable to a limited number of drugs, does not account for differences in PK or PD between the two drugs |
| MABEL | Based on pharmacology rather than an empirical scaling factor; safest approach for high‐risk drug candidates with a high degree of species‐specificity or targeting the immune system | Requires more extensive nonclinical data; unclear which nonclinical model/data is most predictive |
| PK model | Accounts for species differences in PK parameters rather than empirical scaling of dose; ability to calculate safety margins; demonstrated to work well for compounds that are eliminated renally and monoclonal antibodies with linear elimination | Neglects species differences in pharmacology (assume concentration‐effect relationship is the same for animals and humans); dependent on accuracy of nonclinical PK and scaling approach |
| PKPD model | One step further than the PK‐guided approach in that it accounts for species differences in both PK and PD; accounts for pharmacologic activity and can support dose escalation | Requires an experienced modeler and extensive nonclinical data |
FIH, first‐in‐human; MABEL, minimum anticipated biologic effect level; MOA, mechanism of action; MRSD, maximum recommended safe starting dose; PD, pharmacodynamic; PK, pharmacokinetic.
Pharmacokinetic/pharmacodynamic (PK/PD) modeling is increasingly being used in industry and utilizes the concentration‐time course instead of dose for extrapolating from animals to humans. The PK/PD modeling provides a quantitative framework that can support selection of an anticipated therapeutic dose range through simulation, thus identifying an upper dose limit to be studied in the FIH clinical trial and support dose escalation decisions. Last, PK/PD modeling allows for the ability to account for interspecies differences in both PK and PD potentially improving the accuracy of human predictions.
Many publications detailing methods for scaling PK parameters from animals to humans are available.14, 15, 16, 17 Although the basic PK principles apply to all drug candidates, the factors that drive the absorption, distribution, metabolism, and elimination are different for small molecules (<1 kDa) vs. large molecules. Therefore, the methods for interspecies scaling and prediction of human exposures for the two are often different. A comprehensive overview of these methods is outside the scope of this review but the basic principles separated by this classification are discussed below.
Small molecules
In general, there are two approaches used for interspecies scaling: i) physiological‐based pharmacokinetic (PBPK) models, and ii) allometric scaling. The PBPK models provide a more mechanistic method of scaling small molecule PK.18 However, PBPK models require a significant amount of preclinical data to inform the many model parameters and, thus, are not frequently used. The allometric approach is a less complicated empirical approach developed based on the cross species similarities in biology, physiology, and anatomy utilizing a power function correlating physiological parameters with body size.19
The allometric projections have been demonstrated to work well for small molecules, especially those that primarily undergo renal elimination.14 However, when small molecule drugs display high cross‐species variability in PKs (e.g., due to metabolism), this method may not work well. Others have proposed modification to the allometric‐based scaling method to improve predictability. These include the rule of exponents method, which uses maximum life‐span potential and brain weight to correct the exponent,15 liver blood flow,20 and correction for in vitro metabolic clearance.21 The method of choice will depend on the PK characteristics of the drug candidate and the data available for scaling.
The S9 Nonclinical Evaluation for Anticancer Pharmaceuticals published by the ICH provides additional guidance for an empirical method for cytotoxic agents with a very small therapeutic window (i.e., very steep concentration‐safety curve).8 Due to the acceptance of greater safety risk in patients with cancer to achieve a therapeutic benefit, the starting‐dose calculations are based on a dose and regimen that has elicited some toxicity in animals provided it is not a severe, irreversible toxicity. The guidance recommends a starting dose for many small molecules that is one‐tenth the severely toxic dose in 10% of rodents or one‐sixth the highest nonseverely toxic dose in nonrodents is considered an appropriate starting dose. It is important to note that the highest nonseverely toxic dose is in stark contrast to the NOAEL and defined as the highest dose level that does not produce lethal, life‐threatening, or irreversible toxicities when assessing anticancer agents in preclinical toxicity evaluation.
Large molecules
Biodistribution of large molecules are usually limited by polarity, charge, and molecular size.22 Large molecules are not typically substrates of drug metabolizing enzymes or drug transport proteins. Instead, renal excretion and proteolytic degradation to amino acids serve as the primary routes of elimination. Fortunately, both processes are highly conserved across mammalian species; therefore, the methods for allometric scaling can be applied. This is supported by many groups that have demonstrated reasonable accuracy in predicting human clearance and volume of distribution using allometric scaling.23, 24, 25
Methods for predicting human PK of monoclonal antibodies (mAbs), a subclass of large molecule drugs, are mostly dependent on the PK characteristics in nonhuman primates. It has been shown that simple allometric scaling for mAbs that exhibit linear PKs in monkeys can accurately predict human PK within a twofold range.16 However, for mAbs that display nonlinear PK, due to target‐mediated drug disposition, allometric scaling results in an overestimation of exposure when below target‐saturating concentrations. In cases where the relevant target‐mediated drug disposition PK parameters (e.g., target binding affinity constants, baseline target expression levels, and target turnover rate constant) can be informed by experimental and literature data, human predictions have been shown to be successful.17, 26
The guidance documents and methods discussed above are intended for therapies with systemic administration, although similar principles should apply for therapies delivered locally (e.g., topical, inhalation, and intra‐tissue). Although local delivery increases the drug concentrations at the target site, it involves different assumptions for translation of the dose from animals to humans. This is mostly due to the fact that it is often unfeasible to assess the target site PK in humans in order to validate the scaling methods utilized. Therefore, it is critical to evaluate PK in a nonclinical model that is thought to best reflect humans, based on anatomic and physiological considerations, and interspecies scaling of the dose can likely benefit most by utilizing a PBPK approach. A detailed discussion for each delivery route is outside the remit of this article. We direct the reader to these useful articles on translation of dose for topical ophthalmic,27 topical dermatologic,28 and inhalation 29 routes of administration.
Other design considerations
When designing an FIH trial, there are many additional factors to consider in the overall study design, including study size, study population, dose escalation scheme, specification of dose‐limiting toxicities, patient selection, and secondary objectives. Moreover, with the accelerated approval programs, such as the FDA's fast track or breakthrough therapy designations and the EMA's prime status, drug candidates have the potential to be approved with a faster timeline. Thus, it is increasingly important to obtain crucial clinical pharmacology data to provide prescribing physicians with information about the proper use and risks of the product. The FIH studies offer an opportunity to glean such information early on in a development program. Sponsors may incorporate assessment of multiple dosing, food effect, relative bioavailability, QT prolongation, and/or DDI potential as part of the FIH clinical trial. Many elements of the FIH study discussed below, if incorporated, requires “umbrella protocols” that allow an adaptive trial design in which, as data are made available, certain aspects of the study design are modified to maximize learning without undermining the validity and quality of the study. Figure 2 illustrates possible study schema for an FIH study with multiple objectives. To achieve this, processes need to be in place to allow timely access to data on an ongoing basis to enable rapid review and decision making.
Figure 2.
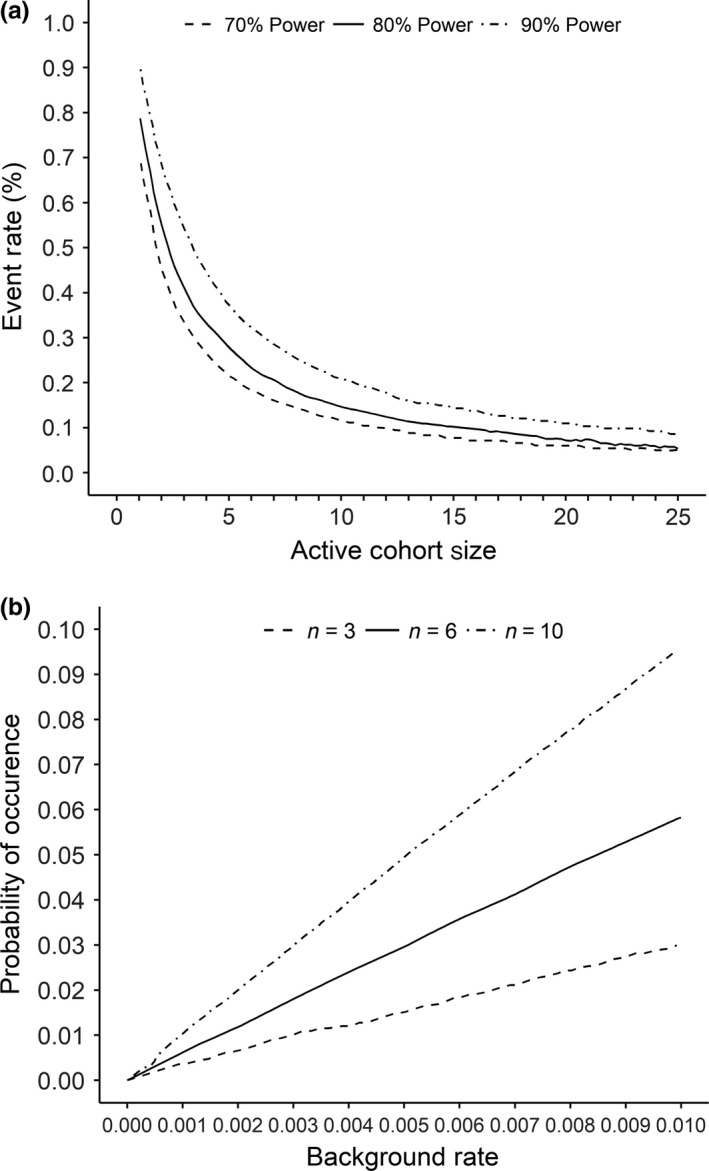
(a) Detectable event rate as a function of active cohort size and power. There is little to gain by increasing the cohort size from 10 to 12 subjects; but an increase from four to six subjects results in an appreciable increase in sensitivity. Reprinted with permission from Buöen et al.30 (b) The relationship between background rate and the probability of the event spontaneously occurring in at least one subject depicted for an active cohort size of three, six, and 10 subjects. Reprinted with permission from Buöen et al.30 DDI, drug‐drug interaction; MAD, multiple ascending dose; MTD, maximum tolerated dose.
Study size and inclusion of placebo subjects
The majority of FIH clinical trials are blinded, placebo‐controlled studies with eight to 10 subjects per cohort randomized in a 3:1 or 4:1 ratio such that six to eight subjects receive the active therapy and two subjects receive placebo. Multiple cohorts are included to receive successively higher doses. However, this design is more anecdotal with sample sizes based on precedence without formal hypothesis testing that is typical in later stage clinical studies.
The ability to detect a safety signal in an FIH study is determined by the total number of subjects in the study as a whole as well as the size of each dose cohort, the actual event rate that the experimental agent would cause if given to a large population and the spontaneous background rate (nondrug‐related toxicity) of a given event. If one assumes a type I error (probability of ascribing an event to a drug when it is not due to the drug) and a type II error (probability of not detecting an event when, in fact, a drug causes a given event), and a range of incidences of events that would be caused by a therapy in which one can develop a curve to show the relationship of cohort size (N) with the ability to observe an event that occurs at a given incidence due to a therapy. As shown in Figure 2 a, the detectable event rate decreases exponentially with increasing cohort size such that increasing the N in the range of one to six subjects results in substantial improvements in the chance of observing a drug‐induced event. The curve is relatively flat above an N of 10, so for the purpose of at least observing an event that is due to a new therapy, a cohort size of between six and 10 subjects is reasonable.30
Only events that occur very commonly due to a drug will be observed in an FIH study. For an event that would occur in about 25% of subjects receiving a new therapy, one has an 80% probability of observing the event at least one time in a cohort of six subjects (Figure 2 a). Although increasing the cohort size above six results in little increase in the probability of observing a given event, the higher the sample size the greater the probability for observing an event that is not due to a study drug but is simply part of the spontaneous event rate for that event. As shown in Figure 2 b, if the background rate of an event is 1%, the probability of observing that event in a cohort of 6 and 10 is about 5% and 9%, respectively. That is, in an FIH study, increasing a cohort size above six gains little in the ability to observe a drug‐induced event and results in observing more events that are not related to the study drug.30, 31, 32
Another aspect that adds to the overall study size is the inclusion of placebo subjects within each dose cohort. The rationale for inclusion of placebo‐dosed subjects is the perceived bias in adverse event (AE) reporting. Despite the small sample size, FIH protocols typically incorporate a pooled within‐study analysis of all placebo subjects to assess differences in AEs between active and placebo treatments. However, one school of thought maintains that inclusion of placebo subjects in FIH trials, which are underpowered, is not justified or useful.33 Most AEs observed in FIH studies are just reported as “probably related” with minimal interpretation of causality and more serious AEs are typically followed with greater diligence and concern. Inclusion of placebo subjects increases the complexity of the study by requiring procedures and documentation to maintain the blind effects on dose escalation decisions by safety monitoring committees as a result of AEs observed with placebo‐dosed subjects, formulation and manufacture of the placebo, and ethical considerations of subjecting placebo‐assigned volunteers to the many potential invasive procedures, such as PK blood draws. One could argue predose and/or time‐matched baseline measurements could be used in lieu of placebo subjects.
Study population
The choice to conduct FIH trials in healthy subjects or patients should be made on a case‐by‐case basis taking into account all factors relevant to the safety of the participants and the value/quality of the data to be generated. This decision is driven by the inherent risk of the drug candidate being investigated and the presence of the drug target in healthy subjects vs. patients. It is most appropriate to enroll patients rather than healthy subjects on the basis of risk‐to‐benefit in the case of higher risk agents (i.e., cytotoxic drugs) targeted at life‐threatening diseases where all existing therapeutic options for the patient have been exhausted, or an invasive dosing route is required for dose administration. On the other hand, it may be more appropriate to enroll healthy subjects when comorbidities or concomitant medications would make it difficult to interpret study results. In some cases, it may be crucial to assess the safety and tolerability of a novel drug candidate prior to testing in compromised patients. It is worth noting, however, that these two choices do not have to be mutually exclusive. Based on information available in the US National Library of Medicine registry (clinicaltrials.gov), many FIH studies include assessment in both healthy volunteers and patients. If healthy volunteers are studied, the upper end of the age range for enrollment is typically capped between 45 and 60 to avoid many comorbidities/concomitant medications commonly found in the elderly population.
Often when a drug candidate is being dosed in an FIH study, full characterization on its reproductive and embryo/fetal development toxicity potential has not been completed. Unless the drug candidate is intended for woman's health, typical FIH studies enroll male volunteers and sometimes include women of nonchildbearing potential, due to concerns about potential adverse embryo/fetal effects of treatment. The FDA guidance published in 1993 left the decision of including women of childbearing potential up to the researchers, IRBs, and women enrolling in the trial.34 In 1998, the FDA provided an updated guidance on the topic mandating sponsors to understand sex differences in PK, safety, and pharmacological response; therefore, it behooves the sponsors to include female subjects in the FIH trials.34 If women of childbearing potential are included in the trials, it should only be in studies short in duration (e.g., 2 weeks) with intensive control of pregnancy risk.6
Dose and dose escalation scheme
To further mitigate risk in an FIH trial the expert scientific group (aka “Duff” report) commissioned after the TGN1412 incident recommended dosing subjects sequentially with an appropriate period of observation between dosing of individual subjects.35 This practice is often referred to as “sentinel dosing” and is more often adopted during early phases of single ascending dose (SAD) trials. For the typical blinded, placebo‐controlled, dose‐escalation study this entails enrolling two subjects at a 1:1 active to placebo ratio followed by the remaining subjects in the respective dose cohort. This is carried out for each dose level for an adequate period of time based on the anticipated AEs and MOA of the drug candidate. The recent EMA guidance emphasized routine use of sentinel dosing within each dosing cohort, for both single and multiple dosing, and suggested that “clear scientific rationale” should be available if this approach is not adopted.2
Decisions regarding dose escalation require balancing safety risks to individual subjects against the risks of failing to define the correct dose range to be used in subsequent studies, or worse yet abandoning development of a potentially useful drug. To avoid the latter, it is important to understand the therapeutic dose range and target saturation, as in most cases it is expected to predefine the expected maximum exposure level to be studied in an FIH trial in healthy volunteers. This is also discussed in the recent EMA guidance in which the maximum exposure in healthy volunteers should be within the estimated human pharmacodynamic range as an MTD approach is not appropriate in healthy volunteers.2 To that end, the EMA recommends implementation of dose‐stopping criteria comprising maximum exposure (either peak plasma concentration (Cmax) or area under the curve (AUC)) in the study design and once the threshold is reached in individual subjects, the stopping criteria is considered to be met.2 This new rule will require sponsors to carefully define a PK/PD relationship of the drug candidates and the range of exposure considered pharmacodynamically relevant.
An important consideration for phase I trials in patients is to avoid unnecessary exposure to subtherapeutic doses while preserving safety and maintaining rapid escalation to a therapeutic dose range. One strategy that is often implemented in dose finding designs is to enroll a single subject at a low dose to assess safety and PK prior to escalation to the next dose level. Additionally, as often the case for oncology drugs, the dose is based on body size in lieu of a fixed dose. The implementation of body size‐based dosing is likely based on the perception that intersubject variability is reduced leading to reduced variability in response. However, recent simulation studies demonstrate that either dosing method results in a similar degree of PK and PD variability suggesting that normalization of dose by body weight is an unnecessary step.36, 37, 38 Regardless of the option taken, the strategy should provide reduced interpatient variability to optimize therapeutic effect coupled with convenience, compliance, and cost‐effectiveness.
The primary goal of FIH trials in oncology is to identify the appropriate dose and dosing interval for testing efficacy in phase II trials (i.e., recommended phase II dose). Therefore, dose escalation methods govern study design and oncology trials typically fall into one of two classifications: rule‐based designs or model‐based designs.39, 40
The rule‐based designs prespecify the dose levels and escalate or de‐escalate (i.e., “up‐and‐down” designs) according to prespecified rules based on the observed target events (e.g., dose‐limiting toxicity (DLT)). The most commonly used design is the traditional 3+3 design that proceeds with cohorts of three patients at prespecified dose levels (Figure 3). The dose escalation continues until at least two patients within a cohort of three to six patients experience a DLT. The MTD or recommended phase II dose is the dose below the level that is too toxic. Other variations of this design have been implemented, including “2+4,” “3+3+3,” and “3+1+1.” Another rule‐based design includes the pharmacologically guided dose escalation, which escalates based on prespecified systemic exposures that correlate with pharmacologic activity (e.g., significant tumor growth inhibition) and/or toxicity.41 Typically, this design converts to the traditional 3+3 design once the target systemic exposure is reached. Last, the accelerated titration designs described by Simon and colleagues42 provided three different designs for intrapatient dose escalation until a dose‐limiting toxicity is observed, after which the design switches to the traditional 3+3 design with 40% dose‐step increments.
Figure 3.
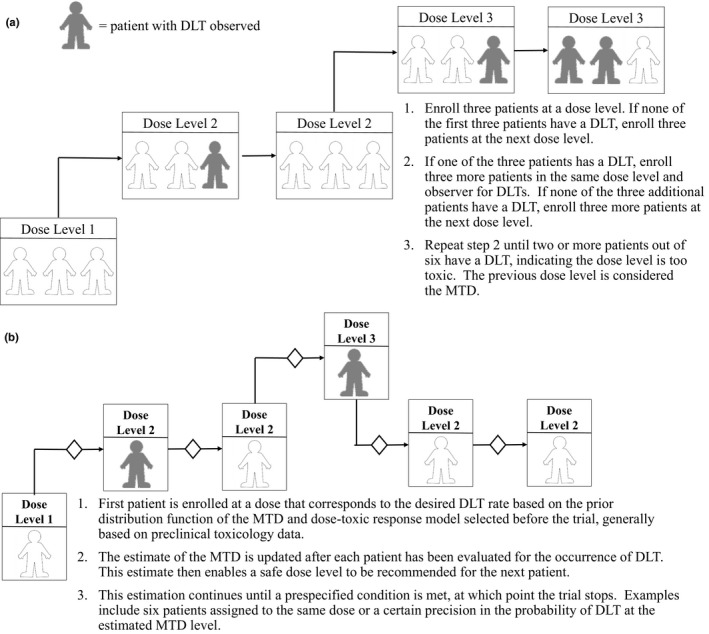
Study design schematics of phase I oncology trials: (a) 3+3 study design and (b) Continual Reassessment Model.
A disadvantage of the rule‐based designs is they are inefficient in establishing the dose. In fact, simulations have shown that only 35% of patients are treated at optimal dose levels.40 Conversely, 55% of patients are treated with an optimal dose utilizing model‐based designs, which use statistical models to assign dose levels based on a prespecified probability of DLT by using data from all enrolled patients to compute a more precise dose‐toxicity curve. Examples of adaptive Bayesian model‐based methods include the continual reassessment method (CRM; Figure 3),43 escalation with overdose control,44 and a modified CRM that utilizes time‐to‐event end points for handling late‐onset or CRMs.45
These study designs were developed with cytotoxic agents in mind where toxicity is the primary decision driver. However, there are many drug candidates with larger therapeutic windows, thus requiring a different mindset than just MTD but perhaps a biologically effective dose predicated on alternative end points besides toxicity. Examples of Bayesian‐based study designs that incorporate both toxicity and efficacy include the EffTox design46 or TriCRM.47 It is also important to consider the PK/PD relationship in elucidating a biologically active dose range as supported by the task force on Methodology for the Development of Innovative Cancer Therapies.48
What else can be considered?
Traditionally, FIH studies are SAD studies, with a separate staggered multiple ascending dose (MAD) study in parallel but lagging behind. Since the concept of adaptive clinical trial design was introduced in the 2004 FDA Strategic Path Initiative, more SAD/MAD combo studies have been conducted, which has been estimated to reduce timelines as much as 12 months compared with separate trials.49 The appeal of the combo design is the advantage of conducting SAD and MAD studies in parallel with the possibility of investigating new doses in adaptive cohorts, although it does demand predefined implementation of restrictive start and stop criteria to ensure safety.
FIH studies provide an opportunity to assess food and formulation effects, which are frequently included in adaptive study designs to guide further clinical development. These assessments can be incorporated into the SAD arm of the study when dose escalation has reached an anticipated therapeutically relevant level (Figure 4), this is particularly important for drugs that exhibit nonlinear PK either based on preclinical PK data in animals or cumulative data from the ongoing trial as clinical PK data become available. Both food and formulation effect assessment should be conducted with a cross‐over design to draw meaningful conclusions.
Figure 4.
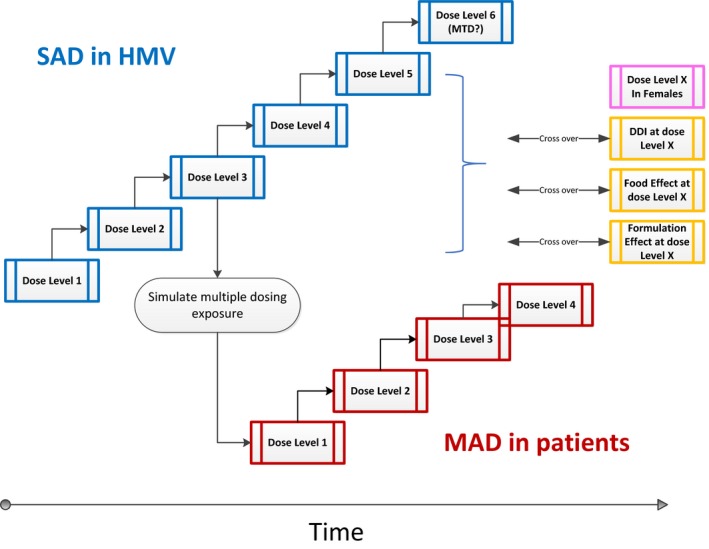
Example study schema for a first‐in‐human trial with multiple objectives. Crossover arms to characterize gender/food/formulation effects at therapeutically relevant dose levels should only be initiated after confirming safety at the same dose level in healthy male volunteers. The drug‐drug interaction assessment at this stage typically aims to identify risk of drug candidates being potent cytochrome P450 inhibitors based on in vitro data. HMV, healthy male volunteers; MAD, multiple ascending dose; MTD, maximum tolerated dose; SAD, single ascending dose.
Based on preclinical metabolism, drug transport, and DDI study results, if the drug candidate shows strong DDI potential,50 in particular associated with key metabolizing enzymes like cytochrome P450 3A4, it would be prudent for sponsors to include DDI assessment in the FIH studies. Design considerations for the DDI arm of the study should follow recent FDA guidance for data to be clinically relevant and informative.51
The FDA guidance Immunogenicity Assessment for Therapeutic Protein Products details the agency's expectation on immunogenicity assessment in FIH trials, in particular for high‐risk protein therapeutics.52 Because animal studies are not predictive of immune responses in humans, FIH studies are the first opportunity for sponsors to gauge immunogenicity potential of a new biologic drug candidate, which carries implications for both clinical safety and efficacy. However, the study population should be kept in mind while interpreting the immunogenicity data, as the immunogenicity profile may or may not be representative of that in the target patient population because many factors, such as immunologic status, may alter the immune response.
Safety monitoring
The monitoring of safety in an FIH study presents a paradox to clinicians and sponsors. The detection of a safety signal with a high degree of certainty is extremely limited by the typically small size of these trials. In addition to the statistical limitations of detecting a safety signal, determining on clinical grounds if an event is due to a new therapeutic modality is fraught with difficulty. Attribution of an AE to an experimental therapy when many events occur spontaneously in the study population is problematic and the dimensions of the problem are magnified in studies conducted in patients rather than healthy volunteers where comorbidities, disease progression, and lingering effects of other therapies can all contribute to a high noise‐to‐signal ratio. Successful signal detection in FIH studies depends on adequate collection of data coupled to a systematic analysis of the data, a determination of the strength of a potential signal and an understanding of the quantitative limitations of typical study designs.
The data collected in an FIH study in most respects are no different from any clinical trial and consists of physical examinations, monitoring of vital signs, laboratory data, including hematology, chemistry and urinalysis, and electrocardiograms (ECGs) collected at baseline and at various times during the conduct of the clinical trial. Depending on preclinical toxicology results, specialized examinations, such as ophthalmologic examinations, psychometric testing, monitoring of suicide scales, or radiographic studies, may also be warranted. If preclinical toxicology suggests that a drug candidate has effects on cardiac conduction or effects on human ether‐a‐go‐go channels, continuous ECG monitoring and triplicate ECGs obtained in conjunction with plasma sampling for PK evaluation would be useful once the dose or range of doses to take forward into further development is determined. In fact, because the FIH study might be the only opportunity to obtain concentration‐response data at supratherapeutic doses for QTc analysis, a negative signal from such studies with high‐quality ECG recording may obviate the need for a dedicated thorough QT prolongation study.53
When a new topical drug candidate is tested systematic collection of localized reactions should be used. When a particular AE of interest is defined based on preclinical information, collecting more detailed information than simply what is required to describe an AE is useful. For example, if toxicology studies found that above certain exposures abnormal hepatic transaminases occurred, and if such abnormalities are also observed in human subjects, a detailed history of recent intake of alcohol and other medications and testing for possible confounding causes would be helpful in determining causality, such as serologic screens for hepatitis C virus, hepatitis type B, human immunodeficiency virus, cytomegalovirus, and indicators of alcohol use.
Certain rare or dramatic events, such as anaphylaxis, Stevens Johnson syndrome, or acute liver failure, are almost always due to an active drug. In addition, drugs that affect multiple signaling pathways or may cause the amplification of immunologic or coagulation cascades or risk the release of cytokines warrant extra caution in monitoring and dosing paradigms.
One of the important outcomes of many FIH studies is to determine the MTD of a new therapeutic entity based on the occurrence of particular AEs above a particular severity grade, referred to as DLT. This is especially true of therapeutic areas in which little or nothing is known about the probable PK/PD relationship and where the intended indication is a disease with dire consequences, such as cancer. In such trials, the goal is to determine the MTD with as few dose levels as possible and to minimize the chance of administering doses that may result in an unacceptable degree of harm.
There is no standardized design to determine MTD based on the occurrence of DLTs but all include a stepwise increase in dose.54 As described above, dose escalation can occur within a subject or dosing by cohort. A critical first step in designing such a trial is to designate what toxicities are important. These can be laboratory tests (such as liver enzymes or serum creatinine) or a set of symptoms, such as severe diarrhea. A preset level of severity is defined based on a standardized grading system, such as the National Cancer Institute's Common Terminology Criteria for Adverse Events grades. It is possible that over the course of the trial an unanticipated DLT could also emerge. The duration of observation also has to be defined during which an event has to occur in order to be considered related to the experimental therapy. In general, it is better to perform a study to determine the MTD in the population of patients for whom the therapy will be intended, because sometimes patients have a different susceptibility to an event compared with healthy people.55 On the other hand, sometimes patients will more often have certain symptoms or findings that are part of their disease, making interpretation difficult. In some settings, the FIH study has to be done in patients because of known or suspected underlying risks of the therapy. In addition, some predefined incidence of a potential DLT has to be set so as to minimize attributing a toxicity to the therapy when in fact it should not be.
It is also helpful to relate PK data to the occurrence of severe or dose‐limiting events. In some cases, if an AE develops in the context of extreme exposures compared with subjects not experiencing the event, one could use this information in future trials to test whether dosing based on exposure is an advantageous approach.
Monitoring of AEs in FIH studies can be done by various ways depending on the perceived risk. If a drug is predicted to have low risk of toxicity, monitoring, and decision making regarding dose advancements based on the occurrence of AEs, it can be done by the sponsor, possibly in conjunction with the trial's investigators. When the risk of severe toxicity is moderate or high, experts with no vested interest in the results of the trial should be enlisted. A completely independent data monitoring committee is not usually warranted, but a joint effort between the sponsor and outside experts in a relevant therapeutic area or clinical discipline is often useful to help determine if a potentially unsafe dose has been reached. However, in some cases, an independent data monitoring committee may be warranted if there is some reason to perform the FIH study in a placebo‐controlled double‐blind manner and if there is a high level of concern that the new agent may be unduly toxic. Such a committee can be privy to unblinded data so that drug‐induced effects can be more readily identified.2, 56 In all cases, it is important to design a safety monitoring plan prior to initiating an FIH study. The purpose of the plan is to set out how and what data will be collected, how and when it will be reviewed, and what are the possible recommendations. For example, in the case where toxicology studies suggest that a particular type of medically serious toxicity is likely to occur, rules should be set out that would require stopping dosing of the study drug in an individual subject or for stopping a study altogether.
Biomarker assessment
The FIH study represents the first opportunity that a biological readout can be obtained in humans. The measurement of biomarkers in conjunction with a drug treatment could be important both during drug development and clinical implementation after marketing approval. Biomarker readouts can help guide drug development: from discovery through target engagement assessment, to diagnostic use, and on to the promise of precision medicine and companion diagnostics in everyday clinical practice. Along this path, the implementation of a biomarker program in the FIH study is a critical step toward biomarker utility, as it can confirm hypotheses from preclinical or observational clinical research studies. Many examples exist of biomarkers that were tested in early clinical studies that have gone on to be used in clinical diagnostics. This is particularly evident in the oncology field with predictive biomarkers commonly used for patient screening and risk assessments, such as BRCA1/2 and prostate‐specific antigen, and prognostic and treatment decisions, such as human epidermal growth factor receptor 2 for trastuzumab treatment. In addition, using a priori selected candidate biomarkers in an FIH study can also support MOA hypotheses that lead to further proof of concept (POC) studies or new targets for specific diseases.57
Biomarkers of pharmacology or disease state are usually thought to be soluble systemic factors that can be easily measured in serum or plasma. However, there are many end points or patient stratification factors that can also be measured as genetic, soluble cell surface, or histological markers. The clinical pharmacologists/scientists should be aware of the literature, as well as in vitro and preclinical pharmacology models to determine if a suitable candidate biomarker should be included in an FIH study. A solid understanding of a drug's pharmacology should be demonstrated with modulation of a candidate biomarker in preclinical animal or in vitro models as well as during safety assessments. The decision to assess a biomarker should include the evaluation of various success factors, such as a thorough literature review, bioanalytical method development, feasibility and validation, clinical sampling strategy, data reduction and analysis strategy, and a plan on how to incorporate biomarker data into the clinical database in a meaningful way. A regulatory assessment should also be conducted. Will data from the FIH only be used to support advancement to phase II POC? On the other hand, is the biomarker data to be used to lay the groundwork for further biomarker studies through late‐phase clinical studies and on to clinical diagnostic tests, such as a licensed screening tool or companion diagnostic? The answers to such questions will help define the fit‐for‐purpose regulatory needs of the biomarker method and data handling. It is important to think holistically when planning biomarker implementation, as one FIH biomarker data set may not only be valuable to a single drug development project, but also useful to a disease indication for which multiple drug candidates will be developed.
Once the need to assess a biomarker in an FIH study is established, the feasibility of performing a biomarker analysis in a clinical trial must be demonstrated. Depending on the biomarker chosen and available information on the methods needed for sampling, measuring, and analyzing the data, this could be a simple paper‐based exercise or might involve significant laboratory work to demonstrate that the proposed method can measure the biomarker adequately. In the latter case, the decision to assess a biomarker will optimally be made long enough in advance to enable the development of a robust bioanalytical method, and this should be taken into account when planning the FIH. If anything is known about the stability of a biomarker, for example, a given protein in serum, then it is possible to plan for a batch analysis at the end of the FIH study and to perform an at‐risk method validation in parallel with the FIH study. Other biochemical measurement methods, such as liquid chromatography‐mass spectrometry or automated clinical analyzers for lipids or other metabolites may be more readily available than developing a custom ligand binding method. Regardless of the final bioanalytical method, there may be challenges in obtaining tissues or samples that are less accessible than serum or plasma. Challenging matrices, such as stool samples, ocular fluids, mucosal secretions, tissue biopsies, and surgical byproducts, can and should be considered for biomarker analyses, but the challenge of accurate analysis is much higher in these cases and feasible sampling as well as analyte extraction should be considered and demonstrated prior to implementation.
Biomarkers sit in a zone of regulatory compliance that allows the clinical development team to decide on what the level of method characterization and documentation is needed. At the very least, a qualification demonstrating precision and accuracy should be performed by testing quality control samples prepared independently in the matrix of interest or a related surrogate matrix. In the best case, an FIH bioanalytical assay for a biomarker should be a fully validated method that includes the a priori determination of the method's quantifiable range and matrix interference followed by a plan‐based assessment of assay precision, accuracy, selectivity, robustness, endogenous levels in healthy and disease state matrix, parallelism, and analyte stability in storage conditions that mimic sample handling.58 The determination of a fit‐for‐purpose compliance level for a given biomarker analysis is best assessed by weighing the goals of the clinical development program and how the biomarker may provide the most value to the program. For example, if the biomarker will be used to assess phase II POC clinical responses to support a phase III decision, then a fully validated method should be implemented. However, if the biomarker in the FIH is aiming to support a program go/no‐go decision for a drug delivery technology, then a qualified assay may be all that is needed. Whatever the purpose, the regulatory and bioanalytical characteristics of the method should fit the needs of the program.
Similar to assessing the assay development needs to fit‐the‐purpose of a given FIH biomarker measurement, biomarker data analysis plan should be defined prospectively. For example, gene expression studies often rely on housekeeping genes to normalize the number of cells in a given sample and matrices, such as tears or urine, differ in concentration or volume due to biologic variation, and as such may require an internal normalization factor, such as total protein or creatinine, respectively. The regulatory requirements may also define how the data should be handled. If the FIH biomarker assessment is exploratory in nature, then simple spreadsheet analyses may be adequate. However, if a regulated development path is intended for the biomarker, then Clinical Data Interchange Standards Consortium compliant data standards would be required. As for the other characteristics of the biomarker assessment discussed above, the intent of the biomarker implementation will guide the rigor required and the formality of the planned analysis.
Global considerations
Many factors contribute to the decision on study site/country selection for FIH trials, including where the sponsors operate, regulatory requirements, ease of enrollment, and distribution of target patient population. Once decided, early engagement and strategically timed interaction with local health authorities are critical to the success of FIH studies. Before such engagement though, a thorough understanding of regulatory agency expectation, if guidance documents are available, will better prepare the sponsors and ensure productive discussions. In the absence of clear guidance, experienced local contract research clinical organizations, clinical development consultants, and key opinion leaders may be tapped to liaise and support the studies.
In the past, regulatory authorities in the ICH countries are among the first to evaluate new chemical entities and new biologic products (World Health Organization (WHO), “Report of a WHO Meeting: The Impact of Implementation of ICH Guidelines in Non‐ICH Countries”).59 In recent years, early phase clinical trials, including FIH studies conducted in non‐ICH countries like Australia, have increased significantly in number.60 Although the ICH‐Good Clinical Practice, which sets the global standard by which clinical trials are run are often adopted by non‐ICH countries,61 the perceived less stringent requirements and faster approval process to initiate FIH trials may be contributing factors to the increase. It is important to note that even though clinical data generated may be accepted by ICH regulatory authorities, as long as the study quality is ensured, data gap in drug quality or preclinical safety may still preclude sponsors to continue clinical development in ICH countries.
CONCLUSION
In conclusion, FIH studies require cross‐functional collaboration to be successful. Adaptive study design allows many questions to be answered, which will benefit downstream clinical development. The monitoring and collection of safety data in FIH studies is similar to the conduct of later‐stage studies, requiring explicit plans to monitor and evaluate the safety data.
Funding.
No funding was received for this work.
Conflict of Interest
The authors declared no competing interests for this work.
References
- 1. Food and Drug Administration, Center for Drug Evaluation and Research . Estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. <https://www.fda.gov/downloads/drugs/guidances/ucm078932.pdf> Cited (2005). Accessed March 2, 2008
- 2. European Medicines Agency . Guideline on strategies to identify and mitigate risks for first‐in‐human and early clinical trials with investigational medicinal products. <http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_001001.jsp&mid=WC0b01ac0580029570> (2018). Accessed March 2, 2018. [DOI] [PMC free article] [PubMed]
- 3. Faqi A. S., ed. A Comprehensive Guide to Toxicology in Preclinical Drug Development. (Academic Press: London, 2013). 885 p. [Google Scholar]
- 4. Gad, S. C . Safety Evaluation of Pharmaceuticals and Medical Devices. (Springer US: Boston, MA, 2011). [Google Scholar]
- 5. Butler, L. D et al Current nonclinical testing paradigms in support of safe clinical trials: an IQ Consortium DruSafe perspective. Regul. Toxicol. Pharmacol. 87, S1–S15 (2017). [DOI] [PubMed] [Google Scholar]
- 6. International Conference on Harmonisation . Guidance on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals M3(R2). <https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidisciplinary/M3_R2/Step4/M3_R2__Guideline.pdf> (2009) Accessed April 12, 2018. [PubMed]
- 7. Preclinical safety evaluation of biotechnology‐derived pharmaceuticals S6(R1). <http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S6_R1/Step4/S6_R1_Guideline.pdf> (2011). Accessed June 19, 2018. [DOI] [PubMed]
- 8. Nonclinical evaluation for anticancer pharmaceuticals S9. <http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S9/Step4/S9_Step4_Guideline.pdf> (2009). Accessed June 29, 2018.
- 9. Guidance on genotoxicity testing and data interpretation for pharmaceuticals intended for human use S2(R1). <http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S2_R1/Step4/S2R1_Step4.pdf> (2011). Accessed April 12, 2018. [PubMed]
- 10. Photosafety evaluation of pharmaceuticals S10. <http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S10/S10_Step_4.pdf> (2013). Accessed April 12, 2018.
- 11. Agoram, B.M. Use of pharmacokinetic/ pharmacodynamic modelling for starting dose selection in first‐in‐human trials of high‐risk biologics. Br. J. Clin. Pharmacol. 67(2), 153–160 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reigner, B.G. & Blesch, K.S. Estimating the starting dose for entry into humans: principles and practice. Eur. J. Clin. Pharmacol. 57(12), 835–845 (2002). [DOI] [PubMed] [Google Scholar]
- 13. Bender, B.C. , Schindler, E. & Friberg, L.E. Population pharmacokinetic‐pharmacodynamic modelling in oncology: a tool for predicting clinical response: population pharmacokinetic‐pharmacodynamic modelling in clinical oncology. Br. J. Clin. Pharmacol. 79(1), 56–71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huh, Y. , Smith, D.E. & Rose Feng, M. Interspecies scaling and prediction of human clearance: comparison of small‐ and macro‐molecule drugs. Xenobiotica 41(11), 972–987 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mahmood, I. & Balian, J.D. Interspecies scaling: predicting clearance of drugs in humans. Three different approaches. Xenobiotica 26(9), 887–895 (1996). [DOI] [PubMed] [Google Scholar]
- 16. Dong, J.Q. et al Quantitative prediction of human pharmacokinetics for monoclonal antibodies: retrospective analysis of monkey as a single species for first‐in‐human prediction. Clin. Pharmacokinet. 50(2), 131–142 (2011). [DOI] [PubMed] [Google Scholar]
- 17. Singh, A.P. et al Quantitative prediction of human pharmacokinetics for mAbs exhibiting target‐mediated disposition. AAPS J. 17(2), 389–399 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bischoff, K.B. Some fundamental considerations of the applications of pharmacokinetics to cancer chemotherapy. Cancer Chemother. Rep. 59(4), 777–793 (1975). [PubMed] [Google Scholar]
- 19. Boxenbaum, H. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J. Pharmacokinet. Biopharm. 10(2), 201–227 (1982). [DOI] [PubMed] [Google Scholar]
- 20. Nagilla, R. & Ward, K.W. A comprehensive analysis of the role of correction factors in the allometric predictivity of clearance from rat, dog, and monkey to humans. J. Pharm. Sci. 93(10), 2522–2534 (2004). [DOI] [PubMed] [Google Scholar]
- 21. Lave, T. et al Interspecies scaling of bosentan, a new endothelin receptor antagonist and integration of in vitro data into allometric scaling. Pharm. Res. 13(1), 97–101 (1996). [DOI] [PubMed] [Google Scholar]
- 22. Tabrizi, M. , Bornstein, G.G. & Suria, H. Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J. 12(1), 33–43 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mordenti, J. , Chen, S.A. , Moore, J.A. , Ferraiolo, B.L. & Green, J.D. Interspecies scaling of clearance and volume of distribution data for five therapeutic proteins. Pharm. Res. 8(11), 1351–1359 (1991). [DOI] [PubMed] [Google Scholar]
- 24. Mahmood, I. Pharmacokinetic allometric scaling of antibodies: application to the first‐in‐human dose estimation. J. Pharm. Sci. 98(10), 3850–3861 (2009). [DOI] [PubMed] [Google Scholar]
- 25. Wang, W. & Prueksaritanont, T. Prediction of human clearance of therapeutic proteins: simple allometric scaling method revisited. Biopharm. Drug Dispos. 31(4), 253‐263 (2010). [DOI] [PubMed] [Google Scholar]
- 26. Luu, K.T. , Bergqvist, S. , Chen, E. , Hu‐Lowe, D. & Kraynov, E. A model‐based approach to predicting the human pharmacokinetics of a monoclonal antibody exhibiting target‐mediated drug disposition. J. Pharmacol. Exp. Ther. 341(3), 702–708 (2012). [DOI] [PubMed] [Google Scholar]
- 27. Durairaj, C. , Shen, J. & Cherukury, M. Mechanism ‐ based translational pharmacokinetic ‐ pharmacodynamic model to predict intraocular pressure lowering effect of drugs in patients with glaucoma or ocular hypertension. Pharm. Res. 31(8), 2095–2106 (2014). [DOI] [PubMed] [Google Scholar]
- 28. Lehman, P.A. , Raney, S.G. & Franz, T.J. Percutaneous absorption in man: in vitro‐in vivo correlation. Skin Pharmacol. Physiol. 24(4), 224–230 (2011). [DOI] [PubMed] [Google Scholar]
- 29. Phillips, J.E. Inhaled efficacious dose translation from rodent to human: a retrospective analysis of clinical standards for respiratory diseases. Pharmacol. Ther. 178, 141–147 (2017). [DOI] [PubMed] [Google Scholar]
- 30. Buöen, C. , Holm, S. & Thomsen, M.S. Evaluation of the cohort size in phase I dose escalation trials based on laboratory data. J. Clin. Pharmacol. 43(5), 470–476 (2003). [DOI] [PubMed] [Google Scholar]
- 31. Iasonos, A. et al The impact of non‐drug‐related toxicities on the estimation of the maximum tolerated dose in phase I trials. Clin. Cancer Res. 18(19), 5179–5187 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sackett, D.L. & Gent, M. Controversy in counting and attributing events in clinical trials. N. Engl. J. Med. 301(26), 1410–1412 (1979). [DOI] [PubMed] [Google Scholar]
- 33. Parasrampuria, D.A. & Benet, L.Z. Inclusion of placebos and blinding for ascending dose first‐in‐human studies and other underpowered phase 1 studies has not been justified and on balance is not useful. Basic Clin. Pharmacol. Toxicol. 117(1), 44–51 (2015). [DOI] [PubMed] [Google Scholar]
- 34. Liu, K.A. , DiPietro Mager, NA . Women's involvement in clinical trials: historical perspective and future implications. Pharm. Pract. 14(1), 708 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Day, M. Duff's report calls for changes in way drugs are tested. BMJ 333(7581), 1240–1242 (2006). [Google Scholar]
- 36. Bai, S. et al A guide to rational dosing of monoclonal antibodies. Clin. Pharmacokinet. 51(2), 119–135 (2012). [DOI] [PubMed] [Google Scholar]
- 37. Wang, D.D. , Zhang, S. , Zhao, H. , Men, A.Y. & Parivar, K. Fixed dosing versus body size‐based dosing of monoclonal antibodies in adult clinical trials. J. Clin. Pharmacol. 49(9), 1012–1024 (2009). [DOI] [PubMed] [Google Scholar]
- 38. Hempel, G. & Boos, J. Flat‐fixed dosing versus body surface area based dosing of anticancer drugs: there is a difference. Oncologist 12(8), 924–926 (2007). [DOI] [PubMed] [Google Scholar]
- 39. Ananthakrishnan, R. & Menon, S. Design of oncology clinical trials: a review. Crit. Rev. Oncol. Hematol. 88(1), 144–153 (2013). [DOI] [PubMed] [Google Scholar]
- 40. Rogatko, A. , Schoeneck, D. , Jonas, W. , Tighiouart, M. , Khuri, F.R. & Porter, A. Translation of innovative designs into phase I trials. J. Clin. Oncol. 25(31), 4982–4986 (2007). [DOI] [PubMed] [Google Scholar]
- 41. Collins, J.M. , Grieshaber, C.K. & Chabner, B.A. Pharmacologically guided phase I clinical trials based upon preclinical drug development. J. Natl. Cancer Inst. 82(16), 1321–1326 (1990). [DOI] [PubMed] [Google Scholar]
- 42. Simon, R. , Freidlin, B. , Rubinstein, L. , Arbuck, S.G. , Collins, J. & Christian, M.C. Accelerated titration designs for phase I clinical trials in oncology. J. Natl. Cancer Inst. 89(15), 1138–1147 (1997). [DOI] [PubMed] [Google Scholar]
- 43. Ivy, S.P. , Siu, L.L. , Garrett‐Mayer, E. & Rubinstein, L. Approaches to phase 1 clinical trial design focused on safety, efficiency, and selected patient populations: a report from the Clinical Trial Design Task Force of the National Cancer Institute Investigational Drug Steering Committee. Clin. Cancer Res. 16(6), 1726–1736 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Babb, J. , Rogatko, A. & Zacks, S. Cancer phase I clinical trials: efficient dose escalation with overdose control. Stat. Med. 17(10), 1103–1120 (1998). [DOI] [PubMed] [Google Scholar]
- 45. Normolle, D. & Lawrence, T. Designing dose‐escalation trials with late‐onset toxicities using the time‐to‐event continual reassessment method. J. Clin. Oncol. 24(27), 4426–4433 (2006). [DOI] [PubMed] [Google Scholar]
- 46. Thall, P.F. & Cook, J.D. Dose‐finding based on efficacy‐toxicity trade‐offs. Biometrics 60(3), 684–693 (2004). [DOI] [PubMed] [Google Scholar]
- 47. Zhang, W. , Sargent, D.J. & Mandrekar, S. An adaptive dose‐finding design incorporating both toxicity and efficacy. Stat. Med. 25(14), 2365–2383 (2006). [DOI] [PubMed] [Google Scholar]
- 48. Booth, C.M. , Calvert, A.H. , Giaccone, G. , Lobbezoo, M.W. , Seymour, L.K. & Eisenhauer, E.A. Endpoints and other considerations in phase I studies of targeted anticancer therapy: recommendations from the task force on Methodology for the Development of Innovative Cancer Therapies (MDICT). Eur. J. Cancer 44(1), 19–24 (2008). [DOI] [PubMed] [Google Scholar]
- 49. Francis, M. , Basque, J. , Legault, E. , Rufiange, M. , Sicard, E. & Lefebvre, M. An integrated first‐in‐human study design involving sequential single and multiple ascending dose cohorts: an emerging trend in phase I clinical trials. Clin. Pharmacol. Ther. 93, S20–S21 (2013). [Google Scholar]
- 50. Food and Drug Administration, Center for Drug Evaluation Research . In vitro metabolism ‐ and transporter‐mediated drug‐drug interaction studies (draft guidance). <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM581965.pdf> (2017) Accessed March 26, 2018.
- 51. Food and Drug Administration, Center for Drug Evaluation and Research . Clinical drug interaction studies — study design, data analysis, and clinical implications (draft guidance). <https://www.fda.gov/downloads/drugs/guidances/ucm292362.pdf> (2017). Accessed March 26, 2018.
- 52. Immunogenicity assessment for therapeutic protein products. <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM338856.pdf> (2014). Accessed June 29, 2018.
- 53. E14 clinical evaluation of qt/qtc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs ‐ questions and answers (R3) guidance for industry. <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073161.pdf> (2017). Accessed April 12, 2018.
- 54. Cutler, N.R. et al Defining the maximum tolerated dose: investigator, academic, industry and regulatory perspectives. J. Clin. Pharmacol. 37(9), 767–783 (1997). [DOI] [PubMed] [Google Scholar]
- 55. Cutler, N.R. & Sramek, J.J. Investigator perspective on MTD: practical application of an MTD definition–has it accelerated development? J. Clin. Pharmacol. 40(11), 1184–1187 (2000). discussion 1202‐1204. [PubMed] [Google Scholar]
- 56. Food and Drug Administration, Center for Drug Evaluation and Research . Guidance for clinical trial sponsors establishment and operation of clinical trial data monitoring committees. <https://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm127073.pdf> (2006). Accessed April 12, 2018.
- 57. Goossens, N. , Nakagawa, S. , Sun, X. & Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 4(3), 256–269 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Arnold, M.E. , Booth, B. , King, L. & Ray, C. Workshop Report: crystal City VI—bioanalytical method validation for biomarkers. AAPS J. 18(6), 1366–1372 (2016). [DOI] [PubMed] [Google Scholar]
- 59. World Health Organization . The impact of implementation of ICH guidelines in non‐ICH countries. <http://apps.who.int/medicinedocs/pdf/h2993e/h2993e.pdf> (2001). Accessed April 12, 2018.
- 60. Frost & Sullivan . Australia: preferred destination for early phase clinical trials. <https://ww2.frost.com/files/6514/7374/3781/Novotech_WP_20160701_v2.1.pdf> (2016). Accessed April 12, 2018.
- 61. Lang, T. A. et al Clinical research in resource‐limited settings: enhancing research capacity and working together to make trials less complicated. (ed King C. H.). PLoS Negl. Trop. Dis.. 4(6), e619 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. International Conference on Harmonisation . Safety pharmacology studies for human pharmaceuticals S7A. <http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S7A/Step4/S7A_Guideline.pdf> (2000). Accessed April 12, 2018.
- 63. International Conference on Harmonisation The non‐clinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) S7B. <http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S7B/Step4/S7B_Guideline.pdf> (2005). Accessed April 12, 2018. [PubMed]
- 64. International Conference on Harmonisation Note for guidance on toxicokinetics: the assessment of systemic exposure in toxicity studies S3A. <http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S3A/Step4/S3A_Guideline.pdf> (1994). Accessed April 12, 2018.
- 65. International Conference on Harmonisation Immunotoxicity studies for human pharmaceuticals S8. <http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S8/Step4/S8_Guideline.pdf> (2005). Accessed April 12, 2018.