

Abstract
Introduction:
Fetoplacental angiogenesis plays a vital role in pregnancy outcome. Vascular endothelial growth factor A (VEGFA) is one major regulator of angiogenesis. It primarily binds to FMS-like tyrosine kinase (FLT1) and kinase insert domain receptor (KDR). In most vascular beds, KDR appears to be the main mediator of angiogenesis. However, the role of both receptors within the human placenta remains unknown.
Methods:
Human fetoplacental ECs were isolated/cultured from placentas of full-term, uncomplicated pregnancies after scheduled Cesarean section. Cells were subjected to RNA interference of either FLT1 or KDR followed by MTT, wound scratch, and tube formation assays. ECs were serum-starved after RNA interference and treated with VEGFA (60 ng/ml), then subjected to western blot to investigate FLT1 or KDR-mediated signaling. All experiments were performed in triplicate utilizing ECs from at least three separate subjects. One-way ANOVA with Tukey post-hoc testing was utilized for statistical analysis.
Results:
Significant knock-down of FLT1 and KDR was confirmed by qPCR (p<0.01) and WB (p<0.0001). KDR knock-down decreased EC metabolic activity (p<0.01), and FLT1 ablation unexpectedly increased EC proliferation (p<0.01). There was no difference in apoptosis regardless of FLT-1 or KDR knock-down. FLT1 knock-down significantly impaired wound scratch closure (p<0.0001) and tube formation (p<0.001). Surprisingly, KDR effects on EC metabolism had no effect on migration, although KDR was important in VEGFA-stimulated Akt and ERK activation. In contrast, FLT1 effects on EC motility were Akt and ERK-independent.
Conclusion:
Human fetoplacental EC migration is primarily regulated by FLT1 but not KDR.
Keywords: Human fetoplacental endothelial cells, FLT1, KDR, endothelial migration
Introduction
During normal pregnancy, a considerable amount of vessel development and vascular growth occur in the human placenta, resulting in a vascular bed that is approximately 550 km in length and 15 m2 in surface area by 40 weeks of gestation [1, 2]. Placental vasculogenesis starts approximately two weeks after conception [3, 4]. A gradual transition to angiogenesis then ensues between 32 days and 9 weeks post-fertilization, with the vast majority occurring from 23 weeks post-conception to term gestation [5, 6].
Several important factors regulate vasculogenesis and angiogenesis. Among these, vascular endothelial growth factor A (VEGFA) plays a central role, starting from embryogenesis and progressing through the lifetime of an organism. VEGFA binds to two key receptors -- (1) FMS-like tyrosine kinase (FLT1), also known as vascular endothelial growth factor receptor 1, and (2) kinase insert domain receptor (KDR), which is also referred to as vascular endothelial growth factor receptor 2. KDR expression is mainly limited to ECs, whereas FLT1 is also expressed in many non-endothelial cell types, including vascular smooth muscle cells and macrophages [7, 8].
FLT1 and KDR are both tyrosine receptor kinases that are phosphorylated under stimulation by ligands such as VEGFA, leading to downstream signaling. Traditionally, KDR has been considered the primary signal transducer of physiologic and pathologic angiogenesis, where its phosphorylation by VEGFA binding results in key angiogenic events, including enhanced EC permeability, migration, proliferation, and survival [9–14]. The role of FLT1 and its downstream signaling on angiogenesis, however, is not as well understood. VEGFA binds to FLT1 with higher affinity than to KDR, but importantly, FLT1 kinase activity is substantially lower and only about one-tenth that of KDR [15–17]. Furthermore, FLT1 overexpression in ECs demonstrated only a minimal increase in proliferation when stimulated with VEGFA, suggesting that the role of FLT1 may be to serve as a “decoy” receptor for ligands, limiting the amount that can interact with KDR [14–16].
Specific to pregnancy and the fetoplacental vasculature, murine studies establish the critical roles of Vegfa, Flt1, and Kdr, where Vegfa−/−, Flt1−/−, and Kdr−/− mice all suffer from embryonic lethality secondary to abnormal blood vessel formation [18–20]. Even mice with only one functional Vegfa allele demonstrate impaired vascular development and embryonic lethality [18]. During human pregnancy, proper angiogenesis of the fetoplacental vascular tree is also vital for successful pregnancy outcome, as supported by the substantially increased risk for adverse pregnancy outcome in growth-restricted pregnancies further complicated by abnormal umbilical artery Doppler velocimetry [21, 22]. However, the specific roles of FLT1 and KDR in the appropriate development of the human fetoplacental vasculature remain unknown. Thus, our objective was to determine the individual contributions of FLT1 and KDR to human fetoplacental EC-mediated angiogenesis in uncomplicated pregnancies, and we hypothesized that EC proliferation and migration are regulated primarily through KDR.
Materials and Methods
Endothelial Cell Isolation and Culture
After approval by the institutional review board at the University of Colorado and subject consent, human fetoplacental villous endothelial cells (ECs) were isolated/cultured from placentas of full-term, uncomplicated, singleton pregnancies immediately after scheduled Cesarean section as previously described with minor modifications [23, 24]. After isolation, ECs were cultured in media supplemented with 5% fetal bovine serum, bovine brain extract with heparin, epidermal growth factor, hydrocortisone, and gentamicin/amphotericin B (Lonza, Walkersville, MD) at 37 °C in a humidified incubator with 5% CO2 enriched air. Consistent with previous data, primarily isolated ECs demonstrated nearly 100 percent purity, and primary cells were used only through the fifth passage to avoid changes in phenotype [23].
siRNA Transfection
For transfection, ECs were seeded in 6-cm dishes and cultured in antibiotic-free media until 50% confluent, then subjected to RNA interference with an RNA oligonucleotide directed against either FLT1 or KDR (Ambion, Carlsbad, CA). A scrambled negative control small interfering RNA (siRNA) was also utilized. Transfection was accomplished by the RNAiMAX lipofectamine-based reagent (Thermo Fisher, Carlsbad, CA) in Opti-Mem I (Thermo Fisher). On the day of transfection, RNAiMAX was combined with 20 nM siRNA duplexes that were diluted in Opti-Mem I and applied to cells, incubated for 6 hrs, then rescued overnight with full media without antibiotics. Cells were then collected for RNA or protein extraction or re-seeded into 12-well plates or 96-well plates for MTT/cell count, wound scratch, or tube formation assays.
RNA extraction, cDNA synthesis, and real-time PCR
Total RNA was extracted using the RNeasy Plus Mini Kit (Qiagen, Germantown, MD) according to the manufacturer’s instructions. RNA was quantified using a Nanodrop ND-2000 UV-Vis Spectrophotometer (Nanodrop Technologies, Wilmington, DE). Two μg of RNA from each treatment were used for synthesis of cDNA. Synthesis of cDNA was conducted using qScript™ cDNA SuperMix (Quanta Biosciences, Beverly, MA) according to the manufacturer’s instructions. Quantitative real-time PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems, Woolston, Warrington, UK) with a QuantStudio 6 Flex Real Time PCR system (Applied Biosystems, Rockford, IL). Primers for all genes were designed using the PrimerQuest tool from Integrated DNA Technologies (Integrated DNA Technologies, Coralville, IA). Primers were designed to have a Tm of 58–60 °C. Primers that spanned exon-exon junctions were chosen for real-time PCR, to prevent binding to genomic DNA. Additionally, a primer directed against ribosomal protein lateral stalk subunit P0 (RPLP0, also known as 36B4) as previously described was used as an endogenous control for gene expression [25]. Real-time PCR assays for each sample were conducted in triplicate wells with all genes including endogenous control on the same plate. Reactions contained Power SYBR Green PCR Master Mix, forward primer (0.5 μM), reverse primer (0.5 μM), and cDNA template made up to a final volume of 10 μl in nuclease-free water. Real-time PCR cycling conditions started with a holding time of 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 sec and 60 °C for 1 min of melting and extension temperatures, respectively. Data were analyzed using the relative CT (2−ΔΔCt) method [26].
Protein Isolation and Western Blotting
Protein extraction from human fetoplacental ECs transfected with siFLT1 or siKDR was performed using M-PER Mammalian Protein Extraction Reagent (Thermo Fisher) with addition of phosphatase and protease inhibitors (Cell Signaling Technologies, Danvers, MA). The supernatant was extracted by centrifugation and protein concentration was determined by BCA protein assay (Thermo Fisher). Ten μg of whole cell lysate was resolved on SDS-PAGE (4–15% precise gels) and then transferred to polyvinylidene difluoride (PVDF) membranes. The following antibodies were used for immunoblotting: monoclonal antibody against FLT1 (1:500, V4262, Sigma-Aldrich), monoclonal antibody against KDR (1:1000, 2479S, Cell Signaling Technology), and monoclonal antibody anti-β-actin (1:2000, A1978, Sigma-Aldrich). Horseradish peroxidase (HRP)-linked anti-rabbit IgG and HRP-linked anti-mouse IgG were used as secondary antibodies. Finally, immunoreactive bands were visualized using an enhanced chemiluminescence detection system (Luminata™ Crescendo Western HRP Substrate, Millipore Corporation, Billerica, MA 01821) using a Konica SRX-101 A medical film processor (Tokyo, Japan).
VEGFA Treatment and Cell Signaling
After cells were transfected with either siFLT1, siKDR, or scramble control siRNA, cells were starved for 1 hr and then treated with VEGFA (R&D, 293-VE, Minneapolis, MN) 60ng/ml for 0, 5, 10, and 60 min in serum- and growth factor-free medium (endothelial basal medium). Protein was extracted after VEGFA treatments, then subjected to western blots as above. The following antibodies were used: polyclonal antibody anti-phopho-Akt (1:1000, 9271S, Cell Signaling Technology), polyclonal antibody anti-Akt (1:1000, 9272S, Cell Signaling Technology), polyclonal antibody anti-phospho-Erk (1:1000, 9101S, Cell Signaling Technology), monoclonal antibody anti-Erk (1:2000, 4696S, Cell Signaling Technology), polyclonal antibody anti-RACK1 (1:1000, 4716S, Cell Signaling Technology), and monoclonal antibody anti-β-actin (1:2000, A1978, Sigma-Aldrich).
Cell proliferation analysis
After siRNA transfection, ECs were rescued overnight, trypsinized and then seeded into 96-well plates in equal numbers per well. As a measure of cellular metabolism as a potential reflection of cell proliferation, MTT assays were performed (BioLegend, San Diego, CA) with cells seeded in 96-well plates, and the OD value was measured. Additionally, cells were seeded into 6-well plates in equal numbers with cell number counted at 72 hours after trypsinization.
Wound scratch assays
After transfection with siFLT1, siKDR, or scramble control siRNA, ECs were rescued overnight, trypsinized, and then seeded into 12-well plates in equal numbers. ECs were cultured within full media until 100% confluence and then scratched with a P200 pipette tip. Cells were put in the Pop IncuCyte scanning machine (Ann Arbor, MI) immediately after scratching, and time-lapse, live images were taken at 0, 4, 8, 14, 16, 20 and 24 hrs within each sample. Images were analyzed with Image J software (https://imagej.nih.gov/ij/). The degree of wound healing, which is correlated to EC migration, was assessed by measuring the remaining cell free (non-closure) area.
Tube formation
Cells were transfected with either siFLT1, siKDR, or scramble control siRNA and rescued overnight in full media. Geltrex™ reduced growth factor basement membrane matrix (Gibco, by Life Technologies, Grand Island, NY) was thawed on ice, and 50 μL was used for coating each well in 96-well plates. The Geltrex™ was then incubated and allowed to polymerize for 30 min at 37 °C. During polymerization, ECs were then trypsinized and stained with 1,1’-dioctadecyl-3,3,3’3’-tetramethylindocarbocyanine perchlorate (DiI; D282, Thermo Fisher), then seeded into coated 96-well plates in equal numbers in full media. Time-lapse, live imaging was then immediately started using the Pop IncuCyte scanning. Live images were taken at every hour from 0 to 16 hrs. Images were analyzed with Image J software, and tube formation was measured by total length and the number of branch points.
Statistical Analysis
All experiments were performed on at least three representative subject samples, with each experiment repeated in triplicate. Representative wound scratch, tube formation, and immunofluorescence images are shown from a single subject, with numerical, graphical, and statistical analysis obtained from combining data from all experiments. Numerical data are reported as means of the replicates performed within all the subjects, with errors representing standard error of the mean (SEM). Data were analyzed using a one-way analysis of variance (ANOVA) with Tukey post-hoc testing for multiple comparisons if the one-way ANOVA demonstrated a statistical difference. Differences where p < 0.05 were considered statistically significant.
Results
RNA interference of FLT1 and KDR in primary human fetoplacental ECs is specific
To investigate the functions of FLT1 or KDR in the process of human placental angiogenesis, we first sought to individually knock down FLT1 or KDR and to ensure specificity of the ablation. FLT1 siRNA transfection significantly inhibited FLT1 mRNA (p<0.01) and protein (p<0.001) expression levels, with no effect on KDR (Figures 1A – 1C). Similarly, the effect of RNA interference on KDR was also confirmed by both real-time PCR and western blotting, where there was significant inhibition of KDR mRNA (p<0.01) and protein (p<0.001) with stable FLT1 expression (Figures 1D – 1F). Supplemental Figures 1A and 1B show the full western blot images from a representative experiment.
Figure 1. RNA interference of FLT1 or KDR is specific.

(A) Real-time PCR demonstrated more than 80% ablation of FLT1 mRNA expression after transfection with siFLT1 (p<0.01), with no effect on KDR expression. (B and C) A representative western blot with graphical representation also confirmed specificity of FLT1 siRNA (p<0.0001). (D) KDR mRNA expression was decreased by approximately 90% after KDR knock-down (p<0.01), with no effect on FLT1 expression. (E and F) A representative western blot with graphical representation after transfection with KDR siRNA also confirmed ablation of KDR expression with no effect on FLT1 expression (p<0.0001). Mock denotes cells exposed solely to exposure to the lipid.
In order to further confirm that there were no unexpected off-target effects to FLT1 or KDR ablation, we also investigated both VEGFA and soluble FLT1 (sFLT1) expression. Transfection with siFLT1 or siKDR showed significant differences in the one-way ANOVA for VEGFA mRNA expression (p<0.05), but post-hoc analysis demonstrated no significant differences in VEGFA expression with siControl, siFLT1, or siKDR knock-down (Supplemental Figure 2). In contrast, we found that FLT1 knock-down resulted in significantly decreased sFLT1 mRNA expression (p<0.0001), using a primer set that spans from exon 13 to the unique C-terminus sequence of sFLT1 (Supplemental Figure 2). This was not an unexpected finding, as the FLT1 siRNA we utilized targeted exon 5, which is proximal to the splice site that would result in expression of sFLT1.
Fetoplacental EC metabolic activity and cell proliferation are differentially regulated by FLT1 and KDR
After confirming specificity of FLT1 and KDR RNA ablation, we wanted to determine the role of each receptor on fetoplacental EC proliferation. We first utilized the MTT assay as a metabolic surrogate for proliferation, and we found that knock-down of KDR significantly decreased EC metabolic activity (p<0.01) when compared to that of control siRNA transfection (Figure 2A). In contrast, there was no effect on MTT OD values after FLT1 ablation.
Figure 2. FLT1 and KDR demonstrate divergent effects on EC metabolic activity and EC proliferation, however, EC apoptosis is not affected by FLT1 or KDR.

(A) MTT assay showed a decrease in EC metabolic activity, which oftentimes is linked to cell proliferation, after KDR knock-down (p<0.01), with no significant effect of FLT1 knock-down on metabolic activity. (B) In contrast, FLT1 ablation resulted in an increase in EC proliferation (p<0.01). Surprisingly, KDR knock-down did not affect EC proliferation. (C) A representative western blot showed that neither FLT1 or KDR ablation led to presence of cleaved caspase-3, a marker of apoptosis. There was also no effect of FLT1 or KDR knock-down on total caspase-3 expression (D).
In order to assess whether the effect of KDR on EC metabolic activity translated into decreased cellular proliferation, we plated equal numbers of cells after FLT1 or KDR RNA interference and assessed total cell number at 24 hours. We were surprised to find no difference in total cell number after KDR ablation (Figure 2B). Furthermore, we found a statistically significant increase in total cell number after FLT1 knock-down, suggesting that enhanced relative VEGFA bioavailability in the setting of low sFLT1 and FLT1 is acting via KDR to increase EC proliferation (p<0.01, Figure 2B).
FLT1 or KDR silencing do not affect EC apoptosis
Because we saw disparate findings between our MTT assay and EC cell count results after FLT1 and KDR knock-down, we then wanted to investigate whether either VEGF receptor affected EC apoptosis. As demonstrated by the absence of cleaved caspase-3 by western blot, neither transfection with FLT1 or KDR siRNA resulted apoptosis (Figures 2C – 2D).
FLT1 is the dominant VEGF receptor that mediates fetoplacental EC migration
Beyond proliferation and apoptosis, other key features of angiogenesis include EC migration. To determine the role of each VEGF receptor on fetoplacental EC migration, we first utilized wound scratch assays. Unlike findings in ECs of other organs, KDR ablation within the human fetoplacental endothelium did not exhibit any effects on wound migration, with essentially 100 percent wound closure at 24 hours (Figure 3). In contrast, we were further surprised to find that FLT1 knock-down resulted in significantly impaired wound closure, with only approximately 65 percent closure at 24 hours, despite enhanced proliferation after FLT1 ablation (p<0.0001, Figure 3).
Figure 3. FLT1 ablation inhibits EC migration and wound closure.

(A) Representative wound scratch images and (B) graphical representation of the findings demonstrated that FLT1 knock-down resulted in more than a 30% decrease in wound closure in comparison to both siControl and siKDR transfection (p<0.0001).
To further assess EC migration and confirm these wound scratch assay findings, we also performed tube formation assays after control siRNA, FLT1 siRNA, or KDR siRNA transfection. Using live-cell imaging, we found that FLT1 knock-down led to deficient tube formation that was seen as early as 2 hours after plating, with impairment being most evident at 12 hours (Figure 4A). More specifically, there were significantly decreased branch points (p<0.001) and total tube length (p<0.001) after FLT1 ablation (Figures 4A – 4C). KDR knock-down, however, did not affect tube formation at any time point. Together, these data demonstrate that FLT1 is the dominant VEGF receptor that mediates fetoplacental EC migration.
Figure 4. FLT1 knock-down results in impaired tube formation.

(A) Representative tube formation images with diI-stained fetoplacental ECs demonstrated impaired tube formation as early as two hours after knock-down of FLT1 that was sustained through 12 hours. Both number of branch points (B) and total tube length (C) were significantly decreased after FLT1 ablation (p<0.001 for both) with no effect of KDR knock-down on tube formation.
FLT1-dependent EC migration is Akt- and ERK-independent
Previous studies on ECs from non-placental vascular beds have found that EC migration and EC stabilization is primarily influenced by KDR phosphorylation that results in downstream Akt and/or ERK signaling [14, 27, 28]. After finding that FLT1 appeared to be the dominant VEGF receptor in fetoplacental EC migration, however, we wanted to investigate whether FLT1 knock-down inhibited Akt and ERK activation. Despite FLT1-mediated EC migration in fetoplacental ECs, we found that there was no effect on VEGFA-stimulated Akt activation after FLT1 ablation (Figures 5A – 5C). Similarly, ERK activation also did not occur in the setting of FLT1 ablation after stimulation with VEGFA (Figures 5D – 5F). Consistent with studies of ECs from vascular beds of other organs, however, KDR knock-down inhibited VEGFA induction of Akt and ERK signaling (Figures 5A – 5F).
Figure 5. Akt and ERK activation are FLT1-independent.

Representative western blot (A) showed Akt activation with VEGFA treatment despite FLT1 ablation, whereas transfection with siKDR led to decreased Akt phosphorylation at both 5 and 10 minutes (A-C). (D – F) Similarly, ERK activation was solely inhibited after KDR knock-down, with no effect on phosphorylation after FLT1 ablation.
FLT1 ablation decreases an anchoring/stabilizing protein of protein kinase C (PKC)
In light of the finding that FLT-mediated fetoplacental EC migration and was Akt-and ERK-independent, we wanted to investigate other potential downstream pathways that could explain our findings. Receptor for activated protein kinase C 1 (RACK1), an anchoring protein for PKC that can both stabilize it in its active form and allow for cellular translocation, has been shown to play a regulatory role in VEGFA/FLT1-dependent cell migration in angioma cells [29]. Within fetoplacental ECs, we found that FLT1 knock-down led to a very slight but technically significant decrease in RACK1 expression after baseline transfection and despite VEGFA stimulation in comparison to siControl transfection (p<0.001, Figure 6). KDR ablation, however, had no effect at all on RACK1 expression, suggesting that RACK1 may be a possible FLT1 downstream mechanism affecting fetoplacental EC migration.
Figure 6. FLT1 ablation decreases expression of RACK1.

RACK1 is a key protein that has been shown to regulate VEGFA-mediated EC migration. (A, B) A representative western blot with graphical depiction demonstrates a small, but statistically significant decrease in RACK1 expression at baseline after FLT1 knock-down (p<0.0001), where there was no effect on RACK1 expression after transfection with siKDR. (A, C) With VEGFA treatment, the decrease in RACK1 expression persists in the setting of FLT1 ablation (p<0.0001).
Discussion
In this study, we sought to determine the individual contributions of FLT1 and KDR to human fetoplacental EC-mediated angiogenesis in uncomplicated pregnancies. We found that within our model of human fetoplacental ECs, FLT1 appears to be the dominant VEGFA receptor involved in mediating EC migration, where FLT1 ablation resulted in significant impairment in wound closure and tube formation. This occurred in spite of down-regulation of the anti-angiogenic factor sFLT1. Furthermore, there was a surprising increase in cell proliferation after FLT1 ablation, demonstrating that the siFLT1-mediated deficiency in EC motility is likely even more profound than what we observed with our wound scratch and tube formation assays.
Our findings were entirely unexpected, primarily because the vast majority of studies on ECs within vascular beds of other organs have shown that EC proliferation, survival, and adherence are primarily regulated through KDR and not FLT1. Within these other ECs, KDR activation results in activation of two intracellular signaling pathways – Akt and ERK -- that are crucial to endothelial biology. We found that similar to other, non-fetoplacental ECs, VEGFA-mediated Akt and ERK phosphorylation occurs through KDR. However, these pathways were unaffected by FLT1 ablation, demonstrating that fetoplacental EC migration occurs independent of Akt and ERK activation.
Despite being widely expressed in a variety of cell types beyond just ECs, FLT1 signaling and its downstream consequences are incompletely understood. Because of its high binding affinity to VEGFA and low kinase activity, it has been suggested that FLT1 may act as a “decoy” receptor by decreasing the amount of available VEGFA to bind to KDR. This is further supported by the in vivo finding that unlike flt1−/− mice, flt1 tyrosine kinase domain-deficient mice (flt1 TK−/−) demonstrated normal angiogenesis and were healthy [30]. If this were the case in our model, however, we would have expected to see an increase in EC proliferation and migration with FLT1 knock-down, presumably through enhanced KDR activation. Instead, not only was EC proliferation increased with FLT1 ablation, but the sole effect of KDR knock-down that we found was a decrease in active metabolism as demonstrated by MTT assay analysis, suggesting that at least in human fetoplacental ECs, the primary role of FLT1 is not to act as a scavenger of VEGFA.
One potential explanation for our findings comes from data demonstrating that subcellular localization of FLT1 is a key factor in determining its activity. In general, intracellular FLT1 stores represent approximately 80 percent of total FLT1, with localization seen both in the Golgi apparatus and the nucleus [31, 32]. In a model of murine hindlimb ischemia, investigators found that membrane surface expression of FLT1 is significantly increased after 10 days of ischemia in comparison to the non-ischemic limb, with no differences in KDR expression [33]. This increase in cell surface expression resulted in an increase in EC proliferation, migration, and angiogenesis [33]. This data, however, is in contrast with others who have found that when the transmembrane domain of FLT1 is localized near the membrane of vascular ECs, it acts as a negative regulator via a “decoy” mechanism [34]. When this domain was visualized within the cytoplasm and far from the plasma membrane, disorganization of blood vessels with embryonic lethality was seen [34]. While discrepant, both studies highlight that subcellular localization may play a key role in the role of FLT1. Additionally, although both studies were performed in mice, the experimental conditions were certainly different, further underscoring the complexity in FLT1 signaling.
Our findings may also be explained by the in vitro nature of our experiments, where FLT1 or KDR protein expression was consistently reduced by more than 95 percent after transfection with siFLT1 or siKDR. This iatrogenically limits the potential interaction and/or heterodimerization of FLT1 and KDR. For instance, it has previously been demonstrated that FLT1/KDR heterodimers inhibit VEGFA-mediated KDR signaling, and this effect was not seen in the setting of FLT1/FLT1 homodimers [35]. Others have also shown that FLT1/KDR heterodimerization inhibits VEGFA-induced ERK activation, mobilization of intracellular calcium, and prostacyclin release in ECs [36]. Together, this demonstrates the potential importance of FLT1 and KDR availability and the need to further investigate their homodimer and heterodimer roles.
Strengths of our study include the use of primarily isolated, human fetoplacental ECs. Additionally, our RNAi efficiency was high, with specificity of each siRNA oligo utilized, which allowed us to delineate the individual roles of FLT1 and KDR in human fetoplacental angiogenesis. However, there are also several limitations to our study. First, while the efficiency of our knock-down was high, this led to nearly complete ablation (greater than 95 percent) of FLT1 or KDR protein expression. Thus, the effects that we saw were likely secondary to FLT1 or KDR homodimerization, and this current study is unable to assess the function of FLT1/KDR heterodimerization. Second, downstream signaling events we investigated were limited to two key pathways that are well-established to mediate EC angiogenesis. Finally, we utilized fetoplacental ECs isolated from uncomplicated, full-term pregnancies. We acknowledge that features of placental angiogenesis are unlikely to be static throughout gestation, and the physiologic trajectory of angiogenesis in full-term ECs may be on the decline. However, in order to obtain ECs from earlier gestational ages, we would have had to utilize placentas from pregnancies that end in preterm delivery, which by definition, are not normal. Similarly, if we obtained tissue from first or second trimester terminations, we would not be able to ascertain that their pregnancy outcome would have been uncomplicated. Thus, we chose to focus this initial investigation on full-term ECs from placentas of uncomplicated, full-term pregnancies in order to establish a baseline role of FLT1 and KDR in normal human fetoplacental endothelium. We further acknowledge that the signaling mechanisms found in our study are not necessarily representative of fetoplacental angiogenesis in normal pregnancies throughout gestation. However, they exemplify fetoplacental ECs that have played a role in achieving a successful, healthy pregnancy outcome.
In conclusion, we have found that human fetoplacental EC proliferation and migration are regulated primarily by FLT1 and not KDR. These findings are important because proper fetoplacental angiogenesis is a key component to healthy pregnancy outcome, and mechanisms underlying impaired fetoplacental EC function and angiogenesis remain incompletely elucidated. Understanding the roles of FLT1 and KDR in normal fetoplacental angiogenesis will better inform future, mechanistic studies that will hopefully allow us to better identify the molecular underpinnings of impaired fetoplacental angiogenesis and its role in adverse pregnancy outcome.
Supplementary Material
Supplemental Figure 1: (A) Full western blot images of the bands shown in Figure 1B. (B) Full western blot images of the bands shown in Figure 1E.
{kind=link}
Supplemental Figure 2: Real-time PCR demonstrates that although there is an overall difference in VEGFA expression with one-way ANOVA analysis (p<0.05), post-hoc testing shows no significant difference in VEGFA expression with siControl, siFLT1, or siKDR knock-down. In contrast, using a primer set that spans from exon 13 to the unique C-terminus region of sFLT1, we found that transfection with siFLT1 results in significantly decreased sFLT1 expression (p<0.0001). This was not unexpected, as the siRNA oligo utilized targets exon 5 of FLT1.
{kind=link}
Supplemental Figure 3: Full western blot images of the bands shown in Figure 5A.
{kind=link}
Supplemental Figure 4: Full western blot images of the bands shown in Figure 5D.
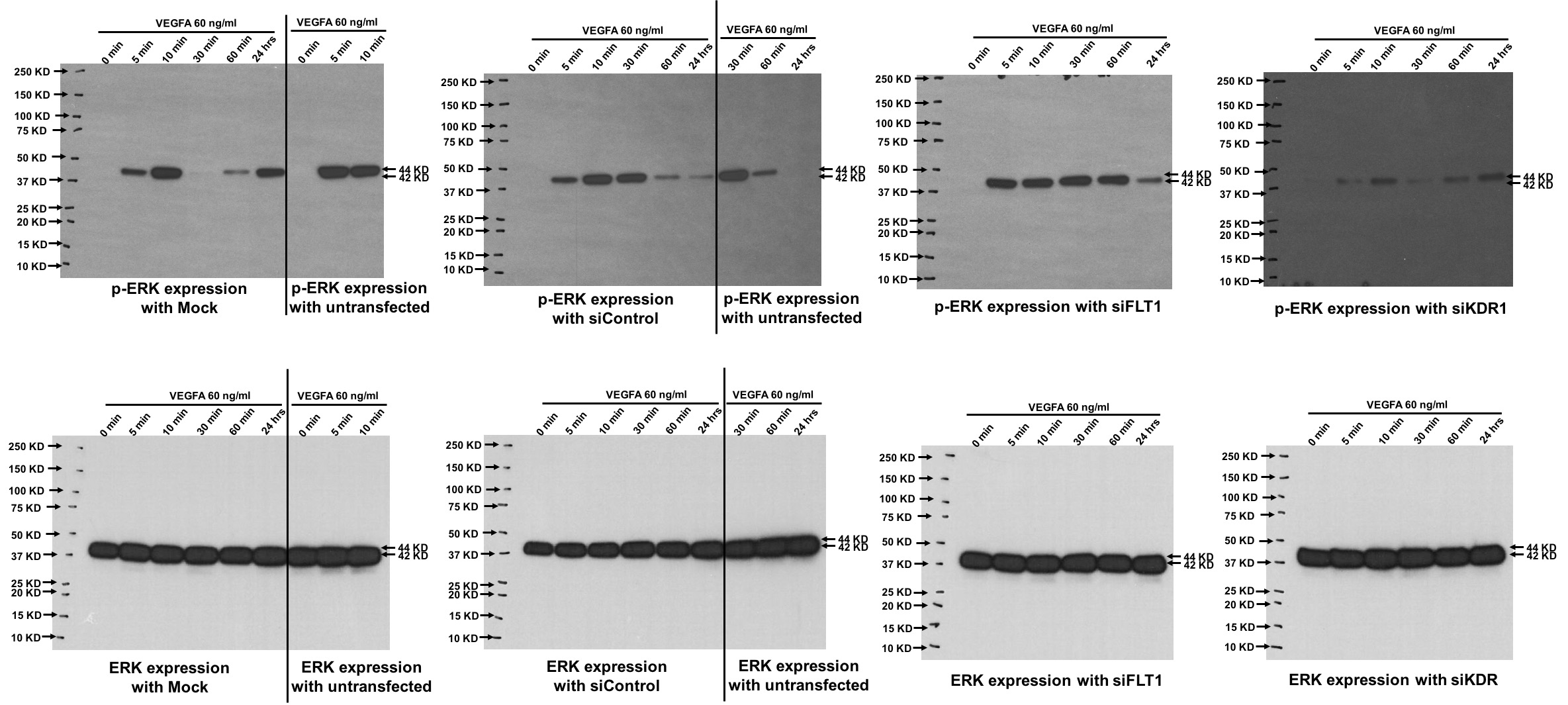{kind=link}
Supplemental Figure 5: Full western blot images of the bands shown in Figure 6A.
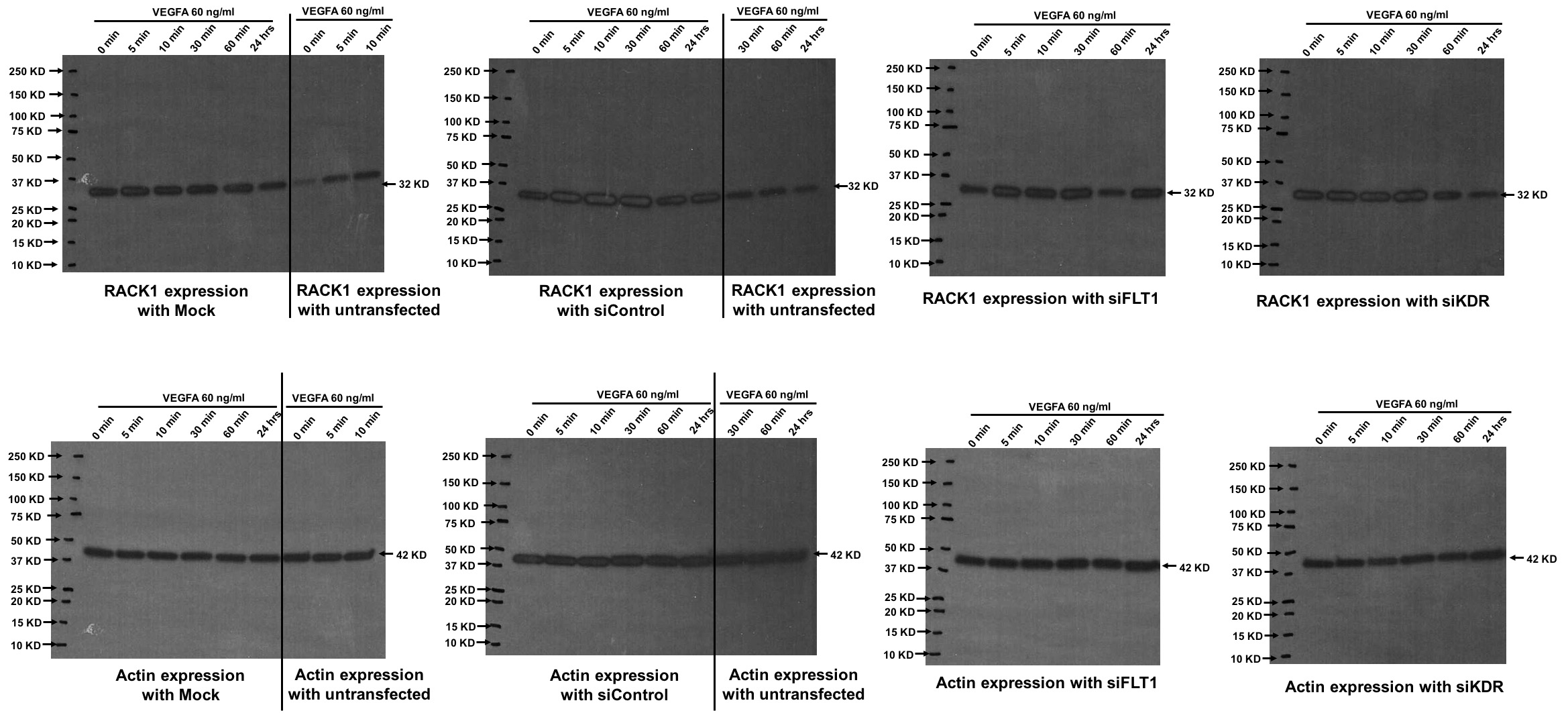{kind=link}
Highlights:
FLT1 ablation significantly impairs wound scratch closure and tube formation in human fetoplacental ECs.
KDR inhibitory effects on EC metabolism had no effect on migration.
FLT1 effects on EC motility are Akt and ERK-independent.
Human fetoplacental EC migration is primarily regulated by FLT1 but not KDR.
Acknowledgements
We are grateful to the Tissue Culture Shared Resource, which is supported by the Cancer Center Support Grant (P30CA046934).
Funding
This work was supported by the National Institutes of Health [HL119846 (EJS)].
References
- [1].Burton GJ, Jauniaux E, Sonographic, stereological and Doppler flow velocimetric assessments of placental maturity, Br J Obstet Gynaecol 102(10) (1995) 818–25. [DOI] [PubMed] [Google Scholar]
- [2].Mayhew TM, Fetoplacental angiogenesis during gestation is biphasic, longitudinal and occurs by proliferation and remodelling of vascular endothelial cells, Placenta 23(10) (2002) 742–50. [DOI] [PubMed] [Google Scholar]
- [3].Demir R, Kaufmann P, Castellucci M, Erbengi T, Kotowski A, Fetal vasculogenesis and angiogenesis in human placental villi, Acta Anat (Basel) 136(3) (1989) 190–203. [DOI] [PubMed] [Google Scholar]
- [4].Sadler TW, Langman’s Medical Embryology, Williams and Wilkins, Baltimore, MD, 1995. [Google Scholar]
- [5].Kaufmann P, Bruns U, Leiser R, Luckhardt M, Winterhager E, The fetal vascularisation of term human placental villi. II. Intermediate and terminal villi, Anat Embryol (Berl) 173(2) (1985) 203–14. [DOI] [PubMed] [Google Scholar]
- [6].Castellucci M, Scheper M, Scheffen I, Celona A, Kaufmann P, The development of the human placental villous tree, Anat Embryol (Berl) 181(2) (1990) 117–28. [DOI] [PubMed] [Google Scholar]
- [7].Niida S, Kondo T, Hiratsuka S, Hayashi S, Amizuka N, Noda T, Ikeda K, Shibuya M, VEGF receptor 1 signaling is essential for osteoclast development and bone marrow formation in colony-stimulating factor 1-deficient mice, Proc Natl Acad Sci U S A 102(39) (2005) 14016–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shibuya M, Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis, J Biochem Mol Biol 39(5) (2006) 469–78. [DOI] [PubMed] [Google Scholar]
- [9].Carmeliet P, Mechanisms of angiogenesis and arteriogenesis, Nat Med 6(4) (2000) 389–95. [DOI] [PubMed] [Google Scholar]
- [10].Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oosthuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert JM, Collen D, Dejana E, Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis, Cell 98(2) (1999) 147–57. [DOI] [PubMed] [Google Scholar]
- [11].Gale NW, Yancopoulos GD, Growth factors acting via endothelial cell-specific receptor tyrosine kinases: VEGFs, angiopoietins, and ephrins in vascular development, Genes Dev 13(9) (1999) 1055–66. [DOI] [PubMed] [Google Scholar]
- [12].Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nube O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P, Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure, Nat Med 5(10) (1999) 1135–42. [DOI] [PubMed] [Google Scholar]
- [13].Nakayama M, Nakayama A, van Lessen M, Yamamoto H, Hoffmann S, Drexler HC, Itoh N, Hirose T, Breier G, Vestweber D, Cooper JA, Ohno S, Kaibuchi K, Adams RH, Spatial regulation of VEGF receptor endocytosis in angiogenesis, Nat Cell Biol 15(3) (2013) 249–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Simons M, Gordon E, Claesson-Welsh L, Mechanisms and regulation of endothelial VEGF receptor signalling, Nat Rev Mol Cell Biol 17(10) (2016) 611–25. [DOI] [PubMed] [Google Scholar]
- [15].Seetharam L, Gotoh N, Maru Y, Neufeld G, Yamaguchi S, Shibuya M, A unique signal transduction from FLT tyrosine kinase, a receptor for vascular endothelial growth factor VEGF, Oncogene 10(1) (1995) 135–47. [PubMed] [Google Scholar]
- [16].Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH, Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor, J Biol Chem 269(43) (1994) 26988–95. [PubMed] [Google Scholar]
- [17].Sawano A, Takahashi T, Yamaguchi S, Aonuma M, Shibuya M, Flt-1 but not KDR/Flk-1 tyrosine kinase is a receptor for placenta growth factor, which is related to vascular endothelial growth factor, Cell Growth Differ 7(2) (1996) 213–21. [PubMed] [Google Scholar]
- [18].Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A, Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele, Nature 380(6573) (1996) 435–9. [DOI] [PubMed] [Google Scholar]
- [19].Fong GH, Rossant J, Gertsenstein M, Breitman ML, Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium, Nature 376(6535) (1995) 66–70. [DOI] [PubMed] [Google Scholar]
- [20].Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC, Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice, Nature 376(6535) (1995) 62–6. [DOI] [PubMed] [Google Scholar]
- [21].Su EJ, Role of the fetoplacental endothelium in fetal growth restriction with abnormal umbilical artery Doppler velocimetry, Am J Obstet Gynecol 213(4 Suppl) (2015) S123–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Maulik D, Mundy D, Heitmann E, Maulik D, Umbilical artery Doppler in the assessment of fetal growth restriction, Clin Perinatol 38(1) (2011) 65–82, vi. [DOI] [PubMed] [Google Scholar]
- [23].Su EJ, Cheng YH, Chatterton RT, Lin ZH, Yin P, Reierstad S, Innes J, Bulun SE, Regulation of 17-beta hydroxysteroid dehydrogenase type 2 in human placental endothelial cells, Biol Reprod 77(3) (2007) 517–25. [DOI] [PubMed] [Google Scholar]
- [24].Su EJ, Lin ZH, Zeine R, Yin P, Reierstad S, Innes JE, Bulun SE, Estrogen receptor-beta mediates cyclooxygenase-2 expression and vascular prostanoid levels in human placental villous endothelial cells, Am J Obstet Gynecol 200(4) (2009) 427e1–8. [DOI] [PubMed] [Google Scholar]
- [25].Frasor J, Stossi F, Danes JM, Komm B, Lyttle CR, Katzenellenbogen BS, Selective estrogen receptor modulators: discrimination of agonistic versus antagonistic activities by gene expression profiling in breast cancer cells, Cancer Res 64(4) (2004) 1522–33. [DOI] [PubMed] [Google Scholar]
- [26].Schmittgen TD, Livak KJ, Analyzing real-time PCR data by the comparative C(T) method, Nat Protoc 3(6) (2008) 1101–8. [DOI] [PubMed] [Google Scholar]
- [27].Claesson-Welsh L, VEGF receptor signal transduction - A brief update, Vascul Pharmacol 86 (2016) 14–17. [DOI] [PubMed] [Google Scholar]
- [28].Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L, Signal transduction by vascular endothelial growth factor receptors, Biochem J 437(2) (2011) 169–83. [DOI] [PubMed] [Google Scholar]
- [29].Wang F, Yamauchi M, Muramatsu M, Osawa T, Tsuchida R, Shibuya M, RACK1 regulates VEGF/Flt1-mediated cell migration via activation of a PI3K/Akt pathway, J Biol Chem 286(11) (2011) 9097–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M, Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice, Proc Natl Acad Sci U S A 95(16) (1998) 9349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mittar S, Ulyatt C, Howell GJ, Bruns AF, Zachary I, Walker JH, Ponnambalam S, VEGFR1 receptor tyrosine kinase localization to the Golgi apparatus is calcium-dependent, Exp Cell Res 315(5) (2009) 877–89. [DOI] [PubMed] [Google Scholar]
- [32].Zhang Z, Neiva KG, Lingen MW, Ellis LM, Nor JE, VEGF-dependent tumor angiogenesis requires inverse and reciprocal regulation of VEGFR1 and VEGFR2, Cell Death Differ 17(3) (2010) 499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Imoukhuede PI, Dokun AO, Annex BH, Popel AS, Endothelial cell-by-cell profiling reveals the temporal dynamics of VEGFR1 and VEGFR2 membrane localization after murine hindlimb ischemia, Am J Physiol Heart Circ Physiol 304(8) (2013) H1085–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hiratsuka S, Nakao K, Nakamura K, Katsuki M, Maru Y, Shibuya M, Membrane fixation of vascular endothelial growth factor receptor 1 ligand-binding domain is important for vasculogenesis and angiogenesis in mice, Mol Cell Biol 25(1) (2005) 346–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cai M, Wang K, Murdoch CE, Gu Y, Ahmed A, Heterodimerisation between VEGFR-1 and VEGFR-2 and not the homodimers of VEGFR-1 inhibit VEGFR-2 activity, Vascul Pharmacol 88 (2017) 11–20. [DOI] [PubMed] [Google Scholar]
- [36].Cudmore MJ, Hewett PW, Ahmad S, Wang KQ, Cai M, Al-Ani B, Fujisawa T, Ma B, Sissaoui S, Ramma W, Miller MR, Newby DE, Gu Y, Barleon B, Weich H, Ahmed A, The role of heterodimerization between VEGFR-1 and VEGFR-2 in the regulation of endothelial cell homeostasis, Nat Commun 3 (2012) 972. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: (A) Full western blot images of the bands shown in Figure 1B. (B) Full western blot images of the bands shown in Figure 1E.
Supplemental Figure 2: Real-time PCR demonstrates that although there is an overall difference in VEGFA expression with one-way ANOVA analysis (p<0.05), post-hoc testing shows no significant difference in VEGFA expression with siControl, siFLT1, or siKDR knock-down. In contrast, using a primer set that spans from exon 13 to the unique C-terminus region of sFLT1, we found that transfection with siFLT1 results in significantly decreased sFLT1 expression (p<0.0001). This was not unexpected, as the siRNA oligo utilized targets exon 5 of FLT1.
Supplemental Figure 3: Full western blot images of the bands shown in Figure 5A.
Supplemental Figure 4: Full western blot images of the bands shown in Figure 5D.
Supplemental Figure 5: Full western blot images of the bands shown in Figure 6A.