

ABSTRACT
Most clinical isolates of Streptococcus pyogenes elaborate a capsular polysaccharide, which is composed of hyaluronic acid, a high-molecular-mass polymer of alternating residues of N-acetyl glucosamine and glucuronic acid. Certain strains, particularly those of the M18 serotype, produce abundant amounts of capsule, resulting in formation of large, wet-appearing, translucent or “mucoid” colonies on solid media, whereas strains of M-types 4 and 22 produce none. Studies of acapsular mutant strains have provided evidence that the capsule enhances virulence in animal models of infection, an effect attributable, at least in part, to resistance to complement-mediated opsonophagocytic killing by leukocytes. The presence of the hyaluronic acid capsule may mask adhesins on the bacterial cell wall. However, the capsule itself can mediate bacterial attachment to host cells by binding to the hyaluronic-acid binding protein, CD44. Furthermore, binding of the S. pyogenes capsule to CD44 on host epithelial cells can trigger signaling events that disrupt cell-cell junctions and facilitate bacterial invasion into deep tissues. This article summarizes the biochemistry, genetics, regulation, and role in pathogenesis of this important virulence determinant.
INTRODUCTION
In her early studies of group A streptococci (Streptococcus pyogenes), Rebecca Lancefield noted an association between virulence and a distinctive appearance of the bacterial colonies on solid media. Isolates that were highly virulent for mice and that grew well in fresh human blood typically formed large colonies with a translucent, liquid appearance (mucoid) or an irregular, collapsed appearance (matte). By contrast, avirulent isolates that grew poorly in human blood formed compact, opaque colonies (glossy). Strains that grew as mucoid or matte colonies usually produced large amounts of M protein, which Lancefield gave the designation “M” because of this association with colony morphology (1, 2). Later work by Armine Wilson demonstrated that the mucoid or matte appearance of such strains was in fact due to elaboration of capsular polysaccharide, not M protein (3). Wilson showed that the mucoid or matte colony type was converted to a nonmucoid or glossy colony by growth on medium containing hyaluronidase which digested the hyaluronic acid capsule, whereas growth on medium containing trypsin, which digested M protein, had no such effect. Furthermore, while many strains that produced abundant capsular polysaccharide also were rich in M protein, expression of the two surface products was not always linked; certain mucoid or highly encapsulated strains produced little or no M protein, and certain strains rich in M protein produced little or no capsule (1, 4).
Both M protein and capsule have since been shown to contribute independently to S. pyogenes virulence. The association of mucoid strains of S. pyogenes with both invasive infection and acute rheumatic fever suggested a role for the capsular polysaccharide in virulence. Anecdotal observations that linked the capsule to pathogenesis of streptococcal disease were supported by a study that characterized the colony morphology of more than 1,000 S. pyogenes clinical isolates received by a streptococcal reference lab in the 1980s. In that study, only 3% of S. pyogenes isolates from patients with pharyngitis had a mucoid colony morphology, whereas 21% of isolates from patients with invasive disease syndromes such as bacteremia, necrotizing fasciitis, or streptococcal toxic shock syndrome and 42% of isolates associated with acute rheumatic fever were mucoid (5). The overrepresentation of mucoid strains among isolates associated with invasive infection and rheumatic fever suggested that the capsule contributes to enhanced virulence. The occurrence of outbreaks of acute rheumatic fever associated with mucoid strains of S. pyogenes at several locations in the United States in the 1980s supported the same inference (6–8). These clinical and epidemiologic observations implicating the capsule in pathogenesis have been corroborated by extensive experimental studies that have demonstrated a definite role of the hyaluronic acid capsule as a virulence factor in S. pyogenes infection.
BIOCHEMISTRY AND GENETICS OF CAPSULE PRODUCTION
Hyaluronic Acid Biosynthesis
The S. pyogenes capsular polysaccharide is composed of hyaluronic acid, a high-Mr (>106 kDa) linear polymer made up of β(1→4)-linked disaccharide repeat units of d-glucuronic acid(1→3)-β-d-N-acetylglucosamine. The polysaccharide is synthesized from the nucleotide sugar precursors UDP-glucuronic acid and UDP-N-acetylglucosamine by a membrane-associated enzyme, hyaluronan synthase. High-Mr hyaluronic acid (or hyaluronan) can be produced by incubation of cell-free membrane extracts of S. pyogenes with the two substrate UDP-sugars in the presence of divalent cations (9–11).
A genetic locus required for hyaluronic acid production in S. pyogenes was located independently by three laboratories using transposon mutagenesis to produce acapsular mutants from a mucoid S. pyogenes strain. Transposon insertions that produced the acapsular phenotype were mapped to a locus that was highly conserved among S. pyogenes strains (12–14). Characterization of this region of the S. pyogenes chromosome revealed a cluster of three genes, hasABC, whose products are involved in hyaluronic acid biosynthesis. The first gene, hasA, encodes hyaluronan synthase, a protein with a predicted Mr of 47.9 kDa that includes at least four predicted membrane-spanning domains, consistent with evidence that the enzyme is membrane associated (15, 16). The hasA gene product shares significant similarity with the NodC protein of Rhizobium meliloti, which is required for synthesis of an N-acetylglucosamine-containing polysaccharide, with chitin synthases, and with hyaluronan synthases from other microbial and higher animal species (17, 18). The S. pyogenes hyaluronan synthase has been expressed in Escherichia coli as a His6-fusion protein. Activity of the purified enzyme is strongly stimulated in the presence of certain phospholipids, particularly cardiolipin (diphosphatidylglycerol), which appears to be associated with the enzyme in S. pyogenes (19). Cardiolipin molecules may contribute to formation of a transmembrane pore for export of the hyaluronic acid polymer, since the hyaluronan synthase itself contains only two relatively small extracellular domains (20).
The second gene, hasB, encodes UDP-glucose dehydrogenase, a protein with a predicted Mr of 45.5 kDa that catalyzes the oxidation of UDP-glucose to UDP-glucuronic acid (21). The third gene in the cluster, hasC, encodes a predicted 33.7-kD protein identified as UDP-glucose pyrophosphorylase (22). The function of this enzyme is to catalyze the condensation of UTP with glucose-1-phosphate to form UDP-glucose. Thus, the reaction catalyzed by the hasC product yields a substrate for UDP-glucose dehydrogenase encoded by hasB, whose reaction product is, in turn, a substrate for hyaluronan synthase encoded by hasA (Fig. 1). While the enzyme protein encoded by hasC is enzymatically active, it is not required for hyaluronic acid synthesis by S. pyogenes. Selective inactivation of hasC resulted in no decrement in hyaluronic acid synthesis by a highly encapsulated strain of S. pyogenes; the implication is that another source of UDP-glucose is available within the cell (23). Furthermore, expression of recombinant hasA and hasB (without hasC) resulted in synthesis of hyaluronic acid in E. coli and Enterococcus faecalis (13, 15). Together, these observations have led to the conclusion that hasA and hasB are the only two genes uniquely required for bacterial synthesis of hyaluronic acid. The three genes hasABC are transcribed as a single message of approximately 4.2 kb from a promoter immediately upstream of hasA (24). The identification of a potential rho-independent terminator at the 3′ terminus of hasC suggests that no additional genes are included in the operon. A paralog of hasB, hasB2, is present at a chromosomal location remote from the has operon. Its product is able to support a low level of hyaluronic acid production in the absence of hasB (25).
FIGURE 1.
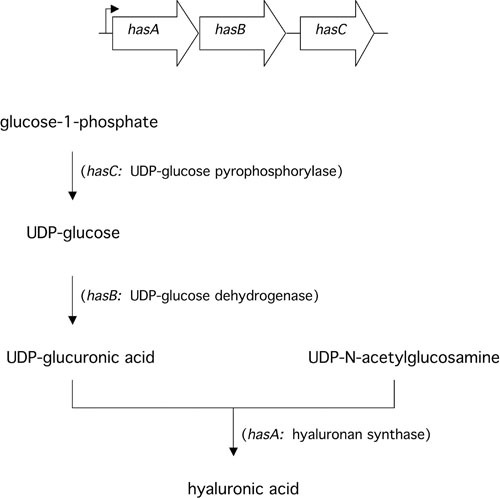
Schematic diagram of the has operon and the function of each gene product in the biosynthetic pathway for synthesis of hyaluronic acid in S. pyogenes.
The simplicity of the capsule gene cluster in S. pyogenes contrasts with the size and complexity of capsule synthesis loci in other encapsulated Gram-positive bacteria, which generally consist of at least 12 genes (26–30). One exception is the type 3 pneumococcus, whose capsule production is strikingly similar to that in S. pyogenes with respect to the structure of the polysaccharide and the capsule synthesis genes; in type 3 pneumococci, capsule synthesis involves three genes that are homologs of hasABC (31, 32). In both S. pyogenes and type 3 pneumococci, it seems likely that the capsule synthesis gene cluster was acquired from another species by horizontal gene transfer. This hypothesis is also suggested by genome comparisons of S. pyogenes with group B Streptococcus: with the exception of the hasABC cluster that is absent in group B Streptococcus, there is a high degree of gene synteny over more than 25 kb of the corresponding chromosomal region in S. pyogenes and group B Streptococcus.
While the has operon is highly conserved, it is not universally present in S. pyogenes clinical isolates. In particular, strains of M types 4 and 22 and some isolates of M 89 lack the capsule synthesis operon and do not produce a hyaluronic acid capsule (33–36). Interestingly, the absence of the capsule synthesis genes in M types 4 and 22 is associated with the presence of enzymatically active hyaluronidase, whereas in serotypes that produce capsule, the chromosomal hylA gene contains a point mutation that renders it catalytically inactive. These divergent patterns of gene expression suggest that S. pyogenes has evolved two mutually exclusive survival strategies: production of an antiphagocytic hyaluronate capsule or production of hyaluronidase, presumably as a spreading factor in host tissues and/or to degrade host hyaluronic acid as a source of nutrition.
Regulation of Hyaluronic Acid Biosynthesis
The has operon is highly conserved among S. pyogenes strains. A study of nine S. pyogenes isolates that came from diverse settings and whose dates of isolation spanned 75 years found only six mutations within a 300-nucleotide central region of the hasA gene (17); all were single nucleotide substitutions, and none resulted in a change in the amino acid sequence of the protein. Despite the high degree of conservation of the hyaluronan synthase gene, individual S. pyogenes strains vary widely in the amount of capsule they produce. Since the has genes are conserved, differences in capsule expression among strains are likely to reflect differences in the regulation of has gene transcription. Experimental support for this hypothesis comes from the observation that has gene transcripts are detected during exponential growth of well-encapsulated strains of S. pyogenes but not in strains that do not produce detectable hyaluronic acid (24). Analysis of the has operon promoter using a reporter system in which the promoter was fused to a promoterless chloramphenicol acetyl transferase (CAT) gene showed that full activity of the has promoter required no more than 12 nucleotides of chromosomal DNA upstream of the promoter’s –35 site (37). This observation is consistent with the finding that many strains of S. pyogenes (10 of 23) contain the insertion element IS1239′ approximately 50 nucleotides upstream of the –35 site without any apparent effect on capsule production (Figure 2) (23).
FIGURE 2.

Map of the region of the S. pyogenes chromosome that includes the has operon encoding enzymes required for hyaluronic acid synthesis. The other genes shown appear not to be involved in capsular polysaccharide synthesis or surface expression. In some strains insertion sequence IS1239′ is present approximately 50 nucleotides upstream of the has operon promoter (adapted from the genome sequence of M1 strain SF370, GenBank accession number AE004092 [93]).
One factor that influences the amount of capsule produced by a particular strain is the structure of the has operon promoter itself. Experiments using CAT reporter gene fusions revealed 3-fold higher activity of the has promoter from a highly encapsulated M type 18 strain than that from a poorly encapsulated M type 3 strain (37). Construction of hybrid promoters by site-directed mutagenesis demonstrated that nucleotide substitutions in two regions accounted for the differences in promoter activity between the two serotypes: three nucleotides in the −35, −10 spacer region and four nucleotides in the +2 to +8 positions relative to the start site of hasA transcription. The characteristic fine structure of the has operon promoter in type 18 strains may account in part for the observation that M18 strains are typically mucoid (i.e., highly encapsulated) since the precise sequence of the has promoter appears to be serotype-specific.
Capsule production varies not only among strains but also in an individual strain under different circumstances. For example, during growth in liquid batch culture, S. pyogenes organisms produce large capsules during the exponential phase and then shed their capsules into the medium during the stationary phase. Studies using a has promoter-CAT fusion integrated into the S. pyogenes chromosome demonstrated a rapid increase in promoter activity to a maximal level during the exponential phase of growth, with a sharp fall as the organisms entered the stationary phase. The production of cell-associated capsular polysaccharide followed an identical but slightly delayed pattern, consistent with the expected lag for changes in the translation of enzyme proteins that are subsequently reflected in the rate of polysaccharide synthesis. These results indicate that the changing levels of encapsulation observed during different phases of growth are due to transcriptional regulation of has operon expression. Further studies of the effect of growth rate on capsule expression in a chemostat continuous culture system found that levels of CAT activity and capsule production were at least three times higher in cells maintained at a constant rapid growth rate (doubling time of 1.4 hours) than in cells maintained at a slow growth rate (doubling time of 11 hours) (S. Albertí, J.L. Levin, and M.R. Wessels, unpublished observations).
Upregulation of capsule production is likely to occur during infection when the organism moves from the external environment into the pharynx or deep tissues, where both temperature and nutrient availability are optimal for bacterial growth. This hypothesis was supported by experiments using a reporter strain of S. pyogenes in which expression of green fluorescent protein was controlled by the has operon promoter. Introduction of the strain into the mouse peritoneum or into the baboon pharynx triggered a rapid increase in fluorescence, reflecting transcription from the has promoter (38). Thus, the regulation of capsule expression may be a useful adaptation to survival in host environments in which the capsule is required to defend the organism against complement-mediated phagocytic killing.
The dynamic changes in S. pyogenes capsule expression with changes in growth rate and the observation that some strains of S. pyogenes appear more highly encapsulated after animal passage have suggested that capsule gene transcription is regulated by cellular mechanisms in addition to those attributable to the intrinsic structure of the has operon promoter. Mga, the trans-acting regulatory protein that influences expression of several S. pyogenes proteins, appears not to regulate capsule expression, at least in most strains of S. pyogenes. Inactivation of Mga was associated with reduced has gene transcription in an M1 strain (39); however, no effect on capsule production and/or has transcription was observed upon Mga inactivation in strains of M6, M18, or M49 (40–42). RofA, a regulatory protein involved in control of protein F expression, also is not known to affect capsule production (43).
Transposon mutagenesis of a poorly encapsulated M3 strain of S. pyogenes resulted in the identification of a novel regulatory locus whose inactivation dramatically increased capsule production (42). The locus consists of two genes, csrR and csrS (also called covR and covS), whose predicted products resemble the regulator and sensor proteins, respectively, of bacterial two-component regulatory systems (Fig. 3). Targeted inactivation of csrR by allelic exchange mutagenesis resulted in a 6-fold increase in capsule production. The increase in hyaluronic acid synthesis was accompanied by a parallel increase in has gene transcription—evidence that CsrR regulates transcription of the capsule synthesis genes. Mutation of csrR appeared not to affect expression of M protein or hemolytic activity, but subsequent work in several laboratories has demonstrated that the system also represses expression of several other virulence factors, including streptokinase (ska), streptolysin S (sag operon), mitogenic factor/streptodornase (speMF), an integrin-like protein/IgG protease (mac or ideS), and in some strains, cysteine protease (speB) (44–47). Microarray transcriptional profiling experiments suggested that CsrRS is involved directly or indirectly in regulation of approximately 10% of S. pyogenes genes (44, 48–50). The membrane-associated CsrS protein is a histidine kinase thought to respond to at least two environmental signals: an elevated extracellular Mg2+ concentration is associated with increased phosphorylation of the cognate regulator CsrR, presumably as a result of stimulating CsrS autokinase activity and/or inhibiting CsrS phosphatase activity for CsrR (51). The human antimicrobial peptide LL-37, at concentrations below those that inhibit S. pyogenes growth, has an effect on CsrR phosphorylation opposite to that of elevated magnesium (52, 53). Phosphorylation of CsrR increases its affinity for binding to the promoter region of the has operon and most other regulated genes to repress transcription (46, 54, 55). The likely importance of this regulatory system in virulence is supported by the markedly greater resistance to in vitro phagocytosis displayed by a (highly encapsulated) csrR mutant than by the wild-type parent strain and a 500-fold increase in the mutant’s virulence for mice after intraperitoneal challenge (42). Up to 40% of invasive S. pyogenes isolates have inactivating mutations in CsrS or CsrR, whereas such mutations are rare among isolates from pharyngitis patients (56–58).
FIGURE 3.
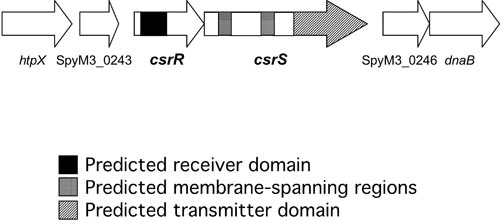
Diagram of the csrRS chromosomal locus encoding a two-component regulatory system that regulates hyaluronic acid synthesis. Sequences corresponding to regions of the predicted proteins with properties characteristic of the response regulator (CsrR) or sensor (CsrS) components are indicated (adapted from the genome sequence of M3 strain MGAS315, GenBank accession number AE014074 [94]).
Capsule production is also controlled by RocA, a protein first identified as a regulator of CsrR expression and later shown to influence phosphorylation of CsrR (59, 60). Mutations in rocA are associated with increased expression of the has operon and of other genes regulated by the CsrRS system. S. pyogenes strains of emm types 18 and 3 each harbor a mutation in rocA that results in expression of a truncated RocA protein and increased capsule production (61, 62).
THE CAPSULE AND HOST IMMUNE DEFENSES
Immunogenicity of Hyaluronic Acid
In 1937, Kendall et al. reported the purification of capsular polysaccharide from mucoid strains of S. pyogenes (63). Their analysis revealed that an apparently identical capsular material was produced by different M types of S. pyogenes. The S. pyogenes polysaccharide contained equal amounts of N-acetylglucosamine and glucuronic acid as exclusive components of monosaccharides; this finding suggested that it might be structurally identical to a polysaccharide isolated from bovine vitreous humor and human umbilical cord. Immunization of rabbits and horses with mucoid S. pyogenes cells failed to elicit antibodies detectable by precipitin reactions with the polysaccharide. Later studies have confirmed that the “serologically inactive” polysaccharide produced by S. pyogenes is hyaluronic acid, a high Mr polysaccharide with a repeating unit structure identical to that of hyaluronic acid isolated from animal sources. Presumably because it is recognized as a “self” antigen, hyaluronic acid is poorly immunogenic in several animal species. Fillit and colleagues evoked antibodies to hyaluronic acid by immunization of rabbits with encapsulated streptococci of group A or C and by immunization of mice with hyaluronidase-treated hyaluronic acid linked to liposomes (64, 65). Such antibodies, however, have not been shown to have any opsonic activity against S. pyogenes in vitro or to confer protection against S. pyogenes infection in vivo.
Capsule and Resistance to Phagocytosis
S. pyogenes, like other Gram-positive pathogens, is resistant to direct bacteriolysis by complement and antibodies. Therefore, clearance of the organisms from the blood or deep tissues depends largely on uptake and killing by phagocytes. This concept was supported by the observations of Todd, Lancefield, and others that virulence of S. pyogenes isolates in both clinical settings and in experimental animal infections was closely paralleled by the capacity of the strains to resist killing by human blood leukocytes. S. pyogenes strains that produced large amounts of M protein multiplied in fresh human blood, while those that lacked M protein did not (66, 67). In addition, however, early studies demonstrated a relationship between surface expression of the hyaluronic acid capsule and resistance to phagocytosis. Mucoid or highly encapsulated strains tended to be resistant, and hyaluronidase treatment increased their susceptibility to phagocytosis in vitro, a result that suggested a protective effect of the capsule (68–70). More recent work with acapsular mutants has provided definitive evidence that the capsule is a major determinant of resistance to phagocytic killing. In several assays measuring complement-dependent phagocytic killing by human blood leukocytes, acapsular mutants of type 18 and type 24 S. pyogenes were significantly more susceptible than the corresponding encapsulated wild-type strains (14, 71, 72).
One mechanism through which capsular polysaccharides may confer resistance to phagocytosis is the inhibition of complement activation. The type III capsular polysaccharide of group B Streptococcus, for example, prevents deposition of C3b on the bacterial surface through inhibition of alternative complement pathway activation by sialic acid residues present in the polysaccharide (73, 74). In S. pyogenes, however, the presence of the capsular polysaccharide appears to have no effect on the amount of C3b deposited on the bacterial surface. Similar amounts of C3 were detected on acapsular mutants and on encapsulated wild-type strains after incubation of the organisms in serum (71). The increased resistance of well-encapsulated S. pyogenes strains to complement-dependent phagocytic killing, therefore, must reflect the capacity of the capsule to block access of neutrophils to opsonic complement components bound to the bacterial surface rather than inhibition of C3b deposition (Fig. 4).
FIGURE 4.
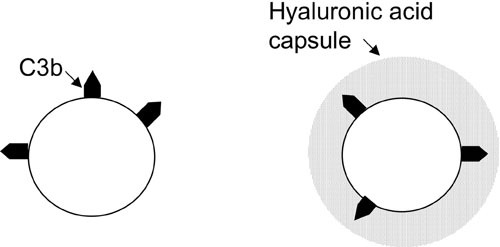
The hyaluronic acid capsule and resistance to complement-mediated phagocytosis. The capsule does not prevent deposition of C3b on the bacterial cell wall but, rather, interferes with the interaction of bound C3b with phagocyte receptors.
Resistance of S. pyogenes to phagocytosis has been assessed most often with the bactericidal test of Lancefield, in which a small inoculum of the test strain is rotated for 3 hours in heparinized fresh human blood (67). Strains that exhibit a significant increase in CFU during rotation in blood are considered resistant. An effect of the capsule is evident in this assay (acapsular mutants show less net growth than encapsulated wild-type strains), but the effect of M protein is more striking (14, 72). M protein-deficient strains not only fail to grow but typically are reduced in number after a 3-hour incubation. By contrast, when resistance to phagocytosis is assessed with a different assay, the effect of capsule is more dramatic than that of M protein. In this second assay system, a larger bacterial inoculum is rotated for 1 hour with human peripheral blood leukocytes in the presence of 10% normal human serum as a complement source (75, 76). In the latter assay, acapsular mutant strains and poorly encapsulated wild-type strains are killed (∼90% reduction in CFU) despite the presence of M protein, while well-encapsulated strains do not change or increase in number. A mutant that is deficient in M protein but that produces wild-type amounts of capsule is killed less efficiently than a capsule-deficient mutant in the same background (72). In a study of isogenic mutants deficient in capsule, M protein, or both in the background of a mucoid type 18 S. pyogenes strain, mouse virulence correlated better with results in the second assay system than with the Lancefield whole-blood assay (72).
CAPSULE AS A VIRULENCE FACTOR
Experimental Infection Models
In the 1940s, investigators attempted to determine experimentally whether the hyaluronic acid capsule contributed to the virulence of S. pyogenes. Hirst found that decapsulating S. pyogenes by treatment with leech extract containing hyaluronidase had no effect on virulence of the bacteria in mice (77). However, Kass and Seastone found reduced virulence of decapsulated S. pyogenes in mice, perhaps because the latter authors administered hyaluronidase to the infected mice at frequent intervals to prevent regeneration of the capsule after challenge (78). Subsequently, more direct proof that the capsule contributes to virulence has come from several studies using acapsular mutant strains of S. pyogenes derived by transposon mutagenesis or by targeted inactivation of gene(s) required for hyaluronic acid synthesis (Table 1).
TABLE 1.
Animal models in which the hyaluronic acid capsule has been shown to enhance virulence of group A streptococci
Because the capsule is required for S. pyogenes resistance to complement-mediated phagocytic killing (see above), it is not surprising that capsule expression enhances virulence in systemic infection models in which the animals succumb to overwhelming bacteremia. In addition, however, the hyaluronic acid capsule contributes in a pivotal fashion to the capacity of the organisms to produce invasive soft tissue infection. A mouse model of invasive S. pyogenes soft tissue infection has been developed in which the bacterial inoculum is delivered by superficial injection just below the skin surface in a small volume and with minimal trauma to the tissues (79, 80). Mice challenged with the highly encapsulated M24 strain Vaughn developed dermal necrosis with underlying purulent inflammation and secondary bacteremia, while mice challenged with the acapsular mutant strain 24-4 developed no lesion at all or minor superficial inflammation and no bacteremia (79). Similar results were observed in the same model with a moderately encapsulated M3 strain of S. pyogenes (originally isolated from a patient with necrotizing fasciitis) which was compared with two isogenic acapsular mutants derived by allelic exchange mutagenesis of the hasA (hyaluronan synthase) gene. In the latter experiments, the majority of animals challenged with the encapsulated M3 parent strain developed spreading, necrotic soft tissue infection and died, whereas the acapsular mutants produced only mild local infection and no deaths (80). The dramatic impact of the capsule on virulence in this model is correlated with equally striking histopathologic findings: In animals challenged with an acapsular mutant, a small focus of neutrophilic inflammation and necrosis is confined by a well-formed abscess. By contrast, after challenge with the encapsulated wild-type strain, acute inflammatory cells extend throughout the subcutaneous tissue and are associated with thrombosis of blood vessels, with infarction and necrosis of the overlying dermis—a histopathologic picture very similar to that in patients with S. pyogenes necrotizing fasciitis. Examination of the histologic sections by immunofluorescence microscopy with an S. pyogenes-specific antibody showed that the acapsular S. pyogenes cells were confined to the abscess cavity, while the encapsulated wild-type organisms were widely dispersed throughout the subcutaneous tissues.
Capsule and Mucosal Colonization
Studies by at least two groups have demonstrated a critical role for the hyaluronic acid capsule in pharyngeal colonization in mice. BALB/c mice inoculated intranasally with the stable (transposon insertion plus chromosomal deletion) acapsular mutant strain 24-4 rapidly cleared the organism from the pharynx, and no mice died (81). In contrast, throat cultures of mice challenged with the revertible (simple transposon insertion) acapsular mutant 24-72 yielded encapsulated revertants, and the rate of persistent throat colonization and lethal systemic infection was similar to that in animals challenged with the encapsulated type 24 parent strain. A study of an acapsular mutant derived from the mouse-virulent M50 strain, B514, yielded similar results: intranasal challenge with the wild-type strain resulted in chronic throat colonization in C57BL/10SnJ mice. After intranasal challenge with an acapsular mutant derived by plasmid insertion mutagenesis of the hasA gene, encapsulated revertants accounted for all S. pyogenes recovered from throat cultures 4 days after challenge (82). Intratracheal inoculation of C3HeB/FeJ mice with the wild-type strain produced pneumonia and secondary systemic infection in approximately 50% of the animals within 72 hours, while only 2 animals challenged with the acapsular mutant became ill, 5 and 6 days after challenge, respectively. In both cases only encapsulated revertants were recovered from cultures of the throat, lungs, and blood (82). The findings in both studies of impaired colonization by the acapsular mutant strains and the rapid emergence of encapsulated revertants together provide strong evidence that encapsulation confers a powerful survival advantage for S. pyogenes in the upper airway. This conclusion was supported also by experiments in a primate model of S. pyogenes pharyngeal colonization: an acapsular mutant derived from an M3 strain was cleared from the pharynx of baboons more quickly than the wild-type parent strain (83). While these results suggest that capsule production enhances initial survival and persistence of S. pyogenes in the pharynx, studies of long-term carriage in a primate model and in human carriers have shown that carriage isolates acquire mutations that result in reduced or absent hyaluronic acid production (84, 85). Taken together, the results of these studies suggest that the hyaluronic acid capsule enhances transmission and initial colonization but that prolonged carriage in human beings may be favored by subsequent downregulation or loss of capsule production.
Capsule and S. pyogenes Attachment to Epithelial Cells
The influence of the hyaluronic acid capsule on the capacity of S. pyogenes to colonize the pharyngeal epithelium may be explained, at least in part, by the protective effect of the capsule in preventing opsonophagocytic killing, although the importance of resistance to opsonophagocytosis at a mucosal site is less well-defined than in blood or deep tissues. In addition, the capsule is an important factor in the process of S. pyogenes attachment to epithelial cells, both by modulating interactions of other bacterial surface molecules with the epithelium and by direct interaction of S. pyogenes capsular hyaluronic acid with eukaryotic cells. The capsule may influence adherence by interfering with the interaction of any of a variety of S. pyogenes surface molecules that have been implicated as adhesins: M and M-like proteins, fibronectin-binding proteins, lipoteichoic acid, and glyceraldehyde-3-phosphate dehydrogenase. In general, acapsular mutant strains of S. pyogenes attach in higher numbers to skin or throat keratinocytes than do the corresponding encapsulated wild-type strains. For type 24 S. pyogenes, the capsule prevents M protein-mediated adherence to keratinocytes, since inactivation of the emm24 gene reduced adherence in the background of an acapsular mutant but not in the background of the same strain expressing wild-type amounts of capsule (86). It is likely that the capsule exerts a similar modulating influence on bacterial adherence mediated by other S. pyogenes surface molecules.
Not only does the capsule influence the binding interactions of other S. pyogenes surface structures, but there is evidence that the capsule itself can serve as a ligand for attachment of S. pyogenes to CD44 on epithelial cells. CD44 is a hyaluronic acid-binding glycoprotein found on a variety of human cells, including epithelial and hematopoietic cells. The interaction of CD44 with hyaluronic acid appears to have important functions in cell migration, organization of epithelia, lymphocyte homing, and tumor metastasis. Because CD44 is expressed on the surface of tonsillar epithelial cells, it may serve as a receptor for binding of hyaluronic acid on the surface of S. pyogenes. This hypothesis was supported by the finding that a monoclonal antibody to CD44 reduced attachment of wild-type strains of S. pyogenes to human pharyngeal keratinocytes by ∼90% but had no effect on attachment of acapsular mutant strains (86). Further evidence that CD44 acts as a receptor for S. pyogenes came from the observation that transfection of K562 cells with cDNA encoding either of two epithelial isoforms of CD44 conferred on the cells the capacity to bind S. pyogenes. Cells expressing CD44 bound a variety of S. pyogenes wild-type strains, including several in which the level of capsule expression was below the limit of detection in a dye-binding assay; however, inactivation of the hyaluronan synthase gene in such a poorly encapsulated S. pyogenes strain completely abrogated binding (86). A study of transgenic mice with defective expression of epithelial CD44 found reduced pharyngeal colonization compared to that in wild type mice after intranasal challenge with S. pyogenes, evidence that CD44 can function as a receptor for S. pyogenes binding to the pharyngeal epithelium in vivo (87).
Capsule and CD44-Mediated Tissue Invasion
CD44 is a transmembrane protein, and its ligation by hyaluronic acid can result in signal transduction and induction of cellular events involved in cell migration. Binding of S. pyogenes to CD44 on human pharyngeal keratinocytes in vitro has been shown to trigger a similar pattern of cellular responses, including the formation of lamellipodia or membrane ruffles at the site of S. pyogenes attachment and disruption of intercellular junctions (88). These events were associated with enhanced translocation of encapsulated S. pyogenes compared to an acapsular mutant across epithelial cell layers in vitro. The results suggested that interaction of the S. pyogenes capsule with CD44 manipulates a host signaling pathway to disrupt epithelial integrity and enable bacterial penetration into underlying tissues.
Capsule and S. pyogenes Entry into Epithelial Cells
Several laboratories have documented the entry of S. pyogenes into human epithelial cells, a process that has been proposed to represent a virulence mechanism—either a step in bacterial invasion into deep tissues or a means of escape of the organism into an intracellular sanctuary shielded from antibiotics and host immune defenses (79, 89–91). However, while there is irrefutable evidence that the capsule plays a key role in invasive infection in vivo, there is equally clear evidence that the capsule prevents S. pyogenes entry into epithelial cells. In studies of S. pyogenes entry into cultured human keratinocytes from pharyngeal epithelium or external skin, poorly encapsulated strains entered cells far more efficiently than well-encapsulated strains, regardless of M type or clinical source of the isolates (79). Acapsular mutants entered more than 1,000 times more efficiently than corresponding encapsulated parent strains. The apparently opposite effects of the capsule on invasive infection in vivo compared to those on epithelial cell “invasion” in vitro suggest that bacterial entry into epithelial cells is not a step in productive infection but rather a host response that limits the spread of poorly encapsulated strains. By contrast, virulent encapsulated strains resist ingestion by eukaryotic cells, both epithelial cells and professional phagocytes, and invade deep tissues by an extracellular route, a process that appears to be facilitated by CD44-mediated disruption of cell-cell junctions. This hypothesis is supported by the finding that clinical isolates from patients with invasive S. pyogenes infection “invade” epithelial cells at much lower rates than throat or skin isolates (92). After internalization, the viability of S. pyogenes within epithelial cells in vitro falls rapidly after bacterial entry (79, 90). It is possible, however, that transient intracellular survival of S. pyogenes, perhaps in a cyclic fashion, contributes to persistent carriage.
CONCLUSIONS
Careful observations by microbiologists and clinicians in the 1920s and 1930s pointed to a link between the hyaluronic acid capsule and S. pyogenes disease pathogenesis. More recent studies have defined the genetic locus that directs hyaluronic acid biosynthesis and have characterized the molecular mechanisms through which the capsule enhances S. pyogenes virulence. It is now appreciated that the has capsule synthesis operon is both highly conserved and widely, but not universally, distributed among S. pyogenes strains, attesting to the adaptive role played by the hyaluronic acid capsule in the coevolution of S. pyogenes with the human host. As a poorly immunogenic “self” antigen, the capsular polysaccharide appears to have persisted in an invariant form in S. pyogenes, presumably because it has not been subject to the selective pressure of an effective, polysaccharide-specific, humoral immune response in the infected host that has led to emergence of multiple capsular types in other pathogenic bacteria. Studies of acapsular mutant strains have demonstrated that the capsule protects S. pyogenes from complement-mediated phagocytic killing and is essential for full virulence in a variety of experimental infection models. The S. pyogenes capsule also influences attachment of the bacteria to human epithelial cells, both by modulating the interaction of M protein and other potential adhesins and by itself serving as a ligand for attachment of S. pyogenes to CD44 on epithelial cells. The latter mechanism may be of particular importance in S. pyogenes colonization of the pharynx, because it appears to participate in the adherence of diverse S. pyogenes strains, including those that produce only small amounts of capsule. Finally, the successful adaptation of S. pyogenes to survival in the human host involves regulation of capsule expression that is dependent on the fine structure of the has operon promoter, on the CsrRS two-component regulatory system, and likely, on additional mechanisms yet to be uncovered.
REFERENCES
- 1.Todd EW, Lancefield RC. 1928. Variants of hemolytic streptococci; their relation to type specific substance, virulence, and toxin. J Exp Med 48:751–767 10.1084/jem.48.6.751. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lancefield RC. 1940. Type specific antigens, M and T, of matt and glossy variants of group A hemolytic streptococci. J Exp Med 71:521–537 10.1084/jem.71.4.521. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson AT. 1959. The relative importance of the capsule and the M-antigen in determining colony form of group A streptococci. J Exp Med 109:257–270 10.1084/jem.109.3.257. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rothbard S, Watson RF. 1948. Variation occurring in group A streptococci during human infection; progressive loss of M substance correlated with increasing susceptibility to bacteriostasis. J Exp Med 87:521–533 10.1084/jem.87.6.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson DR, Stevens DL, Kaplan EL. 1992. Epidemiologic analysis of group A streptococcal serotypes associated with severe systemic infections, rheumatic fever, or uncomplicated pharyngitis. J Infect Dis 166:374–382 10.1093/infdis/166.2.374. [DOI] [PubMed] [Google Scholar]
- 6.Veasy LG, Wiedmeier SE, Orsmond GS, Ruttenberg HD, Boucek MM, Roth SJ, Tait VF, Thompson JA, Daly JA, Kaplan EL, Hill HR. 1987. Resurgence of acute rheumatic fever in the intermountain area of the United States. N Engl J Med 316:421–427 10.1056/NEJM198702193160801. [PubMed] [DOI] [PubMed] [Google Scholar]
- 7.Marcon MJ, Hribar MM, Hosier DM, Powell DA, Brady MT, Hamoudi AC, Kaplan EL. 1988. Occurrence of mucoid M-18 Streptococcus pyogenes in a central Ohio pediatric population. J Clin Microbiol 26:1539–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Westlake RM, Graham TP, Edwards KM. 1990. An outbreak of acute rheumatic fever in Tennessee. Pediatr Infect Dis J 9:97–100 10.1097/00006454-199002000-00007. [PubMed] [DOI] [PubMed] [Google Scholar]
- 9.Markovitz A, Cifonelli JA, Dorfman A. 1959. The biosynthesis of hyaluronic acid by group A Streptococcus. VI. Biosynthesis from uridine nucleotides in cell-free extracts. J Biol Chem 234:2343–2350. [PubMed] [PubMed] [Google Scholar]
- 10.Stoolmiller AC, Dorfman A. 1969. The biosynthesis of hyaluronic acid by Streptococcus. J Biol Chem 244:236–246. [PubMed] [PubMed] [Google Scholar]
- 11.Sugahara K, Schwartz NB, Dorfman A. 1979. Biosynthesis of hyaluronic acid by Streptococcus. J Biol Chem 254:6252–6261. [PubMed] [PubMed] [Google Scholar]
- 12.Dougherty BA, van de Rijn I. 1992. Molecular characterization of a locus required for hyaluronic acid capsule production in group A streptococci. J Exp Med 175:1291–1299 10.1084/jem.175.5.1291. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeAngelis PL, Papaconstantinou J, Weigel PH. 1993. Isolation of a Streptococcus pyogenes gene locus that directs hyaluronan biosynthesis in acapsular mutants and in heterologous bacteria. J Biol Chem 268:14568–14571. [PubMed] [Google Scholar]
- 14.Wessels MR, Goldberg JB, Moses AE, DiCesare TJ. 1994. Effects on virulence of mutations in a locus essential for hyaluronic acid capsule expression in group A streptococci. Infect Immun 62:433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeAngelis PL, Papaconstantinou J, Weigel PH. 1993. Molecular cloning, identification, and sequence of the hyaluronan synthase gene from group A Streptococcus pyogenes. J Biol Chem 268:19181–19184. [PubMed] [Google Scholar]
- 16.Dougherty BA, van de Rijn I. 1994. Molecular characterization of hasA from an operon required for hyaluronic acid synthesis in group A streptococci. J Biol Chem 269:169–175. [PubMed] [PubMed] [Google Scholar]
- 17.DeAngelis PL, Yang N, Weigel PH. 1994. The Streptococcus pyogenes hyaluronan synthase: sequence comparison and conservation among various group A strains. Biochem Biophys Res Commun 199:1–10 10.1006/bbrc.1994.1184. [DOI] [PubMed] [Google Scholar]
- 18.Weigel PH, Hascall VC, Tammi M. 1997. Hyaluronan synthases. J Biol Chem 272:13997–14000 10.1074/jbc.272.22.13997. [PubMed] [DOI] [PubMed] [Google Scholar]
- 19.Tlapak-Simmons VL, Baggenstoss BA, Clyne T, Weigel PH. 1999. Purification and lipid dependence of the recombinant hyaluronan synthases from Streptococcus pyogenes and Streptococcus equisimilis. J Biol Chem 274:4239–4245 10.1074/jbc.274.7.4239. [DOI] [PubMed] [Google Scholar]
- 20.Heldermon C, DeAngelis PL, Weigel PH. 2001. Topological organization of the hyaluronan synthase from Streptococcus pyogenes. J Biol Chem 276:2037–2046 10.1074/jbc.M002276200. [DOI] [PubMed] [Google Scholar]
- 21.Dougherty BA, van de Rijn I. 1993. Molecular characterization of hasB from an operon required for hyaluronic acid synthesis in group A streptococci. Demonstration of UDP-glucose dehydrogenase activity. J Biol Chem 268:7118–7124. [PubMed] [Google Scholar]
- 22.Crater DL, Dougherty BA, van de Rijn I. 1995. Molecular characterization of hasC from an operon required for hyaluronic acid synthesis in group A streptococci. Demonstration of UDP-glucose pyrophosphorylase activity. J Biol Chem 270:28676–28680 10.1074/jbc.270.48.28676. [DOI] [PubMed] [Google Scholar]
- 23.Ashbaugh CD, Albertí S, Wessels MR. 1998. Molecular analysis of the capsule gene region of group A Streptococcus: the hasAB genes are sufficient for capsule expression. J Bacteriol 180:4955–4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crater DL, van de Rijn I. 1995. Hyaluronic acid synthesis operon (has) expression in group A streptococci. J Biol Chem 270:18452–18458 10.1074/jbc.270.31.18452. [PubMed] [DOI] [PubMed] [Google Scholar]
- 25.Cole JN, Aziz RK, Kuipers K, Timmer AM, Nizet V, van Sorge NM. 2012. A conserved UDP-glucose dehydrogenase encoded outside the hasABC operon contributes to capsule biogenesis in group A Streptococcus. J Bacteriol 194:6154–6161 10.1128/JB.01317-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kolkman MAB, Wakarchuk W, Nuijten PJM, van der Zeijst BA. 1997. Capsular polysaccharide synthesis in Streptococcus pneumoniae serotype 14: molecular analysis of the complete cps locus and identification of genes encoding glycosyltransferases required for the biosynthesis of the tetrasaccharide subunit. Mol Microbiol 26:197–208 10.1046/j.1365-2958.1997.5791940.x. [DOI] [PubMed] [Google Scholar]
- 27.Morona JK, Morona R, Paton JC. 1997. Characterization of the locus encoding the Streptococcus pneumoniae type 19F capsular polysaccharide biosynthetic pathway. Mol Microbiol 23:751–763 10.1046/j.1365-2958.1997.2551624.x. [DOI] [PubMed] [Google Scholar]
- 28.Muñoz R, Mollerach M, López R, García E. 1997. Molecular organization of the genes required for the synthesis of type 1 capsular polysaccharide of Streptococcus pneumoniae: formation of binary encapsulated pneumococci and identification of cryptic dTDP-rhamnose biosynthesis genes. Mol Microbiol 25:79–92 10.1046/j.1365-2958.1997.4341801.x. [DOI] [PubMed] [Google Scholar]
- 29.Rubens CE, Haft RF, Wessels MR. 1995. Characterization of the capsular polysaccharide genes of group B streptococci, p 237–244. In Ferretti JJ, Gilmore MS, Klaenhammer TR, Brown F (ed), Genetics of Streptococci, Enterococci, and Lactococci, vol 85. Karger, Basel Switzerland. [PubMed] [Google Scholar]
- 30.Sau S, Bhasin N, Wann ER, Lee JC, Foster TJ, Lee CY. 1997. The Staphylococcus aureus allelic genetic loci for serotype 5 and 8 capsule expression contain the type-specific genes flanked by common genes. Microbiology 143:2395–2405 10.1099/00221287-143-7-2395. [DOI] [PubMed] [Google Scholar]
- 31.Dillard JP, Vandersea MW, Yother J. 1995. Characterization of the cassette containing genes for type 3 capsular polysaccharide biosynthesis in Streptococcus pneumoniae. J Exp Med 181:973–983 10.1084/jem.181.3.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.García E, López R. 1997. Molecular biology of the capsular genes of Streptococcus pneumoniae. FEMS Microbiol Lett 149:1–10 10.1016/S0378-1097(97)00026-8. [PubMed] [DOI] [PubMed] [Google Scholar]
- 33.Flores AR, Jewell BE, Fittipaldi N, Beres SB, Musser JM. 2012. Human disease isolates of serotype m4 and m22 group a streptococcus lack genes required for hyaluronic acid capsule biosynthesis. MBio 3:e00413-12 10.1128/mBio.00413-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friães A, Machado MP, Pato C, Carriço J, Melo-Cristino J, Ramirez M. 2015. Emergence of the same successful clade among distinct populations of emm89 Streptococcus pyogenes in multiple geographic regions. MBio 6:e01780-15 10.1128/mBio.01780-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Henningham A, Yamaguchi M, Aziz RK, Kuipers K, Buffalo CZ, Dahesh S, Choudhury B, Van Vleet J, Yamaguchi Y, Seymour LM, Ben Zakour NL, He L, Smith HV, Grimwood K, Beatson SA, Ghosh P, Walker MJ, Nizet V, Cole JN. 2014. Mutual exclusivity of hyaluronan and hyaluronidase in invasive group A Streptococcus. J Biol Chem 289:32303–32315 10.1074/jbc.M114.602847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turner CE, Abbott J, Lamagni T, Holden MT, David S, Jones MD, Game L, Efstratiou A, Sriskandan S. 2015. Emergence of a new highly successful acapsular group A Streptococcus clade of genotype emm89 in the United Kingdom. MBio 6:e00622 10.1128/mBio.00622-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Albertí S, Ashbaugh CD, Wessels MR. 1998. Structure of the has operon promoter and regulation of hyaluronic acid capsule expression in group A Streptococcus. Mol Microbiol 28:343–353 10.1046/j.1365-2958.1998.00800.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 38.Gryllos I, Cywes C, Shearer MH, Cary M, Kennedy RC, Wessels MR. 2001. Regulation of capsule gene expression by group A Streptococcus during pharyngeal colonization and invasive infection. Mol Microbiol 42:61–74 10.1046/j.1365-2958.2001.02635.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 39.Cleary PP, McLandsborough L, Ikeda L, Cue D, Krawczak J, Lam H. 1998. High-frequency intracellular infection and erythrogenic toxin A expression undergo phase variation in M1 group A streptococci. Mol Microbiol 28:157–167 10.1046/j.1365-2958.1998.00786.x. [DOI] [PubMed] [Google Scholar]
- 40.Perez-Casal J, Caparon MG, Scott JR. 1991. Mry, a trans-acting positive regulator of the M protein gene of Streptococcus pyogenes with similarity to the receptor proteins of two-component regulatory systems. J Bacteriol 173:2617–2624 10.1128/jb.173.8.2617-2624.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Podbielski A, Woischnik M, Pohl B, Schmidt KH. 1996. What is the size of the group A streptococcal vir regulon? The Mga regulator affects expression of secreted and surface virulence factors. Med Microbiol Immunol 185:171–181 10.1007/s004300050028. [DOI] [PubMed] [Google Scholar]
- 42.Levin JC, Wessels MR. 1998. Identification of csrR/csrS, a genetic locus that regulates hyaluronic acid capsule synthesis in group A Streptococcus. Mol Microbiol 30:209–219 10.1046/j.1365-2958.1998.01057.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 43.Fogg GC, Gibson CM, Caparon MG. 1994. The identification of rofA, a positive-acting regulatory component of prtF expression: use of an m gamma delta-based shuttle mutagenesis strategy in Streptococcus pyogenes. Mol Microbiol 11:671–684 10.1111/j.1365-2958.1994.tb00345.x. [DOI] [PubMed] [Google Scholar]
- 44.Federle MJ, McIver KS, Scott JR. 1999. A response regulator that represses transcription of several virulence operons in the group A streptococcus. J Bacteriol 181:3649–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heath A, DiRita VJ, Barg NL, Engleberg NC. 1999. A two-component regulatory system, CsrR-CsrS, represses expression of three Streptococcus pyogenes virulence factors, hyaluronic acid capsule, streptolysin S, and pyrogenic exotoxin B. Infect Immun 67:5298–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller AA, Engleberg NC, DiRita VJ. 2001. Repression of virulence genes by phosphorylation-dependent oligomerization of CsrR at target promoters in S. pyogenes. Mol Microbiol 40:976–990 10.1046/j.1365-2958.2001.02441.x. [DOI] [PubMed] [Google Scholar]
- 47.Lei B, DeLeo FR, Hoe NP, Graham MR, Mackie SM, Cole RL, Liu M, Hill HR, Low DE, Federle MJ, Scott JR, Musser JM. 2001. Evasion of human innate and acquired immunity by a bacterial homolog of CD11b that inhibits opsonophagocytosis. Nat Med 7:1298–1305 10.1038/nm1201-1298. [DOI] [PubMed] [Google Scholar]
- 48.Dalton TL, Collins JT, Barnett TC, Scott JR. 2006. RscA, a member of the MDR1 family of transporters, is repressed by CovR and required for growth of Streptococcus pyogenes under heat stress. J Bacteriol 188:77–85 10.1128/JB.188.1.77-85.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Graham MR, Smoot LM, Migliaccio CA, Virtaneva K, Sturdevant DE, Porcella SF, Federle MJ, Adams GJ, Scott JR, Musser JM. 2002. Virulence control in group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. Proc Natl Acad Sci U S A 99:13855–13860 10.1073/pnas.202353699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gryllos I, Grifantini R, Colaprico A, Jiang S, Deforce E, Hakansson A, Telford JL, Grandi G, Wessels MR. 2007. Mg(2+) signalling defines the group A streptococcal CsrRS (CovRS) regulon. Mol Microbiol 65:671–683 10.1111/j.1365-2958.2007.05818.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 51.Gryllos I, Levin JC, Wessels MR. 2003. The CsrR/CsrS two-component system of group A Streptococcus responds to environmental Mg2+. Proc Natl Acad Sci U S A 100:4227–4232 10.1073/pnas.0636231100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gryllos I, Tran-Winkler HJ, Cheng MF, Chung H, Bolcome R III, Lu W, Lehrer RI, Wessels MR. 2008. Induction of group A Streptococcus virulence by a human antimicrobial peptide. Proc Natl Acad Sci U S A 105:16755–16760 10.1073/pnas.0803815105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tran-Winkler HJ, Love JF, Gryllos I, Wessels MR. 2011. Signal transduction through CsrRS confers an invasive phenotype in group A Streptococcus. PLoS Pathog 7:e1002361 10.1371/journal.ppat.1002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bernish B, van de Rijn I. 1999. Characterization of a two-component system in Streptococcus pyogenes which is involved in regulation of hyaluronic acid production. J Biol Chem 274:4786–4793 10.1074/jbc.274.8.4786. [PubMed] [DOI] [PubMed] [Google Scholar]
- 55.Federle MJ, Scott JR. 2002. Identification of binding sites for the group A streptococcal global regulator CovR. Mol Microbiol 43:1161–1172 10.1046/j.1365-2958.2002.02810.x. [DOI] [PubMed] [Google Scholar]
- 56.Horstmann N, Sahasrabhojane P, Suber B, Kumaraswami M, Olsen RJ, Flores A, Musser JM, Brennan RG, Shelburne SA III. 2011. Distinct single amino acid replacements in the control of virulence regulator protein differentially impact streptococcal pathogenesis. PLoS Pathog 7:e1002311 10.1371/journal.ppat.1002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ikebe T, Ato M, Matsumura T, Hasegawa H, Sata T, Kobayashi K, Watanabe H. 2010. Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog 6:e1000832 10.1371/journal.ppat.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. 2006. Genome-wide analysis of group a streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog 2:e5 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Biswas I, Scott JR. 2003. Identification of rocA, a positive regulator of covR expression in the group A streptococcus. J Bacteriol 185:3081–3090 10.1128/JB.185.10.3081-3090.2003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller EW, Danger JL, Ramalinga AB, Horstmann N, Shelburne SA, Sumby P. 2015. Regulatory rewiring confers serotype-specific hyper-virulence in the human pathogen group A Streptococcus. Mol Microbiol 98:473–489 10.1111/mmi.13136. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lynskey NN, Goulding D, Gierula M, Turner CE, Dougan G, Edwards RJ, Sriskandan S. 2013. RocA truncation underpins hyper-encapsulation, carriage longevity and transmissibility of serotype M18 group A streptococci. PLoS Pathog 9:e1003842 10.1371/journal.ppat.1003842. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lynskey NN, Turner CE, Heng LS, Sriskandan S. 2015. A truncation in the regulator RocA underlies heightened capsule expression in serotype M3 group A streptococci. Infect Immun 83:1732–1733 10.1128/IAI.02892-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kendall F, Heidelberger M, Dawson M. 1937. A serologically inactive polysaccharide elaborated by mucoid strains of group A hemolytic streptococcus. J Biol Chem 118:61–69. [Google Scholar]
- 64.Fillit HM, McCarty M, Blake M. 1986. Induction of antibodies to hyaluronic acid by immunization of rabbits with encapsulated streptococci. J Exp Med 164:762–776 10.1084/jem.164.3.762. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fillit HM, Blake M, MacDonald C, McCarty M. 1988. Immunogenicity of liposome-bound hyaluronate in mice. At least two different antigenic sites on hyaluronate are identified by mouse monoclonal antibodies. J Exp Med 168:971–982 10.1084/jem.168.3.971. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Todd EW. 1927. The influence of sera obtained from cases of streptococcal septicaemia on the virulence of the homologous cocci. Br J Exp Pathol 8:361–368. [Google Scholar]
- 67.Lancefield RC. 1957. Differentiation of group A streptococci with a common R antigen into three serological types, with special reference to the bactericidal test. J Exp Med 106:525–544 10.1084/jem.106.4.525. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rothbard S. 1948. Protective effect of hyaluronidase and type-specific anti-M serum on experimental group A streptococcus infection in mice. J Exp Med 88:325–342 10.1084/jem.88.3.325. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Foley MJ, Wood WB Jr. 1959. Studies on the pathogenicity of group A streptococci. II. The antiphagocytic effects of the M protein and the capsular gel. J Exp Med 110:617–628 10.1084/jem.110.4.617. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stollerman GH, Rytel M, Ortiz J. 1963. Accessory plasma factors involved in the bactericidal test for type-specific antibody to group A streptococci. II. Human plasma cofactor (s) enhancing opsonization of encapsulated organisms. J Exp Med 117:1–17 10.1084/jem.117.1.1. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dale JB, Washburn RG, Marques MB, Wessels MR. 1996. Hyaluronate capsule and surface M protein in resistance to opsonization of group A streptococci. Infect Immun 64:1495–1501. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moses AE, Wessels MR, Zalcman K, Albertí S, Natanson-Yaron S, Menes T, Hanski E. 1997. Relative contributions of hyaluronic acid capsule and M protein to virulence in a mucoid strain of the group A Streptococcus. Infect Immun 65:64–71. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Edwards MS, Kasper DL, Jennings HJ, Baker CJ, Nicholson-Weller A. 1982. Capsular sialic acid prevents activation of the alternative complement pathway by type III, group B streptococci. J Immunol 128:1278–1283. [PubMed] [PubMed] [Google Scholar]
- 74.Marques MB, Kasper DL, Pangburn MK, Wessels MR. 1992. Prevention of C3 deposition by capsular polysaccharide is a virulence mechanism of type III group B streptococci. Infect Immun 60:3986–3993. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baltimore RS, Kasper DL, Baker CJ, Goroff DK. 1977. Antigenic specificity of opsonophagocytic antibodies in rabbit anti-sera to group B streptococci. J Immunol 118:673–678. [PubMed] [PubMed] [Google Scholar]
- 76.Wessels MR, Moses AE, Goldberg JB, DiCesare TJ. 1991. Hyaluronic acid capsule is a virulence factor for mucoid group A streptococci. Proc Natl Acad Sci U S A 88:8317–8321 10.1073/pnas.88.19.8317. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hirst GK. 1941. The effect of a polysaccharide splitting enzyme on streptococcal infection. J Exp Med 73:493–506 10.1084/jem.73.4.493. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kass EH, Seastone CV. 1944. The role of the mucoid polysaccharide (hyaluronic acid) in the virulence of group A hemolytic streptococci. J Exp Med 79:319–330 10.1084/jem.79.3.319. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schrager HM, Rheinwald JG, Wessels MR. 1996. Hyaluronic acid capsule and the role of streptococcal entry into keratinocytes in invasive skin infection. J Clin Invest 98:1954–1958 10.1172/JCI118998. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ashbaugh CD, Warren HB, Carey VJ, Wessels MR. 1998. Molecular analysis of the role of the group A streptococcal cysteine protease, hyaluronic acid capsule, and M protein in a murine model of human invasive soft-tissue infection. J Clin Invest 102:550–560 10.1172/JCI3065. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wessels MR, Bronze MS. 1994. Critical role of the group A streptococcal capsule in pharyngeal colonization and infection in mice. Proc Natl Acad Sci U S A 91:12238–12242 10.1073/pnas.91.25.12238. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Husmann LK, Yung D-L, Hollingshead SK, Scott JR. 1997. Role of putative virulence factors of Streptococcus pyogenes in mouse models of long-term throat colonization and pneumonia. Infect Immun 65:1422–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ashbaugh CD, Moser TJ, Shearer MH, White GL, Kennedy RC, Wessels MR. 2000. Bacterial determinants of persistent throat colonization and the associated immune response in a primate model of human group A streptococcal pharyngeal infection. Cell Microbiol 2:283–292 10.1046/j.1462-5822.2000.00050.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 84.Flores AR, Jewell BE, Olsen RJ, Shelburne SA III, Fittipaldi N, Beres SB, Musser JM. 2014. Asymptomatic carriage of group A streptococcus is associated with elimination of capsule production. Infect Immun 82:3958–3967 10.1128/IAI.01788-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shea PR, Beres SB, Flores AR, Ewbank AL, Gonzalez-Lugo JH, Martagon-Rosado AJ, Martinez-Gutierrez JC, Rehman HA, Serrano-Gonzalez M, Fittipaldi N, Ayers SD, Webb P, Willey BM, Low DE, Musser JM. 2011. Distinct signatures of diversifying selection revealed by genome analysis of respiratory tract and invasive bacterial populations. Proc Natl Acad Sci U S A 108:5039–5044 10.1073/pnas.1016282108. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schrager HM, Albertí S, Cywes C, Dougherty GJ, Wessels MR. 1998. Hyaluronic acid capsule modulates M protein-mediated adherence and acts as a ligand for attachment of group A Streptococcus to CD44 on human keratinocytes. J Clin Invest 101:1708–1716 10.1172/JCI2121. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cywes C, Stamenkovic I, Wessels MR. 2000. CD44 as a receptor for colonization of the pharynx by group A Streptococcus. J Clin Invest 106:995–1002 10.1172/JCI10195. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cywes C, Wessels MR. 2001. Group A Streptococcus tissue invasion by CD44-mediated cell signalling. Nature 414:648–652 10.1038/414648a. [PubMed] [DOI] [PubMed] [Google Scholar]
- 89.LaPenta D, Rubens C, Chi E, Cleary PP. 1994. Group A streptococci efficiently invade human respiratory epithelial cells. Proc Natl Acad Sci U S A 91:12115–12119 10.1073/pnas.91.25.12115. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Greco R, De Martino L, Donnarumma G, Conte MP, Seganti L, Valenti P. 1995. Invasion of cultured human cells by Streptococcus pyogenes. Res Microbiol 146:551–560 10.1016/0923-2508(96)80561-4. [DOI] [PubMed] [Google Scholar]
- 91.Osterlund A, Popa R, Nikkilä T, Scheynius A, Engstrand L. 1997. Intracellular reservoir of Streptococcus pyogenesin vivo: a possible explanation for recurrent pharyngotonsillitis. Laryngoscope 107:640–647 10.1097/00005537-199705000-00016. [PubMed] [DOI] [PubMed] [Google Scholar]
- 92.Molinari G, Chhatwal GS. 1998. Invasion and survival of Streptococcus pyogenes in eukaryotic cells correlates with the source of the clinical isolates. J Infect Dis 177:1600–1607 10.1086/515310. [PubMed] [DOI] [PubMed] [Google Scholar]
- 93.Ferretti JJ, McShan WM, Ajdic D, Savic DJ, Savic G, Lyon K, Primeaux C, Sezate S, Suvorov AN, Kenton S, Lai HS, Lin SP, Qian Y, Jia HG, Najar FZ, Ren Q, Zhu H, Song L, White J, Yuan X, Clifton SW, Roe BA, McLaughlin R. 2001. Complete genome sequence of an M1 strain of Streptococcus pyogenes. Proc Natl Acad Sci U S A 98:4658–4663 10.1073/pnas.071559398. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Beres SB, Sylva GL, Barbian KD, Lei B, Hoff JS, Mammarella ND, Liu MY, Smoot JC, Porcella SF, Parkins LD, Campbell DS, Smith TM, McCormick JK, Leung DY, Schlievert PM, Musser JM. 2002. Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc Natl Acad Sci U S A 99:10078–10083 10.1073/pnas.152298499. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schmidt K-H, Günther E, Courtney HS. 1996. Expression of both M protein and hyaluronic acid capsule by group A streptococcal strains results in a high virulence for chicken embryos. Med Microbiol Immunol 184:169–173 10.1007/BF02456131. [DOI] [PubMed] [Google Scholar]