

Abstract
Engineering physiologically-relevant in vitro models of human organs remains a fundamental challenge. Despite significant strides made within the field, many promising organ-on-a-chip models fall short in recapitulating cellular interactions with neighboring cell types, surrounding extracellular matrix (ECM), and exposure to soluble cues due, in part, to the formation of artificial structures that obstruct >50% of the surface area of the ECM. Here, we report a three-dimensional (3D) cell culture platform based upon hydrophobic patterning of hydrogels that is capable of precisely generating a 3D ECM within a microfluidic channel with an interaction area >95%. In this study, for demonstrative purposes, type I collagen (COL1), Matrigel (MAT), COL1/MAT mixture, hyaluronic acid (HA), and cell-laden MAT were formed in the device. We demonstrate three potential applications, including creating a 3D endothelium model, studying the interstitial migration of cancer cells, and analyzing stem cell differentiation in a 3D environment. Our hydrophobic patterned-based 3D cell culture device provides the ease-of-fabrication and flexibility necessary for broad potential applications in organ-on-a-chip platforms.
1. Introduction
Many 2D cultured systems that have been successfully used for culturing a variety of cell types do not provide a true physiological environment. Consequently, cells cultured on those 2D substrata are morphologically and phenotypically different from those cultured in a 3D environment 1–4. In contrast, 3D cell-culture models have demonstrated the possibility of providing essential 3D cues—from biomechanical cues to cell-cell/ECM interactions—by generating higher levels of cellular differentiation and biologically relevant structural composition 5,6. Nevertheless, current 3D cell-culture models fail to recapitulate accurately specific biological structures and functions, e.g. the exact functional unit-structure of a target organ, the interface between endothelium/epithelium and surrounding ECM/parenchymal cells, and accurate regulation of chemical/oxygen gradients, that are fundamental components for reconstituting physiologically or pathologically relevant conditions. To address these shortcomings, microfluidics-based 3D surrogate models, i.e. “organs-on-a-chip,” have come into the spotlight for their potential to mimic human organs and accurately measure biological responses to an array of physiological and pathological conditions. Examples of the tremendous efforts made to advance existing technologies include models of 3D angiogenesis subject to a concentration gradient of growth factors either from growth medium or neighboring cancer cells, 3D axonal responses under complex gradients, 3D cancer-immune cell interactions via co-culture, and an in vivo-like endothelium on 3D endothelial ECM, gut-on-a-chip, and lung-on-a-chip 7–22. While these models demonstrate the promising future of the organ-on-a-chip, many rely on either a linear array of regularly spaced micro-posts 15–21 or a transwell assay’s porous membrane9,22–25 to pattern cells and/or the ECM within confined structures to replicate 3D organ-specific microenvironments. These structural strategies of cell/ECM patterning do not allow for a large enough effective area of cell-cell/ECM and cell-growth factor/chemokine interactions to supply cells with a sufficient amount of essential nutrients. In most micro-post-based platforms, over 50% of the interface area between the cell channel and ECM channel is blocked by artificial materials, either polydimethylsiloxane (PDMS) or hard plastics9,15–26; the total area of pores on a transwell membrane, which is the effective interaction area, is ~ 5% of the total cell culture area12,22,27,28. Furthermore, the size of these structures is typically smaller than the industrial standard for injection molding; the difficulty of manufacturing these structures at scale thus prevents the widespread transition in industry from non-physiological 2D substrata-based assays to in vivo-like 3D surrogate models29,30. To overcome this issue, pillar-less structure has come into the spotlight. Recently commercialized 3D cell culture platform, “Organoplate”, is capable of constructing 3D hydrogel structure without pillar structure. However, it still uses the structural strategy that consists of stripe protrusions on the bottom, which blocks more than 25% of the channel height31–33. Thus, there is a great need for new design strategies that enable uniform contact with and diffusion between cells and the ECM to better recapitulate in vivo conditions.
Here, we report a simple, yet robust and flexible cell-culture method that enables a variety of quasi-3D ECM hydrogel constructs, including type I collagen (COL1), Matrigel (MAT), COL1/MAT mixture, hyaluronic acid (HA) hydrogel, and cell-laden MAT. Our method is based on patterning thin hydrophobic stripes within which specific hydrogels are contained. A key advantage to this method is that the resulting interaction area between cell-cell/ECM and cell-growth factor/chemokine is >95%. As such, unwanted cell migration due to asymmetrical consumption of growth factors, which plague many 3D microfluidic cell-culture platforms17, is significantly reduced with our method. Overall, the simplicity, biocompatibility, and design flexibility of utilizing continuous thin hydrophobic stripes leads to diverse applications. We describe the patterning, diffusion, wettability, and 3D-liquid-filling characteristics of our method and resulting platform, as well as potential applications, including creating a 3D endothelium model, studying the interstitial migration of cancer cells, and analyzing stem cell differentiation in a 3D environment.
2. Materials and methods
2.1. Fabrication of hydrophobic and hydrophilic patterns
To generate hydroxyl groups onto a glass surface and promote adhesion to a methacrylate group, a glass coverslip (24×24 mm; Corning, USA) is treated with 1M NaOH (Sigma-Aldrich, USA) at room temperature for 1 hr and then rinsed with deionized (DI, MΩ) water. The coverslip is subsequently immersed in 1M HCl (Sigma-Aldrich, USA) at room temperature for 30 min, rinse with DI water, and then dried with pressurized N2 gas. The coverslip is immediately functionalized with methacrylate groups by incubating with 400 μL of a 5:2:3 volume ratio mixture of ethanol (Decon Labs, USA), 3-(trimethoxysilyl)propyl methacrylate (Sigma-Aldrich, USA), and glacial acetic acid (Sigma-Aldrich, USA) at room temperature for 1 hr. The resulting methacrylated glass is thoroughly rinsed with acetone (Sigma-Aldrich, USA) and dried with pressurized N2 gas.
For hydrophobic patterning, a polymerization mixture consisting of 30 wt% of butyl methacrylate (BMA; Sigma-Aldrich, USA), 20 wt% of ethylene dimethacrylate (EDMA; Sigma-Aldrich, USA), 50 wt% of 1-decanol (Sigma-Aldrich, USA), and an additional 1~6 wt% (with respect to total weight of EDMA and BMA) of 2,2-dimethoxy-2-phenylacetophenone (DMPAP; Sigma-Aldrich, USA) is prepared. A pre-polymer solution for hydrophilic patterning is also prepared by mixing 24 wt% of poly(2-hydroxyethylmethacrylate (pHEMA; Sigma-Aldrich, USA), 16 wt% of EDMA, 12 wt% of 1-decanol, 48 wt% of cyclohexanol (Sigma-Aldrich, USA), and an additional 1 wt% of DMPAP. Soft lithography is utilized to create the primary poly(dimethylsiloxane) (PDMS; Dow Corning, USA) slabs for hydrophobic/hydrophilic patterning. Briefly, SU-8 3025 (for 30 μm) and SU-8 3050 (for 50, 70, and 90 μm high channels) resist (MicroChem, USA) are spun onto 3-inch silicon wafers at 3,000 rpm for 30 and 50 µm thickness, 1,900 rpm for 70 µm thickness, or 1,500 rpm for 90 µm thickness. The spin-coated wafers are baked at 95°C for 15~45 min and then exposed to a photomask using 365 nm UV light (150~250 mJ/cm2). Following exposure, the wafers are hard baked at 65°C for 1 min and then at 95°C for 3–5 min. They are then developed with propylene glycol monomethyl ether acetate (PGMEA; MicroChem, USA) for 6~14 min. A prepared mixture of PDMS (1:10 weight ratio of elastomer to curing agent, Dow Corning, USA) is poured onto the patterned silicon wafers and cured at 80°C for at least 1 hr in a dry oven. A slab of PDMS with the embedded microchannels is cut and removed, and inlet and outlet holes are created using a 1-mm biopsy punch for the ECM channel and 4-mm biopsy punch (Integra LifeSciences, USA) for the side channels. The PDMS slab is placed onto the methacrylated coverslip and the microfluidic channels are filled with either hydrophilic (pHEMA-EDMA) or hydrophobic (BMA-EDMA) pre-polymer solution via capillary action. Once filled, the channels are irradiated with UV light (Novacure 2100, 265 nm, 15 mW/cm2; EXFO, USA) for 10 min, with the coverslip facing up toward the light source. The UV-patterned coverslip is then rinsed thoroughly with ethanol twice. To remove unwanted methacrylate groups from the coverslip and to prepare the glass surface for PDMS-glass bonding, the patterned coverslip is immersed in 1M NaOH solution at room temperature for 5 min, washed with DI water, and then dried with pressurized dry N2 gas.
2.2. Fabrication of the hydrophobic pattern-based 3D cell culture device
Like the primary PDMS slab, a secondary PDMS slab is created using soft lithography. Here, SU-8 2100 (MicroChem, USA) is used to create a 150 µm high microchannel negative-relief structure onto which PDMS is cast. Once cured and cut, the secondary PDMS slab is autoclaved at 120°C for 30 min and dried in an oven at 80°C for at least 5 hrs. The PDMS’s patterned side is then exposed to an O2 plasma (Harrick Plasma, USA; 70 mTorr, 80 W, 1 min). The NaOH-treated hydrophobic (BMA-EDMA) or hydrophilic (pHEMA-EDMA) patterned coverslips and the activated secondary PDMS slab are aligned using a stereoscope (SMZ series; Nikon, Japan) and bonded. Afterwards, it is incubated at 80°C for at least 24 hrs to restore PDMS’s intrinsic hydrophobicity. Bonded devices are UV treated in a biosafety cabinet for 1 hr in order to ensure sterility and then stored in a dry cabinet until use.
2.3. Preparation and gelation of type I collagen, Matrigel, the mixture of type I collagen/Matrigel mixture, hyaluronic acid, and cell-laden Matrigel within ECM channel
2.0 mg/mL of type I collagen (COL1) gel solution is prepared by mixing rat tail type I collagen (Corning, USA), 10X phosphate buffered saline (PBS; Lonza, USA), 0.5 N NaOH, and DI water. The pH of the COL1 gel solution is adjusted to pH 7.4 with NaOH. The mixture is injected into the extracellular matrix (ECM) channel of the microfluidic device and gelled for 30 min in a 37°C/5% CO2 incubator. Matrigel (MAT; Corning, USA) solution, thawed on ice, is injected into the ECM channel which had been placed on an ice plate for at least 30 min. The MAT-filled device is then incubated in a 37°C/5% CO2 incubator for 30 min. A COL1 and MAT mixture gel solution (COL1/MAT) is prepared by simply mixing 50% (v/v) COL1 gel solution with 50% (v/v) MAT solution. The COL1/MAT-filled device is then incubated in a 37°C/5% CO2 incubator for 30 min. A hyaluronic acid (HA) gel is obtained as a kit (HyStem-C Cell Culture Scaffold Kit; HyStem, Gelin-S, and Extralink) from Sigma-Aldrich. 4% HA is prepared by mixing 0.4 wt% HyStem, 0.4 wt% Gelin-S, 0.4 wt% Extralink and the concentration is adjusted by DI water. The HA-gel solution is injected into the ECM channel and then incubated in a 37°C/5% CO2 incubator for 30 min. Cell-laden MAT is prepared by mixing 200 µL MAT solution with 20 µL of MCF7/GFP cells (2×106 cells/mL). The cell-laden MAT solution is then injected into the ECM channel and incubated in a 37°C/5% CO2 incubator for 30 min. Ice-cold pipette tips are used when working with any MAT-related process. Upon completion, for each condition, side channels are filled with growth media or Dulbecco’s Phosphate-Buffered Saline (DPBS) and incubated in a 37°C/5% CO2 incubator for cell culture or until imaging. To visualize the hydrogels in the microfluidic device, fluorescent microparticles (5~14 µm; Spherotech, USA) are mixed in each finalized gel solution in a 10 (hydrogel) : 1 (DPBS containing fluorescent microparticles) volume ratio.
2.4. Numerical simulation of pressure distributions around BMA-EDMA and pHEMA-EDMA pattern within the microfluidic device
Pressure distribution around the hydrophobic BMA-EDMA pattern during the hydrogel filling process is calculated by COMSOL Multiphysics 5.0 (COMSOL Inc., Sweden). Water contact angles of BMA-EDMA, PDMS, and glass substrate are set to be 135°, 100° and 28.65°, respectively34–38. The hydrogel solution is assumed to have a dynamic viscosity and density of 1 Pa•s and 1 g/cm3, respectively. A capillary-driven flow model is employed for simulating the hydrogel-filling process. To control for geometry and estimate the structural contribution (5-micron high structure) of the pattern to the hydrogel-filling process, the hydrophilic pHEMA-EDMA is given the same structure as that of the BMA-EDMA pattern. In the pHEMA-EDMA model, the pHEMA-EDMA and glass substrate water contact angles are both assumed to be 28.65°.
2.5. Water contact angle measurement
To characterize water contact angles of the BMA-EDMA and pHEMA-EDMA patterns, a thin film of the BMA-EDMA or the pHEMA-EDMA is created by placing a drop of the appropriate pre-polymer solution onto a methacrylated coverslip and then overlaying a transparency film (3M, USA) for a 10-minute UV blanket exposure (265 nm, 15 mW/cm2; EXFO, USA). The UV-patterned coverslip is subsequently rinsed thoroughly with ethanol twice and incubated at 80°C for at least 1 hr to dry off residual solvent material. Water contact angles are measured in ambient air with a custom apparatus consisting of a 3-axis mechanical stage, optical zoom tube, CCD, light source, and convex lens. For each condition, i.e. the BMA-EDMA (1, 2, 4, and 6% of DMPAP in the pre-polymer mixture), the pHEMA-EDMA thin-film patterns, and the glass surface, 10 µL DI water droplets are placed onto each surface and imaged. Each image is subsequently analyzed with the ImageJ/Contact Angle.jar open-source program (NIH, USA). To demonstrate the hydrophobicity of the BMA-EDMA pattern, a circle-shaped pattern is utilized. Water drops added to the inner area of the circle stayed within the pattern even with greater volumes (over 50 µL) (Supplementary Figure 3). In order to show that the hydrophobicity of the BMA-EDMA pattern is independent from the physical structure of the pattern, the hydrophilic mixture, pHEMA-EDMA, is polymerized with the same primary PDMS slab used to produce the circle-shaped BMA-EDMA pattern.
2.6. Cell preparation
MCF7/GFP cells (Cell Biolabs, USA) were cultured and maintained with Dulbecco’s Modified Eagle Medium (DMEM, high glucose; Lonza, USA) supplemented with 10% fetal bovine serum (FBS; Lonza, USA), 0.1 mM MEM non-essential amino acids (NEAA; Gibco, USA), 2 mM L-glutamine (Gibco, USA), and 1% Pen-Strep (Gibco, USA). Human microvascular endothelial cells (hMVECs, cryopreserved, third passage; Lonza, USA) were expanded with endothelial growth medium (EGM-2 MV; Lonza, USA) until the fifth passage according to standard protocols. Human umbilical vein endothelial cells (HUVECs; ATCC, USA) were expanded with F-12K medium supplemented with 0.1 mg/mL Heparin, 1% endothelial cell growth supplement (ECGS; BD Biosciences, USA), and 10% FBS. Human neural stem cells (hNSCs, H9-derived; Gibco, USA) were cultured on CellStart (Gibco, USA) coated T25 flasks with StemPro NSC SFM (obtained as a kit except for GlutaMAX-I supplement; Gibco, USA) containing 97 mL of KnockOut DMEM/F-12, 1 mL of GlutaMAX-I supplement (Gibco, USA), 2 µg of recombinant hFGF, 2 µg of recombinant hEGF, and 2 mL of StemPro neural supplement. The hNSCs were passaged at 90% confluence and expanded until the fourth passage. Human mesenchymal stem cells (hMSC; Lonza, USA) were expanded with mesenchymal stem cell growth medium (MSCGM BulletKit, Lonza, USA) until the fifth passage. All cells used in this work were dissociated from culture substrates with 0.25% trypsin/ethylenediaminetetraacetic acid (EDTA) (Gibco, USA) for 2 minutes at 37°C, washed with respective growth media, centrifuged at 200 × g (RCF), and resuspended at a concentration of 5 × 105 cells/mL in a T75 flask.
2.7. 3D breast epithelial cancer migration assay
In the previous study, 3D microfluidic cell culture has intensively developed15. We followed this generalized protocol for this study. Two different representative 3D hydrogels, COL1 and COL1/MAT were created in the ECM channel by the aforementioned methods to demonstrate the platform’s utility for a 3D breast epithelial cancer cell migration assay. After gelling each hydrogel, 50 µL of resuspended MCF7/GFP cells (2×106 cells/mL) were seeded onto one side of a prepared device and cultured with prepared media for 4 days. The opposite side of the device was then filled with complete growth media supplemented with 10% FBS to generate an FBS concentration gradient, thereby inducing cells that were attached to the ECM to migrate toward the FBS source. Growth media was replaced daily.
2.8. Generation of a quiescent 3D blood capillary
2×106 cells/mL of harvested hMVECs were seeded into the cell channel of the BMA-EDMA device. The ECM structure for a quiescent 3D blood capillary was created by gelling 3D COL1 within the BMA-EDMA device and coating the 3D COL1 surface with diluted MAT. Briefly, MAT diluted in chilled PBS (200 µg/mL) was introduced in the cell channel to create a thin layer of MAT on the surface of the 3D COL1 gelled within the ECM channel. In order to gel the thin layer of MAT, the microfluidic device was incubated at 37°C under 5% CO2 for 40 min. The hMVECs were cultured with EGM-2 MV for 4 days until a confluent EC monolayer formed on the entire surface of the cell channel. Growth media was replaced daily.
2.9. Diffusion and cell culture characteristics based upon geometric differences between pillar-based and BMA-EDMA-based 3D cell culture assays
To compare our BMA-EDMA device with previously reported pillar-based 3D cell culture platforms, BMA-EDMA stripes were replaced with two arrays of pillars (Figure 4). The pillar-based devices were fabricated via soft lithography. 3D COL1 was prepared in the ECM channels of both the BMA-EDMA and pillar-based devices. 50 µL of 2×106 HUVEC cells/mL in suspension was injected into each device. After cell attachment, the side, cell, and growth medium channels were washed and filled with fresh EGM-2 cells and were cultured for 3 days. On culture day 3, the number of cells that invaded into the 3D COL1 in each assay were counted using 4’,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, USA; 1 µg/mL in DPBS) staining.
Figure 4. Generation of a quiescent 3D blood capillary model using BMA-EDMA-based hydrogel patterning.
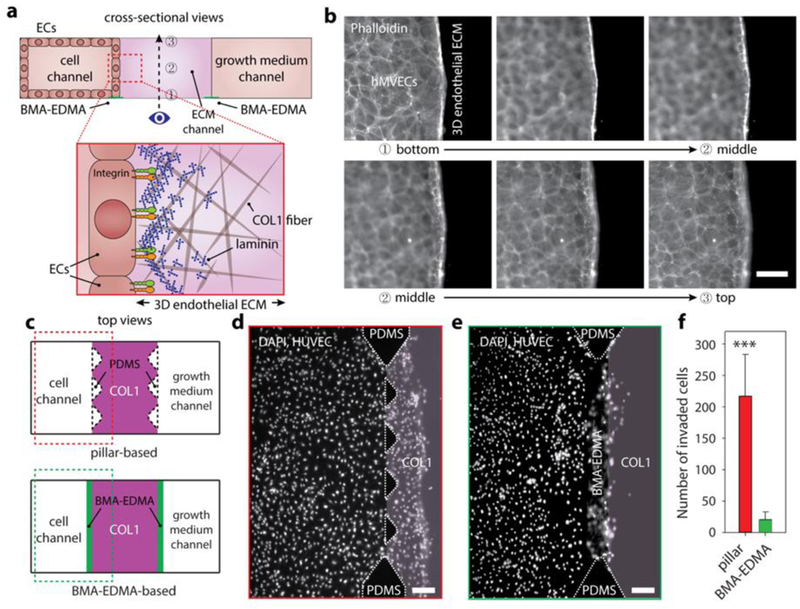
a. Schematic cross-sectional view (not to scale) of the 3D blood capillary model (top). 3D endothelial ECM which consists of COL1 as a 3D constructive ECM and additional thin layer of MAT as a basement membrane (BM) was formed in the ECM channel. Human microvascular endothelial cells (hMVECs) were seeded into the cell channel (left). Bottom illustration depicts a microscopic structural top view of the 3D endothelial ECM (not to scale). Numbers in the top image indicate corresponding focal planes in (b), from bottom (1) to top (3). b. Corresponding fluorescent images of the focal planes shown in (a) of hMVECs cultured (on day4) in the BMA-EDMA device indicate that an hMVEC monolayer was successfully generated without any unwanted angiogenesis/sprouts or migration. Scale bar: 50 µm. c. Schematic top views of pillar-based 3D cell culture assay (top) and our BMA-EDMA-based assay (bottom). The pillar-based assay relies on surface tension generated by sharp edges between each trapezoidal pillar to prevent hydrogel from escaping the ECM area, while our BMA-EDMA-based assay employs an ultra-thin layer of hydrophobic pattern on the glass substrate to generate a sharp change in surface wettability. d. Geometric difference between two assays shown in (c) significantly affected the diffusion profiles of growth factors throughout the cell and ECM channels (see supplementary information for simulated diffusion profiles throughout the channels), resulting in unwanted migration of human umbilical vascular endothelial cells (HUVECs) into the ECM in the pillar-based assay (on day4). Scale bar: 300 µm. e. The larger interface area of the BMA-EDMA-based assay enabled the more efficient supply of growth factors to the cells, which mediated unintended concentration gradient caused by cellular consumption of growth factors relative to the pillar-based assay. Scale bar: 300 µm. f. Quantification of the number of the cells that migrated into the COL1 in the assays shown in (d) and (e). Data are shown as the means ± SD (n = 4). ***p<0.001.
2.10. Numerical simulation of diffusion profiles within the microfluidic device
Diffusion profiles within devices were determined using COMSOL Multiphysics 5.0. The transient (0~24 hrs) diffusion profiles were estimated using Fick’s Second Law, , where C is concentration and χ is the diffusion coefficient. The diffusion coefficient of FBS in the MCF7/GFP growth medium, MCF7/GFP, COL1, and MAT were assumed to be 8×10−11, 1.57×10−12, 5.8×10−11, and 5.76×10−11 m2/s, respectively [18, 21]. The consumption rate of MCF7/GFP was assumed to be 0.02 fg/cell/day.
2.11. Stem cell differentiation
To generate a 3D stem-cell microenvironment, 2×106 cells/mL hNSCs or hMSCs were mixed with a pre-gel solution of MAT and injected into the ECM channel formed by the BMA-EDMA pattern. The device was then incubated in a 37°C/5% CO2 incubator for 30 min. During the gelation process, the device was inverted every 5 min to evenly distribute the cells within the pre-gel solution. Both hNSCs and hMSCs were cultured with their complete growth media, StemPro NSC SFM and MSCGM BulletKit, respectively, for 3 days; growth media was replaced daily. For 2D macro condition, the same seeding density was used.
2.12. Quantitative real-time polymerase chain reaction (qRT-PCR)
RNA extraction was performed via Qiagen’s Quick-Start Protocol. Briefly, hNSCs and hMSCs in the BMA-EDMA device were directly lysed by 300 µL of Buffer RLT (cell-lysis buffer) from RNeasy Mini Kit (Qiagen, USA). As a control, the cells in cell culture flasks were harvested by 0.25% Trypsin/EDTA and washed with DPBS solution. The cell pellets were then lysed with 350 µL of Buffer RLT. Each lysate was loaded into a RNase-free micro-tube and mixed with 350 µL of 70% ethanol. The mixed solutions were transferred to the RNeasy Mini spin column placed in a 2 mL collection tube and centrifuged for 15 seconds at 8,000 × g. The RNeasy silica membrane in the spin column was serially washed with 700 µL of Buffer RW1 for 15 seconds at 8,000 × g, 500 µL of Buffer RPE for 15 seconds at 8,000 × g, and 500 µL of Buffer RPE for 2 min at 8,000 × g. The flow-through at each process was discarded after centrifugation. The RNeasy spin column was then placed in a new 2 mL collection tube and centrifuged at full speed for 1 min to completely dry the membrane. Afterward, the spin column was put in a new 1.5 mL collection tube and 30 µL of RNase-free water was added directly to the membrane. The spin column was then centrifuged for 1 min at 8,000 x g to elute the RNA. The RNA concentration was determined and normalized by measuring the absorbance profile at 260 nm via a NanoDrop spectrophotometer (Thermo Scientific, USA). Reverse transcription (RT) reactions were carried out using the High Capacity RNA-to-cDNA Kit (Invitrogen, USA) under a standard protocol recommended by the manufacturer. qRT-PCR measurements of relative gene expressions were performed by using the Universal PCR Master Mix (Applied Biosystems, USA) and the StepOne Real-Time PCR (Applied Biosystems). Gene expressions in hNSCs cultured in the BMA-EDMA device and a cell culture flask were quantified using TaqMan Gene Expression Assays (Applied Biosystems) for GAPDH (Hs02758991_g1), Nestin (Hs00707120_s1), TUBB3 (Hs00801390_s1), GFAP (Hs00909238_g1), and OLIG2 (Hs00300164_s1). The same gene expression assays, including GAPDH (Hs02758991_g1) and Nestin, were utilized for quantifying those of hMSCs. TaqMan Gene Expression Assays for PPARG (Hs00234592_m1) and RUNX2 (Hs01047973_m1) were employed for evaluating the relative gene expressions. The mRNA levels of each target gene were normalized to that of the endogenous reference gene, GAPDH. The relative gene expression levels were determined by using the comparative Ct method. The normalized expression levels of Nestin and TUBB3 in hNSCs cultured in both 3D- and 2D-conditions were compared. Relative gene expression levels of GFAP and OLIG2 in hNSCs in all culture conditions were normalized to those of the 2D condition. For hMSCs, the gene expression levels of Nestin, PPARG, and RUNX2 were normalized to that of PPARG in 3D.
2.13. Immunocytochemistry
The samples containing MAT were fixed with 2% (v/v) paraformaldehyde (PFA; Sigma-Aldrich, USA) and 0.5% (v/v) glutaraldehyde (GA; Sigma-Aldrich, USA) in DPBS with calcium and magnesium at room temperature for 15 min to prevent auto-fluorescence of MAT. Other samples were fixed with 4% (v/v) PFA at room temperature for 15 min. The specimens were permeabilized with 0.1% Triton-X100 (Sigma-Aldrich, USA) in DPBS at room temperature for 5 min. After immersing the samples in undiluted BlockAid (Life Technologies, USA) at room temperature for at least 30 min to block non-specific binding, primary antibodies for each cell type were mixed in the BlockAid at recommended concentrations. Primary antibodies used in this work were rabbit anti-Nestin (EMD Millipore, USA; 1:500 in BlockAid), rabbit anti-GFAP (EMD Millipore, USA; 1:1000 in BlockAid), mouse anti-beta III Tubulin (EMD Millipore, USA; 1:100 in BlockAid), mouse anti-Oct-4 (EMD Millipore, USA; 20 µg/mL in BlockAid), rabbit anti-RUNX2 (Abcam, USA; 1:1000 in BlockAid), mouse anti-Adiponectin (Abcam, USA; 2.5 µg/mL), and mouse anti-Myogenin (Abcam, USA; 1:50 in BlockAid). The appropriate diluted antibodies were introduced into each device and incubated at room temperature for 1 hr. After washing the samples twice with DPBS, Alexa Fluor 488 goat anti-rabbit (Life Technologies, USA; 10 µg/mL in BlockAid) and/or Alexa Fluor 546 goat anti-mouse (Life Technologies, USA; 4 µg/mL in BlockAid) were added to the samples and incubated for 1 hr. Samples were then washed twice with DPBS. Cell nuclei and actin filaments were counterstained with DAPI and rhodamine Phalloidin (Life Technologies, USA; 200 nM in DPBS), respectively. Stained cells were imaged with either a fluorescence microscope (Eclipse TE2000-e; Nikon, Japan) or a confocal microscope (LSM 880 NLO AxioExaminer with OPO; Zeiss, Germany).
2.14. Statistical analysis
Statistical analysis was performed by Student’s t‐test for two groups (p < 0.05 was considered statistically significant). *p < 0.05, **p < 0.01, ***p < 0.001. Results were shown as mean ± standard deviation (SD). Sample sizes (n) for each statistical analysis were shown in the corresponding figure legends. All analyses were performed with SigmaPlot 12.
3. Results and discussion
3.1. Hydrophobic-based three-dimensional (3D) hydrogel patterning
To create highly-controlled 3D ECMs, we patterned hydrophobic micro-stripes within a microfluidic device. The overall ECM structure was defined by two side channels (1,100 µm wide × 150 µm high) separated by an ECM channel (800 µm wide × 150 µm high) (Fig. 1a). The hydrophobic pattern was fabricated to be less than 5 µm thick on a glass coverslip and combined with a secondary PDMS slab consisting of microchannels so that more than 95% of the interface allowed for interaction among cells, ECM, and biochemical signals (Fig. 1). This process yields a highly versatile 3D ECM structure, which can be employed for various 3D cell culture applications, as we demonstrate below.
Figure 1. Hydrophobic-based three-dimensional (3D) hydrogel patterning.
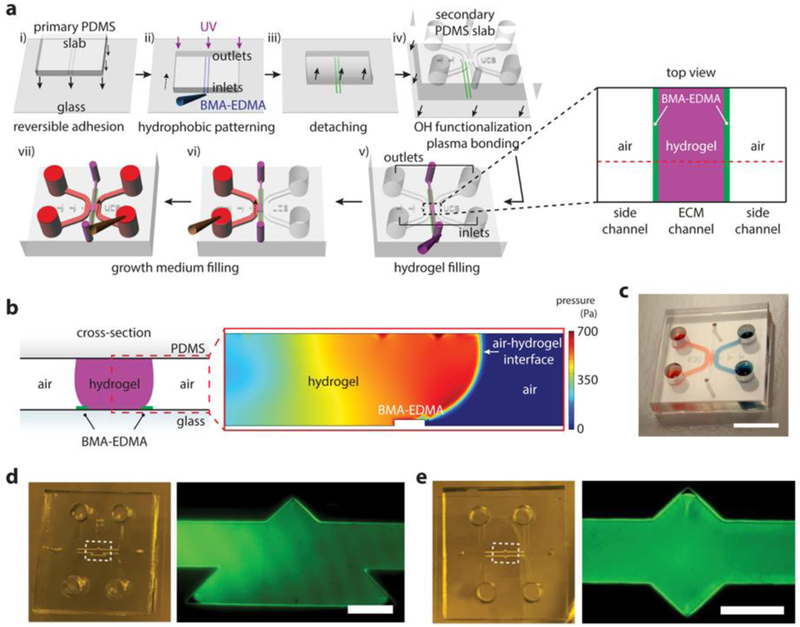
a. Schematic view (not to scale) of hydrophobic butyl methacrylate-ethylene dimethacrylate (BMA-EDMA) patterning and hydrogel filling process. i) Reversible adhesion of the primary poly(dimethylsiloxane) (PDMS) slab to a glass coverslip. ii) Injection of BMA-EDMA solution into the microchannels and UV exposure to polymerize hydrophobic strips on the glass substrate. iii) Detachment of the primary PDMS slab from the glass. iv) Removal of excessive methacrylate groups from the glass and -OH functionalization of the glass. v) Hydrogel filling process for creating a 3D extracellular matrix within a microchannel. Inset shows a schematic top view of the black dotted square. vi-vii) filling growth media after hydrogel gelation. b. (left) Cross-sectional view of the red dotted line from (v) shows 3D hydrogel structure patterned by the BMA-EDMA. (right) COMSOL Multiphysics simulation for the red dotted square in the left schematic shows pressure distribution around the hydrophobic pattern (BMA-EDMA) within the microfluidic channels during the gel filling process, indicating the hydrophobic pattern on the glass substrate is suitable for confining 3D hydrogels. c. A photograph of the gel filled device. Side channels were filled with food dyes. Scale bar, 10 mm. d, e. Demonstration of the versatility of patterning with liquid BMA-EDMA. (left) photograph of the BMA-EDMA patterned device. (right) fluorescence image of the white dotted square in the left image. The ECM channel was filled with 10 µM fluorescein isothiocyanate (FITC)-dextran solution (40 kDa). Scale bar, 250 µm.
To characterize the hydrophobic nature of the BMA-EDMA pattern for 3D hydrogel patterning, we analyzed the pressure distribution around a thin structure of the BMA-EDMA using COMSOL Multiphysics 5.0 (Fig. 1b and Supplementary Fig. 1). The hydrophobicity of the BMA-EDMA structure was the principal factor for precisely confining the hydrogel solution during the gelation process. Since the physical structure of the BMA-EDMA pattern could also affect its water repellency, we created an equivalent structure composed of poly(2-hydroxyethylmethacrylate (pHEMA,; Sigma-Aldrich, USA) and EDMA, and compared pressure distributions within the microchannel using COMSOL Multiphysics 5.0 (Supplementary Fig. 2). The materials used in this study were chosen due to BMA being intrinsically hydrophobic37 and pHEMA being hydrophilic37. The overall internal pressure within a hydrogel solution patterned by the BMA-EDMA was significantly higher than that by the pHEMA-EDMA during the gel filling process, indicating that the hydrophobic BMA-EDMA precisely controlled the position of the liquid, while the internal pressure in the hydrophilic pHEMA-EDMA-based device was sufficient to cause the liquid to flow over the 5-µm pattern, failing to maintain the meniscus of the hydrogel solution at the edge of the pattern (Fig. 1b and Supplementary Fig. 2b).
The apparent hydrophobic characteristics of BMA-EDMA were further investigated by adding water drops continuously onto the inner center of circle-shaped pattern. In contrast to the pHEMA-EDMA pattern, the BMA-EDMA pattern demonstrated superb water repellency (Supplementary Fig. 3). Furthermore, we demonstrated the versatility of liquid patterning BMA-EDMA by creating two different geometric structures (triangular and diamond-shaped), shown in Figure 1d, e. This capability is important for mimicking the 3D structures of different organs.
Patterning BMA-EDMA on a glass surface enables reliable and successful generation of a 3D ECM within a microchannel. Moreover, the structural effect of the overall hydrophobic nature of the BMA-EDMA pattern on the hydrogel filling is negligible for this pattern height (~5 µm).
3.2. Characterizations of the BMA-EDMA photopatterning and hydrogel patterning within the BMA-EDMA patterned microfluidic device
Water contact angles of BMA-EDMA patterns as a function of the amount of photo-initiator in the pre-polymer solution were measured by varying the concentration of the photo-initiator, 2,2-dimethoxy-2-phenylacetophenone (DMPAP). 1wt% of DMPAP was adequate for the highest observed water contact angle (~140°) (Fig. 2a). We also varied the height of the microchannels of the primary PDMS slab to find its optimal range for allowing >95% (resulted pattern height: 5 µm; secondary PDMS channel height: 150 µm) of the interaction area among cells, ECM, and chemicals. The lowest DMPAP concentration (1wt%) and 70 µm high channel conditions yielded the thinnest pattern, while still maintaining hydrophobic characteristics (Fig. 2b). The ultrathin pattern that results is due to the fact that the pre-polymer solution consists mainly of fast evaporating solvents and that the hydrophobic pattern grows from the methacrylate glass surface. As the hydrogel fills the ECM channel, the water-based hydrogel pre-polymer solution contacts glass (bottom), PDMS (top), and the BMA-EDMA pattern (bottom). Since the BMA-EDMA was patterned onto the glass surface, the difference in water contact angle between those surfaces should be sufficient to keep the hydrogel solution within the designated area during the gelation process. Figure 2c shows that the average water contact angle of the BMA-EDMA was significantly greater than that of the glass surface. The intrinsic water contact angle of BMA is known to be hydrophobic39,40. Furthermore, its microscopic structure is also highly porous (Fig. 2d). These two characteristics contribute significantly to the overall water contact angle of the BMA-EDMA pattern. In contrast, although highly porous, pHEMA-EDMA is hydrophilic. As we show, the overall water contact angle of the pHEMA-EDMA pattern was less than that of the BMA-EDMA (Supplementary Fig. 4)40. Scanning electron microscopy (SEM) images reveal the porosity of BMA-EDMA and pHEMA-EDMA, which varies with the amount of solvent (1-decanol and 1-decanol/cyclohexanol mixture, respectively) in the pre-polymer solutions (Fig. 2d and Supplementary Fig. 4). The combination of a highly-developed, hydrophobic, and porous BMA-EDMA material allows us to fabricate nearly superhydrophobic liquid-patterning structures (Fig. 2c, d, and Supplementary Figs. 2 and 4)37.
Figure 2. Characterization of BMA-EDMA and hydrogel patterning within the microfluidic device.
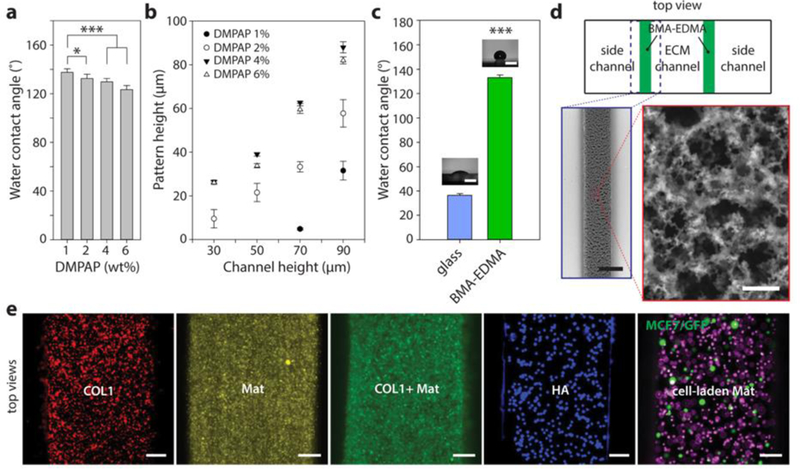
a. Water contact angle (WCA) of hydrophobic pattern as a function of the concentration of photoinitiator, 2,2-dimethoxy-2-phenylacetophenone (DMPAP), in the BMA-EDMA solution. b. Resulting height of polymerized BMA-EDMA patterned using primary PDMS slabs with four different channel heights at four different DMPAP concentrations. n=3. c. Average WCAs of a bare glass coverslip and BMA-EDMA pattern (1% DMPAP, 70 µm channel height for pattern). Images show representative WCA measurements for each condition. Scale bars: 200 µm. ***p<0.001. n=3. d. (counterclockwise from top) Schematic top view of the BMA-EDMA patterned device (not to scale). Scanning electron microscope (SEM) images of the BMA-EDMA pattern show the microporous structure of the pattern which contributes to the hydrophobic property of the pattern. Scale bars: 250 µm (bottom left) and 10 µm (bottom right). All error bars: one standard deviation. e. A variety of hydrogel types, including COL1, MAT, COL1/MAT (1:1 volume ratio), HA, and MCF-7/GFP breast cancer cell-laden MAT were injected into the hydrogel channel of the BMA-EDMA device to demonstrate hydrogel filling. To visualize hydrogels in the device, fluorescent microspheres (5–14 µm; Spherotech, USA) were mixed in each gel solution at a volume ratio of 10 : 1 (hydrogel : DPBS containing fluorescent microsphere (1.0% w/v)). All hydrogels, from the less viscous COL1 (2 mg/mL) to the highly viscous HA (4%), were successfully filled and polymerized without overflowing over the hydrophobic pattern. Scale bars: 100 µm.
Computational simulations (Figure 1b and Supplementary Figure 2a) confirmed that a variety of natural hydrogel pre-polymer solutions, including type I collagen (COL1), Matrigel (MAT), a mixture of COL1 and MAT, hyaluronic acid (HA), and MCF7/GFP human breast cancer cell-laden MAT, are able to fill and remain stable within a BMA-EDMA-confined ECM channel (Fig. 2e).
3.3. 3D breast epithelial cancer cell migration through a fibrous structural extracellular matrix (ECM) and a mixed structure of the fibrous structural ECM and adhesive proteins within the BMA-EDMA device
Cancer cell migration involves multiple, hierarchically-assembled cell types, ECMs, and complex mass transport through the microenvironment. Recapitulating this microenvironment is a crucial component to understanding the fundamental pathophysiology of cancer cell migration in vivo41. However, this complexity is often oversimplified by conventional cell culture assays42,43 or is mismatched by different model species44,45. Here, we demonstrate the 3D interstitial migration of MCF7/GFP cells through either COL1 or COL1/MAT in vitro (Fig. 3). COL1 or COL1/MAT (1:1 ratio) was formed within the ECM channel of a BMA-EDMA device to mimic either a fibrous structural ECM structure or a combination of fibrous structural ECM and adhesive proteins (mostly laminin from MAT), respectively (Fig. 3 b,c). MCF7/GFP cells were initially seeded into the cell channel (Fig. 3a, left). In both cases, growth medium supplemented with 10% FBS was added to the growth medium channel to create an FBS concentration gradient from the growth medium channel (right) through the ECM channel (center) to the cell channel containing 5% FBS (left) (Fig. 3a–c). By day 3 of cell culture, MCF7/GFP cells invaded into the 3D COL1 structure toward the FBS source, indicating that the FBS chemoattractant gradient successfully induced interstitial migration within the COL1 gelled in between hydrophobic stripes (Fig. 3d). Figures 3d and 3e show MCF7/GFP cells migrating individually through COL1. Cells migrated more freely through the COL1 hydrogel than COL1/MAT hydrogel, possibly due to the higher porosity of the structure (Fig. 3). In the previous finding, structural difference in COL1, MAT, and COL1/MAT turned out to be significant, especially for the porosity15. In contrast, cells tended to collectively migrate through the COL1/MAT hydrogel, indicating that the path toward the source was limited by the structure of densely-packed fibrils with MAT; the combined forces of several cells collectively migrating was necessary for movement (Fig. 3f). Furthermore, we found that MCF7/GFP cells could collectively migrate through the COL1/MAT ECM even without leader cells or appropriate matrix metalloproteinases (MMP), which suggests that interstitial migration can occur so long as the minimum pore size is sufficiently large (Fig. 3f).
Figure 3. 3D breast epithelial cancer cell migration through a fibrous structural extracellular matrix (ECM) and a mixture of fibrous structural ECM and adhesive proteins within the BMA-EDMA device.
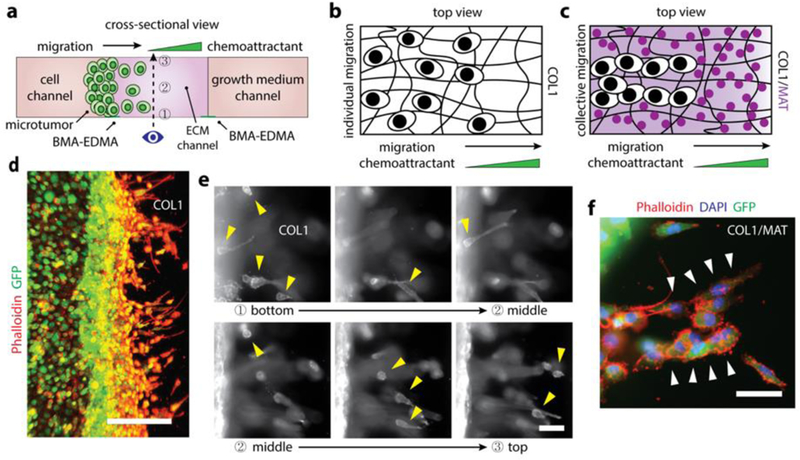
a. Schematic cross-sectional view of the 3D breast epithelial cancer cell migration assay. Two ECM structures, COL1 and COL1/MAT, were formed in the ECM channel (center) created by the BMA-EDMA pattern. MCF-7/GFP cells were seeded in the cell channel (left) to form a 3D microtumor tissue. A complete growth medium containing 5% fetal bovine serum (FBS) was filled into the cell channel after cell attachment, and a complete growth medium containing 10% FBS was filled into the growth medium channel (right). Dotted arrow indicates the imaging axis for the assay. Numbers indicate focal planes corresponding to the images shown in (e), from bottom (1) to top (3). b. An illustration (top view) for the migration behavior of MCF-7/GFP cells within the COL1 fibrous structural ECM. c. Illustrative top view of MCF-7/GFP cells migrating through the COL1/MAT mixture of fibrous structural ECM and adhesive proteins. d. Fluorescent image of the MCF-7/GFP cells (on day 4) migrating through COL1. Actin filaments of the MCF7/GFP cells were immunostained with rhodamine Phalloidin (red) and the cells were stably transfected to express GFP. Scale bar: 300 µm. e. Fluorescent images corresponding to the focal planes in (a), from bottom (1) to top (3). Yellow arrow heads indicate the migrating cells in each focal plane, indicating that the ECM environment created by the BMA-EDMA hydrophobic pattern is truly three-dimensional. f. A fluorescent image shows collectively migrating MCF-7/GFP cells (on day 4) through COL1/MAT as shown in (c), in contrast with COL1 alone, in which cells migrated individually as shown in (b). White arrowheads indicate collectively migrating MCF7/GFP cells. Actin filaments (red) and nuclei (blue) were immunostained with rhodamine Phalloidin and 4’, 6-diamidino-2-phenylindole (DAPI), respectively. Scale bar: 100 µm.
3.4. Generation of a quiescent 3D blood capillary model using BMA-EDMA-based hydrogel patterning
We recently reported a microfluidic cell culture method that enables the formation of a physiological human blood capillary. Using our method, we successfully replicated the structure of an endothelial ECM that consists of 3D COL1 and a basement membrane layer on top of the COL121. Here, we combined the same approach to generate the endothelial ECM with our novel hydrophobic patterning-based platform to increase the interaction/reaction area to >95% between the cells and the 3D ECM/soluble factors (Fig. 4c) while maintaining the quiescent morphology of human microvascular endothelial cells (hMVECs). To create a blood capillary model, hMVECs (Lonza, USA) were seeded into the cell channel, and complete growth medium was added to both side channels (cell channel and growth medium channel) (Fig. 4a). Laminin from the MAT acts as a bridge material between collagen-1 fibers and hMVECs. By day 4 of cell culture in the device, hMVECs formed a perfect 3D-like endothelial monolayer—defects, such as empty areas of the monolayer and unwanted angiogenesis or sprouts, were absent (Fig. 4b).
We also analyzed and compared the diffusion characteristics of our hydrophobic patterning-based device with the more traditional pillar-based platforms using COMSOL Multiphysics 5.0 (Supplementary Fig. 6). In pillar-based platforms, the interaction area between cells and the ECM is largely limited by repeating pillar structures. Because cells continuously consume the vascular endothelial growth factors (VEGFs) that are responsible for their interstitial migration and homeostasis, a VEGF concentration gradient was induced by consumption even in the absence of additional growth factors in the right channels (Supplementary Fig. 6). Importantly, this unwanted gradient is significantly larger in the pillar-based platform than the hydrophobic patterning-based platform, since the surface area available for diffusion per unit volume for the former is smaller than that of the latter. As such, less growth factor enters the cell channel to replenish the growth factor that is consumed (Fig. 4d). In our model, the concentration gradient of 40 kDa VEGF (as a model growth factor) within the COL1 patterned by the PDMS pillars was 3.18× greater than that within COL1 patterned by the BMA-EDMA pattern (Fig. 4d). Interstitial cell migrations could therefore occur even without the addition of growth factors to the opposing side channel, and HUVECs in the pillar-based platform displayed much more aggressive migration toward the source of the growth factors (Fig. 4c–f). In our platform, this unwanted migration was successfully prevented by the larger surface area without compromising design flexibility (Fig. 2c–f). The larger effective interaction area enabled by this thin liquid-patterning component offers a significant improvement over existing microfluidic 3D cell culture models.
3.5. Differentiation characteristics for human neural stem cells (hNSCs) and mesenchymal stem cells (hMSCs) embedded in the 3D ECM created by the BMA-EDMA pattern and comparison between the 3D differentiation vs 2D culture flask-based differentiation
One of the major advantages of our 3D microfluidic cell culture system is that, in addition to generating physiologically-relevant environments, it can also be used to study a variety of biological systems, such as the influence of microenvironments on stem cells. Here, human primary neural stem cells (hNSCs; Gibco, USA) and human mesenchymal stem cells (hMSCs; Lonza, USA) were utilized to demonstrate the feasibility of 3D culturing, differentiation, and analysis of stem cells in micro-scale culture. At the micro-scale, growth factors secreted by stem cells or other neighboring cells can accumulate, creating in vivo-like distributions of the growth factors, whereas soluble factors in a 2D culture flask diffuse through relatively long diffusion lengths that are generally not physiologically relevant [19]. We compared our platform with the conventional method of using a 2D cell culture dish/flask (Fig. 5). For the latter, hNSCs were plated onto a 60-mm cell culture dish (Fig. 5b). For our 3D microfluidic culture platform, hNSCs were initially mixed with MAT and injected into the ECM channel defined by hydrophobic stripes (Fig. 5a). After gelation, both side channels (growth medium channels) were filled with complete growth medium to supply a sufficient amount of nutrients to the hNSCs during culture (Fig. 5a). Importantly, even with the mitogenic growth factors epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF), which are required to maintain the stemness of hNSCs19,46, the hNSCs embedded in the MAT showed significantly lower Nestin (NES) expression when compared to those grown in the 2D cell culture dish. Lower NES expression indicates that the different environmental factors, in particular, the combinational effect of the diffusion length and ECM environment, significantly affect hNSC differentiation characteristics (Fig. 5c). These environmental factors influence hNSC differentiation into neurons (Tuj-1), astrocytes (GFAP), and oligodendrocytes (O4) (Fig. 5c). Interestingly, GFAP expressing cells (astrocytes) dominated in the 3D micro condition even in the presence of hEGF and hFGF, indicating there was a potential difference between 3D micro and 2D macro conditions in the diffusion profiles of growth factors secreted from the hNSCs and/or the differentiated cells, which, in turn, might have affected their differentiation. In the 2D cell culture condition, neuronal differentiation dominated over glial differentiation. However, this trend is reversed in the 3D condition (Fig. 5c), a finding that is consistent with other recent reports19,20,47, indicating that the creation of a 3D microenvironment for the culture of hNSCs as well as incorporation of the effects of a platform’s dimensionality and/or scale in data analysis should be carefully considered in stem cell research.
Figure 5. Differentiation characteristics for human neural stem cells (hNSCs) and mesenchymal stem cells (hMSCs) embedded in the 3D ECM created by the BMA-EDMA pattern and comparison with 2D differentiation.
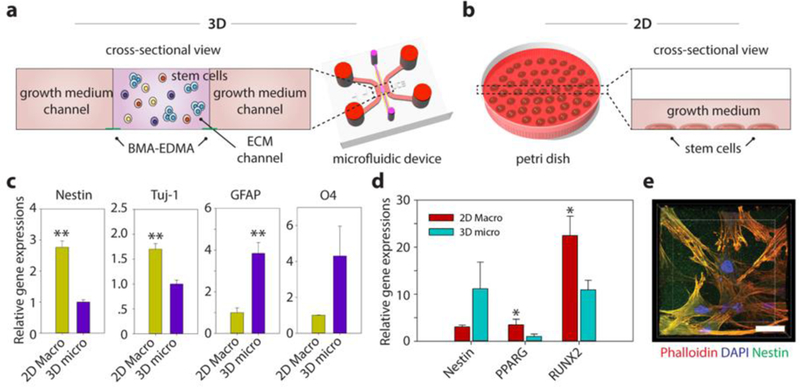
a. Schematic cross-sectional view of the 3D stem cell differentiation assay. A 3D cellular microenvironment was created by placing a mixture of stem cells and pre-gel solution of MAT (final cell density: 2 million cells/mL) within the BMA-EDMA pattern. Both growth medium channels were filled with the appropriate growth media supplemented with growth factors to maintain stemness. b. Illustration depicting 2D stem cell culture in a conventional cell culture dish, for comparison. The normal level for growth media in a culture dish/flask is approximately 1–2 mm. c. Relative gene expressions of hNSCs (on day 3) for Nestin (neural stem cell), neuron-specific Class III ß-tubulin (Tuj-1; neuron), glial fibrillary acidic protein (GFAP; astrocyte), and O4 (oligodendrocyte) in 3D micro (a) and 2D macro (b). Data are shown as the means ± SD (n = 3). **p<0.01. d. Relative gene expressions of hMSCs (on day 3) for Nestin, PPARG (adipocyte), and Runt-related transcription factors 2 (RUNX2; osteocyte) in 3D micro and 2D macro conditions. The differentiation fate of hMSCs was found to be highly dependent on the culture dimension/geometry. Cells cultured in 3D showed lower expression of RUNX2, indicating that the 3D microenvironmental MAT effectively maintained their stemness, while unwanted osteocyte differentiation could not be inhibited in 2D culture. Data are shown as the means ± SD (n = 3). p<0.05. e. A confocal microscope image shows the hMSCs cultured within MAT in the BMA-EDMA device. Actin filaments and nuclei of hMSCs were immunostained with rhodamine Phalloidin (red) and DAPI, respectively. Nestin expression (green) was monitored using a rabbit anti-Nestin antibody and goat anti-Rabbit Alexa Fluor 488 antibody. Scale bar: 20 µm. All error bars: one standard deviation.
Similarly, hMSCs were cultured with the aforementioned culture systems to demonstrate the versatility of our platform for studying 3D stem cell differentiation (Fig. 5d,e). On a 2D surface, hMSCs tended to differentiate into RUNX2-positive cells, losing their stemness even when supplemented with mitogenic growth factors because of the stiff environment, agreeing with previous findings48–50 (Fig. 5d). Additionally, stemness of hMSCs cultured within our platform was better maintained due to the relatively low stiffness of the 3D MAT microenvironment (Fig. 5d,e).
Even though the experimental approaches shown here used simplified conditions, the results demonstrate that our platform has potential applications in generating a physiologically-relevant 3D stem cell microenvironment on chip, while the biocompatibility is comparable to other conventional cell culture platforms36,37. We anticipate that the method developed here will be beneficial for future stem cell research through expansion of our current model and integration of more sophisticated conditions and experiments, such as how differentiation might depend on cell culture period, cell passage, ECM composition, co-culture conditions, cell seeding density, and soluble and mechanical factors within the ECM.
4. Conclusions
In this report, we describe a novel technology to establish 3D cell culture utilizing hydrophobic patterning. We characterized the hydrophobicity of the pattern through water contact angle measurements and optimized the concentration of the photoinitiator, DMPAP. We found that hydrophobic BMA-EDMA patterns successfully confined hydrogels, such as COL1, MAT, COL/MAT, HA, and cell-laden MAT, within a 3D microfluidic channel. In our first application, we observed MCF7/GFP, a human breast epithelial cancer cell line, individually migrate in three dimensions through COL1, as opposed to the collective migration that we observed when the cells moved through COL1/MAT. Furthermore, migrating cells were evenly distributed across the entire surface area of the matrix. In a separate application, we generated a quiescent 3D blood capillary model, which demonstrates that human microvascular endothelial cells can be cultured on our device without any unwanted migration or sprouting/angiogenesis. We also analyzed endothelial cell response to two diffusion profiles generated by two distinctive geometric arrangements. We found that the BMA-EDMA pattern significantly reduced unwanted migration that is commonly found with pillar-based devices. This is a step toward a fully physiologically-accurate platform. Most endothelial/epithelial structures have a similar spatial composition that consists of the lumen space, 3D ECM, and nearby growth factors. By presenting the feasibility of constructing the 3D endothelium with a greater interaction/reaction area, we anticipate that our method could be employed in many 3D endothelium/epithelium applications, such as permeability and angiogenesis assays. As a final application, we demonstrated that dissimilar differentiation characteristics arose for both human neural stem cells and human mesenchymal stem cells when cultured in a 3D vs. 2D environment, providing evidence that the dimensionality of the culture platform plays an important role in stem cell differentiation fates. Our hydrophobic patterning-based microfluidic device is straightforward to implement and use and flexible enough to be used in diverse applications for many cell types.
Supplementary Material
5. Acknowledgements
We thank O. Scheideler for helpful discussions and O. Scheideler, T. Carey, and B. Li for critical reading of this manuscript. This research was funded by National Science Foundation 1509921 (L.L.S), National Institute of Health 1R21CA182375–01A1 (L.L.S), and the Baker Fellows Program (L.L.S). J.K. was funded through the Jungsong Fellowship.
Footnotes
Competing interests
S.H., J.K., R.L., A.M., V.K., and K.L declare that they have no competing interests. L. L. Sohn is a founder, and holds equity in, Nodexus, Inc.
References
- 1.Rimann M & Graf-Hausner U Synthetic 3D multicellular systems for drug development. Current opinion in biotechnology 23, 803–809 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Pampaloni F & Stelzer EH Three-dimensional cell cultures in toxicology. Biotechnology and Genetic Engineering Reviews 26, 117–138 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Harunaga JS & Yamada KM Cell-matrix adhesions in 3D. Matrix Biology 30, 363–368 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edmondson R, Broglie JJ, Adcock AF & Yang L Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay and drug development technologies 12, 207–218 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haycock JW in 3D cell culture 1–15 (Springer, 2011). [DOI] [PubMed] [Google Scholar]
- 6.Koledova Z 3D Cell Culture: Methods and Protocols (Springer, 2017). [Google Scholar]
- 7.Bhise NS et al. Organ-on-a-chip platforms for studying drug delivery systems. Journal of Controlled Release 190, 82–93 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang B et al. Biodegradable scaffold with built-in vasculature for organ-on-a-chip engineering and direct surgical anastomosis. Nature materials 15, 669 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huh D et al. Reconstituting organ-level lung functions on a chip. Science 328, 1662–1668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee JB & Sung JH Organ‐on‐a‐chip technology and microfluidic whole‐body models for pharmacokinetic drug toxicity screening. Biotechnology journal 8, 1258–1266 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Caplin JD, Granados NG, James MR, Montazami R & Hashemi N Microfluidic organ‐on‐a‐chip technology for advancement of drug development and toxicology. Advanced healthcare materials 4, 1426–1450 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Kim S & Takayama S Organ-on-a-chip and the kidney. Kidney research and clinical practice 34, 165–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Selimović Š, Dokmeci MR & Khademhosseini A Organs-on-a-chip for drug discovery. Current opinion in pharmacology 13, 829–833 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Abaci HE & Shuler ML Human-on-a-chip design strategies and principles for physiologically based pharmacokinetics/pharmacodynamics modeling. Integrative Biology 7, 383–391 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shin Y et al. Microfluidic assay for simultaneous culture of multiple cell types on surfaces or within hydrogels. Nature protocols 7, 1247 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shin Y et al. In vitro 3D collective sprouting angiogenesis under orchestrated ANG-1 and VEGF gradients. Lab on a chip 11, 2175–2181 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Jeong GS et al. Sprouting angiogenesis under a chemical gradient regulated by interactions with an endothelial monolayer in a microfluidic platform. Analytical chemistry 83, 8454–8459 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Han S et al. A versatile assay for monitoring in vivo-like transendothelial migration of neutrophils. Lab on a chip 12, 3861–3865 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Han S et al. Three-dimensional extracellular matrix-mediated neural stem cell differentiation in a microfluidic device. Lab on a Chip 12, 2305–2308 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Shin Y et al. Reconstituting vascular microenvironment of neural stem cell niche in three‐dimensional extracellular matrix. Advanced healthcare materials 3, 1457–1464 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Han S et al. Constructive remodeling of a synthetic endothelial extracellular matrix. Scientific reports 5, 18290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HJ & Ingber DE Gut-on-a-Chip microenvironment induces human intestinal cells to undergo villus differentiation. Integrative Biology 5, 1130–1140 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Bhatia SN & Ingber DE Microfluidic organs-on-chips. Nature biotechnology 32, 760 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Brown JA et al. Recreating blood-brain barrier physiology and structure on chip: A novel neurovascular microfluidic bioreactor. Biomicrofluidics 9, 054124 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang YI, Abaci HE & Shuler ML Microfluidic blood–brain barrier model provides in vivo‐like barrier properties for drug permeability screening. Biotechnology and bioengineering 114, 184–194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang B & Radisic M Organ-on-a-chip devices advance to market. Lab on a Chip 17, 2395–2420 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Booth R & Kim H Characterization of a microfluidic in vitro model of the blood-brain barrier (μBBB). Lab on a chip 12, 1784–1792 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Primiceri E et al. Automatic transwell assay by an EIS cell chip to monitor cell migration. Lab on a Chip 11, 4081–4086 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Lim SH & Pavesi A Materials and Methods Culture Cells in Media Channels. BioTechniques 62, 132–133 (2017). [Google Scholar]
- 30.Ruprecht R, Gietzelt T, Müller K, Piotter V & Haußelt J Injection molding of microstructured components from plastics, metals and ceramics. Microsystem Technologies 8, 351–358 (2002). [Google Scholar]
- 31.Trietsch SJ et al. Membrane-free culture and real-time barrier integrity assessment of perfused intestinal epithelium tubes. Nature communications 8, 262 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vulto P et al. Phaseguides: a paradigm shift in microfluidic priming and emptying. Lab on a Chip 11, 1596–1602 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Trietsch SJ, Israëls GD, Joore J, Hankemeier T & Vulto P Microfluidic titer plate for stratified 3D cell culture. Lab on a chip 13, 3548–3554 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Li J et al. Hydrophobic liquid-infused porous polymer surfaces for antibacterial applications. ACS applied materials & interfaces 5, 6704–6711 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Li JS, Ueda E, Nallapaneni A, Li LX & Levkin PA Printable superhydrophilic–superhydrophobic micropatterns based on supported lipid layers. Langmuir 28, 8286–8291 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Levkin PA, Svec F & Fréchet JM Porous polymer coatings: a versatile approach to superhydrophobic surfaces. Advanced functional materials 19, 1993–1998 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geyer FL, Ueda E, Liebel U, Grau N & Levkin PA Superhydrophobic–Superhydrophilic Micropatterning: Towards Genome‐on‐a‐Chip Cell Microarrays. Angewandte Chemie International Edition 50, 8424–8427 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Chuah YJ et al. Simple surface engineering of polydimethylsiloxane with polydopamine for stabilized mesenchymal stem cell adhesion and multipotency. Scientific reports 5, 18162 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu L, Ercan B, Sun L, Ziemer KS & Webster TJ Understanding the role of polymer surface nanoscale topography on inhibiting bacteria adhesion and growth. ACS Biomaterials Science & Engineering 2, 122–130 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Zhang J Switchable and responsive surfaces and materials for biomedical applications(Elsevier, 2014). [Google Scholar]
- 41.Ma Y, Cao X, Feng X, Ma Y & Zou H Fabrication of super-hydrophobic film from PMMA with intrinsic water contact angle below 90. Polymer 48, 7455–7460 (2007). [Google Scholar]
- 42.Zhang YS, Zhang Y-N & Zhang W Cancer-on-a-chip systems at the frontier of nanomedicine. Drug discovery today 22, 1392–1399 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phillips RM, Loadman P & Cronin B Evaluation of a novel in vitro assay for assessing drug penetration into avascular regions of tumours. British journal of cancer 77, 2112 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kramer N et al. In vitro cell migration and invasion assays. Mutation Research/Reviews in Mutation Research 752, 10–24 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Kota J et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 137, 1005–1017 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arap W, Pasqualini R & Ruoslahti E Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 279, 377–380 (1998). [DOI] [PubMed] [Google Scholar]
- 47.Gage FH Mammalian neural stem cells. Science 287, 1433–1438 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Yang K et al. A microfluidic array for quantitative analysis of human neural stem cell self-renewal and differentiation in three-dimensional hypoxic microenvironment. Biomaterials 34, 6607–6614 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Engler AJ, Sen S, Sweeney HL & Discher DE Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Pek YS, Wan AC & Ying JY The effect of matrix stiffness on mesenchymal stem cell differentiation in a 3D thixotropic gel. Biomaterials 31, 385–391 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.