

Abstract
Objective.
Agonism of the protease activated receptor (PAR) 1 by activated protein C (APC) provides neuroprotection and vasculoprotection in experimental neuro-injury models. The pleiotropic PAR1 agonist, 3K3A-APC, reduces neurologic injury and promotes vascular integrity; 3K3A-APC proved safe in human volunteers. We performed a randomized, controlled, blinded, trial to determine the maximally tolerated dose (MTD) of 3K3A-APC in ischemic stroke patients.
Methods.
The NeuroNEXT trial RHAPSODY used a novel continual reassessment method to determine the MTD using tiers of 120, 240, 360 and 540 μg/kg 3K3A-APC. After intravenous tissue plasminogen activator, intraarterial mechanical thrombectomy, or both, patients were randomized to one of the four doses or placebo. Vasculoprotection was assessed as microbleed and intracranial hemorrhage (ICH) rates.
Results.
Between January 2015 and July 2017 we treated 110 patients. Demographics resembled a typical stroke population. The MTD was the highest dose 3K3A-APC tested, 540 μg/kg, with an estimated toxicity rate of 7%. There was no difference in prespecified ICH rates. In exploratory analyses, 3K3A-APC reduced ICH rates compared to placebo from 86.5% to 67.4% in the combined treatment arms (p=0.046), and total hemorrhage volume from an average of 2.1±5.8 mL in placebo to 0.8±2.1 mL in the combined treatment arms (p=0.066).
Interpretation.
RHAPSODY is the first trial of a neuroprotectant for acute ischemic stroke in a trial design allowing thrombectomy, thrombolysis, or both. The MTD was 540 μg/kg for the PAR1 active cytoprotectant 3K3A-APC. A trend toward lower hemorrhage rate in an exploratory analysis requires confirmation.
INTRODUCTION
Activated protein C (APC) is a blood protease with anticoagulant and cell-signaling activities mediated via the protease-activated receptor 1 (PAR1)1. APC and analogs with cell-signaling cytoprotective activities provided beneficial effects in preclinical models of central nervous system (CNS) disorders including stroke, brain trauma, multiple sclerosis (MS), and amyotrophic lateral sclerosis (ALS); systemic disorders such as sepsis, ischemic and reperfusion injury of heart and kidney; liver, pulmonary, kidney and gastrointestinal inflammation; and diabetes and lethal whole-body radiation1, 2. To decrease bleeding risks, the native APC structure was mutated: 3 lysines were replaced by alanines (K191A-K192A-K193A), removing >90% of the anticoagulant activity while preserving multiple cell signaling activities3–5. The resultant molecule, 3K3A-APC, exhibits powerful cytoprotection in rodent stroke models6–8, brain trauma9, ALS10 and MS11. 3K3A-APC acts via the transmembrane G- protein coupled PAR1 to protect neurons, brain endothelial cells and blood brain barrier (BBB) directly in vivo and in vitro8, 12–14 and it also reduces ‘reperfusion injury’ via multiple vasculoprotective effects, 21. 3K3A-APC has satisfied 10/10 of the Stroke Treatment Academic Industry Roundtable (STAIR) suggestions for pre-clinical drug safety and efficacy assessment2, and has an established safety and pharmacokinetic profile in human volunteers15, 16.
The combination of 3K3A-APC with recanalization reduced post-reperfusion hemorrhages in a preclinical model using recombinant tissue plasminogen activator (rt-PA) after embolic stroke12. Additional studies in spontaneously hypertensive rats and aged female mice showed that 3K3A-APC and rt-PA treatment reduced infarction and that 3K3A-APC extended the therapeutic window of t-PA 8. When rt-PA was withheld for 4 hours after ischemia onset it provided no benefit but introduced bleeding; 3K3A-APC provided benefit in the absence of rt-PA, showed the same benefit when given after the ineffective rt-PA, and eliminated rt-PA-associated bleeding.
The NeuroNEXT trial NN104 (RHAPSODY) was a randomized, controlled, blinded dose-escalation safety trial for 3K3A-APC. The primary objective of this Phase 2A trial was to evaluate the safety of ascending intravenous (IV) doses of 3K3A-APC in adult patients presenting with acute ischemic stroke who were eligible for thrombolysis, thrombectomy, or both. There were two secondary objectives: to determine the pharmacokinetic parameters of 3K3A-APC in stroke patients (to be reported elsewhere), and to seek evidence of vasculoprotection, measured as reduced hemorrhage frequency or severity attributable to 3K3A-APC treatment during recanalization therapy.
METHODS
The RHAPSODY trial (A multi-center, Phase 2 trial using a continual reassessment method to determine the safety and tolerability of 3K3A-APC, a Recombinant Variant of Human APC, in combination with t-PA, mechanical thrombectomy or both in moderate to severe acute ischemic stroke, NCT02222714) protocol has been published17. The final version of the RHAPSODY protocol is available from the authors. We conducted a multicenter, prospective, randomized, controlled, double-blinded, Phase 2a trial to evaluate the safety, pharmacokinetics and preliminary efficacy of 3K3A-APC following t-PA or mechanical thrombectomy or both in participants with moderate to severe acute ischemic stroke. Subjects who signed consent (or whose representatives provided surrogate consent) and met all inclusion and exclusion criteria were randomized into the trial using the NeuroNEXT Interactive Web Response System (IWRS). Twenty-two cohorts of four trial participants each were randomized, and each cohort included 3 participants randomized to the study drug dose chosen for that cohort and 1 participant randomized to placebo. Trial inclusion/exclusion criteria are shown in Table One. Randomized subjects who experienced significant neurologic improvement (defined as an NIHSS score < 5) prior to receipt of study drug—‘Early Responders’—and other randomized subjects who did not receive study drug for any reason, were not included in the study, and were replaced in the randomization sequence and CRM cohort. Any subject who received any amount of study drug or placebo was considered “enrolled” and followed to the end of the trial. In response to reported efficacy of intra-arterial thrombectomy (IAT), the protocol was amended on May 1, 2015 to include patients undergoing IAT within 6 hours of symptom onset17. Additional changes extended the upper time window for administration of investigational drug from 90 to 120 minutes; extended the upper age range from 80 to 90 years; and broadened the NIHSS range to >5 (from 7–20 previously)18. Criteria specific to IAT were also added: onset time to arterial puncture time < 6 hours and a baseline CT excluding large core infarctions.
Table One. Inclusion/Exclusion Criteria Used in RHAPSODY.
This table shows an abbreviated version of the inclusion and exclusion criteria. A full version is available from the authors.
| Inclusion | Exclusion |
|---|---|
|
|
The primary outcome of this trial was to establish a maximum tolerated dose (MTD)19 of the study drug using a modified version of the CRM. We defined the MTD as the highest study dose with an estimated dose limiting toxicity (DLT) rate of 10% or less. We assessed for DLTs until 48 hours following the last study drug dose. The pre-defined DLTs included: symptomatic intracerebral cerebral hemorrhage (sICH), defined as blood present on CT or MRI brain image associated with neuroworsening (4 or more point increase on the NIHSS not due to iatrogenic causes such as sedation) that in the opinion of the investigator represented a clinically significant change attributable to the hemorrhage (assumed background rate of 3–6%20–24); an activated partial thromboplastin time (aPTT) at 1 hour after treatment that reached 2x the local upper limit of normal (ULN) defined; findings that meet all three components of Hy’s Law25, 26 ; any other bleeding event classified as serious by the site investigator, or any bleeding that required more than 2 units of packed red blood cells over any two consecutive days; any CTCAE v4.03 Grade 3 laboratory value27 that, in the opinion of the investigator, was related to treatment; or any adverse event that, in the opinion of the investigator, was related to treatment and led to cessation of further dosing. Subarachnoid hemorrhage in subjects who received IAT was NOT considered a DLT.
In a CRM approach, dose cohorts are filled sequentially, with a dose-blinded safety review after each cohort. Based on pre-clinical work and a pilot Phase 1 trial, we selected 4 dose levels of 3K3A-APC to test: 120, 240, 360 and 540 μg/kg. To initiate the CRM process, the following DLT frequencies were pre-defined in each dose tier: 120—5%, 240—7%, 360—9%, and 540—11%. The initial patient cohort was started at the lowest dose level (120 μg/kg). From there, dose escalations were driven by the CRM. After each cohort of 4 patients (1 placebo and 3 treated) was filled, an Internal Medical Monitor (IMM) blinded to dose level reviewed the cohort for adverse events that could be considered a DLT. The IMM or site investigator (also blinded to dose) referred such events to a Safety Review Committee (SRC) who reviewed each proposed event against the protocol definitions of DLT. During the SRC safety review period, the interactive randomization system assigned patients to placebo only, to maintain trial momentum and avoid frequent trial halts and re-starts. Once the blinded safety review was completed, confirmed DLTs were reported to the statistical coordinating center and the CRM re-estimated the DLT rate so that a dose adjustment could be made: increase, decrease or hold. Sites were not informed when the trial was undergoing safety review, so as to maintain trial enthusiasm and momentum. The new dose level was added to the randomization system and the next cohort was opened, again without the sites knowing of the change. After the final group of subjects was enrolled, the final MTD would be the highest dose with estimated toxicity probability ≤10%. The study was to stop once the maximum number of cohorts (22) was observed or one of the pre-specified safety or efficacy stopping guidelines was triggered.
3K3A-APC was administered as a 100-mL intravenous infusion over 15 minutes every 12 hours (± 1 hour) for 5 doses. Drug or visually indistinguishable placebo was prepared by an unblinded pharmacist; all treatment personnel were unaware of the dose and group assignment. Study drug (3K3A-APC or placebo) began no sooner than 30 minutes after the end of the rt-PA infusion, for safety reasons, and no later than 120 minutes following completion of rt-PA infusion or initiation of mechanical thrombectomy (skin puncture), whichever occurred sooner. Within these time limits, the investigators were encouraged to begin infusion as soon as possible after consent. Participants were considered confirmed randomizations, i.e., part of the intent-to-treat (ITT) cohort, only after they received any amount of study drug. Thus, subjects who became ineligible (e.g., rapid responders whose NIHSS dropped to <5) between randomization and initiation of study drug were to be removed from the trial and replaced. Detailed brain imaging was required prior to enrollment but perfusion imaging was not used to select patients for trial enrollment.
Safety evaluations were collected through Day 7 following stroke onset. Subjects were evaluated for DLTs from administration of the first dose to 48 hours following the last dose of treatment. Subjects were seen for assessments on Days 7, 14, 30 and 90. MRI scans, using a common protocol that included gradient echo (GRE) or susceptibility weighted image (SWI) sequences, were obtained at 7, 30 and 90 days after stroke. As with all trials in the NeuroNEXT network, a central Institutional Review Board approved this trial and the consent form. Each trial site IRB reviewed and accepted the cIRB approval and consent form. Informed consent was obtained from every patient, or an authorized caregiver.
Intracerebral hemorrhage volume—including hematomas, petechiae, and cerebral microbleeds (CMB)—was quantified from susceptibility sensitive sequences directly by a central analyst blind to treatment assignment. We used the accepted definition of CMB28. The method used to quantify brain hemorrhage is illustrated in Figure 1. Using Day 30 MRI as the outcome, we considered a scan “positive for hemorrhage” (including hematoma, petechiae, CMB) if the corresponding total hematoma volume was larger than 0.06 mL (5 mm diameter). This value was chosen because it corresponds to the definition of one microbleed and we reasoned that our threshold for detection should be set to miss no more than one CMB. Due to the potential for artifact in the semi-automated image analysis (e.g., calcium, vein), we also performed a sensitivity analysis for the presence of hemorrhage using several volume thresholds for the positive identification of hemorrhage, using the equivalent of 0, 1, 2, or 3 microbleeds (0, 0.06, 0.5, and 1.8ml respectively).
Figure One:
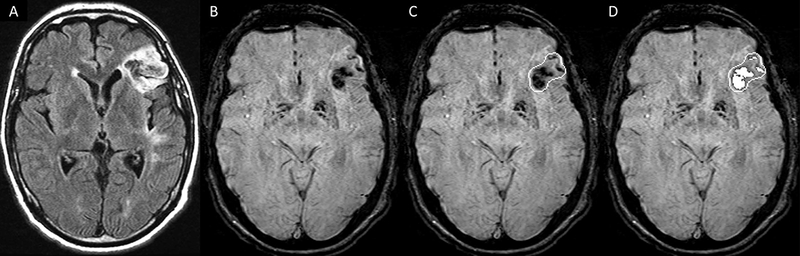
Image Analysis Method for Hemorrhage Volume Quantification. The analyst (unaware of treatment assignment) identified the infarct region using FLAIR (A), then reviewed the susceptibility sequence (B). An object was drawn around abnormal findings (C), and a threshold was applied within the object to outline any hemorrhage (D). The number of pixels lower than the threshold was produced through SPIN software and converted to areas (Spintech Inc., Bingham Farms MI, USA).
The ischemic lesion and infarction territories were identified on MRI as increased signal intensity in various regions of white matter and grey matter. We labeled the lesion and calculated volume using ITK-SNAP version 3.6 based on signal intensity as well as considering neuroanatomical features seen on T2 weighted MRI. We first labeled the lesions in the axial plane as our MRI data were acquired axially and the best resolution was appreciated on this plane. We refined the label shape through the other views (coronal, sagittal) and finally performed volumetry on the primary lesion.
Statistical Analysis plan
Primary endpoint:
For this implementation of a CRM, we sought a single-parameter dose-response model , which is probability of toxicity at dose for some unknown parameter, a, which defines the shape of the dose-toxicity function29. For this study, a hyperbolic tangent dose-response model was used, in which
After each group of subjects was treated, and their status observed as to whether or not each had a DLT, the posterior mean of a was calculated. Based upon that estimate, , the highest dose with estimated toxicity probability, less than or equal to 10% was allocated to the next group of subjects. If this dose was more than one level higher than the previous dose allocated, the dose was to be increased one level only. After the final group of subjects was enrolled, the final MTD was to be defined as the highest dose with estimated toxicity probability less than or equal to the target toxicity level of 10%. The study was to stop once either the maximum number of cohorts (22) was observed or pre-specified safety or efficacy stopping guidelines had been achieved per the study protocol.
Adverse events:
Adverse event data was summarized based on the MedDRA coding system. Data was summarized within MedDRA preferred terms and overall as percent of subjects within treatment groups (both combined treatment group and individual dose levels) that experienced an event. Adverse events were also summarized according to whether the independent medical monitor deemed the event related to study drug and whether the event was expected. Fisher’s exact test was used to compare the percent of subjects with a given event between treatment groups.
Hemorrhage incidence at Day 30:
Analysis of hemorrhage incidence was based on the results of the Day 30 MRI scans. Subjects were considered to have had a hemorrhage if the bleeding volume was greater than 0.06 mL. The percent of subjects that experienced a hemorrhage was tabulated by combined treatment group and also by dose groups. The Pearson chi-square test was used to compare the rates of hemorrhage between the combined treatment group and the placebo group. If any of the expected cell counts were too small to justify the use of the Pearson chi-square test, Fisher’s exact test was used. Additional sensitivity analyses using alternative volume thresholds of > 0 mL, > 0.5 mL, and > 1.8 mL to determine hemorrhage rates were also performed.
Microbleed incidence at Day 30:
The rates of microbleeding within the infarct zone and outside the infarct zone at Day 30 were analyzed separately to compare hemorrhage rates between the two groups. Subjects were considered positive if they experienced one or more microbleeds.
Modified Rankin score at Day 90:
The modified Rankin score was analyzed by comparing the percent of subjects with a score of zero or one between treatment groups. Comparisons were performed using the same approach to compare hemorrhage rates between the two groups.
Barthel index at Day 90:
The Barthel index was analyzed by comparing the percent of subjects with a score of 90 or larger between treatment groups. Comparisons were performed using the same approach to compare hemorrhage rates between the two groups.
RESULTS.
Patients were enrolled between January 2015 and April 2017. Due to the time-compressed nature of acute stroke clinical trials, we screened 2814 patients, obtained consent from 130 patients, and we enrolled 110 patients into the protocol, (Figure 2). Of the 20 consented/not-enrolled patients, 1 patient was found to be ineligible prior to first dose, 7 became clinically unstable, 7 did not receive study drug before expiration of the enrollment window, and 5 cleared symptoms to NIHSS< 5. We filled all planned 22 cohorts of 4 patients, and enrolled 22 additional placebo patients during SRC review periods. Of the 110 enrolled subjects, 44 were randomized to placebo and 66 to 3K3A-APC. To maintain trial enthusiasm and enrollment momentum, the trial remained open during safety reviews—although sites were unaware—thus, more placebo patients were enrolled than would have happened by a strict enrollment ratio. The primary analysis—a Modified Intent-to-Treat—included all subjects who were confirmed randomizations (received at least one dose of study drug) and either: received two or more doses of study drug or suffered a confirmed DLT. For the secondary analyses, we included all patients with an evaluable Day 30 MRI scan. Demographics of the trial groups are shown in Table 2. The distribution of risk factors traditionally associated with adverse events after stroke were relatively well-balanced between placebo and 3K3A-APC treated patients.
Figure Two:
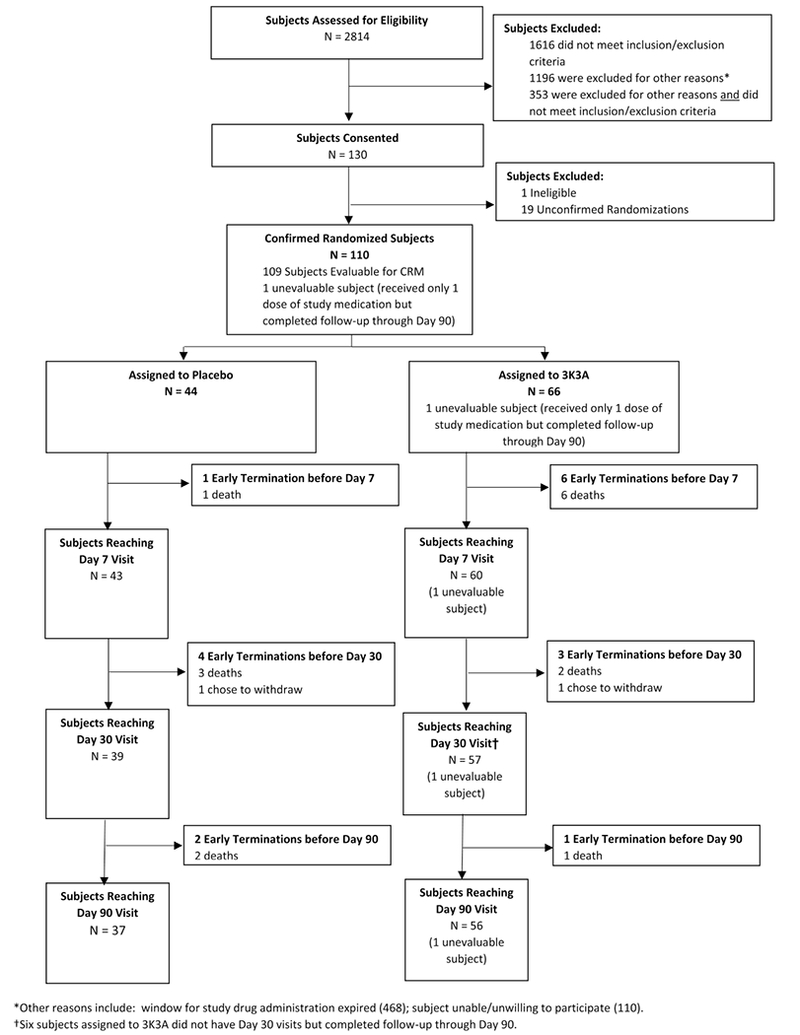
CONSORT Diagram Showing Patients Screened and Then Ultimately Enrolled. Due to the time-compressed nature of acute stroke clinical trials, we screened 2814 patients, obtained consent from 130 patients, and we enrolled 110 patients into the protocol. Of the 20 consented/not-enrolled patients, 1 patient was found to be ineligible prior to first dose, 7 became clinically unstable, 7 did not receive drug before expiration of the enrollment window, and 5 cleared symptoms to NIHSS< 5. These patients were replaced in the randomization scheme. The primary analysis—a Modified Intent-to-Treat—included all subjects who were confirmed randomizations (n=110).
Table Two. Demographic Variables Among 110 Enrolled Patients.
Patients treated with placebo are compared against patients treated with any dose of 3K3A-APC. Likely due to small sample sizes, there were no differences in the distribution of any variable among the 5 dose tiers of 3K3A-APC so these were combined. Continuous variables are shown with standard deviation. * Median (IQR) NIHSS are shown due skewed nature of the observed distributions for these variables. Significance testing was not done due to the small sample sizes and exploratory intent of the trial.
| Placebo (n = 44) |
3K3A-APC (n = 66) |
|
|---|---|---|
| Males | 24 (55%) | 29 (44%) |
| Caucasian | 36 (82%) | 52 (79%) |
| Hispanic/Latino | 7 (16%) | 4 (6%) |
| Age | 64 (12.0) | 64 (15.2) |
| Weight (kg) | 84 (18.0) | 84 (19.1) |
| Platelet Count ≥ 100K | 42 (96%) | 65 (99%) |
| History - Diabetes | 18 (41%) | 18 (27%) |
| History - Hypertension | 33 (75%) | 52 (79%) |
| NIHSS Prior to Recanalization | 13.5 (5 – 30) | 13 (5 – 30) |
| NIHSS Eligibility | 11.5 (5 – 28) | 12 (5 – 40) |
| Modified Rankin - 0 - 1 - ≥ 2 |
41 (93%) 3 (7%) 0 (0%) |
58 (88%) 8 (12%) 0 (0%) |
| Recanalization Therapy - IV t-PA Only - IAT Only - IV t-PA and IAT |
24 (55%) 2 (5%) 18 (41%) |
35 (53%) 3 (5%) 28 (42%) |
| Stroke Onset to First Therapy - Time to t-PA Initiation - Time to First Skin Puncture - Time to t-PA Initiation or First Skin Puncture |
2:07 (1:01) 1:59 (0:44) 5:39 (0:21) 1:53 (0:35) |
2:13 (0:58) 2:16 (0:49) 3:31 (1:37) 2:02 (1:00) |
The primary outcome—the highest dose associated with an estimated toxicity less than 10%–was the 540 μg/kg tier, with an estimated toxicity (DLT rate) of 7%. The final actual DLT rates are shown in Table 3 for all dose tiers compared to placebo. Specifically, DLTs occurred in 4 (9%) placebo treated patients and in 3 (5%) 3K3A-APC treated patients. The CRM model estimated the DLT frequencies in each group: 3% for 120 μg/kg, 4% for 240 μg/kg, 5% for 360 μg/kg and 7% for the 540 μg/kg. Thus, the final MTD estimated in the trial is 540 μg/kg.
Table Three. Adverse events by dose tier.
Patients were enrolled non-sequentially into one of 4 doses of 3K3A-APC or placebo using an implementation of a CRM. Every cohort included 1 placebo and 3 treated patients who all received the dose assigned to that cohort. Extra placebo patients were enrolled during trial pauses for safety review. After filling all 22 planned cohorts, there were 44 placebo-treated and 66 3K3A-APC treated patients assigned to the dose levels shown. Serious adverse events (SAEs) are summarized by dose level as number (percentage) of subjects with any SAE. SAEs were considered ‘related’ if the IMM categorized the event as probably or possibly related to treatment. An event was ‘unanticipated’ if the event was not previously reported or associated with stroke or 3K3A-APC treatment. Neuro-worsening was defined as a 4-or-more point worsening in the NIHSS on 2 occasions not attributable to iatrogenic events such as administration of sedation. Dose limiting toxicities were defined in the protocol (see Methods) and used to drive the CRM. The distribution of events was balanced across all dose tiers.
| N (%) | Total Treated v Total Placebo | ||||||
|---|---|---|---|---|---|---|---|
| Dose (μg/kg) N=number treated |
120 N=12 |
240 N=16 |
360 N=6 |
540 N=9 |
3K3A-APC N=43 |
Placebo N=37 |
p-value |
| Dose Limiting Toxicities (Primary Outcome) | 0 (0) | 2 (8) | 1 (8) | 0 (0) | 3 (5) | 4 (9) | 0.43 |
| Any AE Asymptomatic ICH Symptomatic ICH Headache |
12 (80) 2 (13) 0 (0) 1 (7) |
20 (83) 4 (17) 3 (13) 6 (25) |
9 (75) 4 (33) 0 (0) 2 (7) |
15 (100) 5 (33) 2 (13) 3 (20) |
56 (85) 15 (23) 5 (8) 12 (18) |
41 (93) 17 (39) 2 (5) 10 (23) |
0.24 0.09 0.70 0.63 |
| Any SAE | 8 (53) | 9 (38) | 4 (33) | 9 (60) | 30 (46) | 18 (41) | 0.70 |
| Any Related SAE | 1 (7) | 4 (17) | 1 (8) | 0 (0) | 6 (9) | 4 (9) | 1.00 |
| Any Unanticipated SAE | 1 (7) | 1 (4) | 1 (8) | 2 (13) | 5 (8) | 7 (16) | 0.22 |
| Any Related & Unanticipated SAE | 0 (0) | 0 (0) | 1 (8) | 0 (0) | 1 (2) | 1 (2) | 1.00 |
| Neuroworsening | 2 (13) | 3 (13) | 0 (0) | 3 (20) | 8 (12) | 7 (16) | 0.58 |
Adverse events by dose level are shown in Table 3. Adverse events, SAEs and hemorrhages all occurred with similar frequency in drug and placebo treated groups. Neuroworsening—defined as an increase in the NIHSS by more than 4 points—occurred equally in both groups, 8 (12.1%) 3K3A-APC treated and 8 (15.9%) placebo treated. The mean aPTT after each dose of study drug was not statistically significantly elevated by treatment. There was no evidence of increased hemorrhage rate or volume related to 3K3A-APC, despite its potential anticoagulant property (Table 4). Using the pre-specified volume threshold (0.06ml) for defining “hemorrhage positive” scans, we found hemorrhage in 56% of 3K3A-APC treated vs. 68% of placebo treated patients (p=0.28). In a sensitivity analysis using several volume thresholds for the positive identification of hemorrhage we found less frequent hemorrhage in the 3K3A-APC treated group, although this numerical difference reached statistical significance only using 0 mL threshold (p=0.046, uncorrected for multiple comparisons). We pursued further evidence of an effect on bleeding with post-hoc, exploratory analyses (Table 4): Using quantitative volumetry, there were smaller hemorrhages in the 3K3A-APC treated group, but this difference was not statistically significant (p=0.07). Between the placebo and 3K3A-APC treatment groups, there was no difference in the numbers of microbleeds seen within or outside the infarct zone.
Table Four. Hemorrhage by dose tier.
Intracerebral hemorrhage was identified from 30-day MRI. In 63 cases the imaging was available from Susceptibility Weighted Imaging (SWI) and in 17 cases the Gradient Echo (GRE) sequence was used. There was no difference amongst the groups in the frequency of SWI vs GRE. A central reader unaware of any group assignments first assessed total hemorrhage volume using the method shown in Fig. 1. Then, the number of microbleeds found in the infarct area or remote from the infarct area were counted. The secondary endpoints that were pre-specified in the Statistical Analysis Plan are listed first. Then, we conducted a sensitivity analysis using several volume thresholds for declaring the image “positive” for hemorrhage. Finally, two analyses were performed post-hoc for exploratory purposes only. Since all these analyses are considered hypothesis-generating, there was no correction for multiple comparisons.
| Hemorrhage Type |
Total | Dose (μg/kg) | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| 120 N = 12 |
240 N = 16 |
360 N = 6 |
540 N = 9 |
3K3A-APC N = 43 |
Placebo N = 37 |
|||
| Pre-specified in Statistical Analysis Plan | ||||||||
| Patients with ≥1 Microbleeds in the infarct zone: N (%) | 15 (18.5) |
1 (8.3) |
2 (12.5) |
3 (50.0) |
1 (11.1) |
7 (16.3) |
8 (21.6) |
0.54 |
| Hemorrhage (>0.06 mL) Incidence by Day 30: N (%) | 49 (61.3) |
7 (58.3) |
11 (68.8) |
2 (33.3) |
4 (44.4) |
24 (55.8) |
25 (67.6) |
0.28 |
| Sensitivity Analysis Using Alternative Volume Thresholds | ||||||||
| (> 0 mL): N (%) | 61 (76.3) |
8 (66.7) |
13 (81.3) |
3 (50.0) |
5 (55.6) |
29 (67.4) |
32 (86.5) |
0.046 |
| (>0.5 mL): N (%) | 24 (30.0) |
4 (33.3) |
5 (31.3) |
0 (0.0) |
1 (11.1) |
10 (23.3) |
14 (37.8) |
0.16 |
| (>1.8 mL): N (%) | 13 (16.3) |
1 (8.3) |
2 (12.5) |
0 (0.0) |
1 (11.1) |
4 (9.3) |
9 (24.3) |
0.07 |
| Exploratory Analyses | ||||||||
| Total Hemorrhage Volume Day 30: ml±SD | 1.4±4.3 | 0.9±1.9 | 1.2±2.9 | 0.04±0.1 | 0.5±1.2 | 0.8±2.1 | 2.1±5.8 | 0.07 |
| Microbleeds outside the infarct zone (N±SD) | 0.8±2.7 | 1.8±5.7 | 1±2.6 | 1.8±3.3 | 0.2±0.4 | 1.2±3.6 | 0.4±0.8 | 0.75 |
| Patients with ≥1 Microbleeds outside the infarct zone: N (%) | 20 (25.0) |
3 (25.0) |
4 (25.0) |
2 (33.3) |
2 (22.2) |
11 (25.6) |
9 (24.3) |
0.90 |
Efficacy outcomes were collected to demonstrate feasibility of collection in future efficacy studies. The incidence of favorable outcome (90-day mRS 0 or 1) was not statistically significantly different from placebo, including all dose tiers together (28 (45.2%) treatment vs 27 (62.8% placebo)); due to small numbers, this result was expected. Similarly, the incidence of a favorable 90-day Barthel Index (≥ 90) was not significantly different among groups, 40 (76.9%) treated versus 34 (91.9%) placebo. The median (IQR) 90-day NIHSS was 1.5 (0.0 – 4.0; n=56) in the 3K3A-APC group and 1 (0.0 – 3.0; n=37) in the placebo group (difference not significant). The infarct volume (mean±SD) at 90 days was similar among all dose tiers and the placebo group, 26.2±32.6 ml in 3K3A-APC treated patients (n=56) vs 26.0±42.1 ml in placebo treated patients (n=37).
DISCUSSION.
In the RHAPSODY trial, we found all 3K3A-APC dose tiers were well-tolerated. The highest tolerated dose, 540 μg/kg, was associated with an acceptable DLT rate not statistically different from placebo. Rates of adverse events (serious and non-serious) were acceptable and comparable to placebo at all dose tiers. Bleeding (incidence and volume of intracerebral hemorrhage) was not elevated in the 3K3A-APC treated patients. In exploratory analysis, the hemorrhage rate and size trended lower in the 3K3A-APC group, compared to placebo, but confirmation of this trend will require a larger clinical trial. Although the trial was designed to find the maximally tolerated dose—and the trial succeeded in this—the optimal dose for further study will likely be lower, given the equivalence among doses.
Pharmacologic 3K3A-APC for human use retains the full cytoprotective and regenerative activities of wt-APC with < 10% of its anticoagulant activity3–5. This decrease in the anticoagulant activity of 3K3A-APC relative to wt-APC should reduce the risk of bleeding in patients, which was a serious side effect of wt-APC or DrotAA30. In animal models of stroke12, 13, 31, traumatic brain injury9 and ALS10, 3K3A-APC exerted beneficial effects that were equivalent to, and sometimes greater than those of wt-APC2, 6, 7, 32.
The mechanisms for 3K3A-APC neuroprotection are partly elucidated13, 14. In extensive murine studies, PAR1 activation by APC exerts protective effects throughout the neurovascular unit because the PAR1 receptor functions on neurons, microglia and endothelial cells2. Although PAR1 was initially discovered and then studied for many years as a thrombin receptor33, 34, APC signaling via PAR1—which is key for neuroprotection—differs remarkably from PAR1 signaling caused by thrombin. This striking phenomenon whereby two agonists, thrombin and APC, cause very different cell signaling effects via the same G-protein coupled receptor, PAR1, is termed “biased agonism.”1, 32, 35 Thus, we hypothesize that 3K3A-APC-mediated biased agonism of PAR1 represents a novel strategy which beneficially targets the entire neurovascular unit comprising distinct subsets of cells at the BBB.
Extensive pre-clinical testing was done to assure 3K3A-APC satisfied the consensus statements regarding putative neuroprotective preclinical stroke therapies (STAIR)36. No therapy, however, has yet proven successful in humans, even some that ostensibly satisfied STAIR guidelines. Stroke models in animals replicate some, but not all aspects of human strokes: experimental models typically involve young animals free of co-morbid conditions (e.g., diabetes, hypertension) that impact outcome after stroke, although notably here, the putative compound in the RHAPSODY trial, 3K3A-APC, was effective in aged and in hypertensive animals8. Another critical factor in demonstrating neuroprotection is timing: drugs that show benefit in experimental models when given within minutes or an hour of recanalization have rarely been so tested in clinical trials. In RHAPSODY we required study drug infusion as soon as possible after consent, but no later than 120 minutes after skin puncture (or rt-PA completion). In this way, the protocol was optimized to show benefit since the drug was combined with attempted recanalization, which included thrombectomy in about half the subjects; unfortunately, the protocol did not include post-thrombectomy documentation of recanalization.
Our findings should be interpreted with some limitations in mind. Most importantly, the trial sites in RHAPSODY currently participate in the NINDS sponsored NeuroNEXT clinical trial network and in general were also certified Comprehensive Stroke Centers. The trial was small from the perspective of detecting an effect on hemorrhage or 90-day outcomes. While the reduced frequency and volume of hemorrhages seems to favor 3K3A-APC for vasculoprotection, larger samples will be needed to conclude this with confidence. Finally, although treatment with thrombolysis or thrombectomy or both was required in this trial, successful recanalization was not documented in all cases.
In conclusion, we found the MTD for 3K3A-APC to be 540 μg/kg. The drug was well tolerated over several doses so further dose-finding will be necessary based on activity and efficacy measures. A suggestion of possible vasculoprotection (fewer and smaller hemorrhages) requires confirmation.
Supplementary Material
ACKNOWLEDGEMENTS
The Partners Human Research Affairs for coordination and implementation of the single IRB model.
This trial was funded by the National Institute of Neurological Disorders and Stroke (U01NS088312). The NeuroNEXT Network is supported by the National Institute of Neurological Disorders and Stroke (Clinical Coordinating Center: U01NS077179, Data Coordinating Center: U01NS077352). Research reported in this publication was also supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award numbers of Columbia University Medical Center (UL1TR000040), University at Buffalo/ SUNY (UL1TR001412), Vanderbilt University (ULTR002243) and Washington University at St. Louis (UL1TR000448). Its contents are solely the responsibility of the authors and do not necessarily represent official views of the National Center for Advancing Translational Sciences or the National Institutes of Health. This clinical trial was approved by the NeuroNEXT Executive Committee (Co-Chairs - Christopher Coffey, PhD and Merit Cudkowicz, MD, MSc). The NINDS NeuroNEXT Data and Safety Monitoring Board and Independent Medical Monitors (Harold Adams, MD, Enrique Leira, MD, and Cheryl Bushnell, MD), provided safety and trial oversight.
The authors wish to acknowledge the expertise and guidance of the following: Dixie Ecklund, Marianne Chase, Steven Greenberg, MD, PhD, Mark P. Goldberg, MD, Sara Weymer, Frank del Greco, Pooja Khanolkar, MPH, Samantha King
Footnotes
POTENTIAL CONFLICTS OF INTEREST DISCLOSURES
KP is an employee of ZZ Biotech LLC. HL, TPD and JHG are consultants to ZZ Biotech, LLC. BVZ is scientific founder of ZZ Biotech LLC and chairs its Scientific Advisory Board. The remainder of the authors have no disclosures.
All data was collected independently of the corporate sponsor and stored at the University of Iowa, per Network of Excellence in Neuroscience Trials (NeuroNEXT) Standard Operating Procedures. All analyses were approved by the PI and the senior biostatistician. Although company representatives were present on team phone calls, the investigators retained full control over data, event adjudication, analyses, interpretation and drafting of the manuscript.
Clinical Trial Registration. Clinical Trial Registration-URL: http://www.clinicaltrials.gov. Unique identifier: NCT02222714
References
- 1.Griffin JH, Zlokovic BV, Mosnier LO. Activated protein C: biased for translation. Blood. 2015. May 7;125(19):2898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zlokovic BV G J. Cytoprotective protein C pathways and implications for stroke and neurological disorders. Trends Neurosci. 2011;34(4):198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo H, Zhao Z, Yang Q, et al. An activated protein C analog stimulates neuronal production by human neural progenitor cells via a PAR1-PAR3-S1PR1-Akt pathway. J Neurosci. 2013. April 3;33(14):6181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, Zhao Z, Rege SV, et al. 3K3A-activated protein C stimulates postischemic neuronal repair by human neural stem cells in mice. Nat Med. 2016. September;22(9):1050–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mosnier LO G A, Yegneswaran S, Griffin JH. Activated protein C variants with normal cytoprotective but reduced anticoagulant activity. Blood. 2004;104(6):1740–4. [DOI] [PubMed] [Google Scholar]
- 6.Griffin JH, Fernandez JA, Lyden PD, Zlokovic BV. Activated protein C promotes neuroprotection: mechanisms and translation to the clinic. Thromb Res. 2016. May;141 Suppl 2:S62–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Griffin JH, Mosnier LO, Fernandez JA, Zlokovic BV. 2016 Scientific Sessions Sol Sherry Distinguished Lecturer in Thrombosis: Thrombotic Stroke: Neuroprotective Therapy by Recombinant-Activated Protein C. Arterioscler Thromb Vasc Biol. 2016. November;36(11):2143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Zhao Z, Chow N, et al. Activated protein C analog protects from ischemic stroke and extends the therapeutic window of tissue-type plasminogen activator in aged female mice and hypertensive rats. Stroke. 2013. December;44(12):3529–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walker CT, Marky AH, Petraglia AL, Ali T, Chow N, Zlokovic BV. Activated protein C analog with reduced anticoagulant activity improves functional recovery and reduces bleeding risk following controlled cortical impact. Brain Res. 2010. August 6;1347:125–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong Z, Ilieva H, Hallagan L, et al. Activated protein C therapy slows ALS-like disease in mice by transcriptionally inhibiting SOD1 in motor neurons and microglia cells. J Clin Invest. 2009. November;119(11):3437–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han MH, Hwang SI, Roy DB, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008. February 28;451(7182):1076–81. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Zhang Z, Chow N, et al. An activated protein C analog with reduced anticoagulant activity extends the therapeutic window of tissue plasminogen activator for ischemic stroke in rodents. Stroke. 2012. September;43(9):2444–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo H, Singh I, Wang Y, et al. Neuroprotective activities of activated protein C mutant with reduced anticoagulant activity. Eur J Neurosci. 2009. March;29(6):1119–30. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Guo H, Wang Y, Singh I, et al. Species-dependent neuroprotection by activated protein C mutants with reduced anticoagulant activity. J Neurochem. 2009. April;109(1):116–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams PD, Zlokovic BV, Griffin JH, Pryor KE, Davis TP. Preclinical safety and pharmacokinetic profile of 3K3A-APC, a novel, modified activated protein C for ischemic stroke. Curr Pharm Des. 2012;18(27):4215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lyden P, Levy H, Weymer S, et al. Phase 1 safety, tolerability and pharmacokinetics of 3K3A-APC in healthy adult volunteers. Curr Pharm Des. 2013;19(42):7479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lyden P, Weymer S, Coffey C, et al. Selecting patients for intra-arterial therapy in the context of a clinical trial for neuroprotection. Stroke. 2016. December;47(12):2979–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyden P Using the National Institutes of Health Stroke Scale: A Cautionary Tale. Stroke. 2017. February;48(2):513–9. [DOI] [PubMed] [Google Scholar]
- 19.Goodman SN, Zahurak ML, Piantadosi S. Some practical improvements in the continual reassessment method for phase I studies. Stat Med. 1995. June 15;14(11):1149–61. [DOI] [PubMed] [Google Scholar]
- 20.Hacke W, Kaste M, Bluhmki E, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008. September 25;359(13):1317–29. [DOI] [PubMed] [Google Scholar]
- 21.Goyal M, Demchuk AM, Menon BK, et al. Randomized assessment of rapid endovascular treatment of ischemic stroke. N Engl J Med. 2015. March 12;372(11):1019–30. [DOI] [PubMed] [Google Scholar]
- 22.Campbell BC, Mitchell PJ, Kleinig TJ, et al. Endovascular therapy for ischemic stroke with perfusion-imaging selection. N Engl J Med. 2015. March 12;372(11):1009–18. [DOI] [PubMed] [Google Scholar]
- 23.Berkhemer OA, Fransen PS, Beumer D, et al. A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. 2015. January 1;372(1):11–20. [DOI] [PubMed] [Google Scholar]
- 24.Saver JL, Goyal M, Diener HC, Investigators SP. Stent-Retriever Thrombectomy for Stroke. N Engl J Med. 2015. September 10;373(11):1077. [DOI] [PubMed] [Google Scholar]
- 25.Temple R Hy’s law: predicting serious hepatotoxicity. Pharmacoepidemiol Drug Saf. 2006. April;15(4):241–3. [DOI] [PubMed] [Google Scholar]
- 26.Reuben A Hy’s law. Hepatology. 2004. February;39(2):574–8. [DOI] [PubMed] [Google Scholar]
- 27.Health UDo, Services H. National cancer institute. Common terminology criteria for adverse events (CTCAE).(V4. 03). NIH Publication; 2015(09-5410). [Google Scholar]
- 28.Greenberg SM, Vernooij MW, Cordonnier C, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 2009. February;8(2):165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Quigley J, Pepe M, Fisher L. Continual reassessment method: a practical design for phase 1 clinical trials in cancer. Biometrics. 1990. March;46(1):33–48. [PubMed] [Google Scholar]
- 30.Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001. March 8;344(10):699–709. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Thiyagarajan M, Chow N, et al. Differential neuroprotection and risk for bleeding from activated protein C with varying degrees of anticoagulant activity. Stroke. 2009. May;40(5):1864–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sinha RK, Wang Y, Zhao Z, et al. PAR1 Biased Signaling is Required for Activated Protein C In Vivo Benefits in Sepsis and Stroke. Blood. 2018. January 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000. September 14;407(6801):258–64. [DOI] [PubMed] [Google Scholar]
- 34.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005. August;3(8):1800–14. [DOI] [PubMed] [Google Scholar]
- 35.Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood. 2012. December 20;120(26):5237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fisher M, Feuerstein G, Howells DW, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009. June;40(6):2244–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.