

Abstract
Carbohydrates are structurally complex but functionally important biomolecules. Therefore, they have been challenging but attractive synthetic targets. While substantial progress has been made on advancing chemical glycosylation methods, incorporating enzymes into carbohydrate synthetic schemes has become increasingly practical as more carbohydrate biosynthetic and metabolic enzymes as well as their mutants with synthetic application are identified and expressed for preparative and large-scale synthesis. Chemoenzymatic strategies that integrate the flexibility of chemical derivatization with enzyme-catalyzed reactions have been extremely powerful. Briefly summarized here are our experiences on developing one-pot multienzyme (OPME) systems and representative chemoenzymatic strategies from others using glycosyltransferase-catalyzed reactions for synthesizing diverse structures of oligosaccharides, polysaccharides, and glycoconjugates. These strategies allow the synthesis of complex carbohydrates including those containing naturally occurring carbohydrate postglycosylational modifications (PGMs) and non-natural functional groups. By combining these strategies with facile purification schemes, synthetic access to the diverse space of carbohydrate structures can be automated and will not be limited to specialists.
Keywords: carbohydrate synthesis, chemoenzymatic synthesis, glycolipid, glycosyltransferase, regioselective, enzyme engineering
Graphical Abstract
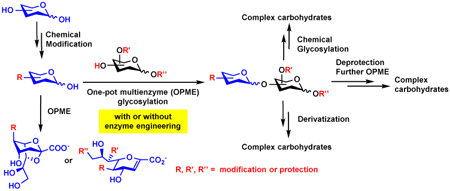
1. Introduction
Carbohydrates are the most abundant and structurally diverse naturally occurring organic compounds. They are presented on the surfaces of all types of cells from different organisms [1]. The cell surface carbohydrate coating (or glycocalyx) can be readily observed under microscopes [2]. Carbohydrates on mammalian cells are involved in numerous biological and pathological processes including homeostasis, cell-cell interaction, cell migration, development, bacterial and viral infection, inflammation, immunology, cancer metastasis, etc. [3, 4]. The variety of these properties are the results of the structural diversity of carbohydrates. Unlike proteins and nucleic acids, carbohydrates are not the products of template-driven biosynthesis but are directly dependent on the expression and substrate specificity of glycosyltransferases as well as the availability of corresponding sugar nucleotides [5]. Diverse monosaccharide building blocks and various stereo- and regiochemistry in glycosidic linkages contribute to the complexity of the linear and branched structures of carbohydrates. Additional modifications, such as epimerization, O-acetylation, O-methylation, O-phosphorylation, and N- or O-sulfation etc., can take place on the carbohydrates. [6, 7]. Most of these modifications occur after the formation of glycosidic linkages and are named as carbohydrate post-glycosylation modifications (PGMs) [6] in analogy to protein post-translational modifications. Therefore, PGMs add another layer of complexity in naturally occurring carbohydrates. To unravel the important functions “coded” by carbohydrates [8, 9], obtaining pure compounds in sufficient amount is necessary. While substantial progress has been made on advancing chemical glycosylation methods, incorporating enzymes into carbohydrate synthetic schemes has become increasingly practical with the increasing availability of carbohydrate-biosynthetic and metabolic enzymes as well as their mutants. Chemoenzymatic strategies that integrate the flexibility of chemical derivatization with enzyme-catalyzed reactions have been extremely powerful. The development of synthetic strategies using exoglycosidases, endoglycosidases, and their mutants in the production of oligosaccharides and glycoconjugates can be found in several recent reviews [10-12]. The current review focuses on one-pot multienzyme (OPME) systems and other representative chemoenzymatic strategies using glycosyltransferase-catalyzed reactions for synthesizing diverse structures of oligosaccharides, polysaccharides, and glycoconjugates.
2. One-pot multienzyme (OPME) chemoenzymatic glycosylation systems
Most carbohydrates in nature are synthesized by Leloir-type glycosyltransferase-catalyzed reactions. These glycosyltransferases require the use of sugar nucleotides as activated sugar donor substrates. Although expensive, the sugar nucleotides of some common human monosaccharide building blocks are commercially available. These compounds can be used directly for enzymatic and chemoenzymatic synthesis of carbohydrates if only small amounts of the desired products are needed. To decrease the synthetic cost and reduce potential inhibition of glycosyltransferases by nucleotide formed, methods for in situ recycling sugar nucleotide have been developed and were reviewed previously [13].
For carbohydrates containing naturally occurring PGMs [6] or non-natural occurring modifications, an efficient strategy is to use one-pot multienzyme (OPME) chemoenzymatic systems (Figure 1) [14]. This strategy relies on in situ production of a sugar nucleotide from a simple monosaccharide or its derivative, which is used as the donor for a glycosyltransferase in the same pot for the synthesis of desired carbohydrates without the isolation of intermediates. This strategy helps to decrease the cost for the synthesis of target carbohydrates in large amounts. More importantly, it allows the introduction of modified monosaccharides as potential precursors for sugar nucleotides for the synthesis of carbohydrates with modifications at specific sites. Multiple OPME systems can be carried out in sequential for producing longer chain oligosaccharides and glycoconjugates.
Figure 1.

Schematic illustration of one-pot multienzyme (OPME) chemoenzymatic reactions for the synthesis of carbohydrates with structural modifications.
2.1. OPME sialylation systems for the chemoenzymatic synthesis of sialosides
An example of OPME glycosylation reactions is shown in Figure 2 for sialylation. Sialyltransferases (SiaTs) are the key enzymes for installing sialic acids. The most common form of sialic acid is N-acetylneuraminic acid (Neu5Ac) [15]. Sialyltransferases use cytidine 5’-monophosphate (CMP)-linked sialic acids such as CMP-N-acetylneuraminic acid (CMP-Neu5Ac) as an activated donor substrate. CMP-Neu5Ac can be prepared by a sugar activation (SA) system SA1 (Figure 2) from its 6-carbon precursor N-acetylmannosamine (ManNAc) involving a sialic acid aldolase-catalyzed aldol-addition reaction from ManNAc and pyruvate to produce Neu5Ac followed by a CMP-sialic acid synthetase (CSS)-catalyzed reaction using cytidine 5’-triphosphate (CTP). SA1 can be combined with a suitable sialyltransferase in one pot to produce sialosides with a desired sialyl linkage from simple ManNAc, pyruvate, CTP, and the acceptor without the isolation of intermediates. Products with different sialyl linkages (α2–3/6/8/9) can be produced by the specificity of different sialyltransferases used.
Figure 2.

OPME sialylation systems for synthesis of sialosides. (Adapted from H. Yu, X. Chen, Org Biomol Chem, 14 (2016) 2809-2818.)
By introducing structural modification at the monosaccharides such as ManNAc or Neu5Ac by chemical synthesis, OPME sialylation systems can be used for direct synthesis of sialosides containing desired structurally modified sialic acid forms, diverse internal glycans, and different sialyl linkages. For synthesizing sialosides containing sialic acid with C5, C7, C8, and/or C9-modifications, the derivatives of sialic acid six-carbon precursors can be chemically synthesized and used as the starting material for one-pot three-enzyme (OP3E) sialylation systems. For sialosides containing C4-modifications at sialic acids, sialic acid derivatives should be used in one-pot two-enzyme (OP2E) systems for the synthesis (Figure 3). Sialic acid analogs with C3-modification can also be introduced by using 3-substituted pyruvate derivative in a sialic acid aldolase-catalyzed reaction. After purification, sialosides containing C3-modified sialic acid residue can be readily formed using this OP2E system [16]. The key for the success of these chemoenzymatic strategies is to access enzymes that have promiscuous substrate specificity to tolerate desired modification on the substrates. Ideally, the enzymes can also be expressed in Escherichia coli in large amounts to allow low cost production of sufficient quantity of catalysts for synthesis.
Figure 3.

OPME chemoenzymatic sialylation systems for synthesizing sialosides with modified sialic acid residues.
Quite remarkably, many wild-type enzymes from bacterial sources have desired properties of high activity, good expression in Escherichia coli, and substrate promiscuity. Using this OPME chemoenzymatic sialylation strategy, libraries of α2–3/6-linked sialosides [17-19] containing naturally occurring sialic acid forms (Figure 4A) and non-natural sialic acid modifications (Figure 4B) have been produced using bacterial sialic acid aldolases (e.g. from Escherichia coli [20] or Pasteurella multocida [21]), CMP-sialic acid synthetases (from Neisseria meningitidis [20] or Pasteurella multocida [22]), and sialyltransferases including Pasteurella multocida α2–3-sialyltransferase 1 (PmSTl) [17], Photobacterium damselae α2–6-sialyltransferase (Pd2,6ST) [18], Photobacterium sp. JT-ISH-224 a2–6-sialyltransferase (Psp2,6ST) [23]. These include sialoside containing modification at C3 [16], C4 [24], C5 [17-19, 25], C7 [19, 26, 27], C8 [19, 28], and/or C9 [17-19, 25, 27, 29, 30] of sialic acid. Similarly, α2–3/6-linked sialosides can be further extended by α2–8-linked sialic acids using a Campylobacter jejuni α2–3/8-sialyltransferase (CjCstII)-containing OPME α2–8-sialylation system (Figure 5) [31]. Typical yields for these OPME sialylation systems were over 50%, many were over 90%, and some reached quantitative yields.
Figure 4.

Sialosides containing naturally occurring sialic acid forms (A) or non-natural modifications (B) that have been synthesized by OPME α2–3/6-sialylation systems.
Figure 5.

Sialosides containing naturally occurring sialic acid forms or non-natural modifications that have been synthesized by OPME α2–3/6- and α2–8-sialylation systems.
2.2. One-pot multienzyme (OPME) systems for the chemoenzymatic synthesis of sialidase inhibitors
The OPME α2–3-sialylation system can be combined with a sialidase for synthesizing sialidase inhibitors or inhibitor precursors. For example, Streptococcus pneumoniae strains collectively have three sialidases [32] SpNanA, SpNanB, and SpNanC. While SpNanA catalyzes the hydrolysis of α2–3/6/8-linked sialosides to produce free monosaccharide Neu5Ac, SpNanB and SpNanC specifically recognize α2–3-linked sialosides as substrates. Furthermore, the product of SpNanB is 2,7-anhydro-N-acetylneuraminic acid (2,7-anhydro-Neu5Ac), a potential prebiotic that can be used as the sole carbon source of a common human gut commensal anaerobic bacterium Ruminoccocus gnavus [33] and the product of SpNanC is 2,3-dehydro-2-deoxy-N-acetylneuraminic acid (Neu5Ac2en or DANA), the general transition state analog inhibitor against common hydrolytic sialidases [32]. By including SpNanB [34] or SpNanC [35] into the OPME scheme, one-pot multienzyme systems for synthesizing sialidase inhibitors or precursors were formed (Figure 6). All enzymes in the systems have some level of tolerance towards substrate modifications. A single mutant of PmST1, PmST1 M144D, was used to replace wild-type PmST1 to decrease its sialoside hydrolysis activity to provide high efficiency in the system. In addition, separating sialoside generation and Sia2en production by SpNanC into two steps was shown to improve the yields for some compounds. The 2,7-anhydro-Neu5Ac and Neu5Ac2en analogs obtained can be further chemically derivatized to generate inhibitors with improved inhibitory activity or improved selectivity [34, 35].
Figure 6.

OPME systems for the synthesis of sialidase inhibitors or sialidase inhibitor precursors 2,3-dehydro-2-deoxy-sialic acids (Sia2ens) and 2,7-anhydro-sialic acids (2,7-anhydro-Sias).
2.3. OPME activation and glycosylation systems for monosaccharides other than sialic acid
OPME glycosylation reactions using bacterial enzymes have also been developed for activating and transferring common monosaccharides in human glycome other than sialic acid (Figure 7 including L-fucose (Fuc), D-galactose (Gal), N-acetylglucosamine (GlcNAc), N acetylgalactosamine (GalNAc), and glucuronic acid (GlcA) [14]. For these systems, the simplest routes are the combination of a glycokinase (GlyK), a nucleotidyltransferase (NucT), and a glycosyltransferase (GlyT). An inorganic pyrophosphatase (PpA) can also be added to breal down the pyrophosphate formed in the nucleotidyltransferase-catalyzed reaction to shift the reaction process towards sugar nucleotide formation. The glycokinase is responsible for the formation of a monosaccharide-1-phosphate from a simple monosaccharide and adenosine 5’ triphosphate (ATP). The nucleotidyltransferase then catalyzes the formation of a suga nucleotide (NDP) such as uridine 5’-diphosphate (UDP)-activated sugars (for Gal, GlcNAc GalNAc, and GlcA) or guanosine 5’-diphosphate (GDP)-activated sugar (for Fuc) Glycosyltransferases are used for synthesizing products with different linkage specificities Similar OPME systems have been developed for synthesizing sugar nucleotides for D-mannose (Man) and N-acetylmannosamine (ManNAc) [14] but have not been coupled with the corresponding glycosyltransferases.
Figure 7.

OPME systems for synthesizing oligosaccharides using glycosyltransferases that require nucleoside diphosphate-sugars as donor substrates. Enzyme abbreviations: GlyK – glycokinase; NucT – nucleotidyltransferase; PpA – inorganic pyrophosphatase; GlyT – glycosyltransferase. (Adapted from H. Yu, X. Chen, Org Biomol Chem, 14 (2016) 2809-2818.)[14]
3. Enzymatically synthesized oligosaccharides as building blocks for chemical glycosylation
Just as chemical synthesis can provide building blocks for enzymatic assembly, OPME reactions can provide building blocks for subsequent chemical glycosylations. For example, glycosyl thiotoluenes are common glycosyl donors for chemical glycosylation. Oligosaccharyl thiotoluene building blocks can be synthesized by enzymatic glycosylation of simpler monosaccharyl thiotoluenes and used for further chemical glycosylation. Such a strategy has been successfully used for chemoenzymatic synthesis of sialyl Lewis × tetrasaccharide (sLexβProN3) [36] and lacto-N-tetraose derivative (LNTβProN3) [37]. For synthesizing sLexβProN3 without a desired β1–4-galactosyltransferase and α1–3-fucosyltransferase, sialylated disaccharyl thiotoluene (Neu5Acα2–3GalβSTol) was chemoenzymatically synthesized from galactosyl thiotolune (GalβSTol), ManNAc, pyruvate, and CTP using a OPME α2–3-sialylation reaction containing Pasteurella multocida sialic acid aldolase (PmAldolase), Neisseria meningitidis CMP-sialic acid synthetase (NmCSS) and PmST1. It was protected and used as a building block for chemical glycosylation processes for the production of the desired tetrasaccharide (Figure 8A) [36]. Similarly, for the synthesis of LNTβProN3 (Figure 8B), disaccharyl thiotolune of lacto-N-biose (Galβ1–3GlcNAcβSTol) was enzymatically synthesized from N-acetylglucosaminyl thiotolune (GlcNAcβSTol), galactose (Gal), and ATP using a one-pot two-enzyme reaction containing Escherichia coli galactokinase (EcGalK) and Bifidobacterium infantis D-galactosyl-β1–3-N-acetyl-D-hexosamine phosphorylase (BiGalHexNAcP). Chemical protection, followed by chemical glycosylation with selectively protected disaccharide acceptor led to the formation of protected tetrasaccharide. After chemical deprotection, acetylation, and global deprotection, the target tetrasaccharide LNTβProN3 was successfully obtained [37].
Figure 8.


Chemoenzymatic synthesis of sLexβProN3 (A), LNTβProN3 (B), iGb3 and Gb3 (C), as well as GM3, blood group H- and A-antigens (D) by chemical glycosylation using building blocks synthesized by OPME reactions.
OPME enzymatic glycosylation followed by chemical protection and activation, chemical glycosylation, and deprotection has also been used for synthesizing glycosphingolipids (GSLs), including globotrihexosylceramide (Gb3) and isoglobotrihexosylceramide (iGb3) (Figure 8C) [38], as well as GM3, blood group antigens H and A GSLs (Figure 8D) [39]. In these systems, isoglobotriaose was synthesized from lactose, glucose-1-phophate (Glc-1-P), and uridine 5’-triphosphate (UTP) by a one-pot three-enzyme system containing UDP-glucose pyrophosphorylase (GalU), UDP-Gal 4-epimerase (GalE), and an α1–3-galactosyltransferase (α1–3GalT). GM3 oligosaccharide was synthesized from lactose, Neu5Ac, and CTP using a one-pot two-enzyme system containing NmCSS and PmSTl M144D. Blood group antigen H oligosaccharide was synthesized from lactose, fucose (Fuc), ATP, and guanosine 5’-triphosphate (GTP) using a one-pot three-enzyme system containing a bifunctional L-fucokinase/GDP-fucose pyrophosphorylase (FKP), PmPpA, and Helicobacter mustelae αl–-fucosyltranferase (Hmα1,2FT). Blood group antigen A oligosaccharide was obtained from blood group antigen H oligosaccharide, N-acetylgalactosamine (GalNAc), ATP, and UTP using a one-pot four-enzyme reaction containing Bifidobacterium infantis N-acetylhexamine kinase (BiNahK), Homo sapiens UDP-GalNAc pyrophosphorylase (hAGX1), PmPpA, and H. mustelae α1–3-GalNAc transferase (HmBgtA). The free oligosaccharides, including globotriaose, were protected by pivoyl or benzoyl groups and activated to form trichloroacetimidate or N-phenyl trifluoroacetimidate glycosyl donors for chemical glycosylation. Reduction, acylation, followed by deprotection led to the formation of desired GSLs.
4. Regioselective enzymatic glycosylation of acceptors containing multiple glycosylation sites
Although OPME systems allow facile synthesis of many oligosaccharides without protection strategies, glycan structures may contain multiple potential glycosylation sites for glycosyltransferases. Regioselectivity in some cases can be achieved by the acceptor substrate specificity of glycosyltransferases. Some mammalian glycosyltransferases may possess the desired selectivity. However, desired regioselectivity may not always be achieved, especially when bacterial glycosyltransferases with higher substrate promiscuous are used. In these cases, several advanced strategies have been developed to generate products with desired regioselectivity. These include chemical and/or enzymatic protection of selected glycosylation sites in acceptor substrates, altering sequences of multiple glycosylation processes, and protein engineering of glycosyltransferases.
4.1. Chemical protection of selected glycosylation sites in acceptor substrates
Partial chemical modification or protection of the acceptor substrates of glycosyltransferases can be used to control the regioselectivity of enzyme-catalyzed reactions. Such a strategy named as “substrate engineering” by Withers et al. has been used to manipulate the substrate regioselectivity of Neisseria meningitidis α1–4-galactosyltransferase NmLgtC and bovine α1–3-galacosyltransferase (α3GalT) by installing a hydrophobic protection group at different locations of monosaccharide acceptors [40]. On the other hand, while some conformationally constrained oligosaccharides may improve catalytic efficiency of certain glycosyltransferases as shown by Boons et al. [41, 42], conformational constraining of acceptor substrates can also be used to block the recognition of glycosyltransferases. For example, Cao et al. used an easy-to-install lactone to protect one of the glycosylation sites of Photobacterium damselae α2–6-sialyltransferase (Pd2,6ST) in the acceptor substrate to allow regioselective sialylation. The lactone can be easily removed after the formation of the desired glycosidic linkage for the synthesis of target ganglioside disialyl tetrasaccharide (Figure 9) [43]. In comparison, without lactone protection, Pd2,6ST-catalyzed sialylation led to the formation of mixtures of disialylated tetrasaccharides and trisialylated pentasaccharide.
Figure 9.

Use of lactone protection as a strategy to provide regioselectivity for chemoenzymatic synthesis of ganglioside disialyl tetrasaccharide. In the absence of lactone protection, mixture of disialyl tetrasaccharides and trisialyl pentasaccharide were formed in Photobacterium damselae α2–6-sialyltransferase (Pd2,6ST)-catalyzed reaction.
Selective O-acetylation or per-O-acetylation of acceptor glycosylation sites has also been used as a strategy to target glycosylations to a single desired site. This has been demonstrated for chemoenzymatic synthesis of polysaccharide analogs containing sialyl galactoside disaccharide repeats (Figure 10A) [44] as well as asymmetric N-glycans (Figure 10B and 10C) [45, 46]. While selected O-acetylation or per-O-acetylation in the glycosyltransferase acceptor substrates for the synthesis of N-glycans [45, 46] was introduced during chemical construction of the compounds, in the synthesis of sialyl galactoside polysaccharide analogs, per-O-acetylated galactose was attached to an azido-modified CMP-sialic acid by Cu(I)-catalyzed azide-alkyne cyclization reaction under mild coupling conditions. The resulting CMP-activated, triazole-containing disaccharide analog was tolerated as a donor substrate for Pd2,6ST-catalyzed formation of longer oligosaccharide. De-acetylation of the resulting oligosaccharide provided an elongated acceptor for another round of chain extension by sialylation [44].
Figure 10.

A) Chemoenzymatic synthesis of disaccharide building block and controlled chemoenzymatic synthesis of size-defined polysaccharides by Pd2,6ST-catalyzed block transfer of disaccharide building blocks. B) Chemoenzymatic synthetic route of asymmetrically multi-antennary glycans using selected O-acetylated acceptor. C) Chemoenzymatic synthesis of modules with selected O-acetylation for N-glycan assembly.
Chemically synthesized acceptors with selective sites protected by monosaccharide caps have also been used as a strategy to control the downstream enzymatic glycosylation at the desired sites. The monosaccharide caps can then be selectively cleaved off by reactions catalyzed by the corresponding glycosidases. For example, β–4-linked mannose, α1–4-linked galactose, and β–4/6-linked GlcNAc were successfully used as the terminal monosaccharide caps on a precursor of complex N-glycans. They were readily removed by the corresponding α-galactosidase, β-mannosidase, and β-GlcNAcidase, respectively, before or after glycosyltransferase-catalyzed reactions taking place at other sites in the acceptor. This strategy allowed multi-dimensional enzymatic derivatization for the generate of a library of N-glycans from a common chemically synthesized precursor [47].
4.2. Enzymatic protection of selected glycosylation sites in acceptor substrates
Pairs of a glycosyltransferase and a glycosidase can also be used as a protection and deprotection strategy for regioselective glycosylation by glycosyltransferase-catalyzed reactions. This has been demonstrated for chemoenzymatic synthesis of an asymmetric human milk oligosaccharide derivative [48]. As shown in Figure 11, terminal LacNAc moiety of the GlcNAcβ1–6-branch of a complex human milk oligosaccharide structure was protected by an α2–6-linked sialic acid residue introduced by an α2–6-sialyltransferase (ST6GAL-I)-catalyzed reaction, preventing the terminal LacNAc from fucosylation by FUT3. Therefore, FUT3 catalyzed the addition of an α1–3-linked fucose selectively to the internal LacNAc residue. The sialic acid residue was removed by Arthrobacter ureafaciens sialidase for the formation of the desired difucosylated oligosaccharide.
Figure 11.

An example by the Boons group of using a glycosyltransferase (ST6Gal-I) and a glycosidase (Arthrobacter ureafaciens sialidase) pair as a protection and deprotection strategy for regioselective glycosylation (fucosylation) by glycosyltransferase (FUT3)-catalyzed reaction.
4.3. Altering the order of multiple glycosylation processes
Carefully choosing reaction sequences provides another solution to selective glycosylation of substrates containing multiple acceptor moieties. For example, lacto-N-neotetraose (LNnT) (Figure 12) can be synthesized from lactose, an inexpensive disaccharide, using GlcNAc activation and transfer OPME system (OPME1) followed by Gal activation and transfer OPME system (OPME2). Direct α2–6-sialylation of the resulting LNnT with a OPME α2–6-sialylation system containing Pd2,6ST (OPME3) led to the addition of sialic acid residues to both Gal moieties [49, 50]. Alternating the sequence of OPME2 and OPME3 reactions led to the formation of monosialylated product with only one sialic acid α2–6-linked to the internal Gal of LNnT moiety. It went through a OPME α2–3-sialylation reaction (OPME4) for the formation of a disialylated analog DS’LNT containing a sialic acid α2-6-linked to the internal Gal and a sialic acid α2–3-linked to the terminal Gal. A similar strategy was used for the synthesis of a monofucosylated and internally α2–6-monosialylated hexasaccharide [51].
Figure 12.

Synthesis of disialylated hexasaccharides DSLNnT and DS’LNnT by sequential OPME reactions with altered sequence of OPME2 and OPME3.
5. Chemoenzymatic synthons
When enzymes involved in the one-pot multienzyme (OPME) glycosylation reactions cannot tolerate the desired substrate modifications, “chemoenzymatic synthons” method can be used. In this strategy, substrates modified with an alternative group are used for OPME enzymatic synthesis of oligosaccharide derivatives which can then be enzymatically or chemically converted to the desired products. Such a strategy has been used for chemoenzymatic synthesis of heparan sulfate oligosaccharides [52]. N-Sulfated analogs of heparan sulfate oligosaccharides [53], N-acyl derivatives of sialidase transition state analog inhibitors [34, 35], and 5,7-di-N-acetyl-legionaminic acid (Leg5,7diNAc)-containing oligosaccharides [27].
Trifluoroacetamido (-NTFA) has been found to be an excellent analog of acetamido (-NAc) group in N-acetylhexosamine-containing substrates for carbohydrate biosynthetic enzymes [54]. For example, N-trifluoroacetylglucosamine (GlcNTFA) can be converted to UDP-GlcNTFA by the combined function of N-acetylhexosamine 1-kinase (NahK) and N-acetylglucosamine-1-phosphate uridylyltransferase (GlmU) [55]. The resulting UDP-GlcNTFA was readily used as a donor substrate of Escherichia coli α1–4-GlcNAc transferase (EcKfiA) [52] or Pasteurella multocida heparosan synthase 2 (PmHS2) [53] for the synthesis of N-TFA-analog of heparan sulfate oligosaccharides or analogs. The N-TFA group can then be easily removed selectively under mild basic conditions to provide an amino group to allow enzymatic [52] or chemical N-sulfation [53]. Derivatization with different acyl groups can also be carried out to form a library of analogs [34, 55].
Another excellent N-sulfate or N-acyl precursor moiety for chemoenzymatic reactions is azido (-N3) group. Substitution of a hydroxyl group by an azido group in the substrate can be tolerated by many carbohydrate biosynthetic enzymes. For example, 6-azido-6-deoxy-N-acetylglucosamine (GlcNAc6N3) can be tolerated by NahK for the synthesis of GlcNAc6N3-1-phosphate. It was converted by Pasteurella multocida N-acetylglucosamine-1-phosphate uridylyltransferase (PmGlmU) to form UDP-GlcNAc6N3, which was used as a donor substrate of PmHS2 for the synthesis of GlcNAc6N3-containing heparosan oligosaccharide analog. The azido group can be chemically reduced to an amino group followed by N-sulfation for the formation of N-sulfated analogs of heparan sulfate oligosaccharides [53]. A similar strategy has been used for the synthesis of N-acyl derivatives of sialidase transition state analog inhibitors where 6-azido-6-deoxyl-N-acetylmannosamine (ManNAc6N3) was used as a starting material in a one-pot multienzyme (OPME) system for the synthesis of 9-azido-2,3-dehydro-2,9-dideoxy-N-acetylneuraminic acid (Neu5Ac9N32en). The azido group was then readily reduced and acylated to form desired derivatives [35]. The strategy was recently used for the synthesis of oligosaccharides containing di-N-acetyllegionaminic acid (Leg5,7Ac2) (Figure 13), a sialic acid analog in lipopolysaccharides of several Gram-negative pathogenic bacteria [27]. In this case, Leg5,7Ac2 itself was not a suitable substrate of Neisseria meningitidis CMP-sialic acid synthetase (NmCSS) for the synthesis of CMP-Leg5,7Ac2. In contrast, its diazido-analog Leg5,7diN3 was readily obtained from chemically synthesized 6-carbon precursor, 2,4-diazido-2,4,6-trideoxymannose (6deoxyMan2,4diN3), by a sialic acid aldolase-catalyzed reaction and was tolerated by NmCSS for the synthesis of CMP-Leg5,7diN3 which was further accepted by bacterial α2–3- and α2–6-sialyltransferases for the synthesis of Leg5,7 diN3-containing oligosaccharides. These compounds can be easily converted to the desired Leg5,7Ac2-containing oligosaccharides [27].
Figure 13.

Application of a chemoenzymatic synthon strategy in the synthesis of Leg5,7Ac2-containing oligosaccharides. A) Leg2,7Ac2 produced from 2,4-diacetamido-2,4,6-trideoxy-D-mannose (6deoxyManNAc4NAc) by PmAldolase-catalyzed reaction cannot be used by NmCSS as the substrate. B) Production of Leg2,7Ac2-containing oligosaccharides by OPME synthesis of Leg2,7diN3-containing glycosides followed by chemical derivatization.
6. Enzyme engineering for altered glycosylation sequence or improved regioselectivity
Many carbohydrate biosynthetic wild-type enzymes from bacterial sources display remarkable promiscuity toward modified substrates, allowing the production of carbohydrates with post-glycosylation modifications using the OPME strategies discussed above. Nevertheless, when efforts to identify an enzyme with suitable activity toward a desired substrate fail, enzyme engineering can provide an alternative [56]. Such effort is greatly facilitated by the availability of protein crystal structures in the presence of substrates or analogs. For example, PmST1 M144D, designed based on crystal structures of PmST1 [57, 58], allows the tolerance of a fucosylated glycan, Lewisx [Lex, Galβ1-4(Fucα1-3)GlcNAcβOR] as an acceptor substrate for efficient synthesis of sialyl Lewisx [sLex, Siaα2-3Galβ1-4(Fucα1-3)GlcNAcβOR] structures, which are well-known tumor-associated carbohydrate antigens and candidates for developing cancer vaccine [59]. In addition, Psp2,6ST A366G has an increased expression level and is more efficient in synthesizing Neu5Acα2-6GalNAcαOSer/Thr STn antigens [60].
Recently, the identification of engineered α2–6-sialyltransferases with selective sialylation activity towards terminal galactose or GalNAc residue in oligosaccharides has been described. Two strategies have been reported to achieve this by protein engineering of CAZy GT80 bacterial sialyltransferases. One strategy was by protein crystal structure-based site-specific saturated mutagenesis to switch PmST1, an α2–3-sialyltransferase, to α2–6-sialyltransferase mutants with desired regioselectivity. PmST1 P34H/M144L was identified as the best candidate which not only has the desired regioselectivity, but also has the highest reported catalytic efficiency for CAZy GT80 α2–6-sialyltransferases. The mutant displayed minor activity toward internal acceptor moieties and required careful reaction monitoring to prevent accumulation of multi-sialylated products. It was successfully used for preparative-scale synthesis of desired monosialylated pentasaccharide Neu5Acα2–6LNnTβProN3 in 90% yield. Disialylated hexasaccharide was also obtained in 5% yield [61]. Another strategy was to engineer the acceptor binding pocket of Pd2,6ST. A double mutant A200Y/S232Y with bulkier amino acid residues was found to successfully block its binding to internal Gal or GalNAc residues while retain its α2–6-sialylation activity towards terminal Gal or GalNAc sites. A number of monosialylated products were successfully obtained in high yields without the formation of multi-sialylated compounds [62].
7. Chemoenzymatic synthesis of glycolipids with facile purification
Considerable efforts have been made recently toward simplifying the purification of oligosaccharide products from chemoenzymatic synthetic reactions [63]. Inspired by using hydrocarbon-conjugated glycans for enzyme activity assays of glycosyltransferases [64], light perfluoroaklyl tags have been used to facilitate the purification of the oligosaccharide products by fluorous solid-phase extraction (FSPE), allowing non-tagged reaction components, such as nucleotides, sugar nucleotides, and monosaccharides to be washed away with water before eluting the tagged product and any remaining glycosyltransferase acceptor with methanol. A triethylene glycol (TEG) or a hexa-ethylene glycol (HEG) linker in C8F17- or the C6F13-tagged glycosyltransferase acceptor was found to improve their compatibility with enzymatic glycosylation reactions [65]. More recently, a sulfo-fluorous tag with a photo-cleavable linker was found to improve the water solubility of fluorous-tagged glycosyltransferase acceptors to achieve high-yields in enzymatic glycosylation reactions while facilitate the product purification by fluorous solid-phase extraction (FSPE) [66].
The hydrophobic tail is an intrinsic component of naturally existing glycolipids. It can be used to facilitate the product purification from chemoenzymatic synthetic reactions without the need to remove it afterwards. Therefore, glycolipids can be readily synthesized by enzyme-catalyzed reactions with facile product purification by C18-cartridges. Such strategy has been successfully applied for the synthesis and purification of GSLs, a class of biologically important glycolipids found in the cell membranes of various organisms. To do this, lactosyl sphingosine (LacβSph) was chemically synthesized from inexpensive starting materials including lactose and phytosphingosine. It was then used readily in one-pot multienzyme (OPME) glycosylation reactions that can be carried out in sequential for building up a large library of glycosyl sphingosines which upon one-step acylation, leading to the formation of desired GSLs. In contrast to glycosylceramide with short glycan chains which have low water solubility, the glycosyl sphingosines are soluble in water and are compatible with enzymatic reactions. The sphingosine component facilitated the facile C18-cartridge-based purification processes (Figure 14). Ganglio- and neolacto-series glycosphingolipids have been successfully obtained using the strategy [67, 68]. Compared to en bloc transfer of oligosaccharides from chemoenzymatically synthesized oligosaccharyl fluorides for the synthesis of glycosphingolipids and glycosphingosines containing a sphingoid base or its derivative using endoglycoceramidase mutants (endoglycoceramidase glycosynthase) [69, 70], the OPME glycosylation of LacβSph strategy is more flexible in accessing a more diverse array of products and also provides an easier C18 cartridge-purification process.
Figure 14.

Efficient chemoenzymatic strategies for synthesizing complex glycosphingolipids by enzymatic extension of lactosyl sphingosine using OPME reactions followed by acylation reaction.
8. Solid phase-assisted chemoenzymatic synthesis and purification strategies
Carbohydrate biosynthetic enzymes have been immobilized on solid supports to improve their stability and allow their reuse in the synthesis [71-75]. To permit facile product purification which is a critical factor to consider for automated synthesis of carbohydrates and glycoconjugates, substrate/product immobilization is a more attractive approach [63]. Substrate/product immobilization on a solid phase support can be done before or after enzymatic reactions. The latter allows enzymatic reactions to be carried out in a solution phase with facile solid-phase purification. Other than attaching a hydrophobic tail similar to the strategies discussed above for the synthesis of fluorous/hydrocarbon tagged-oligosaccharides and glycosphingolipids, the use of thermo-responsive water soluble polymer support [76] and cationic aglycon-containing substrates [77] are attractive approaches. Aspects for consideration towards automated enzymatic synthesis of carbohydrates were detailed recently in an excellent review with discussion on the availability and the choice of enzymes, glycosyltransferase donors, solid-phase supports, linkers and spacers, and instruments [63]. Much progresses have been made and it is expected that chemoenzymatic synthesis of carbohydrates can be automated to allow the access of targets by even non-specialists.
9. Conclusions and future perspectives
Innovations in chemoenzymatic approaches for glycan synthesis are granting access to carbohydrates with increasing structural complexity. The use of OPME systems with chemically modified monosaccharides, strategically designed synthetic routes, protective groups introduced onto acceptor substrates, synthons of glycosyltransferase donor precursors for targets containing exotic sugars, tags to facilitate purifications, and other strategies described here are important for the synthesis of structurally diverse carbohydrates including those containing PGMs, asymmetric branches, and/or repeating motifs. The introduction of selective glycoside hydrolases into OPME systems opens access to the production of defined compounds with demonstrated value as therapeutic sialidase inhibitors and potential prebiotics. Wild-type enzymes can be cloned and characterized from different sources to identify suitable candidates for the synthesis of desired target compounds. Crystal structure-guided mutagenesis and screening is an effective approach to obtain mutants of desired properties if wild-type enzymes with desired functions are not readily available. Chemoenzymatic synthesis of oligosaccharide building blocks can be used for additional chemical glycosylation for the formation of more complex structures. When glycosyltransferases and other carbohydrate biosynthetic enzymes with desired substrate promiscuity are not available, suitable chemically synthesized synthons can be used as the substrates for enzyme-catalyzed version of intermediates which can be converted to the desired targets chemically. Progresses made on strategies to facilitate product purifications can be combined with advances on chemoenzymatic synthesis to access the enormous chemical space of naturally occurring and non-natural derivatives of carbohydrates and glycoconjugates not only by experts in the carbohydrate field but also by non-specialists. Lack of sufficient amounts of glycosyltransferases for the formation of desired glycosidic bonds and sugar nucleotide biosynthetic enzymes for producing nucleotide-activated uncommon sugars remains to be the key challenge for chemoenzymatic synthesis. Methods to improve the expression level and increase the stability of highly active enzymes are in high demand. Continuous efforts are needed for crystal structure studies of synthetic useful enzymes which will help to design synthetic useful mutants with improved properties. A clearer landscape of glycomes is also needed to define the chemoenzymatic synthetic targets. Therefore, combined efforts of glycan analysis, enzyme discovery and characterization, protein crystallography and mutagenesis, synthetic method development, and functional application of carbohydrate products will greatly advance the carbohydrate chemoenzymatic synthesis and also the overall glycoscience field.
Highlights.
Recent glycosyltransferase-dependent chemoenzymatic strategies are summarized
One-pot multienzyme (OPME) glycosylation systems are highly efficient
Enzymatically synthesized building blocks can be used for chemical glycosylation
Identifying suitable enzymes and mutants is the key to success
Strategies for regioselective glycosylation and facile purification are discussed
Acknowledgements
The authors would like to acknowledge the financial supports from the United State National Institutes of Health under Award Numbers U01GM120419, U01GM125288, and R01AI130684. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Varki A, Kornfeld S, in: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, Seeberger PH (Eds.) Essentials of Glycobiology, Cold Spring Harbor (NY: ), 2015, pp. 1–18. [PubMed] [Google Scholar]
- [2].Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG, The endothelial glycocalyx: composition, functions, and visualization, Pflugers Arch. 454 (2007) 345–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Varki A, Biological roles of oligosaccharides: all of the theories are correct, Glycobiology 3 (1993) 97–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hanson S, Best M, Bryan MC, Wong C-H, Chemoenzymatic synthesis of oligosaccharides and glycoproteins, Trends Biochem. Sci 29 (2004) 656–663. [DOI] [PubMed] [Google Scholar]
- [5].Gloster TM, Development of inhibitors as research tools for carbohydrate-processing enzymes, Biochem. Soc. Trans 40 (2012) 913–928. [DOI] [PubMed] [Google Scholar]
- [6].Yu H, Chen X, Carbohydrate post-glycosylational modifications, Org. Biomol. Chem 5 (2007) 865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Muthana SM, Campbell CT, Gildersleeve JC, Modifications of glycans: biological significance and therapeutic opportunities, ACS Chem. Biol 7 (2012) 31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gabius HJ, Biological information transfer beyond the genetic code: the sugar code, Naturwissenschaften 87 (2000) 108–121. [DOI] [PubMed] [Google Scholar]
- [9].Gabius HJ, The sugar code: Why glycans are so important, Biosystems 164 (2018) 102–111. [DOI] [PubMed] [Google Scholar]
- [10].Hayes MR, Pietruszka J, Synthesis of glycosides by glycosynthases, Molecules 22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yamamoto K, Recent advances in glycotechnology for glycoconjugate synthesis using microbial endoglycosidases, Biotechnol. Lett 35 (2013) 1733–1743. [DOI] [PubMed] [Google Scholar]
- [12].Li C, Wang LX, Chemoenzymatic methods for the synthesis of glycoproteins, Chem. Rev 118 (2018) 8359–8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Koeller KM, Wong CH, Synthesis of complex carbohydrates and glycoconjugates: enzyme-based and programmable one-pot strategies, Chem. Rev 100 (2000) 4465–4494. [DOI] [PubMed] [Google Scholar]
- [14].Yu H, Chen X, One-pot multienzyme (OPME) systems for chemoenzymatic synthesis of carbohydrates, Org. Biomol. Chem 14 (2016) 2809–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen X, Varki A, Advances in the biology and chemistry of sialic acids, ACS Chem. Biol 5 (2010) 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chokhawala HA, Cao H, Yu H, Chen X, Enzymatic synthesis of fluorinated mechanistic probes for sialidases and sialyltransferases, J. Am. Chem. Soc 129 (2007) 10630–10631. [DOI] [PubMed] [Google Scholar]
- [17].Yu H, Chokhawala H, Karpel R, Yu H, Wu B, Zhang J, Zhang Y, Jia Q, Chen X, A multifunctional Pasteurella multocida sialyltransferase: a powerful tool for the synthesis of sialoside libraries, J. Am. Chem. Soc 127 (2005) 17618–17619. [DOI] [PubMed] [Google Scholar]
- [18].Yu H, Huang S, Chokhawala H, Sun M, Zheng H, Chen X, Highly efficient chemoenzymatic synthesis of naturally occurring and non-natural alpha-2,6-linked sialosides: a P. damsela alpha-2,6-sialyltransferase with extremely flexible donor-substrate specificity, Angew. Chem. Int. Ed. Engl 45 (2006) 3938–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Song X, Yu H, Chen X, Lasanajak Y, Tappert MM, Air GM, Tiwari VK, Cao H, Chokhawala HA, Zheng H, Cummings RD, Smith DF, A sialylated glycan microarray reveals novel interactions of modified sialic acids with proteins and viruses, J. Biol. Chem 286 (2011) 31610–31622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yu H, Yu H, Karpel R, Chen X, Chemoenzymatic synthesis of CMP-sialic acid derivatives by a one-pot two-enzyme system: comparison of substrate flexibility of three microbial CMP-sialic acid synthetases, Bioorg. Med. Chem 12 (2004) 6427–6435. [DOI] [PubMed] [Google Scholar]
- [21].Li Y, Yu H, Cao H, Lau K, Muthana S, Tiwari VK, Son B, Chen X, Pasteurella multocida sialic acid aldolase: a promising biocatalyst, Appl. Microbiol. Biotechnol 79 (2008) 963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li Y, Yu H, Cao H, Muthana S, Chen X, Pasteurella multocida CMP-sialic acid synthetase and mutants of Neisseria meningitidis CMP-sialic acid synthetase with improved substrate promiscuity, Appl. Microbiol. Biotechnol 93 (2012) 2411–2423. [DOI] [PubMed] [Google Scholar]
- [23].Ding L, Yu H, Lau K, Li Y, Muthana S, Wang J, Chen X, Efficient chemoenzymatic synthesis of sialyl Tn-antigens and derivatives, Chem. Commun 47 (2011) 8691–8693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yu H, Zeng J, Li Y, Thon V, Shi B, Chen X, Effective one-pot multienzyme (OPME) synthesis of monotreme milk oligosaccharides and other sialosides containing 4-O-acetyl sialic acid, Org. Biomol. Chem 14 (2016) 8586–8597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lau K, Yu H, Thon V, Khedri Z, Leon ME, Tran BK, Chen X, Sequential two-step multienzyme synthesis of tumor-associated sialyl T-antigens and derivatives, Org. Biomol. Chem 9 (2011) 2784–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Khedri Z, Li Y, Muthana S, Muthana MM, Hsiao CW, Yu H, Chen X, Chemoenzymatic synthesis of sialosides containing C7-modified sialic acids and their application in sialidase substrate specificity studies, Carbohydr. Res 389 (2014) 100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Santra A, Xiao A, Yu H, Li W, Li Y, Ngo L, McArthur JB, Chen X, A diazido mannose analogue as a chemoenzymatic synthon for synthesizing di-N-acetyllegionaminic acid-containing glycosides, Angew. Chem. Int. Ed. Engl 57 (2018) 2929–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yu H, Cao H, Tiwari VK, Li Y, Chen X, Chemoenzymatic synthesis of C8-modified sialic acids and related alpha2-3- and alpha2-6-linked sialosides, Bioorg. Med. Chem. Lett 21 (2011) 5037–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Khedri Z, Muthana MM, Li Y, Muthana SM, Yu H, Cao H, Chen X, Probe sialidase substrate specificity using chemoenzymatically synthesized sialosides containing C9-modified sialic acid, Chem. Commun 48 (2012) 3357–3359. [DOI] [PubMed] [Google Scholar]
- [30].Li W, Xiao A, Li Y, Yu H, Chen X, Chemoenzymatic synthesis of Neu5Ac9NAc-containing alpha2-3- and alpha2-6-linked sialosides and their use for sialidase substrate specificity studies, Carbohydr. Res 451 (2017) 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yu H, Cheng J, Ding L, Khedri Z, Chen Y, Chin S, Lau K, Tiwari VK, Chen X, Chemoenzymatic synthesis of GD3 oligosaccharides and other disialyl glycans containing natural and non-natural sialic acids, J. Am. Chem. Soc 131 (2009) 18467–18477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xu G, Kiefel MJ, Wilson JC, Andrew PW, Oggioni MR, Taylor GL, Three Streptococcus pneumoniae sialidases: three different products, J. Am. Chem. Soc 133 (2011) 1718–1721. [DOI] [PubMed] [Google Scholar]
- [33].Crost EH, Tailford LE, Monestier M, Swarbreck D, Henrissat B, Crossman LC, Juge N, The mucin-degradation strategy of Ruminococcus gnavus: The importance of intramolecular trans-sialidases, Gut Microbes 7 (2016) 302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xiao A, Slack TJ, Li Y, Shi D, Yu H, Li W, Liu Y, Chen X, Streptococcus pneumoniae Sialidase SpNanB-catalyzed one-pot multienzyme (OPME) synthesis of 2,7-anhydro-sialic acids as selective sialidase inhibitors, J. Org. Chem 83 (2018) 10798–10804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Xiao A, Li Y, Li X, Santra A, Yu H, Li W, Chen X, Sialidase-catalyzed one-pot multienzyme (OPME) synthesis of sialidase transition-state analogue inhibitors, ACS Catal 8 (2018) 43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cao H, Huang S, Cheng J, Li Y, Muthana S, Son B, Chen X, Chemical preparation of sialyl Lewis x using an enzymatically synthesized sialoside building block, Carbohydr. Res 343 (2008) 2863–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yao W, Yan J, Chen X, Wang F, Cao H, Chemoenzymatic synthesis of lacto-N-tetrasaccharide and sialyl lacto-N-tetrasaccharides, Carbohydr. Res 401 (2015) 5–10. [DOI] [PubMed] [Google Scholar]
- [38].Yao Q, Song J, Xia C, Zhang W, Wang PG, Chemoenzymatic syntheses of iGb3 and Gb3, Org. Lett 8 (2006) 911–914. [DOI] [PubMed] [Google Scholar]
- [39].Liu Y, Wen L, Li L, Gadi MR, Guan W, Huang K, Xiao Z, Wei M, Ma C, Zhang Q, Yu H, Chen X, Wang PG, Fang J, A general chemoenzymatic strategy for the synthesis of glycosphingolipids, Eur. J. Org. Chem 2016 (2016) 4315–4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lairson LL, Watts AG, Wakarchuk WW, Withers SG, Using substrate engineering to harness enzymatic promiscuity and expand biological catalysis, Nat. Chem. Biol 2 (2006) 724–728. [DOI] [PubMed] [Google Scholar]
- [41].Galan MC, Venot AP, Glushka J, Imberty A, Boons G-J, α-(2, 6)-Sialyltransferase-catalyzed sialylations of conformationally constrained oligosaccharides, J. Am. Chem. Soc 124 (2002) 5964–5973. [DOI] [PubMed] [Google Scholar]
- [42].Galan MC, Venot AP, Glushka J, Imberty A, Boons G-J, Chemo-enzymatic synthesis of conformationally constrained oligosaccharides, Org. Biomol. Chem 1 (2003) 3891–3899. [DOI] [PubMed] [Google Scholar]
- [43].Meng X, Yao W, Cheng J, Zhang X, Jin L, Yu H, Chen X, Wang F, Cao H, Regioselective chemoenzymatic synthesis of ganglioside disialyl tetrasaccharide epitopes, J. Am. Chem. Soc 136 (2014) 5205–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Muthana S, Yu H, Huang S, Chen X, Chemoenzymatic synthesis of size-defined polysaccharides by sialyltransferase-catalyzed block transfer of oligosaccharides, J. Am. Chem. Soc 129 (2007) 11918–11919. [DOI] [PubMed] [Google Scholar]
- [45].Wang Z, Chinoy ZS, Ambre SG, Peng W, McBride R, de Vries RP, Glushka J, Paulson JC, Boons GJ, A general strategy for the chemoenzymatic synthesis of asymmetrically branched N-glycans, Science 341 (2013) 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Shivatare SS, Chang SH, Tsai TI, Tseng SY, Shivatare VS, Lin YS, Cheng YY, Ren CT, Lee CC, Pawar S, Tsai CS, Shih HW, Zeng YF, Liang CH, Kwong PD, Burton DR, Wu CY, Wong CH, Modular synthesis of N-glycans and arrays for the hetero-ligand binding analysis of HIV antibodies Modular synthesis of N-glycans and arrays for the hetero-ligand binding analysis of HIV antibodies, Nat. Chem 8 (2016) 338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gagarinov IA, Li T, Torano JS, Caval T, Srivastava AD, Kruijtzer JA, Heck AJ, Boons GJ, Chemoenzymatic approach for the preparation of asymmetric bi-, tri-, and tetra-antennary N-glycans from a common precursor, J. Am. Chem. Soc 139 (2017) 1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Prudden AR, Liu L, Capicciotti CJ, Wolfert MA, Wang S, Gao Z, Meng L, Moremen KW, Boons GJ, Synthesis of asymmetrical multiantennary human milk oligosaccharides, Proc. Natl. Acad. Sci. U. S. A. 114 (2017) 6954–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yu H, Lau K, Thon V, Autran CA, Jantscher-Krenn E, Xue M, Li Y, Sugiarto G, Qu J, Mu S, Ding L, Bode L, Chen X, Synthetic disialyl hexasaccharides protect neonatal rats from necrotizing enterocolitis, Angew. Chem. Int. Ed. Engl 53 (2014) 6687–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yu H, Yan X, Autran CA, Li Y, Etzold S, Latasiewicz J, Robertson BM, Li J, Bode L, Chen X, Enzymatic and chemoenzymatic syntheses of disialyl glycans and their necrotizing enterocolitis preventing effects, J. Org. Chem 82 (2017) 13152–13160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chen C, Zhang Y, Xue M, Liu XW, Li Y, Chen X, Wang PG, Wang F, Cao H, Sequential one-pot multienzyme (OPME) synthesis of lacto-N-neotetraose and its sialyl and fucosyl derivatives, Chem. Commun 51 (2015) 7689–7692. [DOI] [PubMed] [Google Scholar]
- [52].Liu R, Xu Y, Chen M, Weiwer M, Zhou X, Bridges AS, DeAngelis PL, Zhang Q, Linhardt RJ, Liu J, Chemoenzymatic design of heparan sulfate oligosaccharides, J. Biol. Chem 285 (2010) 34240–34249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chen Y, Li Y, Yu H, Sugiarto G, Thon V, Hwang J, Ding L, Hie L, Chen X, Tailored design and synthesis of heparan sulfate oligosaccharide analogues using sequential one-pot multienzyme systems, Angew. Chem. Int. Ed. Engl 52 (2013) 11852–11856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sala RF, MacKinnon SL, Palcic MM, Tanner ME, UDP-W-trifluoroacetylglucosamine as an alternative substrate in N-acetylglucosaminyltransferase reactions, Carbohydr. Res 306 (1998) 127–136. [DOI] [PubMed] [Google Scholar]
- [55].Chen Y, Thon V, Li Y, Yu H, Ding L, Lau K, Qu J, Hie L, Chen X, One-pot three-enzyme synthesis of UDP-GlcNAc derivatives, Chem. Commun 47 (2011) 10815–10817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].McArthur JB, Chen X, Glycosyltransferase engineering for carbohydrate synthesis, Biochem. Soc. Trans 44 (2016) 129–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ni L, Sun M, Yu H, Chokhawala H, Chen X, Fisher AJ, Cytidine 5'-monophosphate (CMP)-induced structural changes in a multifunctional sialyltransferase from Pasteurella multocida, Biochemistry, 45 (2006) 2139–2148. [DOI] [PubMed] [Google Scholar]
- [58].Ni L, Chokhawala HA, Cao H, Henning R, Ng L, Huang S, Yu H, Chen X, Fisher AJ, Crystal structures of Pasteurella multocida sialyltransferase complexes with acceptor and donor analogues reveal substrate binding sites and catalytic mechanism, Biochemistry 46 (2007) 6288–6298. [DOI] [PubMed] [Google Scholar]
- [59].Sugiarto G, Lau K, Qu J, Li Y, Lim S, Mu S, Ames JB, Fisher AJ, Chen X, A sialyltransferase mutant with decreased donor hydrolysis and reduced sialidase activities for directly sialylating LewisX, ACS Chem. Biol 7 (2012) 1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ding L, Zhao C, Qu J, Li Y, Sugiarto G, Yu H, Wang J, Chen X, A Photobacterium sp. α2–6-sialyltransferase (Psp2, 6ST) mutant with an increased expression level and improved activities in sialylating Tn antigens, Carbohydr. Res 408 (2015) 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].McArthur JB, Yu H, Zeng J, Chen X, Converting Pasteurella multocida alpha2-3-sialyltransferase 1 (PmST1) to a regioselective alpha2-6-sialyltransferase by saturation mutagenesis and regioselective screening, Org. Biomol. Chem 15 (2017) 1700–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Xu Y, Fan Y, Ye J, Wang F, Nie Q, Wang L, Wang PG, Cao H, Cheng J, Successfully engineering a bacterial sialyltransferase for regioselective α2,6-sialylation, ACS Catal. 8 (2018) 7222–7227. [Google Scholar]
- [63].Wen L, Edmunds G, Gibbons C, Zhang J, Gadi MR, Zhu H, Fang J, Liu X, Kong Y, Wang PG, Toward automated enzymatic synthesis of oligosaccharides, Chem. Rev 118 (2018) 8151–8187. [DOI] [PubMed] [Google Scholar]
- [64].Palcic MM, Heerze LD, Pierce M, Hindsgaul O, The use of hydrophobic synthetic glycosides as acceptors in glycosyltransferase assays, Glycoconj. J. 5 (1988) 49–63. [Google Scholar]
- [65].Hwang J, Yu H, Malekan H, Sugiarto G, Li Y, Qu J, Nguyen V, Wu D, Chen X, Highly efficient one-pot multienzyme (OPME) synthesis of glycans with fluorous-tag assisted purification, Chem. Commun 50 (2014) 3159–3162. [DOI] [PubMed] [Google Scholar]
- [66].Hou K-L, Chiang P-Y, Lin C-H, Li B-Y, Chien W-T, Huang Y-T, Yu C-C, Lin C-C, Water-soluble sulfo-fluorous affinity (SOFA) tag-assisted enzymatic synthesis of oligosaccharides, Adv. Synth. Catal 360 (2018) 2313–2323. [Google Scholar]
- [67].Santra A, Li Y, Yu H, Slack TJ, Wang PG, Chen X, Highly efficient chemoenzymatic synthesis and facile purification of alpha-Gal pentasaccharyl ceramide Galalpha3nLc4betaCer, Chem. Commun 53 (2017) 8280–8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Yu H, Santra A, Li Y, McArthur JB, Ghosh T, Yang X, Wang PG, Chen X, Streamlined chemoenzymatic total synthesis of prioritized ganglioside cancer antigens, Org. Biomol. Chem 16 (2018) 4076–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Vaughan MD, Johnson K, DeFrees S, Tang X, Warren RA, Withers SG, Glycosynthase-mediated synthesis of glycosphingolipids, J. Am. Chem. Soc 128 (2006) 6300–6301. [DOI] [PubMed] [Google Scholar]
- [70].Rich JR, Cunningham AM, Gilbert M, Withers SG, Glycosphingolipid synthesis employing a combination of recombinant glycosyltransferases and an endoglycoceramidase glycosynthase, Chem. Commun 47 (2011) 10806–10808. [DOI] [PubMed] [Google Scholar]
- [71].Nahalka J, Liu Z, Chen X, Wang PG, Superbeads: immobilization in “sweet”chemistry, Chemistry 9 (2003) 372–377. [DOI] [PubMed] [Google Scholar]
- [72].Chen X, Fang J, Zhang J, Liu Z, Shao J, Kowal P, Andreana P, Wang PG, Sugar nucleotide regeneration beads (superbeads): a versatile tool for the practical synthesis of oligosaccharides, J. Am. Chem. Soc 123 (2001) 2081–2082. [DOI] [PubMed] [Google Scholar]
- [73].Dulik DM, Fenselau C, Use of immobilized enzymes in drug metabolism studies, FASEB J. 2(1988)2235–2240. [DOI] [PubMed] [Google Scholar]
- [74].Datta S, Christena LR, Rajaram YR, Enzyme immobilization: an overview on techniques and support materials, 3 Biotech. 3 (2013) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Fiebig T, Litschko C, Freiberger F, Bethe A, Berger M, Gerardy-Schahn R, Efficient solid-phase synthesis of meningococcal capsular oligosaccharides enables simple and fast chemoenzymatic vaccine production, J. Biol. Chem 293 (2018) 953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Huang X, Witte KL, Bergbreiter DE, Wong C-H, Homogenous enzymatic synthesis using a thermo-responsive water-soluble polymer support, Adv. Synth. Catal 343 (2001) 675–681. [Google Scholar]
- [77].Zhu H, Wu Z, Gadi MR, Wang S, Guo Y, Edmunds G, Guan W, Fang J, Cation exchange assisted binding-elution strategy for enzymatic synthesis of human milk oligosaccharides (HMOs), Bioorg. Med. Chem. Lett. 27 (2017) 4285–4287. [DOI] [PubMed] [Google Scholar]