

Abstract
In Alzheimer’s disease (AD), deposition of insoluble amyloid-β (Aβ) is followed by intracellular aggregation of tau in the neocortex and subsequent neuronal cell loss, synaptic loss, brain atrophy, and cognitive impairment. By the time even the earliest clinical symptoms are detectable, Aβ accumulation is close to reaching its peak and neocortical tau pathology is frequently already present. The period in which AD pathology is accumulating in the absence of cognitive symptoms and represents a clinically relevant time window for therapeutic intervention. Sleep is increasingly recognized as a potential marker for AD pathology and future risk of cognitive impairment. Previous studies in animal models and humans have associated decreased non-rapid eye movement (NREM) sleep slow wave activity (SWA) with Aβ deposition. In this study, we analyzed cognitive performance, brain imaging, and cerebrospinal fluid (CSF) AD biomarkers in participants enrolled in longitudinal studies of aging. In addition, we monitored their sleep using a single-channel electroencephalography (EEG) device worn on the forehead. After adjusting for multiple covariates such as age and sex, we found that NREM SWA showed an inverse relationship with AD pathology, particularly tauopathy, and that this association was most evident at the lowest frequencies of NREM SWA. Given that our study participants were predominantly cognitively normal, this suggested that changes in NREM SWA, especially at 1–2 Hz, might be able to discriminate tau pathology and cognitive impairment either before or at the earliest stages of symptomatic AD.
One Sentence Summary:
Slow wave activity during non-rapid eye movement sleep decreases with disease progression in patients with Alzheimer’s disease.
Introduction
Aggregation of amyloid-β (Aβ) into oligomers and fibrils that are present in extracellular Aβ plaques in the brain is a key early step in Alzheimer’s disease (AD) pathogenesis and begins to occur ~15–20 years before the onset of cognitive decline (1). The build-up of insoluble Aβ is followed by the intracellular aggregation of tau and its spread from the medial temporal lobe to different neocortical regions (1, 2). Localized tau aggregation in the medial temporal lobe during normal aging is probably independent of Aβ; however, in AD, its spread to the neocortex appears to be downstream from Aβ build-up and correlates strongly with neuronal cell loss, synaptic loss, brain atrophy, and cognitive impairment. These findings are strongly supported by genetic, pathological, and biomarker data in both sporadic and inherited AD (1, 2). By the time even the earliest clinical symptoms of AD are detectable, Aβ accumulation is close to reaching its peak, and there is almost always some neocortical tau pathology (3). Aβ42, the isoform of Aβ most prone to aggregate in insoluble plaques, decreases in cerebrospinal fluid (CSF) with brain amyloid deposition and correlates with amyloid PET (4). The CSF tau/Aβ42 ratio is related to the dual effect of amyloid and tau pathology and predicts conversion to early symptomatic AD (5–7). Importantly, there is also neuronal and synaptic loss in several brain regions relevant to memory and thinking (3). The period in which AD pathology is accumulating in the absence of cognitive symptoms has been termed “preclinical” AD (8, 9).
A bi-directional relationship between sleep and AD has been proposed based on studies in animal models and humans (10–12). Numerous studies have shown that sleep-wake activity is disturbed in individuals with dementia due to AD (13, 14). Sleep disturbance has been measured via self-report, such as with questionnaires and sleep logs, as well as actigraphy and polysomnography. Increasing evidence also supports sleep disturbance as a marker for AD pathology and future risk of cognitive impairment (15–22). For instance, self-reported sleep disturbances, such as poor sleep quality and short sleep duration, have been associated with increased risk of cognitive impairment (15) and increased Aβ deposition on [11C]-Pittsburgh Compound B (PiB) positron emission tomography (PET) scans (16). Further, excessive daytime sleepiness reported by a cohort of older adults was associated with increased longitudinal Aβ accumulation on PiB-PET scans (17). Sleep logs and actigraphy monitoring have found that reduced sleep efficiency and increased nap frequency in cognitively normal individuals were associated with Aβ deposition (18). Studies with polysomnography have associated increased risk of cognitive impairment in older adults with sleep-disordered breathing (19–21) and periodic limb movements during sleep (PLMS) (22).
In addition to sleep disturbance as a putative marker of AD pathology, evidence also supports the hypothesis that disturbed sleep increases AD risk, at least in part, via an Aβ mechanism (11). We have found that Aβ concentrations in CSF fluctuate with sleep-wake activity in both mice (23) and humans (24). This Aβ cycling pattern has been replicated in multiple studies (25) and assays (26). Aβ concentrations are directly regulated by neuronal activity (27–31) and evidence in mice suggests that decreased interstitial fluid (ISF) during sleep results, at least in part, from altered neuronal/metabolic activity decreasing Aβ production/release. In humans, we have recently shown that targeted slow wave sleep disruption (32) and sleep deprivation (33) will increase overnight CSF Aβ concentrations by 10–30% most likely due to increased Aβ production/release. There is also evidence that Aβ clearance is increased during sleep due to increased ISF bulk flow (for example “glymphatic” clearance) (34).
Studies in animal models using electroencephalography (EEG) to monitor different sleep stages has found changes in sleep parameters and EEG power linked with both Aβ and tau pathology. For instance, Aβ deposition in APPswe/PS1δE9 mice lead to disruption of the sleep-wake cycle (35), whereas increasing tauopathy in P301S tau transgenic mice was associated with decreased time in rapid eye movement (REM) and non-REM (NREM) sleep, increased wakefulness, and decreased NREM slow wave activity (SWA) (36). In humans, atrophy and Aβ accumulation in the medial prefrontal cortex (mPFC) were correlated with both decreased NREM SWA and impaired overnight hippocampus-dependent memory consolidation in cognitively normal older adults (37, 38). Although that cross-sectional study provides associative evidence between Aβ deposition, NREM sleep disruption, and memory impairment, tau pathology was not assessed. Longitudinal studies with AD biomarkers and cognitive evaluations are needed to establish both the sequential links between these events and causation (39), especially in relation to both Aβ and tau.
In this study, we monitored sleep-wake activity in one hundred and nineteen participants enrolled in longitudinal studies of aging at the Knight Alzheimer’s Disease Research Center at Washington University. Sleep-wake activity was monitored over six nights with a single-channel EEG worn on the forehead (Sleep Profiler, Advanced Brain Monitoring), actigraphy (Actiwatch2, Phillips Respironics), and sleep logs. In addition, each participant was assessed for sleep-disordered breathing and periodic leg movements with a home sleep test (Alice PDX, Phillips Respironics). Participants who underwent cognitive testing, apolipoprotein E (ApoE) genotyping, and assessment of AD biomarkers in CSF (Aβ42, tau, phosphorylated tau (p-tau)) or PET scans with [18F]-AV-45 (florbetapir) amyloid and [18F]-AV-1451 (flortaucipir) tau tracers were included in the analyses. Since tau pathology but not Aβ pathology is best associated with cognitive decline in AD, we hypothesized that decreased NREM SWA would be associated with increased tau pathology.
Results
One hundred and nineteen participants aged >60 years old enrolled in longitudinal studies of aging at the Knight Alzheimer’s Disease Research Center at Washington University in St Louis, MO were recruited for the study. Cognitive performance was evaluated by the Clinical Dementia Rating (CDR) (40, 41). Participants also underwent AV-45 amyloid and AV-1451 tau PET imaging and/or lumbar puncture to measure CSF Aβ42, tau, and p-tau concentrations. Sleep monitoring was performed for up to 6 nights with sleep logs, actigraphy, and a single-channel EEG. Average sleep parameters for participants with PET imaging and CSF biomarkers were not substantially different between amyloid-negative vs. amyloid-positive, tau-negative vs. tau-positive, and CDR 0 vs. CDR 0.5 groups regardless of modality used to measure sleep-wake activity (Tables S1 and S2). In participants with PET imaging, REM latency was lower in amyloid-positive participants (t(36)=2.98, p=0.005) but longer in CDR 0.5 individuals (t(36)=−2.49, p=0.018). Amyloid-positive participants had lower wake after sleep onset (WASO) measured by actigraphy (t(35)=2.07, p=0.046) whereas sleep onset latency measured by actigraphy was prolonged in CDR 0.5 individuals (t(35)=−2.33, p=0.026). For participants with CSF, CDR 0.5 individuals were found to have longer REM latency measured by EEG (t(104)=−2.91, p=0.0044) and longer self-reported total sleep time (TST) (t(104)=−2.27, p=0.025). When measured by actigraphy, sleep efficiency was decreased (t(106)=3.40, p=0.0009), sleep onset latency was prolonged (t(106)=−3.86, p=0.0002), and WASO was greater (t(106)=−2.68, p=0.0086) in CDR 0.5 participants compared to CDR 0.
Thirty-eight participants with AV-45 amyloid and AV-1451 tau PET imaging and one hundred and four participants with CSF Aβ42, tau, and p-tau underwent monitoring with the single-channel EEG device. Twenty-seven participants had both PET imaging and lumbar punctures. Characteristics for all participants are provided in Table 1. Of the participants with PET imaging, 52.6% (20/38) of participant were amyloid- and tau-negative with 9 participant amyloid-positive but tau-negative and 8 participants were positive for both amyloid and tau (Table 1). One participant was found to be tau-positive but amyloid-negative. Amyloid negative/positive status was set at a standardized uptake value ratio (SUVR) of 1.19 (42, 43) and tau negative/positive status was set at a SUVR of 1.22 (44). Average amyloid and tau burden on PET are shown in Figures 1 and 2. For participants with CSF, previously published cutoffs for amyloid-positive (CSF Aβ42<1098 pg/ml) and tau-positive (CSF tau>242 pg/ml) were used to define AD pathology (45). Of the participants with CSF, 29.8% were negative for amyloid and tau, 33.7% were amyloid-positive but tau-negative, 25.9% were positive for both amyloid and tau, and 10.6% were amyloid-negative but tau-positive (Table 1).
Table 1:
Participant characteristics
| PET Imaging | CSF | |||
|---|---|---|---|---|
| Mean (SD) / n (%) (N=38) |
Mean (SD) / n (%) (N=104) |
|||
| Age (years) | 73.8 (5.3) | 74.57 (5.20) | ||
| Sex | ||||
| Men | 18 (47.4) | 59 (56.7) | ||
| Women | 20 (52.6) | 45 (43.3) | ||
| Race | ||||
| African-American | 3 (7.9) | 11 (10.6) | ||
| Caucasian | 35 (92.1) | 92 (88.5) | ||
| Asian | 0 (0.0) | 0 (0.0) | ||
| More than one | 0 (0.0) | 1 (1.0) | ||
| CDR | ||||
| 0 | 29 (76.3) | 83 (79.8) | ||
| 0.5 | 9 (23.7) | 21 (20.2) | ||
| ApoE4 | ||||
| Negative | 25 (65.8) | 58 (55.8) | ||
| Positive | 13 (34.2) | 45 (43.3) | ||
| Sleep Medications | ||||
| Yes | 4 (10.5) | 12 (11.5) | ||
| No | 34 (89.5) | 92 (88.5) | ||
| Apnea-Hypopnea Index* (AHI, respiratory events/hour) |
Negative (AHI<5) | 20 (52.6) | 38 (36.5) | |
|
Mild
(AHI 5–15) |
13 (34.2) | 43 (41.4) | ||
| Moderate (AHI 15–30) | 5 (13.2) | 18 (17.3) | ||
| Severe (AHI>30) | 0 (0) | 5 (4.8) | ||
| Periodic Leg Movement Index (PLMI, leg movements/hour) |
Negative (PLMI<15) | 21 (55.3) | 49 (47.1) | |
| Low (PLMI 15–45) | 7 (18.4) | 33 (31.7) | ||
| High (PLMI>45) | 10 (26.3) | 22 (21.2) | ||
| AV-45 PET SUVR | 1.44 (0.61) | ----------- | ||
| AV-1451 PET SUVR | 1.40 (0.49) | ----------- | ||
| Aβ42 (pg/ml) | ---------- | 1012.59 (367.94) | ||
| t-tau (pg/ml) | ---------- | 245.46 (119.69) | ||
| p-tau (pg/ml) | ---------- | 23.66 (13.46) | ||
| AD Pathology | Amyloid neg / Tau neg | 20 (52.6) | 31 (29.8) | |
| Amyloid pos / Tau neg | 9 (23.7) | 35 (33.7) | ||
| Amyloid pos / Tau pos | 8 (21.1) | 27 (25.9) | ||
| Amyloid neg / Tau pos | 1 (2.6) | 11 (10.6) | ||
| Time Interval from Scan/LP to Sleep Study (years) | AV-45 PET | 0.29 (0.48) |
1.00 (2.60) | |
| AV-1451 PET | 0.29 (0.40) |
|||
For the participants who underwent PET imaging (N=38), 4/38 participants used continuous positive airway pressure therapy during sleep monitoring. For participants with CSF (N=104), 12/104 participants used continuous positive airway pressure therapy and 1 participant used lateral position therapy device during sleep monitoring.
SD: standard deviation; CDR: clinical dementia rating; PET: positron emission tomography; CSF: cerebrospinal fluid; SUVR: standardized uptake value ratio; Aβ: amyloid-β; ApoE4: apolipoprotein E4; AD: Alzheimer’s disease; LP: lumbar puncture
Figure 1: Mean amyloid pathology for the 38 participants with PET imaging.
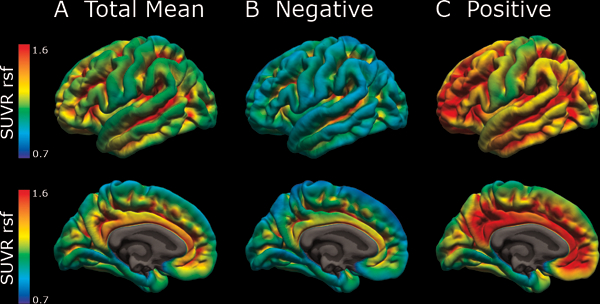
Mean AV-45 amyloid pathology in (A) all, (B) amyloid-negative, and (C) amyloid-positive as measured in standard uptake value ratio (SUVR) units after partial volume correction using a regional spread function.
Figure 2: Mean tau pathology for the 38 participants with PET imaging.
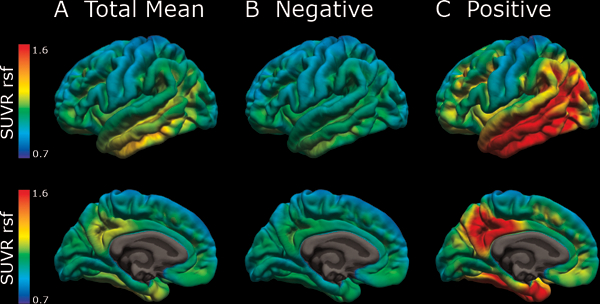
Mean AV-1451 tau pathology in (A) all, (B) tau-negative, and (C) tau-positive subjects as measured in standard uptake value ratio (SUVR) units after partial volume correction using a regional spread function.
Decreased NREM SWA with increased tauopathy
Multiple factors may affect sleep and/or AD pathology including age, sex, and sleep disorders. To assess the relationship between NREM SWA and tau pathology, we performed general linear mixed models of NREM SWA with a mean AV-1451 tau PET composite, age, sex, race, ApoE4 status, CDR, apnea-hypopnea index (AHI), periodic limb movement index (PLMI), and sleep medications. The composite of the mean AV-1451 tau SUVR was determined from the average SUVR of the entorhinal cortex, amygdala, lateral occipital, and lateral temporal regions.
As AV-1451 tau SUVR increased, all-night 1–4.5 Hz SWA was decreased (Table 2). Since previous work found an inverse relationship between the slowest frequencies and amyloid deposition in the mPFC (38), we tested different frequency ranges (1–2 Hz, 2–3 Hz, and 3–4 Hz) in this model and found that this inverse relationship between NREM SWA and tau was maximal in the 1–2 Hz range (Table 2). In our model, time represents longitudinal sleep monitoring over multiple nights. Time was not significant in the model (p>0.05), supporting prior reports that the EEG power measures are stable with small within-subject night-to-night variability (46, 47). Age and sex were also inversely associated with NREM SWA. These findings are not surprising given the well-described decline in NREM SWA with increased age and male sex (48, 49). CDR showed negative association with 1–2 Hz NREM SWA, indicating that 1–2 Hz NREM SWA decreased with worsening tau pathology and cognitive impairment.
Table 2:
Relationship of NREM SWA to AV-1451 tau PET composite after adjusting for age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time
| Dependent Variable |
Co-variate | Estimate | Standard Error |
F statistic (df) |
p-value |
|---|---|---|---|---|---|
| 1–4.5 Hz SWA | Mean AV-1451 tau composite | −8.889 | 2.625 | 11.46 (1,27) | 0.002 |
| Age | −0.670 | 0.243 | 7.59 (1,28) | 0.010 | |
| Sex | −7.850 | 2.636 | 8.87 (1,28) | 0.006 | |
| CDR | −6.882 | 3.312 | 4.32 (1,29) | 0.047 | |
| Race | −9.299 | 4.950 | 3.53 (1,27) | 0.071 | |
| ApoE4 status | +4.002 | 2.891 | 1.92 (1,27) | 0.178 | |
| Sleep medications | +3.225 | 4.468 | 0.52 (1,27) | 0.476 | |
| AHI | +0.141 | 0.166 | 0.73 (1,28) | 0.401 | |
| PLMI | +0.121 | 0.0537 | 5.09 (1,28) | 0.032 | |
| Time | +0.0001 | 0.142 | 0.00 (1,145) | 0.999 | |
| 1–2 Hz SWA | Mean AV-1451 tau composite | −21.477 | 6.123 | 12.30 (1.28) | 0.002 |
| Age | −1.539 | 0.567 | 7.36 (1,28) | 0.011 | |
| Sex | −18.542 | 6.148 | 9.10 (1,28) | 0.005 | |
| CDR | −18.339 | 7.750 | 5.60 (1,29) | 0.025 | |
| Race | −21.620 | 11.537 | 3.51 (1,27) | 0.072 | |
| ApoE4 status | +9.044 | 6.732 | 1.80 (1,27) | 0.190 | |
| Sleep medications | +8.298 | 10.410 | 0.64 (1,27) | 0.432 | |
| AHI | 0.346 | 0.387 | 0.80 (1,28) | 0.379 | |
| PLMI | 0.273 | 0.125 | 4.76 (1,28) | 0.038 | |
| Time | 0.013 | 0.355 | 0.00 (1,146) | 0.970 | |
| 2–3 Hz SWA | Mean AV-1451 tau composite | −5.919 | 1.998 | 8.78 (1,28) | 0.006 |
| Age | −0.509 | 0.186 | 7.51 (1,28) | 0.011 | |
| Sex | −5.577 | 2.009 | 7.71 (1,28) | 0.010 | |
| CDR | −3.654 | 2.500 | 2.14 (1,28) | 0.155 | |
| Race | −6.496 | 3.773 | 2.96 (1,28) | 0.096 | |
| ApoE4 status | +2.925 | 2.210 | 1.75 (1,28) | 0.197 | |
| Sleep medications | +1.731 | 3.410 | 0.26 (1,28) | 0.616 | |
| AHI | +0.085 | 0.126 | 0.46 (1,28) | 0.504 | |
| PLMI | +0.092 | 0.041 | 5.02 (1,28) | 0.033 | |
| Time | −0.013 | 0.095 | 0.02 (1,29) | 0.891 | |
| 3–4 Hz SWA | Mean AV-1451 tau composite | −2.186 | 0.766 | 8.16 (1,27) | 0.008 |
| Age | −0.181 | 0.071 | 6.44 (1,28) | 0.017 | |
| Sex | −2.028 | 0.772 | 6.90 (1,28) | 0.014 | |
| CDR | −1.150 | 0.957 | 1.44 (1,27) | 0.240 | |
| Race | −2.406 | 1.447 | 2.76 (1,27) | 0.108 | |
| ApoE4 status | +1.004 | 0.849 | 1.40 (1,28) | 0.247 | |
| Sleep medications | +0.420 | 1.308 | 0.10 (1,27) | 0.750 | |
| AHI | +0.027 | 0.048 | 0.32 (1,28) | 0.577 | |
| PLMI | +0.035 | 0.016 | 4.84 (1,28) | 0.036 | |
| Time | −0.004 | 0.038 | 0.01 (1,28) | 0.914 |
Linear mixed models were used to calculate the estimates and p-values for the 38 participants with PET imaging. Time: longitudinal sleep monitoring; SWA: slow wave activity; PET: positron emission tomography; NREM: non-rapid eye movement; CDR: clinical dementia rating; AHI: apnea-hypopnea index; PLMI: periodic limb movement index; ApoE4: Apolipoprotein E4
To investigate how regional differences in AV-1451 tau PET were associated with NREM SWA, we performed general linear mixed modeling for NREM SWA in the 1–4.5 Hz, 1–2 Hz, 2–3 Hz, and 3–4 Hz frequency ranges with each region of interest.
Whereas decreased NREM SWA at all slow wave frequencies was associated with increased tau pathology measured by the AV-1451 tau PET composite, regional analyses, uncorrected for multiple comparisons, found that this relationship was most evident in the entorhinal, parahippocampal, inferior parietal, insula, isthmus cingulate, lingual, supramarginal, and orbitofrontal regions (Table S3, Figure S1). After correcting for multiple comparisons, multiple regions on AV-1451 tau PET remained significant for 1–4.5 Hz NREM SWA including the entorhinal, parahippocampal, orbital frontal, precuneus, inferior parietal, and inferior temporal regions (all p<0.05; see Figure 3 and Table S3) This relationship was driven by 1–2 Hz NREM SWA, with only the lingual and medial orbital frontal regions on AV-1451 tau PET significantly associated with 2–3 and 3–4 Hz NREM SWA (all p<0.05). The significance map for 1–2 Hz NREM SWA (Figure 3) shows a similar spatial pattern of tauopathy seen for other changes in AD, such as cortical thickness (44, 50–52).
Figure 3: Relationship between NREM SWA and tau PET varies by region.
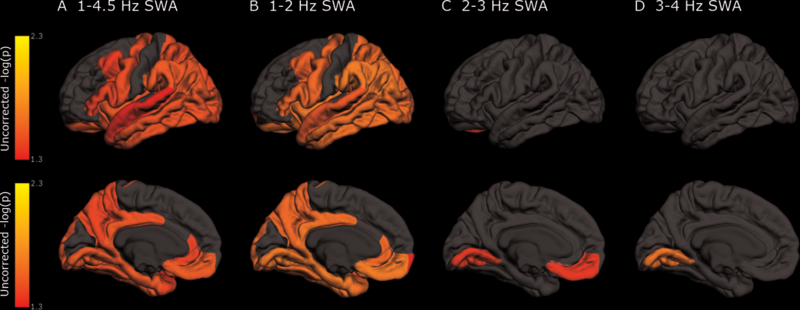
Regional differences in NREM SWA at 1–4.5 Hz (A), 1–2 Hz (B), 2–3 Hz (C), and 3–4 Hz (D) on AV-1451 tau PET after correction for multiple comparisons for the 38 participants with PET imaging. Linear mixed models were performed with NREM SWA as dependent variable and covariates age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time. Each AV-1451 tau PET region was included in the model individually and was corrected for multiple comparisons. The p-value in the model from each region was mapped on a brain image and transformed to a logarithmic scale (p<0.05 = >1.30).
Decreased 1–2 Hz NREM SWA with increased Aβ deposition
Using the same model as for AV-1451 tau PET, we assessed the relationship between NREM SWA and AV-45 amyloid PET. The mean cortical AV-45 amyloid composite was calculated as the average SUVR for the frontal, temporal, and parietal lobes. All night 1–4.5 Hz SWA was not associated with AV-45 amyloid PET (Table 3). Then, we tested NREM SWA in frequency ranges of 1–2 Hz, 2–3 Hz, and 3–4 Hz. There was an inverse relationship between 1–2 Hz NREM SWA and the mean cortical AV-45 amyloid composite (p=0.043, Table 3).
Table 3:
Relationship of NREM SWA to AV-45 amyloid PET composite after adjusting for age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time
| Dependent Variable |
Co-variate | Estimate | Standard Error |
F-statistic (df) |
p-value |
|---|---|---|---|---|---|
| 1–4.5 Hz SWA | Mean cortical AV-45 amyloid composite | −4.926 | 2.422 | 4.14 (1,28) | 0.052 |
| Age | −0.680 | 0.270 | 6.36 (1,28) | 0.018 | |
| Sex | −8.590 | 2.948 | 8.49 (1,28) | 0.007 | |
| CDR | −3.489 | 3.463 | 1.02 (1,29) | 0.322 | |
| Race | −10.003 | 5.567 | 3.23 (1,27) | 0.083 | |
| ApoE4 status | +2.946 | 3.327 | 0.78 (1,27) | 0.384 | |
| Sleep medications | +3.124 | 4.963 | 0.40 (1,27) | 0.534 | |
| AHI | +0.123 | 0.183 | 0.45 (1,28) | 0.507 | |
| PLMI | +0.084 | 0.062 | 1.83 (1,28) | 0.187 | |
| Time | −0.003 | 0.142 | 0.00 (1,145) | 0.982 | |
| 1–2 Hz SWA | Mean cortical AV-45 amyloid composite | −12.017 | 5.676 | 4.48 (1,28) | 0.043 |
| Age | −1.563 | 0.632 | 6.12 (1,28) | 0.020 | |
| Sex | −20.401 | 6.914 | 8.71 (1,28) | 0.006 | |
| CDR | −10.138 | 8.139 | 1.55 (1,29) | 0.223 | |
| Race | −23.326 | 13.041 | 3.20 (1,27) | 0.085 | |
| ApoE4 status | +6.427 | 7.786 | 0.68 (1,27) | 0.416 | |
| Sleep medications | −8.086 | 11.625 | 0.48 (1,27) | 0.493 | |
| AHI | +0.299 | 0.431 | 0.48 (1,28) | 0.493 | |
| PLMI | +0.184 | 0.146 | 1.59 (1,28) | 0.218 | |
| Time | +0.003 | 0.355 | 0.00 (1,146) | 0.994 | |
| 2–3 Hz SWA | Mean cortical AV-45 amyloid composite | −3.239 | 1.814 | 3.19 (1,28) | 0.085 |
| Age | −0.514 | 0.202 | 6.48 (1,28) | 0.017 | |
| Sex | −6.066 | 2.205 | 7.57 (1,28) | 0.010 | |
| CDR | −1.472 | 2.568 | 0.33 (1,28) | 0.571 | |
| Race | −6.936 | 4.167 | 2.77 (1,28) | 0.107 | |
| ApoE4 status | +2.196 | 2.498 | 0.77 (1,28) | 0.387 | |
| Sleep medications | +1.613 | 3.716 | 0.19 (1,28) | 0.668 | |
| AHI | 0.073 | 0.137 | 0.29 (1,28) | 0.598 | |
| PLMI | +0.067 | 0.046 | 2.09 (1,28) | 0.159 | |
| Time | −0.014 | 0.095 | 0.02 (1,29) | 0.886 | |
| 3–4 Hz SWA | Mean cortical AV-45 amyloid composite | −1.350 | 0.682 | 3.91 (1,28) | 0.058 |
| Age | −0.183 | 0.076 | 5.80 (1,28) | 0.023 | |
| Sex | −2.229 | 0.830 | 7.21 (1,28) | 0.012 | |
| CDR | −0.339 | 0.963 | 0.12 (1,27) | 0.727 | |
| Race | −2.645 | 1.565 | 2.86 (1,27) | 0.102 | |
| ApoE4 status | +0.682 | 0.939 | 0.53 (1,28) | 0.474 | |
| Sleep medications | +0.425 | 1.396 | 0.09 (1,27) | 0.763 | |
| AHI | +0.024 | 0.051 | 0.21 (1,28) | 0.647 | |
| PLMI | +0.024 | 0.017 | 1.94 (1,28) | 0.175 | |
| Time | −0.003 | 0.038 | 0.01 (1,27) | 0.928 |
Linear mixed models were used to calculate the estimates and p-values for the 38 participants with PET imaging. Time: longitudinal sleep monitoring; SWA: slow wave activity; PET: positron emission tomography; NREM: non-rapid eye movement; CDR: clinical dementia rating; AHI: apnea-hypopnea index; PLMI: periodic limb movement index; ApoE4: Apolipoprotein E4
Uncorrected for multiple comparisons, decreased NREM SWA at 1–4.5 Hz and 1–2 Hz were associated with increased Aβ deposition in frontal, temporal, and inferior parietal regions, as well as the supramarginal and isthmus cingulate regions (Table S4, Figure S2). At 2–3 and 3–4 Hz, however, this inverse association with Aβ deposition was seen in fewer regions including the inferior parietal, isthmus cingulate, transtemporal, supramarginal, middle frontal, and pars opercularis regions (Table S4, Figure S2). After correcting for multiple comparisons, there were no regions on AV-45 amyloid PET associated with NREM SWA (Table S4).
Decreased NREM SWA linked with increased CSF tau/Aβ42 ratio but not CSF Aβ42
To further assess the relationship between NREM SWA and AD pathology, we analyzed one hundred and four participants with CSF in the same model used in participants with PET imaging except CSF Aβ42 and tau/Aβ42 measurements were used as covariates in place of AV-45 amyloid PET and AV-1451 tau PET, respectively. NREM SWA did not correlate with Aβ42 after adjusting for all covariates whereas CDR, race, ApoE4 status, and sleep medications showed correlation with Aβ42 (Table 4).
Table 4:
Relationship of NREM SWA to Aβ42 after adjusting for age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time
| Dependent Variable |
Co-variate | Estimate | Standard Error |
F statistic (df) |
p-value |
|---|---|---|---|---|---|
| 1–4.5 Hz SWA | Aβ42 | +0.004 | 0.003 | 1.44 (1,10) | 0.257 |
| Age | +0.021 | 0.188 | 0.01 (1,12) | 0.911 | |
| Sex | +0.457 | 2.687 | 0.03 (1,12) | 0.868 | |
| CDR | +8.270 | 3.304 | 6.27 (1,15) | 0.024 | |
| Race | −10.031 | 3.910 | 6.58 (1,10) | 0.029 | |
| ApoE4 status | −6.597 | 2.897 | 5.18 (1,11) | 0.044 | |
| Sleep medications | −17.910 | 6.102 | 8.62 (1,12) | 0.013 | |
| AHI | −0.007 | 0.151 | 0.00 (1,11) | 0.966 | |
| PLMI | +0.055 | 0.041 | 1.83 (1,13) | 0.199 | |
| Time | −0.174 | 0.167 | 1.08 (1,76) | 0.302 | |
| 1–2 Hz SWA | Aβ42 | +0.008 | 0.007 | 1.48 (1,11) | 0.248 |
| Age | +0.077 | 0.447 | 0.03 (1,12) | 0.866 | |
| Sex | +0.900 | 6.394 | 0.02 (1,12) | 0.890 | |
| CDR | +18.082 | 7.873 | 5.28 (1,16) | 0.036 | |
| Race | −21.715 | 9.301 | 5.45 (1,11) | 0.040 | |
| ApoE4 status | −15.565 | 6.894 | 5.10 (1,12) | 0.044 | |
| Sleep medications | −44.695 | 14.524 | 9.47 (1,13) | 0.009 | |
| AHI | −0.056 | 0.360 | 0.02 (1,12) | 0.879 | |
| PLMI | +0.086 | 0.097 | 0.78 (1,14) | 0.393 | |
| Time | −0.489 | 0.401 | 1.49 (1,77) | 0.226 | |
| 2–3 Hz SWA | Aβ42 | +0.003 | 0.002 | 2.44 (1,9) | 0.153 |
| Age | +0.003 | 0.128 | 0.00 (1,11) | 0.980 | |
| Sex | +0.299 | 1.846 | 0.03 (1,11) | 0.874 | |
| CDR | +7.227 | 2.203 | 10.76 (1,14) | 0.005 | |
| Race | −8.654 | 2.739 | 9.98 (1,8) | 0.013 | |
| ApoE4 status | −5.372 | 2.009 | 7.15 (1,10) | 0.024 | |
| Sleep medications | −13.941 | 4.198 | 11.03 (1,11) | 0.007 | |
| AHI | +0.016 | 0.105 | 0.02 (1,10) | 0.884 | |
| PLMI | +0.059 | 0.028 | 4.52 (1,12) | 0.054 | |
| Time | −0.085 | 0.125 | 0.47 (1,16) | 0.504 | |
| 3–4 Hz SWA | Aβ42 | +0.002 | 0.001 | 4.09 (1,12) | 0.066 |
| Age | −0.023 | 0.072 | 0.10 (1,13) | 0.759 | |
| Sex | −0.216 | 1.027 | 0.04 (1,13) | 0.837 | |
| CDR | +2.623 | 1.074 | 5.97 (1,12) | 0.032 | |
| Race | −3.301 | 1.538 | 4.61 (1,12) | 0.053 | |
| ApoE4 status | −1.964 | 1.121 | 3.07 (1,13) | 0.104 | |
| Sleep medications | −6.387 | 2.267 | 7.94 (1,12) | 0.016 | |
| AHI | +0.018 | 0.058 | 0.10 (1,12) | 0.763 | |
| PLMI | +0.024 | 0.015 | 2.72 (1,13) | 0.124 | |
| Time | −0.012 | 0.052 | 0.06 (1,15) | 0.815 |
Linear mixed models were used to calculate the estimates and p-values for the 104 participants with CSF. Time: longitudinal sleep monitoring; SWA: slow wave activity; NREM: non-rapid eye movement; CDR: clinical dementia rating; AHI: apnea-hypopnea index; PLMI: periodic limb movement index; ApoE4: Apolipoprotein E4; Aβ: amyloid-β
Prior work found that tau/Aβ42 ratio is sensitive to early stages of AD pathology and predicts cognitive decline from normal to impaired over several years (5–7). Further, poor sleep has been associated with higher tau/Aβ42 ratios (53). Tau/Aβ42 ratio also controls for the relationship between Aβ42 and tau. In the same model used to investigate the relationship between NREM SWA and tau PET, there was a significant inverse association between NREM SWA and tau/Aβ42 (p<0.05; Table 5) indicating that NREM SWA decreased as the tau/Aβ42 ratio increased (meaning greater AD pathology). CSF tau is marker of neuronal injury and CSF p-tau is a marker for neurofibrillary tangles (54), therefore we also tested the same model with p-tau/Aβ42 ratio and found a similar inverse relationship with NREM SWA as tau/Aβ42 (Table S5). Similar to our findings for tau PET, the relationships between NREM SWA and both tau/Aβ42 and p-tau/Aβ42 ratios were maximal at the lowest 1–2 Hz frequencies. CDR, race, ApoE4 status, and sleep medications were also significantly associated with NREM SWA in the model (all p<0.05; Table 5).
Table 5:
Relationship of NREM SWA to tau/Aβ42 ratio after adjusting for age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time
| Dependent Variable |
Co-variate | Estimate | Standard Error |
F statistic (df) |
p-value |
|---|---|---|---|---|---|
| 1–4.5 Hz SWA | Tau/Aβ42 | −10.788 | 4.673 | 5.33 (1,12) | 0.040 |
| Age | +0.034 | 0.170 | 0.04 (1,12) | 0.846 | |
| Sex | +0.838 | 2.350 | 0.13 (1,12) | 0.728 | |
| CDR | +7.567 | 2.880 | 6.90 (1,14) | 0.020 | |
| Race | −13.860 | 3.818 | 13.18 (1,9) | 0.006 | |
| ApoE4 status | −6.961 | 2.645 | 6.92 (1,11) | 0.024 | |
| Sleep medications | −19.697 | 5.565 | 12.53 (1,12) | 0.004 | |
| AHI | +0.056 | 0.141 | 0.16 (1,11) | 0.699 | |
| PLMI | +0.060 | 0.037 | 2.59 (1,13) | 0.132 | |
| Time | −0.154 | 0.169 | 0.83 (1,70) | 0.365 | |
| 1–2 Hz SWA | Tau/Aβ42 | −25.757 | 11.078 | 5.41 (1,13) | 0.038 |
| Age | +0.106 | 0.403 | 0.07 (1,12) | 0.797 | |
| Sex | +1.571 | 5.571 | 0.08 (1,12) | 0.783 | |
| CDR | +16.425 | 6.807 | 5.82 (1,14) | 0.030 | |
| Race | −31.133 | 9.074 | 11.77 (1,10) | 0.007 | |
| ApoE4 status | −16.216 | 6.278 | 6.67 (1,12) | 0.025 | |
| Sleep medications | −49.386 | 13.196 | 14.01 (1,13) | 0.003 | |
| AHI | +0.103 | 0.333 | 0.10 (1,12) | 0.762 | |
| PLMI | +0.100 | 0.0875 | 1.31 (1,13) | 0.272 | |
| Time | −0.435 | 0.406 | 1.15 (1,74) | 0.288 | |
| 2–3 Hz SWA | Tau/Aβ42 | −11.594 | 3.251 | 12.72 (1,11) | 0.004 |
| Age | +0.008 | 0.122 | 0.00 (1,12) | 0.952 | |
| Sex | +0.343 | 1.680 | 0.04 (1,12) | 0.842 | |
| CDR | +6.431 | 1.765 | 13.27 (1,8) | 0.006 | |
| Race | −12.567 | 2.739 | 21.06 (1,9) | 0.001 | |
| ApoE4 status | −5.995 | 1.889 | 10.07 (1,11) | 0.009 | |
| Sleep medications | −19.944 | 3.810 | 27.40 (1,10) | 0.0004 | |
| AHI | +0.069 | 0.097 | 0.50 (1,10) | 0.496 | |
| PLMI | +0.058 | 0.025 | 5.28 (1,11) | 0.042 | |
| Time | −0.082 | 0.129 | 0.41 (1,17) | 0.533 | |
| 3–4 Hz SWA | Tau/Aβ42 | −5.549 | 1.687 | 10.82 (1,10) | 0.008 |
| Age | −0.019 | 0.064 | 0.09 (1,11) | 0.773 | |
| Sex | −0.001 | 0.878 | 0.00 (1,11) | 0.999 | |
| CDR | +2.223 | 0.911 | 5.95 (1,10) | 0.036 | |
| Race | −5.076 | 1.403 | 13.08 (1,8) | 0.006 | |
| ApoE4 status | −2.104 | 0.978 | 4.63 (1,10) | 0.057 | |
| Sleep medications | −6.784 | 1.965 | 11.92 (1,9) | 0.007 | |
| AHI | +0.040 | 0.050 | 0.64 (1,10) | 0.444 | |
| PLMI | +0.029 | 0.013 | 4.78 (1,10) | 0.053 | |
| Time | −0.019 | 0.053 | 0.13 (1,15) | 0.719 |
Linear mixed models were used to calculate the estimates and p-values for the 104 participants with CSF. Time: longitudinal sleep monitoring; SWA: slow wave activity; NREM: non-rapid eye movement; CDR: clinical dementia rating; AHI: apnea-hypopnea index; PLMI: periodic limb movement index; ApoE4: Apolipoprotein E4; Aβ: amyloid-β
Specific sleep parameters associated with AD pathology
Previous studies have shown a relationship between various sleep parameters and AD pathology (16–18, 53). To investigate the relationship between other sleep parameters in our model, we compared the relationship between sleep parameters measured by the single-channel EEG device, actigraphy, and sleep logs to the mean AV-45 amyloid and AV-1451 tau composites using the same linear mixed model as above (Table S6). Sleep parameters tested in the models included TST, sleep efficiency, sleep latency, REM onset latency, WASO, time in each sleep stage, number of arousals, and time spent napping per day (Table S6). No sleep parameters measured by sleep log or actigraphy were associated with AV-45 amyloid PET. For EEG-derived sleep parameters, REM latency (F(1,30)=12.5, p=0.001) and sleep latency (F(1,29)=4.4, p=0.045) had significant negative relationships with Aβ suggesting that as Aβ deposition increased the time to fall asleep and enter REM sleep decreased.
TST measured by single-channel EEG device (F(1,28)=5.99, p=0.021) and sleep log (F(1,29)=4.80, p=0.037) was positively associated with increased tauopathy in tau PET. In other words, participants slept longer with increased tauopathy. Self-reported time napping on sleep logs was increased with greater tau pathology (F(1,27)=9.28, p=0.005) (Table S6). This suggests that participants with greater tau pathology experienced daytime sleepiness despite increased TST. All other sleep parameters measured by EEG, actigraphy, and sleep log did not show correlation with tauopathy. Using the same model, no sleep parameters measured by EEG, actigraphy, or sleep log were associated with CSF tau/Aβ42 (Table S7).
Discussion
Our study showed that NREM SWA has an inverse relationship with AD pathology measured by PET imaging and CSF biomarkers. In other words, NREM SWA decreased with increased evidence of Aβ deposition and tau accumulation. For PET, this relationship was stronger with tau than with Aβ pathology. We also showed that increased CSF tau/Aβ42 ratio, another marker of AD pathology, was inversely associated with NREM SWA. We observed these associations after adjustment for multiple potential confounders, particularly age, sex, and CDR, supporting a strong relationship independent of these factors. Although AV-45 amyloid PET showed a similar inverse relationship with NREM SWA as AV-1451 tau PET, the estimated magnitude of this association was greater for tau and the findings with CSF tau/Aβ42 suggest that tau is critical for this relationship. Since the study participants were predominantly cognitively normal with the remaining showing only very mild impairment, this suggests that decreased NREM SWA, especially at the lowest 1–2 Hz frequencies, might be associated with tau pathology either before or at the earliest stages of cognitive decline.
Regional analyses of the PET images found that decreases in NREM SWA were most pronounced with Aβ deposition in areas of the frontal, temporal, and parietal lobes. There was no association in our models between NREM SWA and CSF Aβ42. Previous findings associated decreased NREM SWA, particularly at the lowest 0.6–1 Hz frequencies, with Aβ deposition in the mPFC on PiB-PET imaging (38). Since we recorded a single-channel EEG from the forehead, we were unable to localize the NREM SWA more specifically than to the frontal lobes. This location of electrode placement likely contributes to the robust relationship between NREM SWA and tau pathology in the frontal regions. We were also unable to test frequencies in the 0.6–1 Hz range due to hardware limitation of the single-channel EEG device (55, 56). Another recent study reported associations between baseline excessive daytime sleepiness and longitudinal Aβ deposition in the anterior cingulate, posterior cingulate-precuneus, and parietal regions (17). After correcting for multiple comparisons, our study found no association between 1–2 Hz SWA and Aβ deposition in all brain regions analyzed.
For AV-1451 tau PET, regions known to be involved with AD progression showed associations with decreased NREM SWA including the orbitofrontal, entorhinal, parahippocampal, lingual, and inferior parietal regions. These relationships were most evident in the 1–2 Hz range and the association remained valid after correcting for multiple comparisons. This spatial pattern is similar to other imaging changes in AD, such as cortical thickness (50–52). Decreased cortical thickness, however, does not explain our findings since the PET regions of interest were volume-corrected.
We were also able to compare the relationship between PET imaging with AV-45 amyloid and AV-1451 tau tracers, as well as CSF Aβ42 and tau/Aβ42, to sleep parameters measured with different methods such as sleep logs. Although NREM SWA was associated with AD pathology, traditional sleep parameters measured by single-channel EEG-based sleep scoring, actigraphy, or sleep logs generally did not show association in our study. SWA is a measure of sleep homeostasis and may be altered even when other sleep parameters are unchanged (57). Increased TST measured by the single-channel EEG and sleep log were associated with increasing tau pathology on PET, as was self-reported increased time napping. These results, coupled with the NREM SWA findings, suggest that the quality of sleep decreases with increasing tau despite increased sleep time. Further, self-reported napping time per day may be an important question to screen individuals for tauopathy.
A strength of our study is the multiple modalities of both sleep monitoring and biomarkers for AD pathology available from all of our participants. In addition to PET imaging, CSF biomarkers, and different sleep measures, we were able to adjust for multiple variables that affect sleep and AD pathology. Model covariates, such as sex, race, ApoE4, and sleep medications, need further study in other larger cohorts and longitudinal studies. A weakness of this study, however, is that we cannot establish whether or not sleep disturbances preceded or followed the development of AD pathology. Further, this study included only thirty-eight participants in the imaging analyses and therefore a limitation of for these analyses is over-fitting a model with 10 covariates. Another limitation of our study is that stages of AD pathology (for example amyloid-negative/tau-negative) differed between participants with PET imaging and CSF. These limitations are offset by the complementary findings between NREM SWA and AD pathology with both PET imaging and CSF biomarkers. Previous work that reported these associations between sleep parameters and AD pathology generally included larger numbers of participants than our study. However, the fact that we could see robust differences in NREM SWA in relation to tau pathology measured on tau PET and CSF tau/Aβ42 suggests strong relationships between the variables analyzed.
With the rising incidence of AD in an aging population, our findings have potential application in both clinical trials and patient screening for AD to non-invasively monitor for progression of AD pathology. For instance, periodically measuring NREM SWA, in conjunction with other biomarkers, may have utility monitoring AD risk or response to an AD treatment. In order to apply our findings in these settings, further longitudinal studies are needed to confirm that the timing of when NREM SWA decreases in relation of increased Aβ deposition and tauopathy.
Materials and Methods
Study Design
This is an on-going longitudinal observational study to assess the association between sleep parameters and the AD biomarkers in which sleep-wake activity was observed over 6 nights. All sleep data collected by April 3, 2018 were included in the analysis. One hundred and nineteen participants enrolled in longitudinal studies at the Knight Alzheimer’s Disease Research Center at Washington University in St Louis, Missouri were recruited thus far to participate in this study. All participants were >60 years old and assessed clinically with a standard protocol that included obtaining a Clinical Dementia Rating (CDR), which ranged from 0 (no impairment) to 3 (maximal impairment) (40, 41). Participants who completed all assessments were included in the analysis. Of the thirty-eight participants who completed PET imaging, twenty-nine participants (76.3%) had a score of 0 and 9 participants (23.7%) had a score of 0.5. These percentages were similar to one hundred and four participants who underwent lumbar puncture for CSF collection with 79.8% CDR 0s and 20.2% CDR 0.5s. ApoE genotype was obtained from the Knight Alzheimer’s Disease Research Center Genetics Core. Participants also reported if they were taking any of the following medications that could affect sleep: benzodiazepine receptor agonists (zolpidem, zaleplon, eszopiclone), benzodiazepines (triazolam, temazepam, alprazolam), ramelteon, gabapentin, dopamine agonists (ropinirole, pramipexole, rotigotine), doxepin, antihistamines, antidepressants, and narcotics. Participants were listed as on a sleep medication if they were taking at least 1 medication from this list. Participant demographic information is shown in Table 1. The study protocol was approved by the Washington University Institutional Review Board. All participants provided written informed consent and were compensated for their participation in the study.
Sleep Monitoring
Sleep was assessed longitudinally in all participants using three separate measures over 6 nights at home: 1) sleep logs; 2) actigraphy (Actiwatch2, Respironics); 3) a single-channel EEG device worn on the forehead (Sleep Profiler, Advanced Brain Monitoring). Sleep logs and actigraphy were scored as previously reported (18). Single-channel EEG sleep studies were visually scored by registered polysomnographic technologists using criteria adapted from the standard American Academy of Sleep Medicine (AASM) criteria (56). Sleep parameters for time in each sleep stage, sleep latency, sleep efficiency, WASO, and TST were calculated. Nights were excluded if >10% of the recording was artifactual or if the bed and rise times did not match the sleep log and/or actigraphy. All participants needed at least 2 nights from the single-channel EEG device that met these criteria to be included in this analysis.
Due to the increased prevalence of sleep apnea and periodic leg movements during sleep with age, all participants were monitored for one night with a home sleep test (Alice PDX, Respironics). Bed and rise times were confirmed with sleep logs and actigraphy. A minimum of 4 hours artifact-free recording was obtained for all participants. Respiratory events and periodic leg movements were scored by registered polysomnographic technologists using AASM criteria; hypopneas were scored using 4% oxygen desaturation (58). AHI and PLMI were calculated per hour of monitoring time for each participant.
Spectral power analysis
SWA during NREM sleep was calculated from each single-channel EEG study using in Matlab (Mathworks, Natick, MA) as previously described (56, 59, 60). To briefly summarize, the EEG signal was down-sampled to 128 Hz for analysis in order to eliminate processing error. The single-channel EEG device filtered the signal during acquisition with a 0.1–0.6 band-stop filter. We then applied a band-pass (two-way least-squares finite impulse response (FIR)) filter between 0.5 and 40 Hz. Spectral analysis was performed in consecutive 6-sec epochs (Welch method, Hamming window, no overlap). Artifacts were excluded in a semiautomatic method. Power in the 20–30 and 1–4.5 Hz bands for each electrode across all epochs of a recording were displayed. The operator (1st author) then selected a threshold between the 95th and 99.5% threshold of power to remove artifactual epochs. This resulted in fewer than 4% of all epochs being rejected as artifactual.
MRI imaging
T-1 weighted images were acquired using a magnetization prepared rapid gradient-echo sequence on a Siemens Biograph mMR or TIM Trio 3T scanner. Scans had a resolution of either 1 X 1 X 1 mm or 1 X 1 X 1.25 mm. Parcellations of the T1-weighted image into cortical and subcortical regions were performed with FreeSurfer v5.3-HCP (61) for use in the processing of PET data.
PET imaging
Thirty-eight participants underwent positron emission tomography (PET) imaging with both amyloid and tau tracers. Amyloid PET imaging was performed using [18F]-AV-45 (florbetapir). Data from the 50–70 post-injection window were analyzed with an in-house pipeline using FreeSurfer-derived regions of interest (ROIs) (43, 61) (PET Unified Pipeline, https://github.com/ysu001/PUP). Tau PET imaging was completed using [18F]-AV-1451 (flortaucipir). Data from the 80- to 100- minute post-injection window were analyzed. Unprocessed tau PET images were reviewed by a nuclear medicine-trained physician to evaluate for off-target tracer binding; prior to analysis, one potential participant with sleep monitoring and a tau PET scan was excluded due to high bone marrow uptake in the frontal cortex. Regional signal estimates for both tracers were transformed into standardized uptake value ratios (SUVRs) by using cerebellar cortex as the reference region. Data were partial-volume corrected using a regional spread function (RSF) technique (62, 63). ROI PET data were presented as the average across hemispheres for statistical analysis. For global analyses, summary measures of SUVR 1.19 was used for amyloid negative/positive on AV-45 amyloid PET (42, 43) and SUVR 1.22 was used for tau negative/positive on AV-1451 tau PET (44).
CSF biomarkers
CSF was collected under a standardized protocol (45). After fasting overnight, participants underwent a lumbar puncture at 8 AM. Twenty to 30 mls of CSF was collected by gravity drip into a 50 ml conical tube using a 22G atraumatic Sprotte spinal needle, was gently inverted to disrupt potential gradient effects, and centrifuged at low speed to pellet any cellular debris. Samples were aliquoted (500 μl) in polypropylene tubes and stored at −80oC until analysis. CSF Aβ42, total tau, and p-tau181 were measured as previously described using an automated electrochemiluminescence immunoassay (Elecsys on the Cobas e 601 analyzer, Roche) (45, 64).
Statistical analysis
All data were entered into a secure, web-based application designed to support data capture for research studies (Research Electronic Data Capture (REDCap)) (65). Statistical significance for all analyses was set at p<0.05. No methods were used to predetermine sample sizes. All serial sleep monitoring nights were analyzed with general linear mixed models using an unstructured covariance structure to account for the dependencies among the longitudinal measurements (66). For analyses of the participants who completed PET imaging, AV-45 amyloid and AV-1451 tau PET SUVR, mean-centered age (mean age 74.8 years), sex, race, CDR, ApoE4 status (negative/positive), mean-centered AHI (mean AHI 9.8 respiratory events per hour of monitoring time), mean-centered PLMI (mean PLMI 23.2 leg movements per hour of monitoring time), and sleep medication (yes/no) were treated as fixed effects. Analyses of participants with CSF were the same as those with imaging biomarkers and there were no differences in mean-centered age (mean age 74.5 years), mean-centered AHI (mean AHI 9.8 respiratory events per hour of monitoring time), and mean-centered PLMI (mean PLMI 23 leg movements per hour of monitoring time). The time covariate was longitudinal sleep monitoring over multiple nights and was treated as a random effect with random intercepts and slopes used to accommodate individual variation. The normality assumption was verified through residual plots. Statistical analyses for mixed models were performed using Statistical Analysis Software (SAS). Regional analysis of AV-45 amyloid and AV-1451 tau PET was performed using R 3.3.2 (67) with the same model except for individual regions of interest rather than whole brain composite indices. Bonferroni correction was used when comparing between multiple brain regions. Differences in sleep parameters between amyloid negative/positive and tau negative/positive groups were determined by unpaired two-tailed t-test.
Supplementary Material
Figure S1: Relationship between NREM SWA and tau PET varies by region when uncorrected for multiple comparisons. For the 38 participants with PET imaging, uncorrected regional differences in frontal NREM SWA at 1–4.5 Hz (A), 1–2 Hz (B), 2–3 Hz (C), and 3–4 Hz (D) on AV-1451 tau PET. Linear mixed models were performed with NREM SWA as dependent variable and covariates age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time. Each AV-1451 tau PET region was included in the model individually and was not corrected for multiple comparisons. The p-value in the model from each region was mapped on a brain image and transformed to a logarithmic scale (p<0.05 = >1.30).
Figure S2: Relationship between NREM SWA and amyloid PET varies by region when uncorrected for multiple comparisons. For the 38 participants with PET imaging, uncorrected regional differences in NREM SWA at 1–4.5 Hz (A), 1–2 Hz (B), 2–3 Hz (C), and 3–4 Hz (D) on AV-45 amyloid PET. Linear mixed models were performed with NREM SWA as dependent variable and covariates age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time. Each AV-45 amyloid PET region was included in the model individually and was not corrected for multiple comparisons. The p-value in the model from each region was mapped on a brain image and transformed to a logarithmic scale (p<0.05 = >1.30).
Table S1: Group differences in average sleep parameters between amyloid negative/positive, tau negative/positive, and CDR 0/0.5 for participants with PET imaging
Table S2: Group differences in average sleep parameters between amyloid negative/positive, tau negative/positive, and CDR 0/0.5 for participants with CSF
Table S3: Relationship of NREM SWA power to AV-1451 tau PET regions after adjusting for age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time
Table S4: Relationship of NREM SWA to AV-45 amyloid PET regions after adjusting for age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time
Table S5: Relationship of NREM SWA to p-tau/Aβ42 ratio after adjusting for age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time
Table S6: Relationship of sleep parameters to AV-45 amyloid and AV-1451 tau PET after adjusting for age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time
Table S7: Relationship of sleep parameters to tau/Aβ42 ratio after adjusting for age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time
Acknowledgments:
We are indebted to the participants for their contributions to this study. We thank Carlos Cruchaga and Isabelle Gordon for their contributions to this project.
Funding: This study was supported by the following grants from the National Institutes of Health: P01 AG03991; P01 AG026276; P50 AG05681; P30 NS098577; K76 AG054863; UL1 TR000448; P30 CA091842; KL2 TR000450; R01 EB009352. The Ellison Medical Foundation, the Willman Scholar Fund, the Foundation for Barnes-Jewish Hospital, and a Physician Scientist Training Award from the American Sleep Medicine Foundation also supported this study. Precursor for [18F]-AV-1451 (flortaucipir) and technology transfer for production were provided by Avid Radiopharmaceuticals, a wholly owned subsidiary of Eli Lilly. Doses of [18F]-AV45 (florbetapir) and support for acquisition of the scans were provided by Avid Radiopharmaceuticals. The funding sources had no role in the study design, data collection, management, analysis, interpretation of the data, or manuscript preparation.
DMH co-founded and is on the scientific advisory board of C2N Diagnostics. DMH consults for Genentech, AbbVie, Proclara, and Denali. Washington University receives research grants to the lab of DMH from C2N Diagnostics, AbbVie, and Denali.
Neither Dr. Morris nor his family owns stock or has equity interest (outside of mutual funds or other externally directed accounts) in any pharmaceutical or biotechnology company. Dr. Morris is currently participating in clinical trials of antidementia drugs from Eli Lilly and Company and Biogen. He receives research support from Eli Lilly/Avid Radiopharmaceuticals and is funded by NIH grants # P50AG005681; P01AG003991; P01AG026276 and UF01AG032438.
Dr. Benzinger is currently participating in clinical trials of antidementia drugs from Eli Lilly and Company, Biogen, Roche and Jaansen. She receives research support from Eli Lilly/Avid Radiopharmaceuticals (including support for AV-45 and AV-1451 in this work) and is funded by NIH grants # P50AG005681; P01AG003991; P01AG026276; UF01AG032438, R01AG05326, RF1AG053550, R01AG054567–01A1, R01AG052550–01A1.
Dr. Fagan has received research funding from Biogen, Fujirebio and Roche Diagnostics. She is a member of the scientific advisory boards for Roche, Genentech and AbbVie and also consults for Araclon/Griffols and DiamiR.
Footnotes
Competing interests:
BPL, AM, ECL, CDT, JSM, AMZ, LM, and CX: No competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials. These data are available by request the Knight Alzheimer’s Disease Research Center at Washington University in St Louis. [18F]-AV-1451 (flortaucipir) was produced under a material transfer agreement between Washington University and Avid Radiopharmaceuticals.
References and Notes:
- 1.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC, Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 367, 795–804 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ, Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 9, 119–128 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris JC, Price JL, Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. J Molec Neurosci 17, 101–118 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Fagan AM, Mintun MA, Mach RH, Lee S-Y, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM, Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Amyloid-beta-42 in humans. Ann Neurol 59, 512–519 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM, Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol 64, 343–349 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Li G, Sokal I, Quinn J, Leverenz J, Brodey M, Schellenberg G, Kaye J, Raskind M, Zhang J, Peskind E, Montine T, CSF tau/amyloid-beta(42) ratio for increased risk of mild cognitive impairment. Neurology 69, 631–639 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L, Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol 5, 228–234 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Price JL, Morris JC, Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol 45, 358–368 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH, Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia 7, 280–292 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ju Y-E, Lucey BP, Holtzman DM, Sleep and Alzheimer disease pathology-a bidirectional relationship. Nat Rev Neurol 10, 115–119 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lucey BP, Bateman RJ, Amyloid-β diurnal pattern: possible role of sleep in Alzheimer’s disease pathogenesis. Neurobiol Aging 35, S29–S34 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Musiek ES, Holtzman DM, Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 354, 1004–1008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weldemichael DA, Grossberg GT, Circadian rhythm disturbances in patients with Alzheimer’s disease. Int J Alzheimers Dis, 716453 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCurry SM, Logsdon RG, Teri L, Gibbons LE, Kukull WA, Bowen JD, McCormick WC, Larson EB, Characteristics of sleep disturbance in community-dwelling Alzheimer’s disease patients. J Geriatr Psychiatry Neurol 12, 53–59 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Tworoger SS, Lee S, Schernhammer ES, Grodstein F, The association of self-reported sleep duration, difficulty sleeping, and snoring with cognitive function in older women. Alzheimer Dis Assoc Disord 20, 41–48 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Spira AP, Gamaldo AA, An Y, Wu MN, Simonsick EM, Bilgel M, Zhou Y, Wong DF, Ferrucci L, Resnick SM, Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA Neurol 70, 1537–1543 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carvalho DZ, Louis EKS, Knopman DS, Boeve BF, Lowe VJ, Roberts RO, Mielke MM, Przybelski SA, Machulda MM, Petersen RC, Jack CR, Vemuri P, Association of excessive daytime sleepiness with longitudinal β-amyloid accumulation in elderly persons without dementia. JAMA Neurol, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ju Y-E, McLeland JS, Toedebusch CD, Xiong C, Fagan AM, Duntley S, Morris JC, Holtzman DM, Sleep quality and preclinical Alzheimer’s disease. JAMA Neurol 70, 587–593 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yaffe K, Laffan AM, Harrison SL, Redline S, Spira AP, Ensrud KE, Ancoli-Israel S, Stone KL, Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA 306, 613–619 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ancoli-Israel S, Palmer BW, Cooke JR, Corey-Bloom J, Fiorentino L, Natarajan L, Liu L, Ayalon L, He F, Loredo JS, Cognitive effects of treating obstructive sleep apnea in Alzheimer’s disease: a randomized controlled study. J Am Geriatr Soc 56, 2076–2081 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooke JR, Ayalon L, Palmer BW, Loredo JS, Corey-Bloom J, Natarajan L, Liu L, Ancoli-Israel S, Sustained use of CPAP slows deterioration of cognition, sleep, and mood in patients with Alzheimer’s disease and obstructive sleep apnea: a preliminary study. J Clin Sleep Med 5, 305–309 (2009). [PMC free article] [PubMed] [Google Scholar]
- 22.Leng Y, Blackwell T, Stone KL, Hoang TD, Redline S, Yaffe K, Periodic limb movements in sleep are associated with greater cognitive decline in older men without dementia. Sleep 39, 1807–1810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang J-E, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM, Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Y, Potter R, Sigurdson W, Santacruz A, Shih S, Ju Y-E, Kasten T, Morris JC, Mintun M, Duntley S, Bateman RJ, Effects of age and amyloid deposition on Aβ dynamics in the human central nervous system. Arch Neurol 69, 51–58 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lucey BP, Gonzales C, Das U, Li J, Siemers ER, Slemmon JR, Bateman RJ, Huang Y, Fox GB, Claassen JA, Slats D, Verbeek MM, Tong G, Soares H, Savage MJ, Kennedy M, Forman M, Sjögren M, Margolin R, Chen X, Farlow MR, Dean RA, Waring JF, An integrated multi-study analysis of intra-subject variability in cerebrospinal fluid amyloid-β concentrations collected by lumbar puncture and indwelling lumbar catheter. Alzheimers Res Ther 7, 53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lucey BP, Mawuenyega KG, Patterson BW, Elbert DL, Ovod V, Kasten T, Morris JC, Bateman RJ, Associations Between β-Amyloid Kinetics and the β-Amyloid Diurnal Pattern in the Central Nervous System. JAMA Neurol 74, 207–215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee J-M, Holtzman DM, Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci 14, 750–756 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cirrito JR, Kang J-E, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM, Endocytosis is required for synaptic activity-dependent release of Amyloid-beta in vivo. Neuron 58, 42–51 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM, Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron 48, 913–922 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Verges DK, Restivo JL, Goebel WD, Holtzman DM, Cirrito JR, Opposing synaptic regulation of Amyloid-beta metabolism by NMDA receptors in vivo. J Neurosci 31, 11328–11337 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R, APP processing and synaptic function. Neuron 37, 925–937 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Ju Y-ES, Ooms SJ, Sutphen C, Macauley SL, Zangrilli MA, Jerome G, Fagan AM, Mignot E, Zempel JM, Claassen JA, Holtzman DM, Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain 140, 2104–2111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lucey BP, Hicks TJ, McLeland JS, Toedebusch CD, Boyd J, Elbert DL, Patterson BW, Baty J, Morris JC, Ovod V, Mawuenyega KG, Bateman RJ, Effect of sleep on overnight CSF amyloid-β kinetics. Ann Neurol 83, 197–204 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Illiff JJ, Takano T, Deane R, Nedergaard M, Sleep drives metabolite clearance from the adult brain. Science 342, 373–377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, Holtzman DM, Disruption of the sleep-wake cycle and diurnal fluctuation of amyloid-β in mice with Alzheimer’s disease pathology. Sci Transl Med 4, 150ra122 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holth JK, Mahan TE, Robinson GO, Rocha A, Holtzman DM, Altered sleep and EEG power in the P301S tau transgenic mouse model. Ann Clin Transl Neurol 4, 180–190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mander BA, Rao V, Lu B, Saletin JM, Lindquist JR, Ancoli-Israel S, Jagust W, Walker MP, Prefrontal atrophy, disrupted NREM slow waves and impaired hippocampal-dependent memory in aging. Nat Neurosci 16, 357–364 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mander BA, Marks SM, Vogel JW, Rao V, Lu B, Saletin JM, Ancoli-Israel S, Jagust WJ, Walker MP, β-amyloid deposition in the human brain disrupts NREM slow wave sleep and associated hippocampus-dependent long-term memory. Nat Neurosci 18, 1051–1057 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lucey BP, Holtzman DM, How amyloid, sleep, and memory connect. Nat Neurosci 18, 933–934 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morris JC, The clinical dementia rating (CDR): current version and scoring rules. Neurology 43, 2412–2414 (1993). [DOI] [PubMed] [Google Scholar]
- 41.Berg L, Miller JP, Storandt M, Duchek J, Morris JC, Rubin EH, Burke WJ, Coben LA, Mild senile dementia of the Alzheimer type: 2. Longitudinal assessment. Ann Neurol 23, 477–484 (1988). [DOI] [PubMed] [Google Scholar]
- 42.Benzinger TL, Raichle M, Koudelis D, Butler T, Hornbeck R, Pulizos C, Knight ADRC Research Imaging (KARI) Methods and Definitions DR14 v1.1. (Washington University in St Louis, St Louis, MO, 2018). [Google Scholar]
- 43.Su Y, D’Angelo GM, Vlassenko AG, Zhou G, Snyder AZ, Marcus DS, Blazey TM, Christensen JJ, Vora S, Morris JC, Mintun MA, Benzinger TL, Quantitative analysis of PiB-PET with FreeSurfer ROIs. PLoS ONE 8, e73377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mishra S, Gordon BA, Su Y, Christensen J, Friedrichsen K, Jackson K, Hornbeck R, Balota DA, Cairns NJ, Morris JC, Ances BM, Benzinger TL, AV-1451 PET imaging of tau pathology in preclinical Alzheimer disease: defining a summary measure. Neuroimage 161, 171–178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schindler SE, Gray JD, Gordon BA, Xiong C, Batrla-Utermann R, Quan M, Wahl S, Benzinger TL, Holtzman DM, Morris JC, Fagan AM, Cerebrospinal fluid biomarkers measured by Elecsys assays compared to amyloid imaging. Alzheimers Dement, 1–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tan X, Campbell I, Feinberg I, Internight reliability and benchmark values for computer analyses of non-rapid eye movement (NREM) and REM EEG in normal young adult and elderly subjects. Clin Neurophysiol 112, 1540–1552 (2001). [DOI] [PubMed] [Google Scholar]
- 47.Zheng H, Sowers M, Buysse DJ, Consens F, Kravitz HM, Matthews KA, Owens JF, Gold EB, Hall M, Sources of variability in epidemiological studies of sleep using repeated nights of in-home polysomnography: SWAN sleep study. J Clin Sleep Med 8, 87–96 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fukuda N, Honma H, Kohsaka M, Kobayashi R, Sakakibara S, Kohsaka S, Koyama T, Gender difference of slow wave sleep in middle aged and elderly subjects. Psychiatry Clin Neurosci 53, 151–153 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Cauter EV, Leproult R, Plat L, Age-related changes in slow wave sleep and REM sleep and relationship with growth hormone and cortisol levels in healthy men. JAMA 284, 861–868 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Johnson KA, Schultz A, Betensky RA, Becker JA, Sepulcre J, Rentz D, Mormino E, Chhatwal J, Amariglio R, Papp K, Marshall G, Albers M, Mauro S, Pepin L, Alverio J, Judge K, Philiossaint M, Shoup T, Yokell D, Dickerson B, Gomez-Isla T, Hyman B, Vasdev N, Sperling R, Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol 79, 110–119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xia C, Makaretz SJ, Caso C, McGinnis S, Gomperts SN, Sepulcre J, Gomez-Isla T, Hyman BT, Schultz A, Vasdev N, Johnson KA, Dickerson BC, Association of in vivo [18F]AV-1451 tau PET imaging results with cortical atrophy and symptoms in typical and atypical Alzheimer disease. JAMA Neurol 74, 427–436 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brier MR, Gordon B, Friedrichsen K, McCarthy J, Stern A, Christensen J, Owen C, Aldea P, Su Y, Hassenstab J, Cairns NJ, Holtzman DM, Fagan AM, Morris JC, Benzinger TL, Ances BM, Tau and Aβ imaging, CSFmeasures, and cognition in Alzheimer’s disease. Sci Transl Med 8, 338–366 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sprecher KE, Koscik RL, Carlsson CM, Zetterberg H, Blennow K, Okonkwo OC, Sager MA, Asthana S, Johnson SC, Benca RM, Bendlin BB, Poor sleep is associated with CSF biomarkers of amyloid pathology in cognitively normal adults. Neurology 89, 445–453 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buerger K, Ewers M, Pirttila T, Zinkowski R, Alafuzoff I, Teipel SJ, DeBernardis J, Kerkman D, McCulloch C, Soininen H, Hampel H, CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain 129, 3035–3041 (2006). [DOI] [PubMed] [Google Scholar]
- 55.(Advanced Brain Monitoring).
- 56.Lucey BP, McLeland JS, Toedebusch CD, Boyd J, Morris JC, Landsness EC, Yamada K, Holtzman DM, Comparison of a single-channel EEG sleep study to polysomnography. J Sleep Res 25, 625–635 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chinoy ED, Frey DJ, Kaslovsky DN, Meyer FG, Wright KP, Age-related changes in slow wave activity rise time and NREM sleep EEG with and without zolpidem in healthy young and older adults. Sleep Med 15, 1037–1045 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.C. Iber, The AASM Manual for Scoring of Sleep and Associated Events: Rules, Terminology and Technical Specifications: version 2.1. (American Academy of Sleep Medicine, Westchester, IL, 2014). [Google Scholar]
- 59.Landsness EC, Crupi D, Hulse BK, Peterson MJ, Huber R, Ansari H, Coen M, Cirelli C, Benca RM, Ghilardi MF, Tononi G, Sleep-dependent improvement in visuomotor learning: a causal role for slow waves. Sleep 32, 1273–1284 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Landsness EC, Goldstein MR, Peterson MJ, Tononi G, Benca RM, Antidepressant effects of selective slow wave sleep deprivation in major depression: a high-density EEG investigation. J Psychiat Res 45, 1019–1026 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fischl B, FreeSurfer. Neuroimage 62, 774–781 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Su Y, Blazey TM, Snyder AZ, Raichle ME, Hornbeck RC, Aldea P, Morris JC, Benzinger TL, Quantitative amyloid imaging using image-derived arterial input function. PLoS ONE 10, e0122920 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rousset OG, Ma Y, Evans AC, Correction for partial volume effects in PET: Principle and Validation. J Nucl Med 39, 904–911 (1998). [PubMed] [Google Scholar]
- 64.Bittner T, Zetterberg H, Teunissen CE, Ostlund RE, Militello M, Andreasson U, Hubeek I, Gibson D, Chu DC, Eichenlaub U, Heiss P, Kobold U, Leinenbach A, Madin K, Manuilova E, Rabe C, Blennow K, Technical performance of a novel, fully automated electrochemiluminescence immunoassay for the quantitation of beta-amyloid (1–42) in human cerebrospinal fluid. Alzheimers Dement 12, 517–526 (2016). [DOI] [PubMed] [Google Scholar]
- 65.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG, Research electronic data capture (REDCap)-a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 42, 377–381 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hedeker D, in Encyclopedia of statistics in behavioral science, Everitt BS, Howell DC, Eds. (John Wiley & Sons, Ltd, Chichester, 2005), pp. 729–738. [Google Scholar]
- 67.R Foundation for Statistical Computing, R. C. Team, Ed. (Vienna, Austria, 2013).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Relationship between NREM SWA and tau PET varies by region when uncorrected for multiple comparisons. For the 38 participants with PET imaging, uncorrected regional differences in frontal NREM SWA at 1–4.5 Hz (A), 1–2 Hz (B), 2–3 Hz (C), and 3–4 Hz (D) on AV-1451 tau PET. Linear mixed models were performed with NREM SWA as dependent variable and covariates age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time. Each AV-1451 tau PET region was included in the model individually and was not corrected for multiple comparisons. The p-value in the model from each region was mapped on a brain image and transformed to a logarithmic scale (p<0.05 = >1.30).
Figure S2: Relationship between NREM SWA and amyloid PET varies by region when uncorrected for multiple comparisons. For the 38 participants with PET imaging, uncorrected regional differences in NREM SWA at 1–4.5 Hz (A), 1–2 Hz (B), 2–3 Hz (C), and 3–4 Hz (D) on AV-45 amyloid PET. Linear mixed models were performed with NREM SWA as dependent variable and covariates age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time. Each AV-45 amyloid PET region was included in the model individually and was not corrected for multiple comparisons. The p-value in the model from each region was mapped on a brain image and transformed to a logarithmic scale (p<0.05 = >1.30).
Table S1: Group differences in average sleep parameters between amyloid negative/positive, tau negative/positive, and CDR 0/0.5 for participants with PET imaging
Table S2: Group differences in average sleep parameters between amyloid negative/positive, tau negative/positive, and CDR 0/0.5 for participants with CSF
Table S3: Relationship of NREM SWA power to AV-1451 tau PET regions after adjusting for age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time
Table S4: Relationship of NREM SWA to AV-45 amyloid PET regions after adjusting for age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time
Table S5: Relationship of NREM SWA to p-tau/Aβ42 ratio after adjusting for age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time
Table S6: Relationship of sleep parameters to AV-45 amyloid and AV-1451 tau PET after adjusting for age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time
Table S7: Relationship of sleep parameters to tau/Aβ42 ratio after adjusting for age, sex, race, CDR, ApoE4 status, AHI, PLMI, sleep medications, and time