

Abstract
Stable isotope labeling by amino acids in cell culture (SILAC) has become very popular as a quantitative proteomic method since it was firstly introduced by Matthias Mann’s group in 2002. It is a metabolic labeling strategy in which isotope-labeled amino acids are metabolically incorporated in vivo into proteins during translation. After natural (light) or heavy amino acid incorporation, differentially labeled samples are mixed immediately after cell lysis and before any further processing, which minimizes quantitative errors caused by handling different samples in parallel. In this chapter, we describe protocols for basic duplex SILAC, triplex SILAC for use in non-dividing cells such as neurons, and for measuring amounts of newly synthesized proteins.
Keywords: SILAC, BONLAC, protein synthesis and proteomics
Introduction
In SILAC, light or heavy isotope-containing amino acids are incorporated into proteins during protein synthesis and then all the proteins are analyzed by mass spectrometry for relative quantitation (Ong et al., 2002). Cells should undergo at least five doublings in the SILAC media to ensure label incorporation of at least 97% (Hoedt, Zhang, & Neubert, 2014; Zhang & Neubert, 2009). However, a variety of primary cells do not divide in culture or some cells are not stable in SILAC media for long term culture. Zhang et al. reported the use of a multiplex SILAC labeling strategy for nondividing primary neurons by introducing two different sets of heavy amino acids in 2011 (Zhang, Deinhardt, Chao, & Neubert, 2011). In that case, the two cell populations are always equally labeled and accurate SILAC quantitation can be performed by comparing only medium and heavy labeled peptides from the partially labeled cells. SILAC can also be used to measure newly synthesized proteins by using two different heavy labels (Schwanhausser et al., 2011). However, measuring the synthesis of new proteins over a relatively short period of time in a high background of pre-existing proteins can be difficult. Bioorthogonal noncanonical amino acid tagging (BONCAT) and SILAC (combined as BONLAC) can be combined to measure the expression of newly synthesized proteins (Bowling et al., 2016; Eichelbaum, Winter, Berriel Diaz, Herzig, & Krijgsveld, 2012; Zhang, Bowling, et al., 2014). BONCAT involves the tagging of nascent peptides with a methionine analog, which then is detected using a tagged-alkyne probe by a Huisgen chemistry reaction called cycloadditon, and SILAC employs stable isotopically-labeled, differentially weighted (medium or heavy) amino acids that are incorporated into newly synthesized proteins to provide a mass spectrometry (MS) profile. In 2010, super-SILAC was introduced to tissue analysis by Matthias Mann’s group (Geiger, Cox, Ostasiewicz, Wisniewski, & Mann, 2010). Libraries of super-SILAC mixes are created using different cell tumor lines. By combing super-SILAC mix and the tissue sample at a 1:1 ratio, super-SILAC can be used as internal standard to quantify proteins accurately in the tissue samples (Geiger et al., 2010; Neubert & Tempst, 2010). All these approaches are quite useful in quantitative proteomics and different methods are applied based on the experimental needs.
This unit describes SILAC labelling of proteins and how to facilitate their identification and quantification with MS–based proteomics. In this chapter, we describe the SILAC protocols for (1) global phosphoproteomic analysis in dividing cells, and (2) proteomics analysis in differentiated, non-dividing cell types such as primary neurons, and (3) comparative proteomic analysis of nascent protein synthesis using BONLAC.
In summary, metabolic labeling of cultured cells is described in basic protocol 1. Protein digestion and amino acid incorporation testing are outlined in basic protocol 2. Then, an application of phosphoproteomics using three-plex SILAC is provided in basic protocol 3. Finally, a SILAC application in primary neurons is provided in alternate protocol 1 and an application of BONLAC for de novo protein synthesis is provided in alternate protocol 2.
BASIC PROTOCOL 1
Cell Culture, SILAC Labeling
In SILAC experiments, lysine and arginine are the most commonly used labeling amino acids because trypsin specifically cleaves at lysine and arginine residues. Thus all tryptic peptides except those at the carboxyl termini of proteins are quantifiable, with a single labeled amino acid (Zhang, Annan, Carr, & Neubert, 2010). In this protocol, we describe the preparation of SILAC multiplex media (light, medium and heavy), cell culturing and labeling.
Materials
Dulbecco’s Modified Eagle Medium (DMEM) deficient in lysine and arginine (Specialty Media, Millipore)
SILAC amino acid stock solution for labeling (see recipes)
Penicillin and streptomycin
Sterile phosphate buffered saline (PBS)
Cell lysis buffer (see recipes)
Sterile trypsin/EDTA
Dialyzed serum (Invitrogen)
0.22-um filter flasks
Note: sterile reagents and equipment should be used for cell culturing.
Prepare 1,000× stock solutions for the light, medium and heavy labeling amino acids by dissolving the amino acids in PBS. Dilute the light, medium and heavy amino acids 1,000-fold with DMEM deficient in the amino acid of choice, making ‘light’, ‘medium’ and ‘heavy’ media.
-
Add supplements to media to attain their working concentrations: diluting penicillin/streptomycin to 1× concentration. Filter the three culture media with 0.22-um filter flasks.
Save some of the serum free medium for the purpose of cell starvation prior to stimulation if needed.
Add 10% dialyzed fetal bovine serum to the ‘light’, ‘medium’ and ‘heavy’ media. These media are now ready for cell culture.
-
Split a dish of cells into three separate dishes, each containing ‘light’, ‘medium’ or ‘heavy’ SILAC medium respectively. When the cells are >80% confluent, rinse the cells with PBS, detach the cells with trypsin/EDTA and split the cells to new dishes.
After SILAC labeling is complete, culture the cells in serum-free SILAC media for 24 h to (a) synchronize the cells and to (b) lower the basal phosphorylation levels.
The media should contain light, medium or heavy amino acids consistent with previous labeling. Stimulation can be done in this step after cell starving. In a biological replicate experiment, the SILAC labeling is reversed between replicates by stimulating cells that are labeled with amino acids of different mass (light/medium/heavy label switching) in each experiment. We recommend three or more biological replicates for each experiment.
-
Remove media from the dishes, wash twice with ice cold PBS. Tilt the dishes to facilitate complete removal of PBS. To each 10 million cells add 100 ul cell lysis buffer.
Fresh phosphatase and protease inhibitors should be added just before use.
-
Sonicate the cells to reduce viscosity. Save 2 ul for protein concentration measurement and mix equal amount of light, medium and heavy lysates based on total protein amount. Store the protein in −80 °C before use.
Save 5ul of the medium or heavy lysate to measure the label incorporation if necessary. Bradford protein assay is used to measure protein concentration.
BASIC PROTOCOL 2
In-Solution Digestion and Measurement of Incorporation Rates of the SILAC Amino Acids
After harvesting cells from cultured media and cell lysis, in-solution digestion of the cell lysate is performed. Trypsin is usually the first choice for digestion of proteins into peptides that have convenient size for MS analysis. This protease is also preferred for its high specificity and activity. At the same time, it is important to check the incorporation efficiency of the SILAC amino acids especially when the light group is needed for quantitation analysis, because incomplete labeling will skew the SILAC ratio in favor of the light proteins.
Material
Dithiothreitol (DTT)
Iodoacetamide
Ammonium bicarbonate
Trypsin Gold, mass spectrometry grade (Promega Corporation)
Trifluoroacetic acid (TFA)
HPLC grade Acetonitrile (ACN)
Washing buffer: 2 % ACN and 0.1 % TFA
Elution buffer: 80 % ACN and 0.1 % TFA.
Sep-Pak tC18 1 cc Vac Cartridge (100 mg Sorbent, 5mg capacity) (Waters)
C18 StageTips (200ul tip, 10ug capacity) (Millipore Sigma)
EASY-nLC 1000 Liquid Chromatography System (ThermoFisher Scientific)
Q Exactive™ Hybrid Quadrupole-Orbitrap Mass Spectrometer (ThermoFisher Scientific)
Self-packed column (New Objective) (C18-AQ 3 ¼m resin- Dr. Maisch, 20 cm length × 75 μm ID)
DTT is added to samples that have been prepared as described in Basic Protocol 1 to yield a final concentration of 5 mM using a 500 mM DTT stock solution. Incubate at 50 °C for 20 min with mixing at 700 RPM in ThermoMixer.
-
Add 1M iodoacetamide to a final concentration of 14 mM. Incubate at room temperature for 20 min in the dark.
Iodoacetamide should be freshly made.
-
Add additional 1M DTT to a final concentration of 5 mM and incubate at room temperature for 20 min.
This step is used to quench unreacted iodoacetamide.
Dilute the solution with 4x volume of 25 mM ammonium bicarbonate to make sure urea final concentration is no more than 1.6 M.
-
Add 1 M CaCl2 to a final concentration of 1 mM.
Calcium may improve the activity of trypsin but is not essential.
-
Add trypsin to the liquid (enzyme:substrate 1:200) and incubate at 37 °C overnight in the ThermoMixer.
Make sure trypsin final concentration is more than 5 ng/ul to ensure good digestion efficiency.
-
Cool to room temperature and add 10% TFA to a final concentration of 0.4%.
Make sure the PH value is below 2 for the following desalting step. Pellet can be expected at this step. Centrifuge at 20,000 × g for 5 min at room temperature and discard the pellet.
-
Activate the Sep-Pak tC18 1 cc Vac cartridge with 1 mL 100% ACN, then equilibrate the cartridge with 1 mL 0.1% TFA. Add the peptide sample to the cartridge and allow the liquid to pass through. Wash the cartridge with 2 mL 0.1% TFA. Elute peptides with 1 mL 80% ACN/0.1% TFA. Dry the eluate by vacuum centrifugation and dissolve the peptides in 2% ACN/0.1% FA (Rappsilber, Ishihama, & Mann, 2003).
Sep-Pack and StageTips have different capacities. The correct one should be chose depending on the sample amount. StageTips can be used for desalting if the protein amount is less than 5 ug.
-
Analyze label incorporation of samples using LC-MS/MS.
An EASY-nLC 1000 coupled with Q Exactive system is used in this chapter. Top 10 data-dependent acquisition (DDA) is used for proteomics experiment here.
-
Analyze data with MaxQuant software.
The data analysis was performed with MaxQuant software (Version 1.5.2.8, Max Planck Institute of Biochemistry) (Cox & Mann, 2008). Peptides were identified by setting the heavy-labeled amino acids as possible variable mass modifications in Maxquant. The ‘re-quantify’ box is unchecked when doing the incorporation test. The ‘peptides.txt’ file is inspected to calculate the distribution of incorporated medium or heavy amino acids. Briefly, “Reverse” and “Potential contamination” are filtered out. For each row in each sample, there are three different intensity values for ‘light’, ‘medium’ and ‘heavy’ channels. The percentage of incorporated medium (or heavy) amino acid for each peptide was then calculated using the intensity of medium (or heavy) channel divided by the sum intensity of ‘light’, ‘medium’ and ‘heavy’ channels. The distribution graph is made based on all the filtered peptides.
-
After ensuring full incorporation (in most cases greater than 97%- incomplete labeling will affect relative quantitation of peptides according to the incompleteness of labeling) of medium and heavy amino acids, analyze mixture samples using the same method above. The minimum acceptable heavy label incorporation should be 97% ((1- (1/2)5)) after five cell doublings (Ong et al., 2002; Ong & Mann, 2006)
The gradient length should be adjusted based on the loading amount, sample complexity and desired depth of analysis. The ‘re-quantify’ box is checked when doing the sample analysis.
BASIC PROTOCOL 3
Studying Protein Phosphorylation with Triplex SILAC
An important step in analyzing signaling networks is the identification and quantitative characterization of global phosphorylation sites (Dephoure, Gould, Gygi, & Kellogg, 2013).
Here, triple SILAC labeling with Arg0Lys0-Arg6(13C6)Lys4(D4)-Arg10(13C615N4)Lys8(13C615N2) is used for a three-condition (three-plex) experiment. The workflow is shown in Figure 1.
Figure 1:
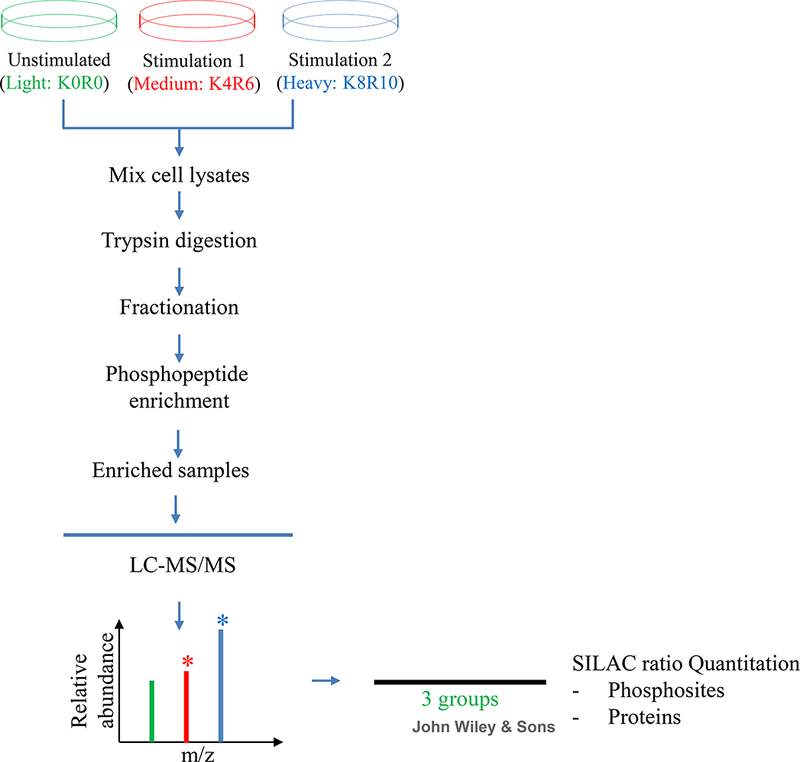
Schematic diagram for studying protein phosphorylation with triple SILAC.
Cells are cultured in SILAC media (unstimulated-K0R0, stimulation 1-K4R6 and stimulation 2-K8R10) and harvested after complete labeling. The equally mixed cell lysates are digested using trypsin, followed by HILIC fractionation. After phosphopeptide enrichment, samples are analyzed using LC-MS/MS. Data processing and statistical analysis are then performed.
Material
3T3L1 cells
Lysis buffer (see recipe)
Agilent 1100 Series HPLC System
TSKgel Amide-80 column packed with 5 μm spherical silica particles (Millipore Sigma)
HILIC buffers: mobile phase A, 0.1 % TFA; mobile phase B, 0.1 % TFA, 99.9 % ACN
Titansphere TiO2 10 μm beads (GL Sciences)
Solid Phase Extraction Disk with octyl group (C8) (Millipore Sigma)
Phosphorylation enrichment buffers: conditioning buffer A, 0.1 % TFA and 99.9 % ACN; conditioning buffer B, 3 % TFA and 60 % ACN; washing buffer A, 3 % TFA and 30 % ACN; washing buffer B, 0.1 % TFA and 80 % ACN; elution buffer A, 3 % NH3·H2O; elution buffer B, 1.5 % NH3·H2O and 50 % ACN.
-
Prepare sufficient numbers of light, medium and heavy labeled cells in SILAC media as described in Basic Protocol 1.
For example, prepare approximately 2×107 3T3L1 cells (two six-well plates) for each labeling condition, corresponding to 4 mg total protein. However, the number of cells needed depends on the cell line used, the protein expression level and pSTY phosphorylation level of the proteins of interest, and the sensitivity of the LC-MS instrument used.
-
After SILAC labeling is complete, culture the cells in serum free SILAC media (containing light, medium or heavy amino acids consistent with previous labeling) for 12~24 h. Stimulate the cells appropriately for the three conditions.
Starving time or stimulation condition varies depending on the different cell types and different experimental design.
Harvest cells in three different conditions separately as described in Basic Protocol 1, and measure the protein concentration using the Bradford protein assay.
Check the labeling efficiency of the ‘medium’ and ‘heavy’ labeled samples as described in Basic Protocol 2. Figure 2 shows one example of the incorporation frequency test.
Mix equal amount of ‘light’, ‘medium’ and ‘heavy’ lysates based on total protein content (labeled as ‘mixture’).
Digest the mixture using trypsin and desalt the peptides using Sep-Pak as described in Basic Protocol 2.
Dry the desalted peptides by vacuum centrifugation and resuspend dried peptides from the mixed samples in 100 μL 20 % mobile phase A and 80 % mobile phase B. Sonicate the sample for 5 minutes to help dissolve the peptides. Centrifuge at 20,000 × g for 5 min at room temperature and discard the pellet.
-
Fractionate the peptide mixture using a HILIC column. Load the sample into the sample loop and start the gradient (0–5 min 80% B, 40 min 60% B, 55 min 0% B, 65 min 0% B). Collect 4 minute fractions. Dry the fractions by vacuum centrifugation and use them for phosphopeptide enrichment step.
Loading amount varies according to the capacity of the column in use and how many fractions are collected depends on the experimental purpose- if sufficient sample is available, more fractionation usually provides greater depth of proteome coverage.
-
Enrich fractions for phosphopeptides using Titansphere TiO2 beads.
Efficient purification and clean-up can be achieved by making a titanium column using a plug of Empore Disk C8, followed by TiO2 beads packed in a 10 ul pipette tip (Fang-Ke Huang, Guoan Zhang, & Neubert, 2015).
Dry the eluted peptides after TiO2 enrichment by vacuum centrifugation and dissolve them in 0.1% FA for LC-MS analysis as described in Basic Protocol 2.
-
Data are analyzed using Maxquant as described in Basic Protocol 2.
A summary of numbers of all identified proteins, peptides and phosphosites is shown in Table 1. Each comparison was done using two conditions, resulting in 3 binary comparisons. A SILAC ratio distribution should be checked to determine whether it is skewed in favor of the light labeled proteins because of incomplete labeling. Figure 3 shows some examples of the ratio distributions including ‘Heavy/Light’, ‘Medium/Light’ and ‘Heavy/Medium’. The plots show a ratio very close to 1:1 for each protein, as would be expected after only 10 minutes of growth factor stimulation.
Figure 2:
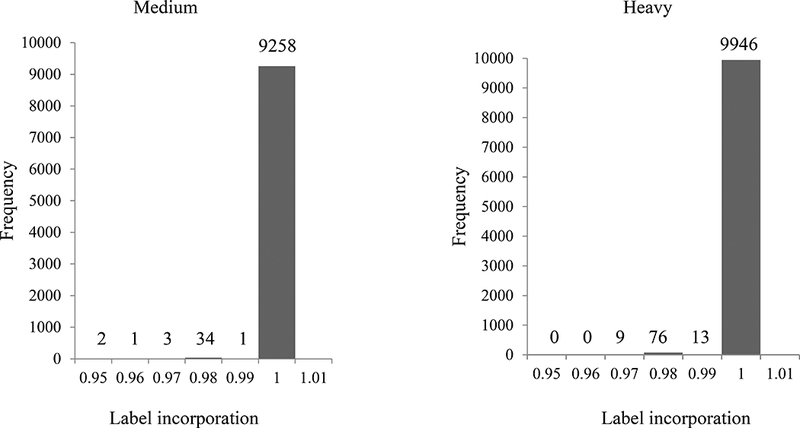
Label incorporation for medium and heavy groups.
Label incorporation is calculated for all identified peptides and distributions are shown in histograms. The numbers of peptides in each % labeling bin are shown above the bars. No peptides contained less than 95% label.
Table 1:
A summary of numbers for all identified proteins, peptides and phosphosites and label incorporation rates is also shown.
| Protein | Peptide | Phosphosites |
|---|---|---|
| 6100 | 41890 | 15286 |
Figure 3:
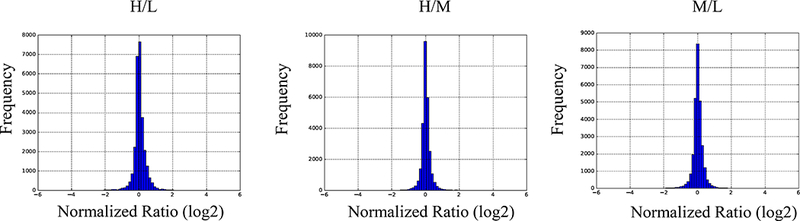
Normalized ratios of different experimental protein groups. Distribution of ratios (log2) of all identified peptides from heavy vs light (H/L), heavy vs medium (H/M) or medium vs light (M/L) are shown.
ALTERNATE PROTOCOL 1
SILAC for Cultured Primary Neurons
Conventional SILAC is difficult to apply to non-dividing primary neurons because it requires nearly complete metabolic labeling of proteins (Zhang, Deinhardt, & Neubert, 2014). Two different sets of heavy stable isotope-containing amino acids (designated as ‘medium’ and ‘heavy’) are introduced for labeling neuronal cells. Because the two identical cell populations grown under the same conditions incorporate medium and heavy amino acids at the same rate, they are always equally labeled. The unlabeled peptides can be ignored during quantitation, and the amounts of the medium and heavy labeled peptides are directly compared. Experimental replicates are performed with reversed labels to rule out any possible isotope effects or other, incidental differences between batches of labeled amino acids.
Prepare primary neurons and split cells equally into two culturing plates (Zhang, Deinhardt, et al., 2014).
Prepare the culture medium as described in Basic Protocol 1 and grow neurons for about 10 days before proceeding to cell treatment/lysis/LC-MS analysis as described in Basic Protocol 2. In non-dividing primary neurons, we have found that most proteins are >90% labeled after 10 days in culture (Zhang et al., 2011).
ALTERNATE PROTOCOL 2
BONLAC for Studying Stimulus-induced Alterations in the Expression of New Proteins
BONCAT selectively isolates de novo proteins synthesized within a specified temporal window (Landgraf, Antileo, Schuman, & Dieterich, 2015). By combining BONCAT with SILAC (BONLAC), the newly synthesized protein can be accurately quantified despite a vast excess of previously made proteins.
-
Incubate cells in a culture medium without methionine, lysine, and arginine and expose them to DMEM medium supplemented with AHA and heavy labeled amino acids.
The SILAC amino acid concentrations are the same as those in regular DMEM. Stimulation and control treatments are also introduced in this step for the differential analysis. Two different sets of heavy amino acids are used for comparison since the labeling period is usually short and incomplete. After the addition of the heavy amino acids, the cells are incubated for 2–4 hours before harvesting. Even though most proteins incorporate only about 1% label per hour, after BONCAT enrichment the newly synthesized (labeled) proteins are highly enriched, and most are over 50% labeled (Bowling et al., 2016; Zhang, Bowling, et al., 2014). Using pulse labeling with heavy amino acids, it was established that some proteins had a half-life of 4.5 hours (eg., Rrm2) whereas others, like Hist1h1c, had a half-life of 62.1 hours (Schwanhausser et al., 2011).
Lyse cells and combine equal amount of treated/untreated cells as described in Basic Protocol 1.
-
Enrich AHA-labeled proteins using a kit (Click-iT Protein Enrichment Kit, Invitrogen Life Science Technologies, ThermoFisher Scientific).
AHA-containing nascent proteins are cross-linked covalently to alkyne agarose beads using reagents provided by the kit. Beads are transferred to a SDS wash buffer (1% SDS, 100 mM Tris, pH 8, 250 mM NaCl, and 5 mM EDTA), and proteins on beads are then reduced with DTT at 70 °C and alkylated with iodoacetamide at room temperature. The beads are then washed sequentially with 100 column volumes of the following three buffers to remove non-specific binding proteins: (1) SDS wash buffer, (2) 8 M urea, and (3) 20% acetonitrile.
All the enrichment steps including cell lysis and tryptic digestion are performed using the Click-iT kit from ThermoFisher Scientific; if unconstituted, the components are stable indefinitely when kept refrigerated and in the dark. Once reconstituted, the components used for catalysis can be aliquoted and stored at −200C for up to 1 year.
Digest immobilized proteins on-resin, with trypsin at 37 °C overnight in 25 mM ammonium bicarbonate.
Desalt the resulting tryptic peptides using StageTips and dry under vacuum in a SpeedVac device.
Load the samples for LC-MS analysis as described in Basic Protocol 2.
DATA ANALYSIS TO IDENTIFY AND QUANTIFY PROTEINS AND PHOSPHORYLATED PEPTIDES
Data analysis is performed as follows:
-
Process raw files using the MaxQuant computational proteomics platform version 1.5.2.8 for peptide identification and quantitation.
The fragmentation spectra are searched against the protein database, which can be downloaded from UNIPROT. MaxQuant default parameters are used, with a minimum ratio count of 2, allowing up to two missed tryptic cleavages. Carbamidomethylation of cysteine is set as a fixed modification, and oxidation of methionine and protein N-terminal acetylation, D4-lysine, 13C6-arginine, 13C6–15N2-lysine and 13C6–15N4-arginine are used as variable modifications for the database search. MaxQuant parameters are set as below: MS1 first search-20 ppm, MS1 main search-4.5 ppm, and fragment mass tolerances-20 ppm. Both peptide and protein identifications are filtered at 1% false discovery rate (FDR).
-
Use the Perseus software platform (http://www.perseus-framework.org) for interpreting peptide/protein quantification or post-translational modification.
For peptide phosphorylation analysis, the ‘phospho(STY)Sites.txt’ file is imported to Perseus. MaxQuant normalized ratios (such as H/M, heavy vs. medium isotopes) are used for quantitative analysis. Total intensities are calculated as the ion intensities (the sum of heavy and medium in the previous case), as reported by MaxQuant. The statistical significance of the log-2 transformed ratios and total intensities is calculated by the Perseus software (v.1.5.5.3) using the Significance_B option and the Benjamini-Hochberg correction to account for multiple hypothesis testing.
For visual inspection and manual calculation, use narrow mass widths (depending on the accuracy of the mass spectrometer used, approximately 5–7 ppm for a well-calibrated Q Exactive MS for example) to extract ion chromatograms for ‘light’, ‘medium’ and ‘heavy’ peptides and use these values to determine relative abundance of the phosphorylated peptides.
REAGENTS AND SOLUTIONS
SILAC amino acid stock solutions
Prepare 1,000× stock solutions for the light, medium and heavy labeling amino acids by dissolving the amino acids in PBS. Store up to 1 year at −80 °C. Prepare the 1x working solutions fresh on day of use. The working concentrations of light arginine and lysine are 84 and 146 mg/L respectively. The working concentration of (medium) L-arginine 13C6 hydrochloride and L-lysine D4 are 86.5 and 148.7 mg/L respectively; the working concentration of (heavy) L-arginine 13C615N4 hydrochloride and L-lysine 13C615N2 are 88.0 and 151.3 mg/L respectively.
Note that because the molecular weights of heavy amino acids are different from their light counterparts, their concentrations (in mg/L) are different.
Cell lysis buffer
9 M urea with 5mM NaF, 1 mM Na3VO4, 50 mM Tris, pH 8.2, and protease inhibitors (Complete tablet; Roche, Mannheim, Germany). Prepare fresh before use.
COMMENTARY
Background Information
SILAC allows highly accurate protein quantitation through metabolic encoding of whole cell proteomes using stable isotope labeled amino acids, which are incorporated into proteins during in vivo protein synthesis (Hoedt et al., 2014). The most commonly used labeling amino acids in SILAC are lysine and arginine because trypsin is the most commonly used enzyme in proteomics, so that each tryptic peptide carries a single label at its C-terminus. In general, cells need to be maintained in SILAC medium for at least five divisions/doublings to ensure virtually complete labeling of proteins and exchange of amino acids.
Conventional SILAC is difficult to apply to non-dividing cells like primary neurons because of the incomplete labeling. SILAC variants are designed to meet different experimental needs. By introducing two different sets of heavy amino acids for labeling, SILAC quantitation using partially labeled cells can be successfully applied to non-dividing cells with a high resolution MS instrument (Zhang et al., 2011; Zhang, Deinhardt, et al., 2014). Straightforward and accurate SILAC quantitation can be performed (Zhang et al., 2011). BONLAC, which is BONCAT combined with SILAC, can separate newly synthesized proteins with minimal contamination from pre-existing proteins, and thus be applied in comparative proteomic analyses of new protein synthesis in cell lines and an intact brain circuit (Bowling et al., 2016; Howden et al., 2013; Zhang, Bowling, et al., 2014). In 2004, John Yates and colleagues reported the first metabolic labeling of mammals for relative proteomic quantitation by feeding the rat 15N-containing food (Wu, MacCoss, Howell, Matthews, & Yates, 2004). In 2008, Matthias Mann’s group developed a mouse SILAC diet using 13C6-lysine that can do completely labeling of the F2 generation, which was also extended to other in vivo animal models afterwards (Kruger et al., 2008; Shenoy & Geiger, 2015; Zanivan, Krueger, & Mann, 2012). However, complete labeling of model organisms is costly and time-consuming. In 2010, Super-SILAC was introduced by Tamar Geiger in Matthias Mann’s group to accurately quantify human tumor proteomes by combining SILAC-labeled cell lines with human tumor tissues and using SILAC as a spike-in standard to analyze the unlabeled tissue samples (Geiger et al., 2010; Neubert & Tempst, 2010; Shenoy & Geiger, 2015). These SILAC labeling strategies have greatly facilitated the application of SILAC to different models that are not suitable for quantitation with conventional SILAC workflows.
Critical Parameters and Troubleshooting
In the triple SILAC experiment shown in this chapter, complete labeling is extremely important because the light condition is also used for comparison and quantitation. The level of incomplete incorporation from unlabeled amino acid in ‘medium’ or ‘heavy’ sample will contribute to the signal in ‘light’ sample, which, in turn, will adversely affect quantitative accuracy. Some supplements in the cultured medium may contain free (unlabeled) amino acids, which can cause incomplete labeling. Therefore it is important to use dialyzed fetal bovine serum (FBS), instead of conventional serum in SILAC culture. In dialyzed FBS, free amino acids have been removed because of the possible incomplete labeling. Table 2 lists troubleshooting suggestions for dealing with some possible problems.
Table 2:
Troubleshooting for possible issues.
| Possible issues | Troubleshooting |
|---|---|
| Cells do not grow or die when dialyzed serum is used |
Add purified growth factors or a small percentage of normal serum to the medium (Gehrmann, Hathout, & Fenselau, 2004) |
| Ratios of observed total H/L is not equal to 1.0 |
If incomplete labeling leads to the problem, culture the cells in vitro at least seven generations to ensure complete incorporation (Kleifeld et al., 2011); or use medium and heavy amino acids instead of light and heavy amino acids; or correct the SILAC ratio for incomplete labeling by monitoring the label incorporation of every protein (Spellman, Deinhardt, Darie, Chao, & Neubert, 2008) |
| Ratios for peptides within a protein are not similar |
Use the ‘proteinGroups.txt’ file to find the ratios for specific protein |
| Quantitation errors based on only 1 or 2 peptides quantitation errors caused by shared (razor) peptides |
Filter the raw data using criteria as shown in Figure 4 or filter the data with at least 3 peptides identified |
| Media needed for cells of interest is not available |
Use modified media with supplements(Gehrmann et al., 2004) |
| Solid tissues | Use super-SILAC method (Geiger et al., 2010) |
Statistical Analyses
In triple SILAC, MaxQuant normalized H/M or H/L ratios (heavy vs. medium isotopes or heavy vs. light isotopes) are used for quantitative analysis. Both peptide and protein identifications were filtered at 1% false discovery rate (FDR). Ratios are inverted for experiments with reversed isotopic labeling. The statistical significance of the log-2 transformed ratios and total intensities is calculated by Perseus software using the Significance_B option and the Benjamini-Hochberg correction to account for multiple hypothesis testing. Only proteins measured at least twice and those that were consistently up or down regulated each time, are selected (Bowling et al., 2016; Zhang, Bowling, et al., 2014). Criteria can be chosen to increase confidence in the results, including the peptide score and localization probability. Only the candidates that met all these criteria were used for quantitation.
Interpreting Results
In the triple SILAC experiment, a total of 6,100 proteins, 41,890 peptides, and 15,286 phosphorylated peptides were identified (summarized in Figure 2 upper panel). The ‘peptides.txt’ file is used to calculate the distribution of incorporated medium or heavy amino acids. Figure 2 lower panel shows the histograms of SILAC incorporation of medium or heavy labeled peptides. X-axis indicates the incorporation efficiency of peptides and Y-axis indicates the average intensity of the peptide. The histograms indicate both medium and heavy amino acids are completely labeled. Figure 3 shows the distribution plots of the average normalized log2 fold change vs average intensity of peptides. All identified peptides are used and the SILAC ratio is not skewed in favor of the light protein, further indicating the complete labeling efficiency.
The ‘phospho(STY)Sites.txt’ file was used to show the scatter plots to indicate the significant changes for each comparison group. The phosphorylated peptides were filtered using score > 80, and localization probability > 0.75, resulting in 7,158 peptides for Significance_B analysis (Figure 4) with a Benjamini-Hochberg FDR < 1% (Cox & Mann, 2008). The Y-axis shows average intensity of the peptide and x-axis indicates the average normalized log2 fold change (H/L, H/M and M/L). For example, in the first panel, phosphorylated peptides whose expression increased in the heavy condition are on the positive side of x-axis, while those that decreased are on the negative side of the x-axis. The data points are colored by their ‘significance B’, with blue crosses having values > 0.05, yellow squares between 0.05 and 0.01, green diamonds between 0.01 and 0.001 and red circles < 0.001.
Figure 4:
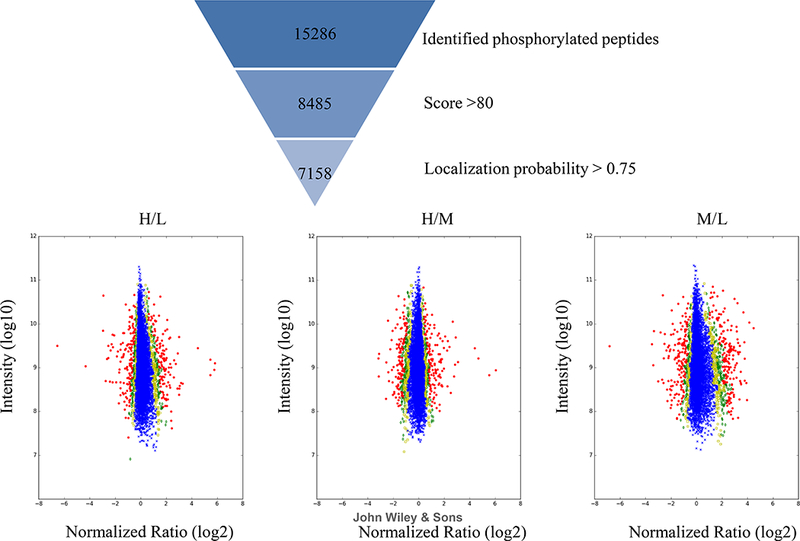
Proteome-wide quantitation accuracy and significance.
Phosphosite data are filtered with score >80 and localization probability >0.75. Normalized phosphosite ratios are plotted against summed intensities. The data points are colored by their ‘significance B’ values, with blue crosses having values >0.05, yellow squares between 0.05 and 0.01, green diamonds between 0.01 and 0.001 and red circles < 0.001.
Time Considerations
Cell culture time depends on the cell types, the protein amount needed and different experimental purposes. Additional time may be needed for large scale experiments for deep proteome coverage. Typically, cells take about 5 to 6 days to adapt and grow in SILAC media. Harvesting, digestion of proteins and incorporation efficiency test will require two or three days. Sample fractionation by HILIC and phosphorylated peptide enrichment will need two days. Analysis of proteins by MS typically takes 3 to 4 days, depending on the LC gradient and number of fractions. If necessary, adapted cells can be maintained in SILAC medium and a second round of experiments can be performed as soon as sufficient cell numbers are available.
Significance Statement:
SILAC is a powerful technology for identification and relative quantitation of proteins in complex samples. This mass spectrometry based, high throughput approach is accurate and robust, making it a very useful tool for quantitative proteomics.
ACKNOWLEDGEMENTS
We gratefully acknowledge support from NIH NINDS grants P30 NS050276 and Shared Instrumentation Grant 1S10RR027990 to Thomas Neubert and R37NS034007 to Eric Klann.
LITERATURE CITED
- Bowling H, Bhattacharya A, Zhang G, Lebowitz JZ, Alam D, Smith PT, . . . Klann E. (2016). BONLAC: A combinatorial proteomic technique to measure stimulus-induced translational profiles in brain slices. Neuropharmacology, 100, 76–89. doi: 10.1016/j.neuropharm.2015.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, & Mann M (2014). Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics, 13(9), 2513–2526. doi: 10.1074/mcp.M113.031591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N, Gould KL, Gygi SP, & Kellogg DR (2013). Mapping and analysis of phosphorylation sites: a quick guide for cell biologists. Mol Biol Cell, 24(5), 535–542. doi: 10.1091/mbc.E12-09-0677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichelbaum K, Winter M, Berriel Diaz M, Herzig S, & Krijgsveld J (2012). Selective enrichment of newly synthesized proteins for quantitative secretome analysis. Nat Biotechnol, 30(10), 984–990. doi: 10.1038/nbt.2356 [DOI] [PubMed] [Google Scholar]
- Fang-Ke Huang, Guoan Zhang, & Neubert TA. (2015). Phosphorylation Site Profiling of NG108 Cells Using Quadrupole-Orbitrap Mass Spectrometry. Neuromethods, 114, 127–141. doi: 10.1007/7657_2015_89 [DOI] [Google Scholar]
- Gehrmann ML, Hathout Y, & Fenselau C (2004). Evaluation of metabolic labeling for comparative proteomics in breast cancer cells. J Proteome Res, 3(5), 1063–1068. doi: 10.1021/pr049906k [DOI] [PubMed] [Google Scholar]
- Geiger T, Cox J, Ostasiewicz P, Wisniewski JR, & Mann M (2010). Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat Methods, 7(5), 383–385. doi: 10.1038/nmeth.1446 [DOI] [PubMed] [Google Scholar]
- Hoedt E, Zhang G, & Neubert TA (2014). Stable isotope labeling by amino acids in cell culture (SILAC) for quantitative proteomics. Adv Exp Med Biol, 806, 93–106. doi: 10.1007/978-3-319-06068-2_5 [DOI] [PubMed] [Google Scholar]
- Howden AJ, Geoghegan V, Katsch K, Efstathiou G, Bhushan B, Boutureira O, . . . Acuto O. (2013). QuaNCAT: quantitating proteome dynamics in primary cells. Nat Methods, 10(4), 343–346. doi: 10.1038/nmeth.2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleifeld O, Doucet A, Prudova A, auf dem Keller U, Gioia M, Kizhakkedathu JN, & Overall CM (2011). Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates. Nat Protoc, 6(10), 1578–1611. doi: 10.1038/nprot.2011.382 [DOI] [PubMed] [Google Scholar]
- Kruger M, Moser M, Ussar S, Thievessen I, Luber CA, Forner F, . . . Mann M. (2008). SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell, 134(2), 353–364. doi: 10.1016/j.cell.2008.05.033 [DOI] [PubMed] [Google Scholar]
- Landgraf P, Antileo ER, Schuman EM, & Dieterich DC (2015). BONCAT: metabolic labeling, click chemistry, and affinity purification of newly synthesized proteomes. Methods Mol Biol, 1266, 199–215. doi: 10.1007/978-1-4939-2272-7_14 [DOI] [PubMed] [Google Scholar]
- Neubert TA, & Tempst P (2010). Super-SILAC for tumors and tissues. Nat Methods, 7(5), 361–362. doi: 10.1038/nmeth0510-361 [DOI] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, & Mann M (2002). Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics, 1(5), 376–386. [DOI] [PubMed] [Google Scholar]
- Ong SE, & Mann M (2006). A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat Protoc, 1(6), 2650–2660. doi: 10.1038/nprot.2006.427 [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, & Mann M (2003). Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem, 75(3), 663–670. [DOI] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, . . . Selbach M. (2011). Global quantification of mammalian gene expression control. Nature, 473(7347), 337–342. doi: 10.1038/nature10098 [DOI] [PubMed] [Google Scholar]
- Shenoy A, & Geiger T (2015). Super-SILAC: current trends and future perspectives. Expert Rev Proteomics, 12(1), 13–19. doi: 10.1586/14789450.2015.982538 [DOI] [PubMed] [Google Scholar]
- Spellman DS, Deinhardt K, Darie CC, Chao MV, & Neubert TA (2008). Stable isotopic labeling by amino acids in cultured primary neurons: application to brain-derived neurotrophic factor-dependent phosphotyrosine-associated signaling. Mol Cell Proteomics, 7(6), 1067–1076. doi: 10.1074/mcp.M700387-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, MacCoss MJ, Howell KE, Matthews DE, & Yates JR 3rd. (2004). Metabolic labeling of mammalian organisms with stable isotopes for quantitative proteomic analysis. Anal Chem, 76(17), 4951–4959. doi: 10.1021/ac049208j [DOI] [PubMed] [Google Scholar]
- Zanivan S, Krueger M, & Mann M (2012). In vivo quantitative proteomics: the SILAC mouse. Methods Mol Biol, 757, 435–450. doi: 10.1007/978-1-61779-166-6_25 [DOI] [PubMed] [Google Scholar]
- Zhang G, Annan RS, Carr SA, & Neubert TA (2010). Overview of peptide and protein analysis by mass spectrometry. Curr Protoc Protein Sci, Chapter 16, Unit16 11. doi: 10.1002/0471140864.ps1601s62 [DOI] [PubMed] [Google Scholar]
- Zhang G, Bowling H, Hom N, Kirshenbaum K, Klann E, Chao MV, & Neubert TA (2014). In-depth quantitative proteomic analysis of de novo protein synthesis induced by brain-derived neurotrophic factor. J Proteome Res, 13(12), 5707–5714. doi: 10.1021/pr5006982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Deinhardt K, Chao MV, & Neubert TA (2011). Study of neurotrophin-3 signaling in primary cultured neurons using multiplex stable isotope labeling with amino acids in cell culture. J Proteome Res, 10(5), 2546–2554. doi: 10.1021/pr200016n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Deinhardt K, & Neubert TA (2014). Stable isotope labeling by amino acids in cultured primary neurons. Methods Mol Biol, 1188, 57–64. doi: 10.1007/978-1-4939-1142-4_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, & Neubert TA (2009). Use of stable isotope labeling by amino acids in cell culture (SILAC) for phosphotyrosine protein identification and quantitation. Methods Mol Biol, 527, 79–92, xi. doi: 10.1007/978-1-60327-834-8_7 [DOI] [PMC free article] [PubMed] [Google Scholar]