

Abstract
Activation of the Hedgehog (Hh) pathway is causative of virtually all sporadic and Gorlin syndrome-related basal cell carcinomas (BCC), with loss of function of Patched1 (Ptc1) being the most common genomic lesion. Sporadic BCCs also overexpress desmoglein-2 (Dsg2), a desmosomal cadherin normally found in the basal layer. Using a mouse model of Gorlin syndrome (Ptc1+/lacZ mice), we found that overexpressing Dsg2 in the basal layer (K14-Dsg2/Ptc1+/lacZ) or the superficial epidermis (Inv-Dsg2/Ptc1+/lacZ mice) resulted in increased spontaneous BCC formation at 3 and 6 months, respectively. The tumors did not show loss of heterozygosity of Ptc1, despite high levels of Gli1 and phosphorylated Stat3. A panel of sporadic human BCCs showed increased staining of both Dsg2 and P-Stat3 in 9/9 samples. Overexpression of Dsg2 in ASZ001 cells, a Ptc1−/− BCC cell line, induced Stat3 phosphorylation and further increased Gli1 levels, both in an autocrine and paracrine manner. Three different Stat3 inhibitors reduced viability and Gli1 expression in ASZ001 cells, but not in HaCaT cells. Conversely, stimulation of Stat3 in ASZ001 cells with IL-6 increased Gli1 expression. Our results indicate that Dsg2 enhances canonical Hh signaling downstream of Ptc1 to promote BCC development through the activation of P-Stat3 and regulation of Gli1 expression.
INTRODUCTION
The Hh signaling pathway has an essential role in skin development, homeostasis, and hair follicle cycling. Sonic Hedgehog (Shh) signaling in the dermal papilla stimulates bulge stem cells proliferation and growth into the dermis during anagen of the hair follicle cycle, and also controls papillary fibroblast activation and matrix remodeling (Chiang et al., 1999; Lichtenberger et al., 2016). Shh signals by binding to its receptor Patched1 (Ptc1 in mice, PTCH1 in humans). In the absence of Shh, Ptc1 localizes to the primary cilium and prevents activation of Smoothened (Smo), a GPCR family member that regulates activation of the Gli family of transcription factors (Gli1, 2 and 3). Binding of Shh to Ptc1 alleviates Smo repression and allows its accumulation at the primary cilium, inhibiting Gli3 processing into a transcriptional repressor and promoting full activation and nuclear localization of full length Gli2 and Gli3. These constitutively expressed Gli family members then induce Gli1 and Ptc1 transcription, which are widely used markers of canonical Hh pathway activity (Riobo et al., 2007).
Basal cell carcinoma (BCC) is the most commonly diagnosed cancer in the United States, with an estimated 4 million new cases reported annually (Rogers et al., 2015). Most cases are sporadic and linked to UV light exposure, but a small percentage of patients are diagnosed with Gorlin syndrome, an autosomal dominant disease. Gorlin syndrome patients are heterozygous for PTCH1, and not only develop BCCs at high frequency, but also medulloblastoma, benign jaw cysts, and ovarian and cardiac fibromas. The BCC of Gorlin patients arise by somatic loss of heterozygosity (LOH) and constitutive activation of Smo (Athar et al., 2014). While 90% of sporadic BCCs present loss-of-function mutations of PTCH1, almost 10% are driven by gain-of-function mutations in Smo (Epstein, 2008). The mouse model for Gorlin syndrome (Ptc1lacZ/+mice) are susceptible to BCC formation after UV or γ-radiation, or when crossed into a p53 null background (Aszterbaum et al., 1999; Pazzaglia, 2006; So et al., 2006). However, Ptc1lacZ/+ mice only develop sporadic lesions at an advanced age and at low frequency (Pazzaglia, 2006; Aszterbaum et al., 1999). In this model, expression of LacZ serves as a reporter of canonical Hh pathway activation and tissue X-gal staining is highly specific in the tumors of these animals.
We recently reported that Dsg2, a desmosomal cadherin, is upregulated in human BCCs (Brennan and Mahoney, 2009). Current research suggests a role for Dsg2 as a modulator of cell signaling through activation of Akt, ERK, and Stat3 pathways in keratinocytes in addition to cell adhesion (Brennan et al., 2007; Brennan et al., 2012a; Overmiller et al., 2016). Transgenic expression of Dsg2 in the superficial epidermis under the involucrin promoter (Inv-Dsg2) activates growth and survival signaling in keratinocytes and increases susceptibility to squamous cell carcinoma (SCC) induction (Brennan et al., 2007). Since the signals activated by Dsg2 can potentiate Hh signaling (Riobo et al, 2006a; Riobo et al., 2006b; Gu et al., 2012), we generated compound Inv-Dsg2/Ptc1+/lacZ mice to study the effect of Dsg2 overexpression on skin tumorigenesis in a Ptc1+/− background. Inv-Dsg2/Ptc1+/lacZ mice developed earlier squamous lesions in response to DMBA-TPA than wild-type (WT) or Inv-Dsg2 animals and, surprisingly, also developed BCCs during the chemical carcinogenesis study (Brennan-Crispi et al., 2015a). In this study, we investigated the effect of overexpressing Dsg2 in the superficial epidermis vs. basal cell layer on spontaneous formation of BCC in Ptc1+/lacZ mice, comparing Ptc1+/lacZ; Inv-Dsg2 mice to a cross with newly engineered K14-Dsg2 mice (Cooper et al., 2018) to generate Ptc1+/lacZ; K14-Dsg2 mice.
Here, we report that Dsg2 expression dramatically enhances the rate of spontaneous BCC formation in both animal models, suggesting a non-cell autonomous mode of action that could be explained by increased paracrine signaling. The spontaneous BCC show no evidence of LOH of Ptc1, as opposed to BCCs induced by radiation (Aszterbaum et al., 1999), and have in common increased P(Tyr705)Stat3 staining. In vitro studies showed that Dsg2 potentiates canonical Hh signaling and Stat3 activation in ASZ001 BCC cells both in an autocrine and paracrine manner. Inhibition of Stat3 reduced Gli1 expression and induced cell death in an additive manner to the Smo inhibitor vismodegib. In summary, our data revealed that Dsg2 induces a paracrine mediator that stimulates the canonical Hh signaling through Stat3.
RESULTS
Altered skin homeostasis phenotype by compartment-specific Dsg2 expression is exacerbated by Ptc1 haploinsufficiency.
We first compared the backskin morphology of 3 months-old Ptc1+/lacZ, K14-Dsg2; Ptc1+/lacZ, and Inv-Dsg2; Ptc1+/lacZ mice, which have variable expression domains of transgenic Dsg2-Flag (Fig. 1A). Inv-Dsg2/Ptc1+/lacZ mice showed extensive elongation and crowding of basal keratinocytes (palisading) together with significant epidermal hyperproliferation (Fig. 1A and B). In contrast, the skin of Ptc1+/lacZ and K14-Dsg2/Ptc1+/lacZ mice was grossly normal (Fig. 1A and B). Palisading of keratinocytes and cytokeratin 17 (CK17) expression are both markers of BCC preneoplastic changes (Markey et al., 1992; Grachtchouk et al., 2003). In Ptc1+/lacZ mice, CK17 staining was only observed in hair follicles (Fig. 1A, arrows), as previously reported (McGowan and Coulombe, 1998). In contrast, Inv-Dsg2;Ptc1+/lacZ mice showed strong CK17 immunostaining in the superficial epidermis (Fig. 1A). The overlap of CK17 expression in palisading basal cells suggests that they exhibit characteristics of early BCC. Despite the normal skin morphology, K14-Dsg2/Ptc1+/lacZ mice also showed increased CK17 staining, albeit less than Inv-Dsg2; Ptc1+/lacZ skin, particularly in localized regions of the interfollicular epidermis (IFE), suggesting the occurrence of preneoplastic changes as well (Fig. 1A).
Figure 1. Overexpression of Dsg2 results in spontaneous BCC formation.
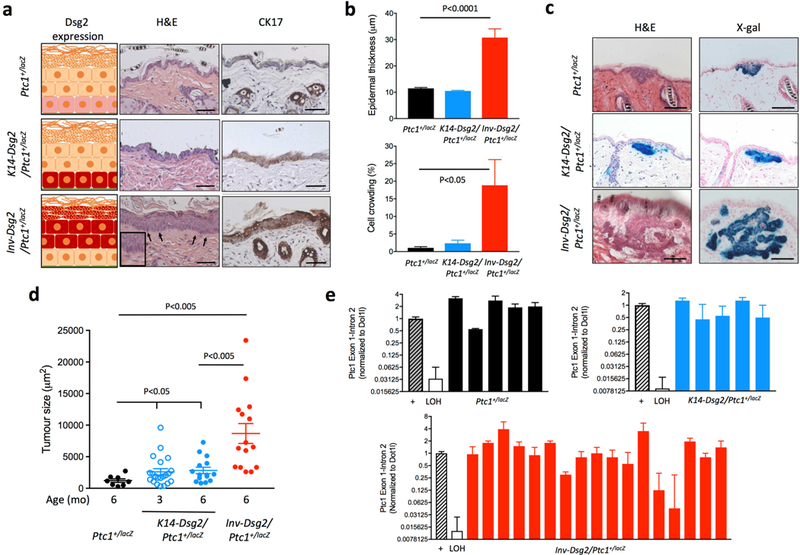
(a) Left side: cartoons showing the endogenous expression level of Dsg2 (pink) or transgene overexpression (red) in the epidermis of the different mouse genotypes. Formalin-fixed, paraffin-embedded skin sections from 3-month old Ptc1+/lacZ, K14-Dsg2/Ptc1+/lacZ and Inv-Dsg2/Ptc1+/lacZ mice were stained with haematoxylin and eosin (center) or were immunostained for cytokeratin 17 (CK17) (right). Scale bars = 50 microns. (b) Quantification of epidermal thickness and percentage of cell crowding (palisading) in backskin of 3-month old mice of the three genotypes (n=4–8 animals per group). (c) Representative examples of spontaneous BCCs from 6-month old Ptc1+/lacZ, Inv-Dsg2/Ptc1+/lacZ and K14-Dsg2/Ptc1+/lacZ mice stained for H&E and X-Gal. Scale bar = 50 microns. (d) Quantification of tumor size in Ptc1+/lacZ, Inv-Dsg2/Ptc1+/lacZ, and K14 Dsg2/Ptc1+/lacZ mice at 3 and 6 months of age. Each data point represents one BCC (n=9–21), some animals developed several tumors. (e) LOH status of the wild-type ptc1 allele in individual laser-capture microdissected BCCs of the indicated genotypes (n=5–17). Medulloblastomas from Ptc1+/lacZ mice positive and negative for LOH served as internal controls.
Ectopic expression of Dsg2 increases spontaneous BCC tumorigenesis in Ptc1+/lacZ mice.
We next examined the skin of 3- and 6-month old mice for signs of BCC, based on histology and positive staining for X-gal in consecutive sections, an indicator of activation of the canonical Hh pathway. Ptc1+/lacZ mice had no evidence of tumours at 3 months, but develop small BCCs by 6 months (Fig. 1C and Table 1). In striking contrast, K14-Dsg2/Ptc1+/lacZ mice developed spontaneous, microscopic BCCs at 3 months of age, which did not appear to grow to a larger size at 6 months (Fig. 1C and 1D and Table 1). BCC-like tumors were prominent and large in Inv-Dsg2/Ptc1+/lacZ mice at 6 months of age, but were undetectable at 3 months, suggesting a retarded tumor initiation compared to K14-Dsg2/Ptc1+/lacZ, but more rapid growth (Fig. 1C and 1D, and Table 1). No signs of BCC were detected in WT, Inv-Dsg2, or K14-Dsg2 at 6 months of age, indicating that Ptc1 haploinsufficiency is necessary for BCC formation. While the study was not designed to determine the cell of origin of these tumors, many BCCs were physically associated with hair follicles, as expected with the Ptc1+/lacZ model. Taken together, these data suggest that Dsg2 strongly enhances spontaneous BCC development in a Ptc1 heterozygous background in a cell autonomous and non-cell autonomous manner.
Table 1.
Quantitative analysis of spontaneous BCC characteristics in all mouse genotypes at 3 and 6 months of age.
| Genotype | Age (mo.) |
% Incidence (n) |
Mean BCCs per mouse |
Mean BCCs/cm2 |
Mean BCC burden (μm2/cm2) |
Mean BCC size (μm2) |
|---|---|---|---|---|---|---|
| Ptc1+/lacZ | 3 | ND | N/A | N/A | N/A | N/A |
|
K14-Dsg2; Ptc1+/lacZ |
3 | 81 % (9/11) | 1.9 | 0.2 | 544 | 2,981 |
|
Inv-Dsg2; Ptc1+/lacZ |
3 | ND | N/A | N/A | N/A | N/A |
| Ptc1+/lacZ | 6 | 56 % (5/9) | 0.8 | 0.2 | 251 | 1,139 |
|
K14-Dsg2; Ptc1+/lacZ |
6 | 67 % (8/12) | 1.1 | 0.4 | 1,175 | 2,827 |
|
Inv-Dsg2; Ptc1+/lacZ |
6 | 100 % (4/4) | 3.75 | 0.7 | 6,501 | 8,681 |
Most Dsg2-mediated BCC tumors do not show loss of heterozygosity of Ptc1.
Since spontaneous BCC formation in Gorlin syndrome patients (Hahn et al., 1996), as well as BCC formation after irradiation in the Ptc1+/lacZ mouse model have loss of heterozygosity (LOH) of the WT Ptc1 allele (Aszterbaum et al., 1999), we sought to determine Ptc1 LOH status in the spontaneous BCC in our Ptc1+lacZ, Inv-Dsg2/Ptc1+lacZ and K14-Dsg2/Ptc1+lacZ animals. To this end, we dissected X-gal+ BCC lesions using laser capture microdissection, isolated genomic DNA, and assessed the status of the WT allele of Ptc1 by RT-PCR using primers that specifically recognize the Ptc1 wild type allele (Mille et al., 2014). Surprisingly, none of the spontaneous BCCs from Ptc1+/lacZ and K14-Dsg2/Ptc1+lacZ mice had LOH of Ptc1 (Fig. 1E). Only 2 of 17 Inv-Dsg2/Ptc1+lacZ tumors showed signs of possible LOH (Fig. 1E). Since we could not perform whole exon sequencing of Ptc1, it could be possible that some BCCs have LOH undetectable by this method. However, we used medulloblastomas from Ptc1+/lacZ mice that were either positive or negative for LOH by other methods as internal controls. Our findings suggest that Ptc1 LOH could be dispensable for BCC initiation and increased growth in Dsg2-driven BCC tumorigenesis.
Stat3 activation is a common feature of BCCs derived from both animal models.
We next set out to determine if BCCs share similar characteristics in both animal models. The tumors and normal skin of Inv-Dsg2/Ptc1+/lacZ and K14-Dsg2/Ptc1+/lacZ were analyzed for expression of the Flag-tagged Dsg2 (Fig. 2A, top panels). The Dsg2 transgene was not expressed within the tumors of Inv-Dsg2/Ptc1+/lacZ genotype, but was detected in the adjacent superficial epidermis. However, K14-Dsg2/Ptc1+/lacZ tumors showed diffuse, cytoplasmic expression of Dsg2-Flag within the tumor mass (Fig. 2A, top panel), while the IFE showed barely detectable levels of Flag staining, consistent the transgene expression in the parental K14-Dsg2 line (Cooper et al., 2018). The lack of transgene expression in the BCCs form Inv-Dsg2/Ptc1+/lacZ mice further supports a non-cell autonomous effect. In order to identify the potential molecular drivers of Dsg2-mediated BCC formation, we analyzed ERK and Stat3 activation, common to both Dsg2 and Hh signaling. Tumors in K14-Dsg2/Ptc1+/lacZ and Inv-Dsg2/Ptc1+/lacZ mice were largely negative for P-ERK1/2 staining (not shown). However, nuclear P-Stat3(Tyr705) staining was detected in the BCCs of both Inv-Dsg2/Ptc1+/lacZ and K14-Dsg2/Ptc1+/lacZ mice (Fig. 2A, bottom panels), suggesting that Stat3 activation could be related to spontaneous BCC development. In addition, the normal IFE of Inv-Dsg2/Ptc1+/lacZ showed higher percentage of positive plus highly positive P-Stat3(Tyr705) staining compared to K14-Dsg2/Ptc1+/lacZ and Ptc1+/lacZ animals (Fig. 2B), which could be related to the faster growth of BCCs in the Inv-Dsg2/Ptc1+/lacZ mice. Moreover, we analyzed a small set of human sporadic BCCs (n=9), and found high P-Stat3(Tyr705) staining overlapping with high diffuse Dsg2 staining in 100% of the tumors, in comparison with the adjacent normal tissue (Fig. 2C).
Figure 2. Dsg2 increased Stat3 phosphorylation in murine and in human sporadic BCCs.
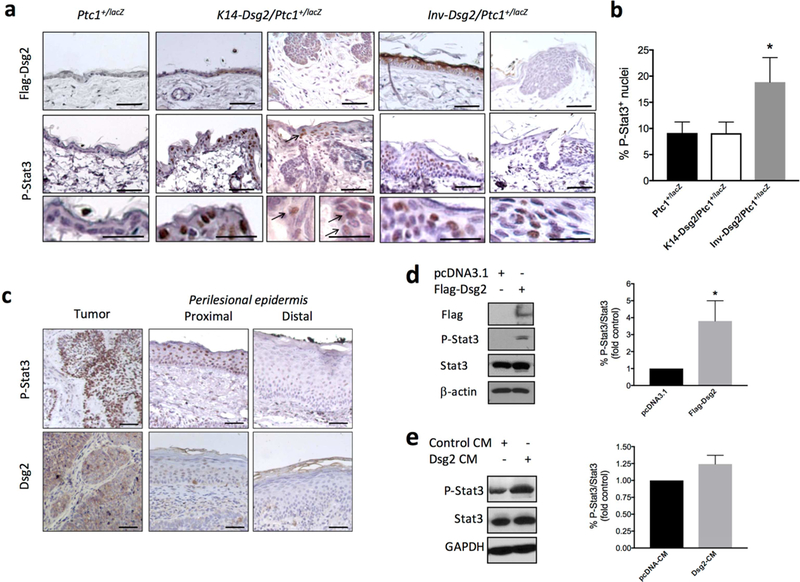
(a) Formalin-fixed, paraffin-embedded skin sections from 6-month old Ptc1+/lacZ and Inv-Dsg2; Ptc1+/lacZ and 3-month old K14-Dsg2/Ptc1+/lacZ mice were immunostained for Flag (top panels), and P-Stat3 (bottom panels). Scale bars = 50 microns, inset scale bar = 100 microns. Representative images from n=3–5 animals. (b) Quantification of positive and highly positive P-Stat3+ nuclei in the interfollicular epidermis (not tumor areas) of the indicated mouse genotypes. Graph represents mean ± SEM (n=3–5; *P<0.05) (c) Representative example of human BCC immunostained for Dsg2 and P-Stat3. IHC staining of tumor, and both proximal and distal perilesional skin are shown. (d) Representative Western blot analysis of Dsg2.Flag, P-Stat3 (Tyr705), Stat3, and β-actin in ASZ001 transfected with empty plasmid (pcDNA3) or with pcDNA3 encoding Flag-tagged Dsg2 (mDsg2) (n=3; *P<0.05) (e) Naïve ASZ001 were stimulated for 30 min with conditioned medium from cells transfected as in (d), followed by lysis and western blot analysis of P-Stat3(Tyr705) and total Stat3 levels (n=3; P=0.068).
Dsg2 stimulates Stat3 in BCC cells in vitro.
ASZ001 cells, established from a BCC of a UV-irradiated Ptc1+/lacZ mouse, have constitutive activation of the Hh pathway, evidenced by high levels of Gli1 (So et al., 2006). In agreement with the tumor immunostaining data, transfection of ASZ001 cells with Dsg2 increased Stat3(Tyr705) phosphorylation by ~4-fold (Fig. 2D). Next, we evaluated the effect of conditioned medium (CM) from ASZ001 cells transfected with Dsg2-Flag or an empty plasmid onto naïve ASZ001 cells. The CM of Dsg2 transfected cells increased Stat3 phosphorylation by ~25 % after 30 min stimulation when compared to the CM of pcDNA-transfected cells (Fig. 2E). These results suggest that Dsg2 overexpression can stimulate Stat3 phosphorylation in a cell autonomous and non-cell autonomous manner.
Dsg2 upregulates Gli1 expression in vitro
Despite the high Hh pathway activity under basal conditions, introduction of Dsg2 into ASZ001 cells increased Gli1 mRNA expression further by ~2-fold (Fig. 3A). Since ASZ001 cells have LOH of Ptc1, this suggests that Dsg2 potentiates Hh signaling downstream of Ptc1. We also evaluated the effect of stable Dsg2 expression in LIGHT2 cells, which increased Gli-luciferase activity when the canonical Hh pathway is active (Taipale et al., 2000). LIGHT2-Dsg2 cells had a ~2-fold larger increase in Gli-luciferase in response to purmorphamine (a Smo agonist) than LIGHT2-pcDNA cells (Fig. 3B). These in vitro data recapitulate our in vivo findings by showing that Dsg2 potentiates canonical Hh signaling.
Figure 3. Stat3 mediates Dsg2-induced Gli1 upregulation and is essential for BCC cells viability.
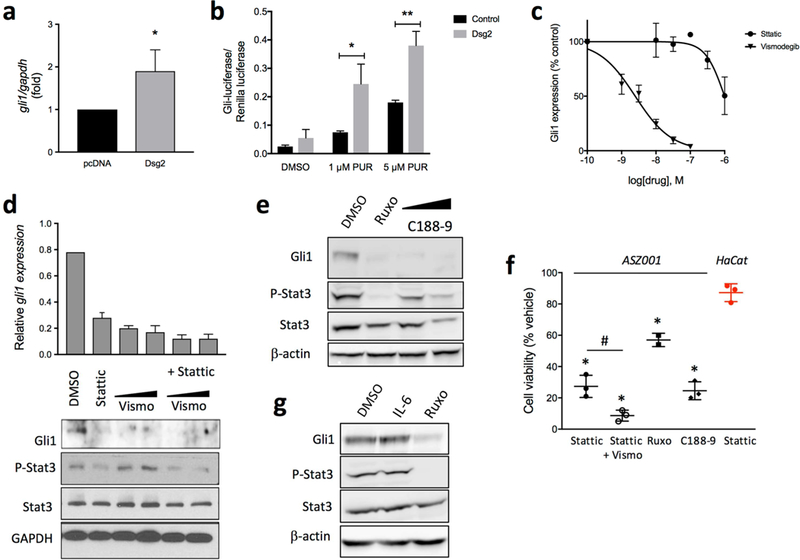
(a) gli1 mRNA expression by qPCR of ASZ001 cells transiently transfected with empty plasmid (pcDNA3) or with pcDNA3 encoding Flag-tagged Dsg2 (mDsg2) (n=3) *p<0.05; Student’s t-test. (b) NIH3T3-LIGHT2 cells stably transfected with empty vector (black) of a pcDNA-Dsg2 (grey) and serum starved for 24 h in the presence of the Smo agonist purmorphamine (PUR). Representative results of Gli-luciferase activity (normalized to Renilla luciferase) from an experiment performed in triplicate (n=3). * P<0.05 **P<0.01, one-tailed Student’s t-test. (c) Inhibition of gli1 expression, by qPCR, in ASZ001 cells treated with increasing concentrations of Stattic or vismodegib for 24 h (n=3). (d) qPCR and Western blot analysis of Gli1 expression in cells treated with 1 μM Stattic, 50–100 nM vismodegib, or a combination of both for 48 h. All treatments are significantly reduced compared to control (p<0.001). n=3, *p<0.05, Student’s t-test. Efficacy of Stattic is shown below by changes in P-Stat3 (Tyr705). (e) Western blot analysis of Gli1 expression and Stat3 phosphorylation in ASZ001 cells treated with 20 μM ruxolitinib (Ruxo) or 1–3 μM C188–9 for 48 h. (f) Gli1 expression and Stat3 phosphorylation in ASZ001 cells stimulated with 10 ng/ml IL-6 or IL-6 plus 20 μM ruxolitinib (Ruxo) (n=3). (g) ASZ001 cells were incubated for 48 h with 1 µM Stattic, 1 µM Stattic plus 100 nM vismodegib, 20 μM ruxolitinib, or 3 μM C188–9. Cell viability was determined with the WST-1 assay and expressed as % of viability of cells treated with DMSO (vehicle). Student’s t-test, #p<0.05; *p<0.0001. In red, viability of HaCaT cells incubated with 1 µM Stattic for 48 h (n=3–4).
Inhibition of Stat3 downregulates Gli1 expression and reduces ASZ001 cell viability.
We treated ASZ001 cells with increasing concentrations of Stattic, a novel Stat3 inhibitor that targets its SH2 domain (Schust et al., 2006) or the FDA-approved Smo inhibitor Vismodegib for 48 h. Vismodegib reduced gli1 mRNA levels in a dose-response manner, with an apparent IC50 = 2.5nM, as expected (Fig. 3C). Remarkably, Stattic also reduced gli1 mRNA levels in a dose-response manner, with IC50 < 1 µM (Fig. 3C). The exact IC50 of Stattic could not be determined because of massive cell death with concentrations >3 µM. Co-incubation of the cells with 1 µM Stattic and 50 nM or 100 nM vismodegib resulted in a more profound reduction of Gli1 mRNA and protein levels compared to either inhibitor alone (Fig. 3D). To confirm the efficacy of Stattic to inhibit Stat3, we measured phosphorylation of Stat3 at Tyr705 in all the tested conditions. As expected, Stattic treatment for 48 h reduced Stat3 phosphorylation by 47%, while Stattic and Vismodegib together resulted in a further inhibition of Stat3 phosphorylation (65%) (Fig. 3D and Fig. S1). A JAK1/2 inhibitor (Ruxolitinib) and a second Stat3 inhibitor (C188–9) also reduced Gli1 expression in ASZ001 and phospho-Stat3 by 98% and 42%, respectively (Fig. 3E and Fig. S1), confirming the essential role of Stat3 on Gli1 expression in this BCC cell line. Furthermore, stimulation of Stat3 in ASZ001 cells with IL-6 increased the expression of Gli1 by 20% (Fig. 3F) Remarkably, all three Stat3 inhibitory drugs reduced ASZ001 viability, with the JAK1/2 inhibitor being the least potent, while HaCaT cells were not significantly affected by Stattic (Fig. 3G). Altogether, these findings support the notion that Dsg2 stimulates Gli1 expression via paracrine activation of Stat3 signalling.
DISCUSSION
In this study, we report that overexpression of Dsg2 enhances spontaneous BCC formation in a Ptc1+/lacZ background, using two different animal models, one overexpressing Dsg2 in the superficial epidermis (Inv-Dsg2;Ptc1+/lacZ) and the other in the proliferative basal cell layer (K14-Dsg2;Ptc1+/lacZ), from which BCCs are thought to arise. This study agrees with our previous report that Inv-Dsg2;Ptc1+/lacZ mice were more susceptible to chemical-induced SCC and BCC formation (Brennan-Crispi et al., 2015; Youssef et al., 2010; Wang et al., 2011; Peterson et al., 2015). Our findings firmly establish that Dsg2 increases Stat3 phosphorylation and promotes Hh-mediated BCC development by cell autonomous and non-cell autonomous mechanisms. Moreover, we show that Stat3 is a potential therapeutic target for human BCC since Stat3 inhibition is cytotoxic even in BCC cells that completely lacked Ptc1 like most BCC lesions in patients. Of note, overexpression of Dsg2 in either epidermal compartment (basal or superficial) was insufficient to induce BCC carcinogenesis (data not shown), despite evoking a hyperproliferative phenotype in the Inv-Dsg2 skin, demonstrating that haploinsufficiency of Ptc1 is still necessary for BCC formation.
Interestingly, while Inv-Dsg2;Ptc1+/lacZ mice exhibited preneoplastic changes consistent with early BCC formation by three months of age and developed larger spontaneous BCCs at six months, the K14-Dsg2;Ptc1+/lacZ mice exhibited multiple small BCCs as early as three months of age. This significantly earlier onset may be due to the presence of the Dsg2 transgene within the tumors and the cells of origin in K14-Dsg2;Ptc1+/lacZ mice. Though there were no overt epidermal changes in these mice, there is significant activation of various signaling cascades within the skin of K14-Dsg2 animals (Cooper et al., 2018), suggesting that Dsg2 overexpression in the basal layer may prime the cells for malignant transformation.
Although the strict requirement of Ptc1 heterozygosity could suggest that tumors were secondary to LOH of Ptc1, most of the tumors appear to retain the WT allele of Ptc1, although our analysis cannot exclude missense mutations or frameshifts in the more 3’ regions of the Ptc1 gene. However, point missense and frameshift mutations are typical of UV exposure and not of copy loss that is characteristic of tumors without UV irradiation. Our findings suggest that LOH of Ptc1 is not necessary to drive early tumor formation in Dsg2-transgenic mice, leading us to investigate alternative mechanisms. A commonality between the two Dsg2 overexpression models is the increase in active (phosphorylated) Stat3 in the tumor mass and sometimes in the adjacent epidermis. In a BCC cell culture model, introduction of Dsg2 increased Stat3 phosphorylation and doubled gli1 expression. This is remarkable given that those BCC cells have LOH of Ptc1 and are thought to exhibit basal maximal activation of the Hh pathway. Additionally, inhibition of Stat3 with three different pharmacological inhibitors lead to a strong reduction of Gli1 expression, suggesting that Dsg2 may increase gli1 expression through Stat3. In support, it was very recently reported that IL-6/Stat3 signaling is necessary for BCC formation and that Stattic or depletion of IL-6R strongly reduces BCC growth in vivo and in vitro (Sternberg et al., 2018). Moreover, it was previously reported that Stat3 is necessary for SmoM2-induced BCC formation as a mediator of paracrine cytokine signaling (Gu et al., 2012).
Since Dsg2 expression regulates inflammatory gene networks (Gupta et al., 2015), future studies elucidating a link between Dsg2 and cytokine expression will provide further mechanistic insight. Overexpression of Dsg2 in HaCaT keratinocytes resulted in the upregulation of the urokinase-type plasminogen activator receptor (uPAR) and IL-6 receptor (IL6R) (Cooper et al., 2018). Since both uPAR and IL6R are able to activate Stat3 (Shushakova et al., 2005), both cytokine receptors are candidate mediators of Dsg2-induced BCC formation (Fig. 4). The non-cell autonomous effect of Dsg2 could be explained by increased uPA and/or IL-6 production or by transfer of the upregulated receptors by extracellular vesicles or exosomes by Dsg2-expressing cells (Fig. 4). In this regard, we have previously described that Dsg2 can alter the number and protein content of extracellular vesicles released by keratinocytes (Overmiller et al., 2017).
Figure 4. Proposed model for the autocrine/paracrine action of Dsg2 on basal keratinocytes.
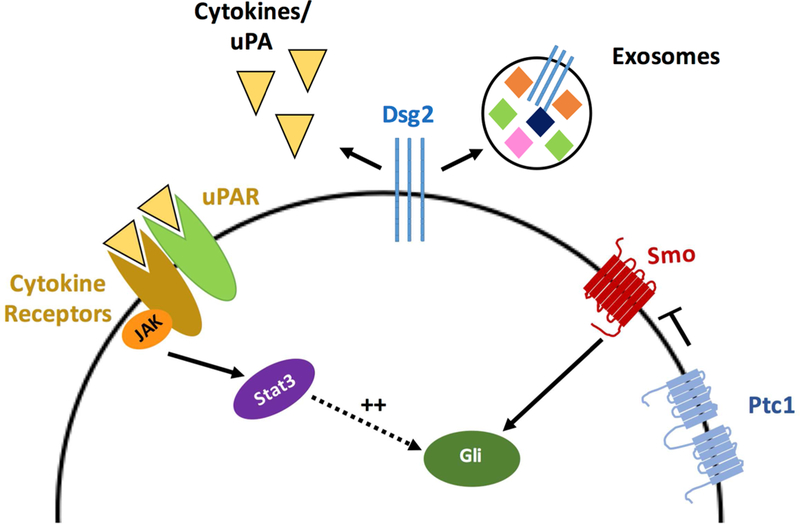
Overexpression of Dsg2 (shown in the same cell, but can be in a different cell) promotes secretion of cytokines, upregulation of receptors like uPAR and IL6R, and increased exosome secretion, leading to increased phosphorylation of Stat3 in target cells, which potentiates Gli1 expression.
The only specific therapy for advanced BCC, the Smo inhibitor vismodegib, is highly efficacious early in disease but its side effects are not well tolerated, leading to therapy withdrawal by many patients and tumor relapse (Tang et al., 2012). In addition, some patients develop resistance to vismodegib by at least two mechanisms: novel mutations in Smo and Smo-independent activation of Gli1 by atypical protein kinase C-ι/λ (Atwood et al., 2013; Atwood et al., 2015). Identifying additional therapeutic targets that achieve inhibition of Gli-dependent transcription is paramount. Of importance, our findings show that inhibition of Stat3 can abrogate Gli1 expression and that simultaneous inhibition of Smo and Stat3 additively reduces cell viability. Therefore, Stat3 may be a pharmacological target in vismodegib-resistant BCC tumors worth pursuing in translational studies. In addition, combinatorial therapy of naïve BCC with lower doses of vismodegib and a Stat3 inhibitor might be an effective alternative with lower magnitude of vismodegib side effects.
In summary, we demonstrate that Dsg2 activates Stat3 and promotes Hh-driven BCC tumorigenesis, and that targeting both Smo and Stat3 is an effective means of inhibiting Gli1 expression and reducing tumor cell viability. These results may also have implications for other forms of cancer in which both Hh and Stat3 signaling is deregulated.
MATERIALS AND METHODS
Transgenic and knock-in mouse models
All animal studies were in compliance with the Institutional Animal Care & Use Committee approvals at Thomas Jefferson University. Ptc1+/LacZ mice (Ptch1tm1Mps/J) were from the Jackson Laboratory (Bar Harbor, ME). The Inv-Dsg2 and K14-Dsg2 mice were previously described in detail (Brennan et al., 2007) (Cooper et al., 2018). All animals were maintained under AAALAC approved conditions. Ptc1+/LacZ and Inv-Dsg2 mice were crossed to yield WT, Ptc1+/LacZ, Inv-Dsg2, and Inv-Dsg2/Ptc1+/LacZ mice. Ptc1+/LacZ and K14-Dsg2 mice were crossed to yield WT, Ptc1+/LacZ, K14-Dsg2, and K14-Dsg2/Ptc1+/LacZ mice. Animals were sacrificed at indicated time points and tissues collected for analysis as either formalin fixation, paraffin embedding (FFPE) or frozen in cryopreservation compound (OCT) samples.
Determination of loss-of-heterozygosity (LOH) in tumors
OCT embedded samples were cut into 12 µm-thick cryo sections on Super Frost slides (Fisher). Consecutive sections were also cut and stained for histology and X-gal to identify tumors. BCCs were microdissected using an Arcturus Laser Capture microscope (Life Technologies). After dehydrating the tissue, the areas of interest were captured on CapSure High Sensitivity (HS) caps using the IR laser. Genomic DNA (gDNA) was isolated from the lesions using a DNA Pico Pure Isolation kit (Life Technologies); a final volume of 15µl containing the gDNA was obtained per BCC lesion.
qPCR was performed on gDNA obtained from each dissected lesion. The levels of the WT allele of Ptc1 relative to Dot1l were used to determine whether the Ptc1 WT allele was retained or lost in BCC lesions using primers recognizing Ptch1 Exon1-Intron1/2, which is deleted in the Ptc1 mutant allele (Goodrich et al., 1997). The internal granule cell layer of the normal cerebellar tissue of Ptc1+/lacZ mice was used to establish the levels of one copy of Ptc1, and advanced medulloblastoma tissues displaying Ptc1 LOH were used as positive control (Mille et al., 2014). qPCR results were obtained using the ΔΔCT method and SybrGreen reagents on a Viia7 system (Life Technologies); reactions were carried in triplicate using 1.5µl of the gDNA extract per reaction. Primer sequences: Dot1l-F: TAG TTG GCA TCC TTA TGC TTC ATC; Dot1l-R: GCC CCA GCA CGA CCA TT; Ptch1 ex1 int1/2-F: CCT TCG CTC TGG AGC AGA TT; Ptch1 ex1 int1/2-R: GGA TCC CAA GGA GGA AGA AGA.
Supplementary Material
ACKNOWLEDGEMENTS
This research was supported by the National Cancer Institute of the National Institutes of Health (Brennan-Crispi, F31CA171680; Mahoney, AR056067), the Canadian Institutes of Health Research (Charron), and a Caldas fellowship from Colciencias, Colombia (Tamayo-Orrego).
The CK17 antibody was gifted by Pierre Coloumbe (Johns Hopkins University) and ASZ001 cells by Ervin H. Epstein (Children’s Hospital Oakland Research Institute).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- Aszterbaum M, Epstein J, Oro A, Douglas V, LeBoit PE, Scott MP, et al. Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice. Nat Med 1999; 5:1285–91. [DOI] [PubMed] [Google Scholar]
- Athar M, Li C, Kim AL, Spiegelman VS, Bickers DR. Sonic hedgehog signaling in Basal cell nevus syndrome. Cancer Res 2014; 74: 4967–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood SX, Li M, Lee A, Tang JY, Oro AE. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature 2013; 494:484–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015; 27:342–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan-Crispi DM, Hossain C, Sahu J, Brady M, Riobo NA, Mahoney MG. Crosstalk between Desmoglein 2 and Patched 1 accelerates chemical-induced skin tumorigenesis. Oncotarget 2015a; 6:8593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan-Crispi DM, Mahoney MG, Riobo NA. Methods for Detection of Ptc1-Driven LacZ Expression in Adult Mouse Skin. Methods Mol Biol 2015b; 1322:167–85. [DOI] [PubMed] [Google Scholar]
- Brennan D, Hu Y, Joubeh S, Choi Y, Ehitaker-Menezes D, O’Brien T, et al. Suprabasal Dsg2 expression in transgenic mouse skin confers a hyperproliferative and apoptosis resistant phenotype to keratinocytes. J Cell Sci 2007; 120:758–71. [DOI] [PubMed] [Google Scholar]
- Brennan D, Mahoney M. Increased expression of Dsg2 in malignant skin carcinomas: A tissue-microarray based study. Cell Adh Migr 2009; 3:148–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan D, Peltonen S, Dowling A, Medhat W, Green K, Wahl J, et al. A role for caveolin-1 in desmoglein binding and desmosome dynamics. Oncogene 2012a; 31:1636–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Swan RZ, Grachtchouk M, Bolinger M, Litingtung Y, Roberton EK, et al. Essential role of Sonic Hedgehog during hair follicle morphogenesis. Dev Biol 1999; 205:1–9. [DOI] [PubMed] [Google Scholar]
- Cooper F, Overmiller A, Loder A, Brennan-Crispi DM, McGuinn KP, et al. Enhancement of cutaneous wound healing by Dsg2 augmentation of uPAR secretion. J Invest Dermatol 2018. (in press). [DOI] [PMC free article] [PubMed]
- Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer 2008; 8:743–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997; 277:1109–13. [DOI] [PubMed] [Google Scholar]
- Grachtchouk V, Grachtchouk M, Lowe L, Johnson T, Wei L, Wang A, et al. The magnitude of hedgehog signaling activity defines skin tumor phenotype. EMBO J 2003; 22:2741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu D, Fan Q, Zhang X, Xie J. A role for transcription factor STAT3 signaling in oncogene smoothened-driven carcinogenesis. J Biol Chem 2012; 287:38356–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Nitoiu D, Brennan-Crispi D, Addya S, Riobo NA, Kelsell DP, et al. Cell cycle- and cancer-associated gene networks activated by Dsg2: evidence of cystatin a deregulation and a potential role in cell-cell adhesion. PLoS One 2015; 10:e0120091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996; 85:841–51. [DOI] [PubMed] [Google Scholar]
- Infante P, Alfonsi R, Ingallina C, Quaglio D, Ghirga F, D’Acquarica I, et al. Inhibition of Hedgehog-dependent tumors and cancer stem cells by a newly identified naturally occuring chemotype. Cell Death Dis 2016; 7:e2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenberger BM, Mastrogiannaki M, Watt FM. Epidermal b-catenin activation remodels the dermis via paracrine signalling to distinct fibroblast lineages. Nat Commun 2016; 7:10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markey AC, Lane EB, Macdonald DM, Leigh IM. Keratin expression in basal cell carcinomas. Br J Dermatol 1992; 126:154–60. [DOI] [PubMed] [Google Scholar]
- McGowan KM, Coulombe PA. Onset of keratin 17 expression coincides with the definition of major epithelial lineages during skin development. J Cell Biol 1998; 143:469–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mille F, Tamayo-Orrego L, Levesque M, Remke M, Korshunov A, Cardin J, et al. The Shh receptor Boc promotes progression of early medulloblastoma to advanced tumors. Dev Cell 2014; 31:34–47. [DOI] [PubMed] [Google Scholar]
- Overmiller AM, McGuinn KP, Roberts BJ, Cooper F, Brennan-Crispi DM, Deguchi T, et al. c-Src/Cav1-dependent activation of the EGFP by Dsg2. Oncotarget 2016; 7:37536–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overmiller AM, Pierluissi JA, Wermuth PJ, Sauma S, Martinez-Outschoorn U, Tuluc M, et al. Desmoglein 2 modulates extracellular vesicle release from squamous cell carcinoma keratinocytes. FASEB J 2017; 31:3412–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazzaglia S Ptc1 heterozygous knockout mice as a model of multi-organ tumorigenesis. Cancer Lett 2006; 234:124–34. [DOI] [PubMed] [Google Scholar]
- Peterson SC, Eberl M, Vagnozzi AN, Belkadi A, Veniaminova NA, Verhaegen ME, et al. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell 2015; 16:400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riobo NA, Haines GM, Emerson CP Jr. Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res 2016a; 66:839–45. [DOI] [PubMed] [Google Scholar]
- Riobo NA, Lu K, Ai X, Haines GM, Emerson CP Jr. Phosphoinositide-3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc Natl Acad Sci USA 2006b; 103:4505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riobo NA, Manning DR. Pathways of signal transduction employed by vertebrate Hedgehogs. Biochem J 2007; 403:369–79. [DOI] [PubMed] [Google Scholar]
- Rogers HW, Weinstock MA, Feldman SR, Coldiron BM. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the U.S. population, 2012. JAMA Dermatol 2015; 151:1081–6. [DOI] [PubMed] [Google Scholar]
- Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecular inhibitor of STAT3 activation and dimerization. Chem Biol 2006; 13:1235–42. [DOI] [PubMed] [Google Scholar]
- Shushakova N, Tkachuk N, Dangers M, Tkachuk S, Park JK, Hashimoto K, et al. Urokinase-induced activation of the gp130/Tyk2/Stat3 pathway mediates a pro-inflammatory effect in human mesangial cells via expression of the anaphylatoxin C5a receptor. J Cell Sci 2015; 118: 2743–53. [DOI] [PubMed] [Google Scholar]
- So PL, Langston AW, Daniallinia N, Hebert JL, Fujimoto MA, Khaimskiy Y, et al. Long-term establishment, characterization and manipulation of cell lines from mouse basal cell carcinoma tumors. Exp Dermatol 2006; 15:742–50. [DOI] [PubMed] [Google Scholar]
- Sternberg C, GRuber W, Eberl M, Tesanovic S, Stadler M, Elmer DP, et al. Synergistic cross-talk of Hedgehog and Interleukin-6 signaling drives growth of basal cell carcinoma. Int J Cancer 2018. (in press). [DOI] [PMC free article] [PubMed]
- Tang JK, Ally MS, Chanana AM, Mackay-Wiggan JM, Aszterbaum M, Lindgren JA, et al. Inhibition of the hedgehog pathway in patients with basal-cell nevus syndrome: final results form the multicentre, rendomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 2016; 17:1720–31. [DOI] [PubMed] [Google Scholar]
- Tang JY, Mackay-Wiggan JM, Aszterbaum M, Yauch RL, Lindgren J, Chang K, et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N Engl J Med 2012; 366: 2180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovich L, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cylopamine. Nature 2000; 406:1005–9. [DOI] [PubMed] [Google Scholar]
- Wang GY, Wang J, Mancianti ML, Epstein EH Jr. Basal cell carcinomas arise from hair follicle stem cells in Ptch1(+/−) mice. Cancer Cell 2011; 19:114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef KK, Van Keymeulen A, Lapouge G, Beck B, Michaux C, Achouri Y, et al. Identification of the cell lineage at the origin of basal cell carcinoma. Nat Cell Biol 2010; 12: 299–305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.