

Abstract
UVB wavelengths of light induce the formation of photoproducts in genomic DNA that are potentially mutagenic and detrimental to epidermal cell function. Interestingly, the mineralocorticoid and androgen receptor antagonist spironolactone (SP) was recently identified as an inhibitor of UV photoproduct removal in human cancer cells in vitro via its ability to promote the rapid proteolytic degradation of the DNA repair protein XPB (xeroderma pigmentosum group B). Using normal human keratinocytes in vitro and skin explants ex vivo, we find that SP rapidly depleted XPB protein in both systems and abrogated two major responses to UVB-induced DNA damage, including the removal of UV photoproducts from genomic DNA and the activation of the ATR/ATM DNA damage kinase signaling. These effects were also correlated with both mutagenesis and a predisposition to UVB-induced cell death but were unique to SP, as neither the SP metabolites canrenone and 7α-thiomethylspironolactone nor the more specific mineralocorticoid receptor antagonist eplerenone impacted XPB protein levels or the UVB response. Our findings provide an approach for studying XPB and its roles in the UVB DNA damage response in human skin ex vivo and indicate that SP may increase UVB mutagenesis and skin cancer risk in certain individuals.
INTRODUCTION
UVB wavelengths of sunlight induce the formation of cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6-4) pyrimidone photoproducts [(6-4)PPs] in genomic DNA of epidermal keratinocytes (Cadet, Grand and Douki, 2015). These UV photoproducts block the movement of RNA and DNA polymerases and therefore have the potential to induce cell death and mutagenesis (Batista, Kaina, Meneghini et al, 2009). The sole mechanism for removing UV photoproducts from genomic DNA is nucleotide excision repair (NER) (Sancar, 2016, Reardon and Sancar, 2005, Wood, 1997), and along with the DNA damage response kinases ATM (ataxia telangiectasia mutated) and ATR (ATM and Rad3-related) (Sancar, Lindsey-Boltz, Unsal-Kacmaz et al, 2004, Kemp and Hu, 2017, Ciccia and Elledge, 2010, Saldivar, Cortez and Cimprich, 2017), these core regulatory systems allow proliferating cells to properly respond to and survive DNA damage induced by UVB. Defects in these processes lead to photosensitivity and an elevated risk of skin carcinogenesis, and both phenotypes are commonly observed in patients with the disease xeroderma pigmentosum (XP) (DiGiovanna and Kraemer, 2012).
Using a high throughput assay for monitoring (6-4)PP removal in HeLa cells, a recent screen of FDA-approved small molecule drugs identified the classical mineralocorticoid receptor antagonist spironolactone (SP) as a strong inhibitor of nucleotide excision repair (NER) (Alekseev, Ayadi, Brino et al, 2014). SP was further shown to promote the rapid and specific proteosomal degradation of the XPB (XP group B) protein subunit of TFIIH (transcription factor II-H) (Compe and Egly, 2016), which is known to be mutated in a small number of humans with the disorders XP, XP with Cockayne Syndrome (XP/CS), and trichiothiodystrophy (TTD) (Oh, Khan, Jaspers et al, 2006, Natale and Raquer, 2017, Fassihi, Sethi, Fawcett et al, 2016). Patients with XPB mutations have all been reported to readily sunburn after minimal sun exposure, and approximately half have developed basal and/or squamous cell carcinomas. Fibroblasts from XPB patients display lower levels of the XPB protein, exhibit defects in NER, and show increased sensitivity to UV radiation (Oh, Khan, Jaspers et al, 2006, Fassihi, Sethi, Fawcett et al, 2016).
Because SP is used to treat hypertension, congestive heart failure and cirrhotic ascites as well as various dermatological conditions, including adult acne in women (Endly and Miller, 2017, Canavan, Chen and Elewski, 2016), we examined the effects of SP on XPB protein levels and UVB DNA damage responses in normal human keratinocytes in vitro and in human skin explants ex vivo. We observed that XPB became rapidly depleted from both keratinocytes in vitro and skin epidermis ex vivo following treatment with high concentrations of SP. This loss of XPB was correlated with an inhibition of CPD and (6-4)PP removal by NER, a failure to properly phosphorylate and activate a subset of ATR/ATM DNA damage response kinase substrates, and a sensitization to the lethal effects of UVB. Interestingly, these effects were unique to SP, as its major metabolites canrenone and 7α-thiomethylspironolactone did not impact XPB levels or affect UVB responses. Nonetheless, treatment of UVB-irradiated HaCaT keratinocytes with low, physiologically relevant concentrations of SP resulted in increased mutagenesis at the HPRT locus. Thus, these results indicate that SP could be mutagenic and contribute to skin carcinogenesis in certain individuals. Our findings also suggest that SP is a convenient pharmacological tool to mimic XP patient skin in UV photobiology studies that employ human skin specimens ex vivo.
RESULTS
Spironolactone-mediated XPB depletion abrogates UVB DNA damage responses in human keratinocytes in vitro
To determine whether SP affects XPB expression in human skin keratinocytes, telomerase-immortalized neonatal foreskin keratinocytes (N-TERTs) (Dickson, Hahn, Ino et al, 2000, Kemp, Spandau, Simman et al, 2017) were treated for 3 hr with increasing concentrations of SP and then harvested for immunoblot analysis. As shown in Figure 1a, XPB protein levels decreased in a SP dose-dependent manner. Time course analyses using 10 μM SP showed that XPB protein was depleted by more than 95% within 2 hr of SP treatment (Figure 1b). Because SP was identified as an inhibitor of (6-4)PP removal in a high-throughput screen in UVC-irradiated HeLa cells (Alekseev, Ayadi, Brino et al, 2014), we next examined whether SP affected the removal rate of UV photoproducts from genomic DNA. These experiments showed that whereas nearly 90% of CPDs were removed from cells within 24 hr in vehicle-treated N-TERTs, this repair was inhibited to a significant extent by SP treatment (Figure 1c). A similar inhibition of (6-4)PP removal was also observed (Supplementary Figure 1a), and the extent of repair inhibition was found to be SP dose-dependent (Supplementary Figure 1b). One of the roles of XPB-containing TFIIH in NER is to unwind DNA around the lesion, and we observed that the accumulation of the single-stranded DNA binding protein RPA (Replication Protein A) on chromatin following UVB exposure was partially abrogated by SP treatment (Figure 1d). Similar to the earlier report in HeLa cells (Alekseev, Ayadi, Brino et al, 2014), XPB protein levels in N-TERT keratinocytes were uniquely impacted by SP treatment (Supplementary Figure 2). Consistent with the current model that XPB is dispensable for transcription of genes by RNA polymerase II (Alekseev, Nagy, Sandoz et al, 2017), SP treatment did not affect basal transcription in N-TERTs but did negatively affect the resumption of transcription after UVB exposure (Supplementary Figure 3).
Figure 1. Spironolactone depletes XPB protein and abrogates DNA damage responses in UVB-irradiated, telomerase-immortalized (N-TERT) keratinocytes in vitro.

(a) Immunoblot analysis of XPB protein levels following treatment with the indicated concentration of spironolactone (SP) (n=2). Molecular weight markers and a cross-reacting band (*) are indicated. (b) Time course analysis of XPB loss following SP treatment (n=2). (c) Immunodot blot analysis of CPD removal from genomic DNA (n=3). Blots were probed sequentially with anti-CPD and anti-DNA antibodies. (d) Confluent N-TERTs grown for 24 hr in basal medium lacking growth factors were treated as indicated and harvested 0.5 hr after exposure to 100 J/m2 UVB to enrich for chromatin-associated proteins (n=2). (e) Cells were treated as in (d) except that cells were harvested 1 hr after UVB exposure and cell lysates were analyzed for ATR/ATM kinase substrate protein phosphorylation on the indicated protein residue (n=4). (f) Cell survival analysis 2 day following exposure to the indicated fluence of UVB (n=3). All graphs show the average ± SEM, and the asterisks (*) indicate significant differences (p<0.05).
In addition to DNA repair, the protein kinases ATR and ATM are also activated by several different mechanisms in UV-irradiated cells (Kemp and Hu, 2017, Kemp and Sancar, 2016, Wakasugi, Sasaki, Matsumoto et al, 2014, Derheimer, O’Hagan, Krueger et al, 2007), where the kinases are thought to regulate DNA repair, cell fate, and other aspects of the cellular DNA damage response. We therefore monitored the phosphorylation of several ATR/ATM substrates, including the heterochromatin regulatory protein KAP1 and the tumor suppressor protein p53. SP treatment partially abrogated the phosphorylation of both proteins following UVB exposure (Figure 1e), and this aberrant signaling was not due to alterations in either ATR or ATM protein expression (Supplementary Figure 2). SP also partially inhibited the UVB-dependent phosphorylation of the canonical ATM substrate CHK2 in asynchronously growing N-TERTs (Supplementary Figure 4). In contrast, the major ATR substrate CHK1 was not affected by SP treatment (Supplementary Figure 4), which is likely due to the different mechanisms by which the ATR kinase is activated to phosphorylate specific substrates in replicating and non-replicating cells (Kemp, 2017, Kemp and Hu, 2017). Nonetheless, SP induced the rapid loss of XPB in both proliferating and differentiated N-TERT keratinocytes (Supplementary Figure 5) and completely inhibited UVB-induced KAP1 phosphorylation in growth-arrested, non-replicating HaCaT cells (Supplementary Figure 6). Correlated with these defects in both UV photoproduct removal and ATR/ATM kinase signaling, we observed that SP treatment sensitized N-TERTs to the acute and lethal effects of UVB irradiation (Figure 1f).
We next examined whether the negative effects of SP on UVB responses are also observed in normal, non-immortalized, primary human keratinocytes. Keratinocytes were therefore isolated and cultured from adult female human skin discarded during routine surgical procedures. We found that SP treatment rapidly promoted the loss of XPB protein in keratinocytes from three different donors (Figure 2a). As expected, SP treatment also inhibited UVB-induced ATR/ATM kinase signaling (Figure 2b) and CPD removal from genomic DNA (Figure 2c). Lastly, SP sensitized all three sets of primary keratinocytes to the lethal effects of UVB treatment (Figure 2d).
Figure 2. Spironolactone negatively impacts XPB protein levels and UVB DNA damage responses in primary adult human keratinocytes in vitro.
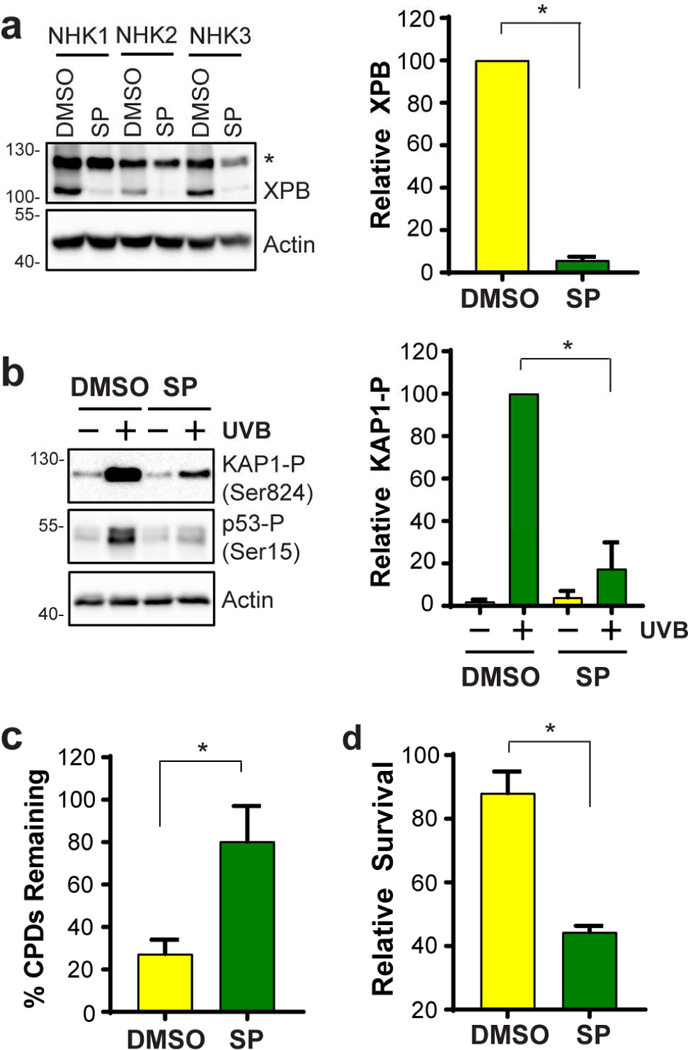
(a) Normal, primary, non-immortalized keratinocytes from three different skin donors (NHK1, 2, 3) were treated for 2 hr with DMSO or 10 μM SP, and cell lysates were analyzed by immunoblotting. (b) ATR/ATM substrate protein phosphorylation was analyzed as in Fig. 1d. (c) Analysis of CPD removal in normal keratinocytes was performed as described in Fig. 1c. (d) Cell survival 2 days after exposure to 100 J/m2 of UVB. All graphs show the average ± SEM, and the asterisks (*) indicate significant differences (p<0.05).
Spironolactone depletes the XPB protein from human skin epidermis ex vivo
To determine whether SP treatment affects XPB protein levels and the UVB response in intact human skin tissue, we used an ex vivo skin culture system in which small skin punch biopsies from multiple donors were placed in a hanging well insert of a cell culture plate such that only the dermis of the biopsy was in contact with culture medium (Backvall, Wassberg, Berne et al, 2002). We then supplemented the medium below the biopsies with either DMSO or 20 μM SP. The treatment was repeated with fresh media and drug on 3 sequential days, and the epidermis was then isolated from the biopsy for immunoblot analysis of XPB protein levels. As shown in Figure 3a, treatment with SP led to a remarkable depletion of epidermal XPB protein levels, with an average reduction of greater than 80% among 9 different donor samples. To determine whether this effect on XPB was specific to SP and independent of mineralocorticoid receptor signaling, we exposed skin biopsies to eplerenone (EP), which is structurally similar to SP and shows greater specificity towards the mineralocorticoid receptor (Kolkhof and Barfacker, 2017) Consistent with previous reports that EP does not affect XPB protein levels in HeLa and primary endothelial cells (Alekseev, Ayadi, Brino et al, 2014, Elinoff, Chen, Dougherty et al, 2018), we found that EP did not significantly affect XPB protein levels in human skin epidermis (Figure 3a). Additional experiments monitoring the dose- and time-dependent loss of XPB from human skin epidermis ex vivo revealed that micromolar concentrations of SP were necessary to significantly impact XPB protein levels (Figure 3b) and that significant reductions in XPB protein levels could be observed after only 1 day of treatment (Supplementary Figure 7).
Figure 3. Spironolactone depletes the XPB protein and modulates DNA damage responses in UVB-irradiated human skin ex vivo.

(a) Skin explant culture medium was supplemented on 3 sequential days with DMSO or 20 μM SP or eplerenone (EP). Epidermal lysates were analyzed by immunoblotting (n=5-9). (b) Skin biopsies were treated with increasing concentrations of SP as in (a) (n=3). (c) Skin biopsies treated for 3 days with DMSO or SP were exposed to 700 J/m2 UVB. Genomic DNA was purified from the epidermis and analyzed by immunodot blotting (n=4-6). (d) Epidermal lysates were prepared 1 hr after UVB irradiation and analyzed by immunoblotting (n=5). (e) Skin biopsies were treated as in (c), fixed in formalin, and stained with H&E (scale bar = 0.1 mm). Apoptotic “sunburn” cells (indicted by white arrows) counted (n=6). All graphs show the average ± SEM, and the asterisks (*) indicate significant differences (p<0.05).
We next examined whether this loss of XPB protein was associated with an inhibition of CPD removal following UVB exposure. Skin biopsies cultured as described above were therefore treated for 3 days with DMSO or 20 μM SP before exposure to 700 J/m2 of UVB. Genomic DNA was then purified either immediately or 48 hr later and analyzed for CPD content by immunodot blot analysis. As shown in Figure 3c, SP treatment was associated with a significant inhibition in CPD removal. We next monitored the activation of ATR/ATM kinase signaling following UVB exposure, which typically occurs within minutes to hours following UV irradiation. In the epidermis of control skin samples, UVB-dependent induction of KAP1 and p53 phosphorylation were readily observed 1 hr after UVB exposure (Figure 3d). In contrast, these phosphorylation events were significantly abrogated by SP treatment. Thus, we conclude that just as in keratinocytes in vitro, SP treatment also depletes XPB protein and inhibits DNA damage responses to UVB radiation in human skin epidermis ex vivo.
Because SP treatment was shown to sensitize keratinocytes to the lethal effects of UVB light in vitro (Figures 1 and 2), we next examined whether epidermal cells of SP-treated human skin are more prone to undergo cell death following UVB exposure ex vivo. Hematoxylin and eosin-staining of SP-treated skin 48 hr after UVB exposure revealed an increased number of characteristic “sunburn” cells defined by pyknotic nuclei and eosinophilic cytoplasm (Figure 3e). Thus, as we observed with keratinocytes in vitro, epidermal cells of human skin depleted of XPB via SP treatment ex vivo are more sensitive to UVB radiation.
Spironolactone metabolites do not impact XPB or the response to UVB
SP is rapidly metabolized in humans in vivo to the two major metabolites canrenone (Can) and 7α-thiomethylspironolactone (TMS), which also inhibit the mineralocorticoid receptor and are found at much higher levels than the parent compound in the serum of human subjects taking orally administered SP (Gardiner, Schrode, Quinlan et al, 1989). We therefore examined whether these metabolites similarly affect XPB protein levels in N-TERT keratinocytes. Interestingly, neither Can or TMS negatively impacted XPB protein levels, whereas the mineralocorticoid receptor antagonist EP led to a modest but statistically significant increase in XPB protein (Figure 4a). Consistent with the lack of effect of Can, TMS, and EP on XPB protein levels, neither EP nor the SP metabolites negatively affected CPD removal in UVB- irradiated N-TERTs (Figure 4b) or cell survival following UVB exposure (Figure 4c).
Figure 4. Spironolactone metabolites and the mineralocorticoid receptor antagonist eplerenone do not impact XPB protein levels or the UVB DNA damage response.

(a) N-TERTs were treated for 3 hr with DMSO or with 10 μM spironolactone (SP), canrenone (Can), 7α-thiomethylspironolactone (TMS), or eplerenone (EP). Cell lysates were analyzed by immunoblotting (n=3-5). (b) N-TERTs treated as in (A) were irradiated with 100 J/m2 UVB and harvested for analysis of CPD levels by immunodot blotting (n=3). (c) Cells treated as in (a) were analyzed for cell survival (n=3-6). (d) HaCaT cells were treated for 2 hr with DMSO or 300 nM SP, Can, or EP before exposure to 100 J/m2 UVB. The number of 6-thioguanine-resistant clones (per million plated cells) was quantified two weeks later (n=2-3). All graphs show the average ± SEM, and the asterisks (*) indicate significant differences (p<0.05).
Spironolactone increases UVB mutagenesis
Previous measurements of SP levels in human subjects detected a peak serum concentration of approximately 80 ng/ml (~190 nM) (Gardiner, Schrode, Quinlan et al, 1989), which is 50-100 times lower than the 10-20 μM (4.2-8.3 μg/ml) concentrations that we used here to maximally deplete XPB from keratinocytes and skin epidermis. Nonetheless, lower, more physiologically relevant concentrations of SP that do not completely deplete XPB nor cause increased cell lethality could still have important effects on the cellular response to UVB in human keratinocytes, including on the risk of gene mutagenesis. We therefore carried out an assay in HaCaT keratinocytes to monitor mutagenesis at the HPRT locus by examining the emergence of UVB-irradiated cells that become resistant to the cell killing effects of the HPRT substrate 6-thioguanine (Johnson, 2012). Treatment of HaCaT cells with low doses of SP led to a modest reduction in XPB protein levels and cell growth following UVB exposure (Supplementary Figure 8). As shown in Figure 4d, we observed that treatment of cells with 300 nM SP prior to UVB irradiation led to a 2.7-fold increase in the number of HPRT-mutant cells. In contrast, treatment with Can or EP did not affect mutagenesis (Figure 4d). Thus, we conclude that physiological concentrations of SP that only modestly impact XPB protein levels can promote mutagenesis in UVB-irradiated human keratinocytes.
DISCUSSION
Using both primary and telomerase-immortalized keratinocytes in vitro and human skin explants ex vivo, we find that the commonly used therapeutic drug SP can be used to deplete cells and tissues of XPB and abrogate important cellular responses to UVB-induced DNA damage. As a component of the multi-subunit enzyme TFIIH (Compe and Egly, 2016), XPB has long been known to be important for the removal of UV photoproducts by nucleotide excision repair. XPB was also recently shown to regulate ATR kinase activation in non-replicating cells in response to genotoxins that impede transcription (Kemp, 2017). Thus, these results further validate an important role for XPB in the DNA damage response and suggest that SP may be a convenient pharmacological tool for depleting the protein from both cells in vitro and human skin tissue ex vivo. This approach may therefore be useful in mimicking some of the clinical properties of skin from patients with mutations in XPB (DiGiovanna and Kraemer, 2012) or with other NER defects in ex vivo skin culture systems.
The significant reductions in XPB protein levels observed here following treatment with high concentrations of SP clearly sensitize cells to the lethal effects of UVB irradiation. The few human patients that have been identified with mutations in XPB display a partial reduction (2258%) in XPB protein levels in their cultured fibroblasts in vitro (Oh, Khan, Jaspers et al, 2006), and all patients have been reported to readily sunburn after minimal sun exposure (Oh, Khan, Jaspers et al, 2006, Fassihi, Sethi, Fawcett et al, 2016). To our knowledge, there is no evidence that human subjects taking SP are particularly photosensitive or prone to sunburn, and thus we expect that patients taking standard doses of SP likely exhibit only small reductions in epidermal XPB protein levels. However, our observation that even a low concentration of SP significantly elevates UVB mutagenesis argues that even small changes in XPB protein levels could nonetheless have important biological effects, particularly in sun-exposed areas of skin. Because the HaCaT keratinocytes we used in this assay already have mutations in both p53 alleles as well as other chromosomal abnormalities (Boukamp, Petrussevska, Breitkreutz et al, 1988, Lehman, Modali, Boukamp et al, 1993), the promotion of UVB mutagenesis by SP might require additional, previously incurred genetic changes in skin keratinocytes in vivo. Interestingly, the treatment of colon cancer cell lines with SP was shown to upregulate the expression of NKG2D ligands (Leung, Vong, Lin et al, 2013), which facilitate tumor cell elimination by natural killer cells. Thus, in principle, SP may simultaneously increase mutagenesis and promote the elimination of mutant cells by the immune system.
Epidemiological studies of human subjects have found no evidence that SP increases the risk of any cancer, including melanoma and non-melanoma skin cancers (Mackenzie, Morant, Wei et al, 2017). Our findings here that the major SP metabolites canrenone and 7α- thiomethylspironolactone do not affect XPB protein levels is therefore clinically relevant and supports existing evidence that SP is generally considered to be a safe medication. Standard oral doses of SP (up to 100 mg/day) lead to sub-micromolar concentrations of SP (~190 nM) in the blood within a few hours (Gardiner, Schrode, Quinlan et al, 1989). Though doses of up to 100 mg/day of SP are frequently used for its anti-androgen effects in adult women with acne, it should be noted that increased doses of SP (up to 400 mg/day) are used in hormone regimens in transgender persons (Hembree, Cohen-Kettenis, Gooren et al, 2017, Wesp and Deutsch, 2017). Thus, it is possible that high doses of SP or altered SP metabolism may lower XPB protein levels in skin epidermis in specific subsets of patients.
There has also been interest in developing topical formulations of SP (Kelidari, Saeedi, Akbari et al, 2015) to limit the systemic and potentially endocrine-disrupting effects of orally administered SP. Moreover, the topical use of SP and related mineralocorticoid receptor antagonists have been shown in clinical trials to counteract epidermal atrophy and the inhibition of wound healing caused by glucocorticoid treatment (Nguyen, Farman, Maubec et al, 2016, Maubec, Laouenan, Deschamps et al, 2015, Boix, Nguyen, Farman et al, 2018). Chronic treatment with topical SP formulations, especially as proposed for use in damaged skin, would likely result in higher levels of SP than those associated with systemic treatment. Thus, topical treatments with SP could potentially be problematic in areas of sun-exposed skin. Because our data show that neither eplerenone nor the SP metabolites affect XPB protein levels or the UVB DNA damage response but effectively inhibit the mineralocorticoid receptor (Kolkhof and Barfacker, 2017), we suggest that these other mineralocorticoid receptor antagonists may be safer for potential topical application on human skin.
MATERIALS AND METHODS
Cell culture and treatments
Telomerase-immortalized keratinocytes (N-TERTs) from neonatal foreskins, primary human keratinocytes from discarded human skin, and HaCaT cells were maintained at 37°C in a 5% CO2 humidified incubator and were cultured using routine procedures. Cells were exposed to the indicated fluences of UVB radiation using a Philips F20T12 broadband UVB light source at a dose rate of 5 J/m2/sec. Treatments with the indicated compounds involved direct addition to the cell culture medium. Cell survival was determined 2 days after treatment by crystal violet staining, as previously described (Kemp and Sancar, 2016).
Skin explant culture
Punch biopsies (8 mm) from de-identified human skin discarded during panniculectomy procedures were placed in hanging Millicell™ cell culture inserts (Millipore) in wells of a 24-well plate containing culture medium, essentially as previously described (Backvall, Wassberg, Berne et al, 2002). Patient consent for experiments was not required because de-identified, leftover surgical human tissue is considered to be discarded material by our institution is therefore not considered and thus the studies were except. Explants were kept in a 5% CO2 humidified incubator at 37°C for up to 6 days after isolation. Experiments were initiated 1 day after beginning ex vivo culture, and medium containing the indicated compounds was changed daily. At the indicated time points, biopsies were snap frozen in liquid nitrogen or fixed in formalin. Sections were stained with H&E to identify apoptotic, “sunburn” epidermal cells (Sheehan and Young, 2002).
Immunoblotting
Cell and tissue protein lysates were prepared in RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 2 mM EDTA, 1% triton X-100, 1% sodium deoxycholate, 0.1% SDS) containing a 1:200 dilution of protease inhibitor cocktail (Sigma), 1 mM DTT, 0.1 mM PMSF, 10 mM NaF, 1 mM Na2VO3, and 10 mM glycerophosphate and analyzed by immunoblotting using the antibodies indicated in Supplementary Table 1.
CPD immunodot blot assay
Genomic DNA was purified from cells and tissues using the GenElute Mammalian Genomic DNA Miniprep Kit (Sigma), quantified using PicoGreen fluorescence (Invitrogen), and immunodot blotted essentially as described (Gaddameedhi, Kemp, Reardon et al, 2010, Kemp, Spandau, Simman et al, 2017).
HPRT mutagenesis assay
The HPRT (hypoxanthine phosphoribosyltransferase) mutagenesis assay was performed essentially as described (Johnson, 2012) and involved selection of UVB-irradiated HaCaT cells for 2 weeks with 6-thioguanine and the counting of resistant clones following crystal violet staining. Additional details are provided in the Supplementary Material.
Statistical analyses
For each experimental approach, the number of independent experiments that were performed is provided in the appropriate figure legend. Two-tailed, paired or unpaired Student’s t-tests were carried out where indicated to determine whether differences between treatment samples reached statistical significance (p<0.05).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the WSU Proteome Analysis Laboratory and Center for Genomics Research for the use of equipment to carry out this work. This work was supported by the grants from the National Institute of Environmental Health Sciences (ES020855 to J.B.T.), the National Institute on Aging (AG048946 to J.B.T.), and by the Veterans Administration (1101CX000809 to J.B.T.).
Abbreviations:
- SP
spironolactone
- ATM
ataxia telangiectasia-mutated
- ATR
ATM- and Rad3-related
- XP
xeroderma pigmentosum
- XPB
XP group B
- CPD
cyclobutane pyrimidine dimer
- NER
nucleotide excision repair
- Can
canrenone (Can)
- TMS
7α-thiomethylspironolactone
- EP
eplerenone
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alekseev S, Ayadi M, Brino L, Egly JM, Larsen AK, Coin F (2014). A small molecule screen identifies an inhibitor of DNA repair inducing the degradation of TFIIH and the chemosensitization of tumor cells to platinum. Chem Biol 21: 398–407. [DOI] [PubMed] [Google Scholar]
- Alekseev S, Nagy Z, Sandoz J, Weiss A, Egly JM, Le May N, et al. (2017). Transcription without XPB establishes a unified helicase-independent mechanism of promoter opening in eukaryotic gene expression. Mol Cell 65: 504–14.e4. [DOI] [PubMed] [Google Scholar]
- Backvall H, Wassberg C, Berne B, Ponten F (2002). Similar UV responses are seen in a skin organ culture as in human skin in vivo. Exp Dermatol 11: 349–56. [DOI] [PubMed] [Google Scholar]
- Batista LF, Kaina B, Meneghini R, Menck CF (2009). How DNA lesions are turned into powerful killing structures: Insights from UV-induced apoptosis. Mutat Res 681: 197–208. [DOI] [PubMed] [Google Scholar]
- Boix J, Nguyen VT, Farman N, Aractingi S, Perez P (2018). Mineralocorticoid receptor blockade improves glucocorticoid-induced skin atrophy but partially ameliorates anti-inflammatory actions in an irritative model in human skin explants. Exp Dermatol 27: 185–7. [DOI] [PubMed] [Google Scholar]
- Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE (1988). Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 106: 761–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet J, Grand A, Douki T (2015). Solar UV radiation-induced DNA bipyrimidine photoproducts: Formation and mechanistic insights. Top Curr Chem 356: 249–75. [DOI] [PubMed] [Google Scholar]
- Canavan TN, Chen E, Elewski BE (2016). Optimizing non-antibiotic treatments for patients with acne: A review. Dermatol Ther (Heidelb) 6: 555–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ (2010). The DNA damage response: Making it safe to play with knives. Mol Cell 40: 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compe E, Egly JM (2016). Nucleotide excision repair and transcriptional regulation: TFIIH and beyond. Annu Rev Biochem 85: 265–90. [DOI] [PubMed] [Google Scholar]
- Derheimer FA, O’Hagan HM, Krueger HM, Hanasoge S, Paulsen MT, Ljungman M (2007). RPA and ATR link transcriptional stress to p53. Proc Natl Acad Sci US A 104: 12778–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, et al. (2000). Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol 20: 1436–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiovanna JJ, Kraemer KH (2012). Shining a light on xeroderma pigmentosum. J Invest Dermatol 132: 785–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinoff JM, Chen LY, Dougherty EJ, Awad KS, Wang S, Biancotto A, et al. (2018). Spironolactone-induced degradation of the TFIIH core complex XPB subunit suppresses NF- kappaB and AP-1 signalling. Cardiovasc Res 114: 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endly DC, Miller RA (2017). Oily skin: A review of treatment options. J Clin Aesthet Dermatol 10: 49–55. [PMC free article] [PubMed] [Google Scholar]
- Fassihi H, Sethi M, Fawcett H, Wing J, Chandler N, Mohammed S, et al. (2016). Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect. Proc Natl Acad Sci US A 113: E1236–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaddameedhi S, Kemp MG, Reardon JT, Shields JM, Smith-Roe JL, Kaufmann WK, et al. (2010). Similar nucleotide excision repair capacity in melanocytes and melanoma cells. Cancer Res 70: 4922–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner P, Schrode K, Quinlan D, Martin BK, Boreham DR, Rogers MS, et al. (1989). Spironolactone metabolism: Steady-state serum levels of the sulfur-containing metabolites. J Clin Pharmacol 29: 342–7. [DOI] [PubMed] [Google Scholar]
- Hembree WC, Cohen-Kettenis PT, Gooren L, Hannema SE, Meyer WJ, Murad MH, et al. (2017). Endocrine treatment of gender-dysphoric/gender-incongruent persons: An endocrine society clinical practice guideline. J Clin Endocrinol Metab 102: 3869–903. [DOI] [PubMed] [Google Scholar]
- Johnson GE (2012). Mammalian cell HPRT gene mutation assay: Test methods. Methods Mol Biol 817: 55–67. [DOI] [PubMed] [Google Scholar]
- Kelidari HR, Saeedi M, Akbari J, Morteza-Semnani K, Gill P, Valizadeh H, et al. (2015). Formulation optimization and in vitro skin penetration of spironolactone loaded solid lipid nanoparticles. Colloids Surf B Biointerfaces 128: 473–9. [DOI] [PubMed] [Google Scholar]
- Kemp MG (2017). DNA damage-induced ATM- and rad-3-related (ATR) kinase activation in non-replicating cells is regulated by the XPB subunit of transcription factor IIH (TFIIH). J Biol Chem 292: 12424–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp MG, Hu J (2017). PostExcision events in human nucleotide excision repair. Photochem Photobiol 93: 178–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp MG, Sancar A (2016). ATR kinase inhibition protects non-cycling cells from the lethal effects of DNA damage and transcription stress. J Biol Chem 291: 9330–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp MG, Spandau DF, Simman R, Travers JB (2017). Insulin-like growth factor 1 receptor signaling is required for optimal ATR-CHK1 kinase signaling in ultraviolet B (UVB)-irradiated human keratinocytes. J Biol Chem 292: 1231–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolkhof P, Barfacker L (2017). 30 YEARS OF THE MINERALOCORTICOID RECEPTOR: Mineralocorticoid receptor antagonists: 60 years of research and development. J Endocrinol 234: T125–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman TA, Modali R, Boukamp P, Stanek J, Bennett WP, Welsh JA, et al. (1993). P53 mutations in human immortalized epithelial cell lines. Carcinogenesis 14: 833–9. [DOI] [PubMed] [Google Scholar]
- Leung WH, Vong QP, Lin W, Janke L, Chen T, Leung W (2013). Modulation of NKG2D ligand expression and metastasis in tumors by spironolactone via RXRgamma activation. J Exp Med 210: 2675–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IS, Morant SV, Wei L, Thompson AM, MacDonald TM (2017). Spironolactone use and risk of incident cancers: A retrospective, matched cohort study. Br J Clin Pharmacol 83: 653–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maubec E, Laouenan C, Deschamps L, Nguyen VT, Scheer-Senyarich I, Wackenheim-Jacobs AC, et al. (2015). Topical mineralocorticoid receptor blockade limits glucocorticoid-induced epidermal atrophy in human skin. J Invest Dermatol 135: 1781–9. [DOI] [PubMed] [Google Scholar]
- Natale V, Raquer H (2017). Xeroderma pigmentosum-cockayne syndrome complex. Orphanet J Rare Dis 12: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VT, Farman N, Maubec E, Nassar D, Desposito D, Waeckel L, et al. (2016). Re- epithelialization of pathological cutaneous wounds is improved by local mineralocorticoid receptor antagonism. J Invest Dermatol 136: 2080–9. [DOI] [PubMed] [Google Scholar]
- Oh KS, Khan SG, Jaspers NG, Raams A, Ueda T, Lehmann A, et al. (2006). Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): Xeroderma pigmentosum without and with cockayne syndrome. Hum Mutat 27: 1092–103. [DOI] [PubMed] [Google Scholar]
- Reardon JT, Sancar A (2005). Nucleotide excision repair. Prog Nucleic Acid Res Mol Biol 79: 183–235. [DOI] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, Cimprich KA (2017). The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18: 622–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A (2016). Mechanisms of DNA repair by photolyase and excision nuclease (nobel lecture). Angew Chem Int Ed Engl 55: 8502–27. [DOI] [PubMed] [Google Scholar]
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S (2004). Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73: 39–85. [DOI] [PubMed] [Google Scholar]
- Sheehan JM, Young AR (2002). The sunburn cell revisited: An update on mechanistic aspects. Photochem Photobiol Sci 1: 365–77. [DOI] [PubMed] [Google Scholar]
- Wakasugi M, Sasaki T, Matsumoto M, Nagaoka M, Inoue K, Inobe M, et al. (2014). Nucleotide excision repair-dependent DNA double-strand break formation and ATM signaling activation in mammalian quiescent cells. JBiol Chem 289: 28730–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesp LM, Deutsch MB (2017). Hormonal and surgical treatment options for transgender women and transfeminine spectrum persons. Psychiatr Clin North Am 40: 99–111. [DOI] [PubMed] [Google Scholar]
- Wood RD (1997). Nucleotide excision repair in mammalian cells. J Biol Chem 272: 23465–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.