

Abstract
The inflammatory bowel diseases (IBD), which include Crohn’s disease (CD) and ulcerative colitis (UC), are multifactorial, chronic conditions of the gastrointestinal tract. While IBD has been associated with dramatic changes in the gut microbiota, changes in the gut metabolome -- the molecular interface between host and microbiota -- are less-well understood. To address this gap, we performed untargeted LC-MS metabolomic and shotgun metagenomic profiling of cross-sectional stool samples from discovery (n=155) and validation (n=65) cohorts of CD, UC, and non-IBD control subjects. Metabolomic and metagenomic profiles were broadly correlated with fecal calprotectin levels (a measure of gut inflammation). Across >8,000 measured metabolite features, we identified chemicals and chemical classes that were differentially abundant (DA) in IBD, including enrichments for sphingolipids and bile acids, and depletions for triacylglycerols and tetrapyrroles. While >50% of DA metabolite features were uncharacterized, many could be assigned putative roles through metabolomic “guilt-by-association” (covariation with known metabolites). DA species and functions from the metagenomic profiles reflected adaptation to oxidative stress in the IBD gut, and were individually consistent with previous findings. Integrating these data, however, we identified 122 robust associations between DA species and well-characterized DA metabolites, indicating possible mechanistic relationships that are perturbed in IBD. Finally, we found that metabolome- and metagenome-based classifiers of IBD status were highly accurate and, like the vast majority of individual trends, generalized well to the independent validation cohort. Our findings thus provide an improved understanding of perturbations of the microbiome-metabolome interface in IBD, including identification of many potential diagnostic and therapeutic targets.
Introduction
Inflammatory bowel disease (IBD) is a chronic inflammatory condition of the gastrointestinal tract that results from altered interactions between gut microbes and the intestinal immune system1, 2. There are two main IBD subtypes, ulcerative colitis (UC) and Crohn’s disease (CD), which localize in the large and small intestines, respectively, and are characterized by unique microbial signatures3. Previous studies have shown major shifts in the gut microbial composition of patients with IBD2, 4–8. Likewise, microbial composition can shape the environment in the colon as metabolites they produce can be involved in signaling, immune system modulation or have antibiotic activity9–11. It is less clear, however, how specific microbes and the small molecules they modulate may interact to cause, sustain, mitigate, or predict inflammatory conditions such as IBD.
Broadly, gut metabolite profiles are jointly derived from diet, modified human metabolites and microbially-derived compounds that shape the microbiota-host interactions9. For example, short chain fatty acids (SCFAs) such as butyrate, acetate and propionate are produced by gut bacteria when they break down dietary fiber. SCFAs can affect host cells by modulating histone deacetylase (HDAC) inhibitory activity, gene expression, cell proliferation, and immune response7, 12. In addition, butyrate can protect against colitis by regulating Treg cell production and enhancing antibacterial activity of macrophages13, 14. In stool from patients with IBD there is a decrease in butyrate, a SCFA that is important in modulating the immune system along with a decrease in butyrate-producing bacteria2, 15.
Commensal microbes can also alter pools of available metabolites thereby modifying host-generated signaling molecules. Untargeted serum metabolomics of germ-free versus conventional mice showed that a large number of serum metabolites arise due to commensal microbes16. For example, tryptophan metabolism is largely affected by the presence of gut bacteria, as microbial tryptophan decarboxylases (among other enzymes) convert tryptophan from the diet into tryptamine and other molecules. Microbially-derived tryptophan metabolites alter host physiology not only by decreasing the available tryptophan (which can in turn perturb serotonin production, and by extension, behavior17), but also by producing indole derivatives that activate the aryl hydrocarbon receptor18. There is a decrease in tryptophan metabolism genes in microbiome samples from patients with Crohn’s disease4. A mouse study recently found that animals lacking one of the IBD susceptibility genes, CARD9, had altered microbial metabolism of tryptophan and were more susceptible to colitis19.
Some previous studies have identified differences in fecal metabolites in IBD20–25. However, these studies have tended to rely on small cohorts or 16S rRNA amplicon-based profiles of the associated IBD microbiota (i.e. lacking shotgun metagenomic information). In a study of healthy individuals, untargeted fecal metabolomics correlated better with 16S-based microbiome composition than targeted metabolomics26. IBD-associated taxa were also highly correlated with metabotype in a study of inactive pediatric IBD. In the same study, healthy first-degree relatives displayed a similar microbiome and metabotype as relatives with inactive disease25. Both CD and UC gut microbiomes exhibit general decreases in taxonomic diversity relative to healthy gut microbiomes, along with Phylum-level decreases in Firmicutes and increases in Proteobacteria3, 25, 27. In CD specifically, proportions of the Clostridia family are altered: the Roseburia and Faecalibacterium genera of the Lachnospiracae and Ruminococcaceae families are decreased, whereas Ruminococcus gnavus increases5, 28, 29. Together, these findings suggest that yet-to-be characterized molecules in the gut metabolome, linked to inflammation and ultimately IBD, may be largely microbially derived or modified.
In this work, we took an unbiased approach to identify gut metabolites, microbial species, and microbial enzymes that were differentially abundant (DA) in IBD relative to non-IBD controls. To that end, we performed untargeted LC-MS metabolomic profiling and shotgun metagenomic sequencing of stool samples from a 153-member discovery cohort and a 65-member validation cohort, each containing a cross-sectional sampling UC, CD, and control subjects. While metagenomic findings were largely in agreement with previous studies, metabolomic profiles revealed >2,700 DA metabolites in IBD, including 224 that were significantly elevated in both UC and CD. IBD-elevated metabolites were enriched for sphingolipids and bile acids (among other chemical classes), as well as many uncharacterized metabolites of potential microbial origin. Indeed, many DA metabolites participated in robust associations with DA microbial species and enzymes: suggestive of biological mechanisms relating their abundances. Finally, the vast majority of IBD associations from the discovery cohort replicated in the independent validation cohort, thus making our findings a useful resource for the study of microbiome and metabolic perturbations in IBD.
Results
To characterize the gut metabolic profile and microbiome composition in IBD, we collected and analyzed stool samples from a cross-sectional cohort of individuals enrolled in PRISM (the Prospective Registry in IBD Study at MGH). This cohort included 155 subjects: 68 with Crohn’s disease (CD), 53 with ulcerative colitis (UC), and 34 non-IBD controls (Fig. 1A). Each stool sample was subjected to metagenomic sequencing followed by profiling of microbial community taxonomic composition and functional potential. In addition, each sample was analyzed by four liquid chromatography tandem mass spectrometry (LC-MS) methods measuring polar metabolites, lipids, free fatty acids, and bile acids, respectively. LC-MS metabolomic profiling was carried out using sensitive, high-resolution mass spectrometers in non-targeted modes, thus capturing large numbers of known and uncharacterized metabolites, including those of potential microbial origin.
Figure 1. IBD is associated with broad changes in subjects’ gut multi’omic profiles.
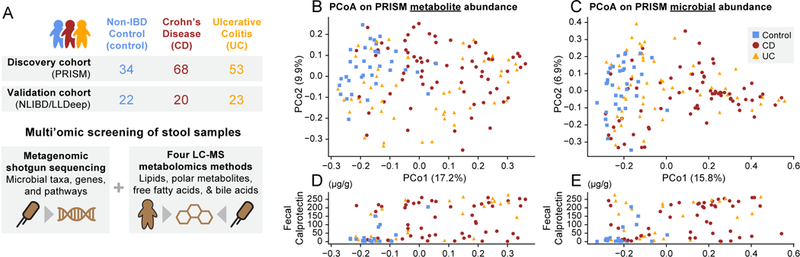
(A) We collected and profiled stool metagenomic and metabolomic data from two IBD cohorts: a 155-member discovery cohort (PRISM) and a 65-member validation cohort (NLIBD/LLDeep). (B) Principal coordinates analysis (PCoA) of PRISM cohort subjects based on gut metabolomic profiles (Bray-Curtis distance). (C) The same subjects ordinated on Bray-Curtis distances between gut metagenomic species profiles. (D, E) Subject fecal calprotectin (FC) levels (μg/g) plotted against the first PCoA axes from panels B and C, respectively. Note that FC measurements were not available for all subjects.
3,829 metabolite features (43% of total) were assigned to putative molecular classes based on comparisons with HMDB30. 466 features (representing 346 unique compounds) were annotated as standards through comparison with reference data generated from an in-house compound library (see Supplementary Datasets 1 and 2). Shotgun metagenomic and metabolomic data were then analyzed 1) to identify IBD- and disease-subtype specific changes in subject microbial and metabolic profiles, 2) to describe associations between microbial and metabolite features, and 3) to assess the power of these features to classify IBD status and subtype across populations. Relationships discovered in the PRISM cohort were validated against an independent cohort of 20 CD patients, 23 UC patients and 22 population controls from the Netherlands. These analyses are expanded below, with additional technical detail provided in Methods.
Broad metabolic shifts in IBD correlate with host inflammation
The major patterns of variation in the 155 Boston PRISM subjects’ measurements of >8K metabolite features largely separated non-IBD control versus CD subjects, indicative of broad metabolic differences between these two phenotypes (Fig. 1B). Such differences could result from a combination of sources, including the effects of disease activity in host tissues, the activity of an IBD-altered microbiome, and differences in subject diet and medication use. UC subjects’ metabolic profiles were more broadly distributed, with roughly half resembling non-IBD control subjects’, and the remainder more similar to CD subjects’ metabolomes (Supplementary Fig. 1). Similar patterns of variation among disease phenotypes were apparent in microbial taxonomic profiles from the subjects’ corresponding metagenomes (Fig. 1C; see also Supplementary Fig. 2 and Supplementary Dataset 4). Indeed, the first axes of ordination for the two datasets were well correlated (Spearman’s r=0.664, two-tailed p<10−20), consistent with strong coupling of gut metabolic profile, microbial community composition, and disease status.
We hypothesized that broad variation in metabolic profile across subjects, especially within the UC subjects, might be explained in part by subjects’ levels of active inflammation. We evaluated this by comparing the first axis of metabolic variation with subjects’ levels of fecal calprotectin (FC): a biomarker for severity of inflammation in IBD31. Across 93 subjects with FC measurements, the first axis of metabolic variation correlated in a reasonably strong and highly statistically significant manner with FC (Spearman’s r=0.486, two-tailed p<10−6; Fig. 1D). This correlation was driven in part by the tendency of control subjects to have very low FC levels (mean=35 μg/g) and CD patients very high levels (mean=130 μg/g). However, the correlation remained strong and significant when evaluated on UC subjects only (n=25, r=0.565, two-tailed p=0.003). We observed a similar trend between FC measurements and the first axis of metagenomic variation (Fig. 1E), leading us to conclude 1) that our UC subjects vary from control-like levels of inflammation to more active inflammation, and 2) that this variation may contribute to UC subjects’ more heterogeneous metabolic and metagenomic profiles.
The first axes of metabolomic and taxonomic variation were also significantly associated with Shannon diversity (Supplementary Fig. 3). Consistent with previous findings, more inflamed, IBD-like samples (toward the right in Fig. 1C) tended to have markedly lower Shannon diversity (Spearman’s r=−0.572, two-tailed p<10−14, n=155). A similar, albeit weaker, trend was observed for metabolite profiles (r=−0.321, p<10−4, n=155), which exhibited less overall variation in within-sample diversity. Notably, these mutual associations with diversity were not sufficient to explain the strong coupling between the first axes of metabolomic and taxonomic variation, which remained significant after subtracting diversity effects using linear regression (residual correlation analysis, r=0.364, p<10−5, n=155; see Methods).
The 68 CD subjects in the PRISM (discovery) cohort were sub-classified according to disease localization: L1 (ileal, n=14), L2 (colonic, n=22), L3 (ileocolonic, n=29), L1+L4 (ileal + upper GI, n=1), and unknown (n=2). Compared to the strong separations we observed between CD and non-IBD subjects in the metabolomic and metagenomic data, we observed little to no stratification by disease localization among CD subjects (Supplementary Fig. 4). More formally, overall diagnosis (CD/UC/non-IBD) explained statistically significant fractions of the distance variation among subjects’ metabolomic and metagenomic profiles (permutational analysis of variance, p<10−4; Methods), while disease localization did not have a significant effect among CD subjects (p=0.22 and p=0.35), possibly due to the established nature of IBD within the PRISM cohort. As a consequence of this finding, we treated CD as a single diagnosis in subsequent analyses.
Metabolite enrichments in IBD versus control phenotypes
To dissect metabolic changes in IBD at greater resolution, we applied a multivariable linear model to each metabolic feature to test association with IBD phenotype while controlling for other covariates (age and medication use; Methods and Supplementary Dataset 3). Nominal p-values for UC- and CD-specific effects were subjected to multiple hypothesis testing correction using the Benjamini-Hochberg32 method with a False Discovery Rate (FDR) threshold of 0.05. Despite this strict filtering procedure, 2,729 metabolite features (31%) were significantly differentially abundant (DA) in IBD, including 200 matched against 151 unique standards. Out of all DA metabolites, the majority (1,931; 71%) were significantly depleted in IBD (CD or UC) relative to non-IBD controls; 224 (8%) were significantly elevated in both CD and UC; 505 (19%) were specifically elevated in CD; and only 69 (3%) were specifically elevated in UC (a possible consequence of UC subjects’ more heterogeneous metabolic profiles). The large number of individually DA metabolites is consistent with the broad changes in metabolite profiles of IBD subjects described above in the context of overview ordination (see Fig. 1B).
We performed enrichment analysis (rank-based Wilcoxon tests; Methods) to identify broad classes of compounds that were significantly over- or under-abundant in IBD phenotypes (ranking metabolite features by their CD- and UC-specific effect sizes). We defined metabolite classes based on HMDB annotations and focused on the 97 metabolite classes with at least 10 putative members in our dataset. Across these classes, we searched for enrichments in IBD or non-IBD controls that were statistically significant after correction for multiple hypothesis testing (Benjamini-Hochberg FDR q<0.05). Eight of the 97 molecular classes were significantly over-abundant in CD, with the strongest effects observed among sphingolipids, carboximidic acids, and bile acids (Fig. 2A). Seven of these classes were additionally significantly over-abundant in UC, while phenylacetamides were elevated, but not to a statistically significant degree (Fig. 2B). No molecular classes were specifically over-abundant in UC.
Figure 2. Metabolic enrichments in IBD versus control phenotypes.
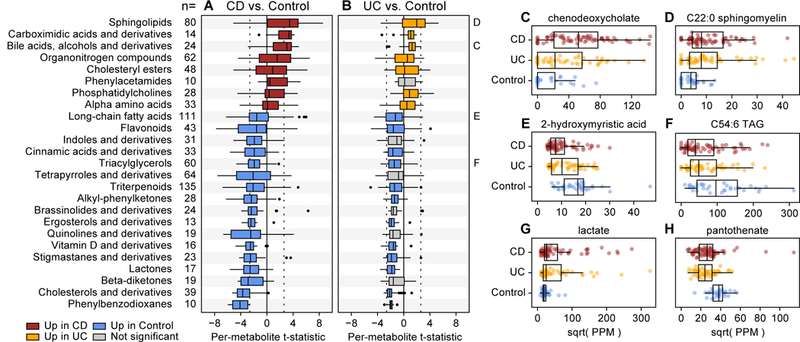
We applied Wilcoxon rank-sum tests to metabolites’ individual differential abundance trends (t-statistics from the linear models) to identify classes of molecules that were broadly enriched in IBD. Focusing on classes of molecules with at least 10 putative members (see the “n=“ column), (A) eight were significantly (FDR q<0.05) positively enriched in CD, meaning that their members tended to be more abundant in CD, and 17 classes were significantly negatively enriched, meaning that their members tended to be more abundant in controls (nominal p-values were two-tailed). (B) A subset of these trends were similarly significant in comparisons between UC and controls, with the remainder (gray) tending to trend in the same direction as CD vs. control comparisons. The dotted line indicates the significance threshold for an individual metabolic feature [abs(t)>2.61]. Panels C through H highlight examples of individually differentially abundant standards measured across 68 CD, 53 UC, and 34 non-IBD control subjects. Metabolites highlighted in panels C, D, E, and F are representatives of broader classes analyzed in A and B. Abundances are in units of parts per million (PPM) after separately sum-normalizing within each LC-MS method; values are square-root scaled for visualization. Boxplot “boxes” indicate the first, second, and third quartiles of the data. Boxplot “whiskers” indicate the inner fences of the data, with points outside the inner fences plotted as outliers.
IBD-enriched bile acids included cholate (q=0.003) and chenodeoxycholate (q=0.0002; Fig. 2C). In the healthy gut, these primary bile acids aid in digestion of lipids, and are deconjugated by microbes to secondary bile acids. We observed complementary depletions for the secondary bile acids lithocholate and deoxycholate in CD, but the changes did not meet our threshold for FDR significance (q=0.06 and 0.13 respectively). The relative over-abundance of primary bile acids in IBD subjects’ guts is consistent with disruption of bile acid transformation activities in the IBD microbiome33. Sphingolipids, another of the overabundant classes in IBD, play multiple roles in the healthy gut, including 1) as structural components of intestinal cell membranes and 2) as signaling molecules involved in cell fate decisions34. In addition to their presence in the membranes of human cells, sphingolipids are prevalent in the membranes of Bacteroidetes, and these microbially-derived sphingolipids modulate the invariant natural T cell population35. Previous work suggests that sphingolipid metabolism may be disrupted in IBD, resulting in an accumulation of specific sphingolipid compounds that promote an inflammatory state36–38. Two of these compounds, ceramide and sphingomyelin (Fig. 2D), were significantly over-abundant in both CD and UC (q<0.02 in all comparisons).
Many more molecular classes were significantly depleted in CD and UC relative to controls (see Fig. 2 A and B). Triterpenoids and long-chain fatty acids (LCFAs; including 2-hydroxymyristic acid, Fig. 2E) were the most numerous depleted classes (total n=135 and 111), while phenylbenzodioxanes and cholesterols (including cholestenone) were the most consistently depleted classes (the majority of their members were individually significantly depleted in CD). Phenylbenzodioxanes are primarily derived from fruits, which reinforces that some of the detected metabolic changes are a result of variation in subjects’ diets. Triacylglycerols (TAGs), including C54:6 TAG (Fig. 2F), were additionally enriched in controls relative to CD and UC subjects. This change, coupled with the above-mentioned enrichments for LCFAs and cholesterols, is consistent with previously suggested perturbations of fatty acid metabolism in IBD39.
While their molecular classes were not generally differentially enriched in IBD, other notable DA metabolites included lactate (up in IBD; Fig. 2G) and pantothenate (down in IBD; Fig. 2H). Lactate has been previously reported as elevated in CD and UC patients40, and is notable for being produced by members of the IBD gut microbiome, including lactobacilli, enterococci, and pediococci. Pantothenate (vitamin B5) is a precursor for coenzyme A, which is notable here for being involved in fatty acid metabolism. Moreover, pantothenate is produced by the healthy gut microbiota, and so (like lactate) its differential abundance in IBD subjects may indicate a perturbation of microbe-metabolite relationships in the gut: a topic we explore in detail in a later section. While not statistically significant in this cohort, the SCFAs butyrate and propionate were decreased in both UC and CD relative to controls (Supplementary Dataset 3).
Modules of chemically-related compounds are perturbed in IBD
To further explore biological patterns underlying the 2,729 DA metabolites, we clustered the DA metabolites based on the similarity of their residuals from the above-described linear modeling approach. Metabolites co-clustered by this method will therefore tend to co-vary independently of their relationship with IBD phenotype, age, and medication use. A total of 1,403 such clusters were identified with intra-cluster Spearman correlation of 0.7 (note that these unsupervised clusters, listed in Supplementary Dataset 1, are distinct from the HMBD-defined molecular classes used above during enrichment analysis). Each cluster was assigned a representative metabolite: the cluster centroid, or the standard metabolite closest to the centroid (where applicable). The 50 largest clusters accounted for 780 DA features (29% of total): consistent with a smaller number of biological signals explaining many DA metabolites.
Clusters of co-varying metabolites can arise by a variety of mechanisms, including 1) chemical modification of a common parent metabolite, 2) metabolites interrelated by a biochemical pathway, 3) metabolites co-produced by a specific microbe, and 4) metabolites co-contributed from a specific dietary source. Biological signals suggested by metabolite co-variation, especially those arising from inter-conversion of metabolites, can be used to transfer knowledge from annotated metabolites to their unannotated partners. This “guilt-by-association” principle also arises in gene co-expression data, where it has been applied to identify modules of functionally related genes41 and predict gene function assignments42. Following this logic, we found that co-clustered metabolites were 2.7x more similar in retention time, 3.0x more similar in mass/charge ratio, and 15x more likely to belong to the same chemical class relative to random metabolite pairs (Methods). Clusters are thus enriched for similar physicochemical properties, and cluster co-membership may be predictive of such properties.
The largest metabolite cluster enriched in IBD (and second-largest overall) contained 39 metabolite features, all of them enriched in CD (with one additionally enriched in UC; Fig. 3A). This cluster contained 12 putative bile acids, including matches to cholate, chenodeoxycholate, and their structural variants. This cluster also contained 17 unlabeled metabolites, which may also be related to bile acid metabolism via guilt-by-association logic. The largest cluster contained 62 metabolite features: all of them elevated among controls (Fig. 3B). 11 features in this cluster were annotated as putative tetrapyrroles, but the cluster contained no validated standard metabolites, thus making it a promising target for further characterization. The previously described control-like and CD-like subdivisions of the UC population were also readily apparent in these individual clusters.
Figure 3. Clusters of chemically related, IBD-perturbed metabolites revealed by abundance covariation.
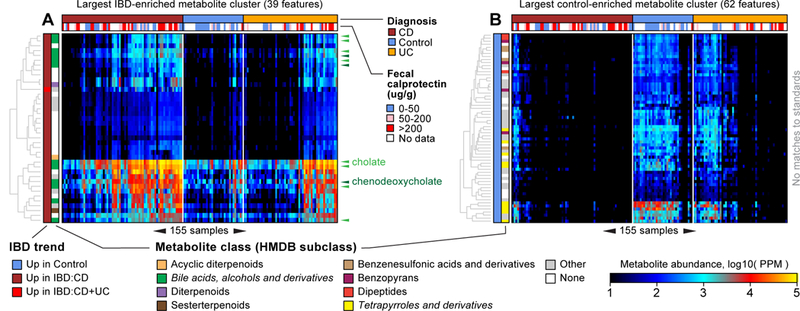
We clustered differentially abundant (DA) metabolites after regressing out the effects of diagnosis, subject age, and medication use (Methods). A small number of (large) clusters explained many of the DA metabolites. (A) The second-largest cluster contained 39 metabolite features, all of them significantly elevated among CD patients (and one in UC patients as well). This cluster was enriched for putative bile acids and derivatives. Multiple variants of the standards cholate (light green triangles) and chenodeoxycholate (dark green triangles) occur in this cluster. (B) The largest cluster contained 62 metabolite features, all of them significantly elevated in non-IBD controls. This cluster was enriched for putative tetrapyrroles and derivatives. The 155 samples (columns) are ordered the same way in both panels according to Bray-Curtis similarity (and phenotype) of overall metabolic profile (as established in Supplementary Fig. 1). Note the control-like versus CD-like substructure among UC subjects.
Other clusters of interest included the 3rd-largest cluster (33 members), which was consistently elevated among non-IBD controls, and contained a variety of triacylglycerol metabolites (Supplementary Fig. 5). Cluster 13 (18 members) was uniquely elevated in CD subjects and enriched for organonitrogen compounds, including the standards linoleoyl ethanolamide, palmitoylethanolamide, and N-oleoylethanolamine (Supplementary Fig. 6). Clusters 23 and 25 were elevated in CD and UC subjects and contained a variety of long-chain fatty acids, including the standards arachidonic acid, adrenic acid, docosapentaenoic acid, and eicosatrienoic acid (Supplementary Fig. 7). Notably, the 99 clusters (7%) containing standards were more the exception than the rule: most clusters remain largely uncharacterized, allowing the potential for many previously undescribed, IBD-associated metabolites of microbial origin.
Species-level changes in IBD microbiome community composition
As introduced above, taxonomic profiling of subjects’ gut microbiomes showed that the largest source of variation corresponded with separation of non-IBD control versus CD phenotypes, while UC subjects were more heterogeneous (see Fig. 1B). To further dissect this trend, we applied the linear modeling approach introduced above to the abundances of 195 species-level clades (from 67 genera) that were present in at least five samples at 0.1% relative abundance (Supplementary Dataset 5). A total of 50 species were differentially abundant (DA) in one or more phenotypes, of which 35 were elevated in controls relative to IBD (Supplementary Fig. 2). Roseburia hominis, Dorea formicigenerans, and Ruminococcus obeum were among the species exhibiting the strongest enrichments in non-IBD controls. The fact that these and many other species were significantly depleted in IBD relative to controls is consistent with the general trend toward loss of species diversity in the IBD microbiome2, 3, 27 and with specific previous taxonomic enrichment studies4, 5, 43, 44. Unclassified Roseburia species were significantly elevated in both CD and UC subjects, while Bifidobacterium breve and Clostridium symbiosum were uniquely DA and enriched in UC. Twelve species were uniquely DA and enriched in CD, including Ruminococcus gnavus, Escherichia coli, and Clostridium clostridioforme. Many of these species-specific enrichments and depletions were in line with previous studies as cited here and discussed in the Introduction.
Putative mechanistic associations between IBD-linked microbes and metabolites
The multi’omic nature of this dataset enables identification of microbial features and metabolites that 1) are mutually differentially abundant in IBD and 2) which covary independently of their mutual covariation with disease. Such relationships are consistent with a mechanism relating the abundance of the species and metabolite which is then perturbed during IBD pathogenesis. For example, a positive association between a metabolite and species could indicate that the metabolite promotes the growth of that species, or that the species produces that metabolite. To identify such relationships, we performed large-scale association discovery between differentially abundant (DA) metabolites and species, focusing on representative DA metabolites and species from the clustering approach described above (notably, most species clustered alone by this approach). More importantly, we performed association discovery on metabolite and species residual abundances from the above-described linear modeling approach, which will tend to de-emphasize associations driven purely by mutual association with disease status. This revealed a total of 15,679 FDR-significant (q<0.05) associations between representative DA metabolites and species. Among these was a positive association between lactic acid and Pediococcus acidilactici (Spearman’s r=0.23): one of the expected microbe-metabolite relationships alluded to above (see Fig. 2G). To further enrich for putatively mechanistic relationships that are perturbed in disease, we specifically focused on the subset of associations that were nominally significant (p<0.05) and in the same direction when considering raw metabolite and species abundances from non-IBD controls only (we refer to these associations as “confirmed in controls”).
This filtered dataset encompassed 2,279 associations between DA metabolites and species (Supplementary Fig. 8), including 122 associations involving standards and characterized species (Fig. 4A). Associations covered 901 metabolite clusters representing 1,878 DA metabolites. 46 of 50 DA species were represented in at least one association. However, of the large number of possible associations between these metabolites and species, only 6% were statistically significant and confirmed in controls. This implies that, although many metabolites are associated with one or more species, they tend not to associate mechanistically with most species (and vice versa). The largest group of associations were positive associations between metabolites and species that were both elevated in controls (1,398 associations; 61% of all significant associations). These associations were representative of a general pattern of “concordance” with disease, resulting (for example) when a species produces a protective metabolite. Discordant associations (e.g. negative associations between metabolites and species that both increased in disease) accounted for only ~2% of total associations. In these cases, while the species and metabolite may be mechanistically linked, the mechanism does not appear to directly aggravate IBD pathogenesis.
Figure 4. Potentially mechanistic associations between IBD-linked microbes and metabolites.
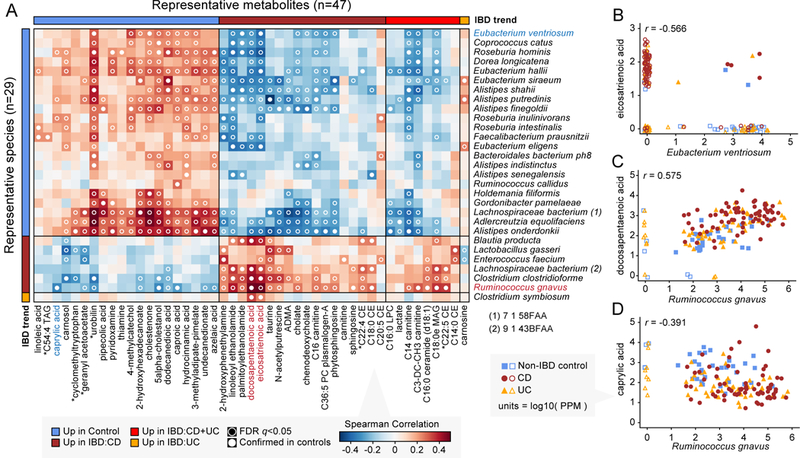
(A) Covariation between microbes and small molecules DA in IBD, specifically those linking FDR-significant, confirmed-in-controls metagenomic species and metabolites matched against standards (Spearman correlation with two-tailed nominal p-values). When multiple metabolomic features matched the same standard, the feature with the highest mean absolute correlation was selected for plotting. Starred (*) metabolites indicate a match to a standard with isomeric forms that could not be differentiated. The standard L-1,2,3,4-Tetrahydro-beta-carboline-3-carboxylic acid is listed as “cyclomethyltryptophan.” (B), (C), and (D) highlight examples of individual correlations across 68 CD, 53 UC, and 34 non-IBD control subjects (see text). Metabolites and species in these examples are colored in panel A. Values plotted are raw measurements (not residuals) normalized to parts per million (PPM) units and then log10-transformed. Values <1 PPM (including 0s) were set to 1 PPM for plotting; corresponding points are shown without fill and jittered (all other points have solid fill).
The CD-associated compounds eicosatrienoic (ETA) and docosapentaenoic (DPA) acid were involved in negative associations with control-associated species and positive associations with IBD-associated species. ETA and DPA are polyunsaturated long-chain fatty acids (PUFAs), and are examples of omega-3 and omega-6 fatty acids, respectively. ETA and DPA are important constituents of eukaryotic cell membranes, and their elevation in the IBD-afflicted gut may be explained by higher rates of host cell death/turnover or reduced absorption from diet. In addition to roles in immune and inflammatory signaling, PUFAs possess bactericidal activity by virtue of their hydrophobic nature and potential to disrupt bacterial cell membranes45. This activity is particularly consistent with the negative correlations involving ETA (e.g. with Eubacterium ventriosum; Fig. 4B), several of which had a “mutually exclusive” character (i.e. when ETA was present, the corresponding species tended to be absent, and vice versa). Conversely, DPA associated positively with IBD-associated species, most notably Ruminococcus gnavus (Fig. 4C). This suggests that DPA encourages the growth of these species, possibly through disrupting the growth of health-associated species.
Caprylic acid, also known as octanoic acid, is a medium-chain fatty acid (MCFA) with antibacterial and antiviral properties46. Caprylic acid was enriched in non-IBD controls in our dataset, consistent with previously observed patterns of MCFA depletion in IBD22. Like short-chain fatty acids (SCFAs), MCFAs may occur in the gut as a breakdown product from anaerobic fermentation of fiber, though dietary contributions are perhaps more abundant. Consistent with this idea, caprylic acid was (weakly) positively correlated with a number of health-associated gut anaerobes, including Alistipes shahii, A. putredinis, and A. finegoldii. On the other hand, caprylic acid was significantly negatively associated with the abundance of Ruminococcus gnavus. Such a negative relationship would be consistent with possible uptake and metabolism of caprylic acid by R. gnavus (in which case, as the species’ abundance increases, more caprylic acid is used up). Alternatively, and more consistent with its aforementioned antibacterial properties, caprylic acid may have an inhibitory effect on growth of R. gnavus.
To experimentally validate the potential for IBD-associated metabolites to exert growth effects on an IBD-associated species, we cultured R. gnavus in the presence of eight molecules with which it was observed to associate in the preceding analysis (see Methods). Among four predicted negative associations, caprylic acid did indeed inhibit the growth of R. gnavus at high concentrations, as hypothesized above (Supplementary Fig. 9). Among four predicted positive associations, taurine and docosapentaenoic acid were confirmed to enhance growth, while phytosphingosine exhibited a paradoxical inhibitory effect. Given the many factors that could impact the results of growth assays (e.g. strain specificity and molecular concentrations) and the potential for mechanisms of association beyond direct effects on growth (e.g. production as byproduct), these results provide promising initial support for the usefulness of our multi’omic association framework in focusing downstream experiments.
IBD-associated changes in microbial function and their metabolic associations
To understand the functional consequences of microbial community changes in IBD, we first functionally profiled gene families in all metagenomes using HUMAnN2, and then summed their abundances according to Enzyme Commission (EC) number annotations (see Methods and Supplementary Dataset 6). We applied the above-described linear modeling approach to this enzyme abundance data, revealing 568 enzymes that were differentially abundant (DA; FDR-corrected q<0.05) in CD, UC, or both (Supplementary Fig. 10 and Supplementary Dataset 7). However, examining species-level functional attribution data, it was clear that many of these DA enzymes could be explained by a single IBD-associated species dominating contributions of the enzyme to the community. More specifically, when defining “dominating” as “explaining >50% of enzyme copies in >50% of samples,” then E. coli alone dominated 220 DA enzymes, owing in part to that species’ strong enrichment in IBD and exceptionally thorough functional annotations. While some enzymes in this category may indeed have mechanistic connections to IBD, others may simply expand (or shrink) in copy number alongside their source genomes (whose abundance is changing for reasons unrelated to encoding of that particular enzyme).
246 DA enzymes were not dominated by any single species, suggesting that their enrichment in controls (or IBD) was better explained by a community-level shift in functional potential, and therefore of greater mechanistic significance. For example, magnesium-importing ATPase (EC 3.6.3.2) was enriched in both CD and UC subjects relative to controls (Fig. 5A). Magnesium deficiency has been described as a known side-effect of IBD47: one which could be explained in part by sequestration of the ion by the IBD-associated microbiome. Ethanolamine ammonia-lyase (EC 4.3.1.7) was similarly enriched in the IBD gut (Fig. 5B). This enzyme is involved in the production of glycerophospholipids, one of the most significantly enriched classes of metabolite in CD and UC subjects (see Fig. 2A). A final example of an IBD-enriched enzyme was glutathione-disulfide reductase (“GR”; EC 1.8.1.7; Fig. 5C). GR catalyzes the production of glutathione: a compound involved in resistance to oxidative stresses. Oxidative stress is a hallmark of inflammation in the IBD-afflicted gut48, thus giving species encoding GR a selective advantage in that environment.
Figure 5. IBD-associated changes in microbial function and their metabolic associations.
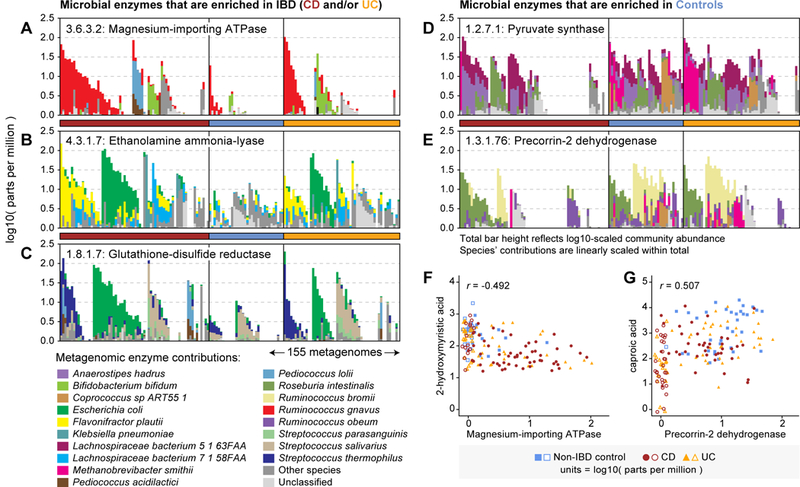
(A) - (E) highlight examples of metagenomically contributed enzymes that were differentially abundant in IBD, annotated by their taxonomic contributors (A - C are enriched in IBD; D and E are depleted). In each case, the enzyme was contributed by a mixture of species across the cohort, and not dominated by a single species. Each set of stacked bars represents one of the 155 PRISM metagenomes (arrayed on horizontal axes). Community enzyme abundance (log10-transformed parts per million) is represented by the top of each stack of bars; contributions from major species are linearly scaled within the total bar height. Samples are first sorted according to the dominant contributor to a function and then grouped by phenotype (sample ordering differs between panels). (F) and (G) illustrate correlations between community-total enzyme abundance and IBD-associated metabolites across 68 CD, 53 UC, and 34 non-IBD control subjects. Values plotted are raw measurements (not residuals) normalized to parts per million (PPM) units and then log10-transformed. Values <1 PPM (including 0s) were set to 1 PPM for plotting; corresponding points are shown without fill and jittered (all other points have solid fill). The given r values indicate Spearman correlation.
Additional examples of DA enzymes were reflective of transitions from a more obligate anaerobic to facultative anaerobic microbiome in IBD. For example, pyruvate synthase (EC 1.2.7.1), an anaerobic enzyme that catalyzes pyruvate/acetyl-CoA interconversion, was enriched in controls and completely undetected in a subpopulation of CD patients (Fig. 5D). Enzymes involved in the synthesis of cobalamin (vitamin B12) were also enriched in controls, including precorrin-2 dehydrogenase (EC 1.3.1.76; Fig. 5E). While vitamin B12 (a tetrapyrrole-containing structure) is too large to be captured by the LC-MS methods used here, its derivatives and associated compounds may be among the putative tetrapyrroles that were enriched in the largest cluster of IBD-depleted metabolite features (see Fig. 3B).
To more formally evaluate potential mechanistic links between DA enzymes and metabolites, we repeated the clustering and association procedures described above in the context of metabolite-species associations. Metabolite-enzyme associations followed many of the same patterns observed for species and enzymes: association density was low (3%), suggesting that most metabolites associated with only a few enzymes (and vice-versa), and the vast majority of interactions (95%) were concordant with IBD pathogenesis (Supplementary Fig. 11). Several associations occurred between standard metabolites and the enzymes discussed above. For example, magnesium-importing ATPase was strongly negatively associated with 2-hydroxymyristic acid (a control-enriched compound; Spearman’s r=−0.492; Fig. 5F). Conversely, precorrin-2 dehydrogenase was positively associated with caproic acid (another control-enriched compound; r=0.507). While such relationships are consistent with compounds acting as enzyme substrates and products (respectively), this does not appear to be the case for these specific enzyme:compound pairs, suggesting that other factors likely mediate their associations (e.g. encoding by/interaction with subsets of IBD-associated species).
Most IBD trends replicate in an independent validation cohort
We evaluated the generality of the differentially abundant (DA) metabolite features and microbial species identified above in an independent cohort of 20 CD, 23 UC, and 22 non-IBD control subjects from the Netherlands (see Methods). Of 2,456 metabolite features that were DA in CD (up or down) in the PRISM cohort, 2,300 (94%) trended in the same direction in the Netherlands cohorts, of which 959 (39%) were also FDR-significant (Supplementary Fig. 12). Of 1,049 metabolite features that were DA in UC (most of which were also DA in CD), 865 (82%) trended in the same direction, of which 117 (11%) were FDR-significant. Similar patterns were observed for DA microbial species: 36 of 38 species that were DA in CD among Boston PRISM subjects trended in the sample direction among Netherlands subjects, with 13 achieving statistical significance. All 15 UC-significant species from the Boston PRISM subjects trended in the same direction among Netherlands subjects, with 3 achieved statistical significance. Hence, the majority of IBD-associated changes identified in the PRISM cohort generalized in sign to the Netherlands cohorts. Statistical significance was not as consistently replicated, which we can attribute in part to loss of power in the Netherlands cohorts from smaller sample size (total n=65 versus 155 for PRISM).
Multi’omic signatures differentiate IBD subtypes across cohorts
To evaluate if differences in metabolite or microbial composition could be used to classify subjects according to IBD phenotype, we trained random forest (RF) classifiers on subjects’ metabolic and microbial species profiles (separately and combined). Classification performance was evaluated within the PRISM cohort (using five-fold cross-validation) and between cohorts by training on the entire PRISM cohort and validating on the independent Netherlands cohort. In both of these approaches, classifiers are trained on one set of samples and then tested on another (non-overlapping) set, meaning that testing performance does not benefit from potential overfitting of classifiers to their training data.
All classifiers performed considerably better than random in the task of distinguishing IBD and non-IBD controls, with AUC values ranging from 0.86 to 0.92 (AUC values close to 1.0 indicate that a classifier attained a high sensitivity at a very low false positive rate, while a value of 0.5 is expected at random; Fig. 6A and Supplementary Fig. 13). Cross-validation results (AUC 0.90–0.92) were only marginally better than independent validation results (AUC 0.86–0.89), indicating that the PRISM-trained classifier generalized well to the Netherlands cohort, which is consistent with the feature-level concordance described above. Classifiers trained on metabolite features versus microbial species performed similarly, despite the metabolite feature space being considerably larger (1,000s vs. 10s of features). The integration of metabolite and microbial species data did not produce a marked improvement in classification accuracy relative to metabolite features alone, which is consistent with a high degree of shared information between the gut’s microbial and metabolomic profiles.
Figure 6. Predicting IBD status and subtype from gut microbiome multi’omic features.
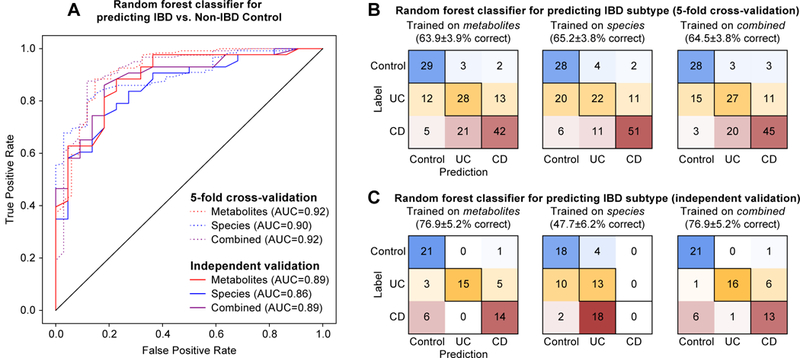
We trained random forest classifiers on metabolites, microbial species, and their combination to identify IBD patients and IBD subtypes. Training/testing was carried out within the PRISM cohort using five-fold cross-validation, in addition to models trained on the full PRISM cohort and then tested (validated) on the independent Netherlands cohorts. (A) ROC curves depict trade-offs between classifiers’ true positive rates (TPRs) and false positive rates (FPRs) as classification stringency varies. The area under the curve (AUC) statistic is a summary measure of classifier performance: AUC values close to 1 indicate that a high TPR was achieved with low FPR (ideal performance), while AUC values close to 0.5 indicate random performance. (B) “Confusion matrix” evaluations of IBD subtype classifiers within the Boston PRISM cohort. The number in row i and column j indicates how many samples were labeled as subtype i but assigned to subtype j. A perfect subtype classifier (100% accuracy) would have 0 counts for all non-diagonal entries (i.e. no misclassified samples). Matrix cells are shaded within-row in proportion to their value (red for CD, orange for UC, and blue for non-IBD control). (C) Confusion matrix evaluations of IBD subtype classifiers trained on the Boston PRISM cohort and tested on the independent Netherlands cohorts. Accuracy values in B and C indicate the fraction of correctly classified instances; error values reflect the standard error of a proportion.
Predicting IBD subtype (summarized simply as CD and UC) was comparatively more challenging. Within the PRISM cohort, metabolites, species, and their combination predicted UC, CD, or non-IBD control labels correctly 64–65% of the time: less successful than case/control predictions, but still considerably greater than random (i.e. 33% correct; Fig. 5B). The most common source of classification error was labeling UC subjects as non-IBD or CD. This is not surprising, given that the distribution of UC subjects overlapped with the (largely distinct) CD and non-IBD populations (see Fig. 1B). Comparatively, Non-IBD subjects were rarely classified into one of the two IBD subtypes, while CD subjects were sometimes erroneously classified as UC. More distinction among the input data types was observed when applying the PRISM-trained IBD subtype classifier to the Netherlands cohorts (Fig. 5C). While the metabolite-incorporating classifiers performed reasonably well (77% correct classification), the species-based classifier performed considerably worse (48% correct classification), largely due to marked misclassification of CD subjects as UC. This suggests that, although many IBD-varying species trended similarly in the Netherlands cohort, subtype-informative details of their abundance distributions (as learned from the PRISM cohort) were less conserved.
Discussion
This study represents one of the first efforts to discover and validate IBD-associated changes in the human gut metabolome and microbiome in an integrated multi’omic framework. Many of the individually differentially abundant species and metabolic classes identified and validated here (e.g. bile acids and sphingolipids) are in agreement with previous findings, while others (e.g. dicarboxylic acids) are, to the best of our knowledge, unique to this study. More generally, we observed that metabolites and metabolite classes were frequently depleted in IBD patients relative to non-IBD controls. This pattern is suggestive of a loss of “metabolic diversity” among IBD subjects that is analogous to the loss of taxonomic (ecological) diversity observed in the IBD microbiome. This diversity is likely to be inclusive of a large number of previously undescribed, microbially-derived metabolites that were unclassified or putatively classified in our comparisons with reference databases.
The ability of untargeted metabolomics approaches to quantify vast numbers of uncharacterized metabolites is both a strength and limitation relative to targeted approaches26. While uncharacterized metabolites no doubt encompass previously undescribed microbe- and disease-associated molecules of biological interest, they also include non-biological adducts and fragments of sample molecules, and are generally more challenging to interpret. We approached these challenges using a combination of methods: 1) experimentally validating metabolites against a standard compound library (a precise but resource-intensive process), 2) approximate annotation of metabolites to broad chemical classes, and 3) clustering of metabolites according to residual covariation across samples. Covariation-based clusters were found to be enriched for metabolites of similar physicochemical properties: a form of “guilt-by-association” that complements existing network-based approaches49 to metabolite characterization. The covariation-based approach suggested potential roles for many unlabeled metabolites that clustered with known standards (as in Fig. 3A). On the other hand, the process also revealed clusters of wholly uncharacterized, IBD-associated metabolites (as in Fig. 3B). Such clusters may represent microbial metabolites with pro- or anti-inflammatory effects, and are prime candidates for additional experimental characterization.
Computational methods also provide a guide to downstream experimental validation and characterization of mechanisms relating the IBD microbiome and metabolome (including identification of microbiome-derived metabolites). By prioritizing associations between microbial species and enzymes that associate with metabolites independently of disease status, we enrich for potential mechanistic associations that may become perturbed in IBD. Many details of these associations remain to be determined. For example, a positive association between a microbial taxon and metabolite could be explained by 1) the metabolite representing a preferred carbon source that promotes the species’ growth, 2) the metabolite occurring as a by-product of the species’ metabolism, or 3) the metabolite selectively inhibiting the growth of other species (or otherwise interacting ecologically). These options can be disentangled computationally by analysis of genome and metagenome annotations, when available, as well as experimentally by growing microbial species in the presence of their associated metabolites and/or profiling their metabolic output in monoculture. Naturally, further in vivo experiments (e.g. in mouse models of IBD) will be required to confirm that validated microbe-metabolite associations play a causal role in IBD pathogenesis. Such efforts are laborious, and hence the computationally derived subset of putative associations uncovered here will be a critical aid, as will further bioinformatic prioritization based on meta’omic profiling.
The vast majority of IBD-associated species and metabolites discovered in the PRISM cohort agreed in directionality with an independent validation cohort. Statistical significance was not as consistently replicated, in part due to power limitations of the smaller validation study (hence, further replication in a larger cohort could be warranted in the future). At the same time, integrating individual microbial and metabolomic signals was sufficient to build accurate classifiers for case/control status that generalized to new subjects. Surprisingly, combining signals of both types (microbes and metabolites) did not boost classification performance markedly. This result is suggestive of tight coupling of the IBD gut metabolome and microbiome, which may result from a combination of 1) both profiles changing in response to disease, 2) an altered microbiome perturbing the metabolome, or 3) an altered metabolome perturbing the microbiome (with potential feedback therein). Mechanisms underlying this coupling will be naturally expanded through experimental validation of targeted microbe-metabolite associations, as described above.
Predicting IBD subtype (UC vs. CD) proved challenging, though this result was not surprising in light of other findings from the study. While CD patients did not stratify strongly by disease localization (see Supplementary Fig. 4), CD patients as a whole separated well from non-IBD controls (see Fig. 1). The same could not be said for UC patients, which were dispersed into inflamed/CD-like and non-inflamed/control-like subpopulations, consistent with previous reports of high variability among UC microbiomes25. Many features that were individually differentially abundant in UC were also differentially abundant in CD, while the converse was not true. This is typical of the UC microbiome in general, and it suggests that IBD-linked perturbations may be divided into at least two modules: 1) perturbations that are associated with inflammation in general, and 2) perturbations that are specific to CD. The first module underlies the general association between multi’omic profiles and inflammation status (as measured here by fecal calprotectin level), and provides a basis for classifying case/control status. The second module can also aid in classifying case/control status, in that it is informative for CD subtype specifically. However, the absence of a strong UC-specific signal, coupled with heterogeneity among the UC subpopulation, hindered the predictability of UC status. That being said, a small number of molecules, including ethyl 9-hexadecenoate (see Fig. 2F), were individually differentially abundant in UC, and make promising targets for further study.
A number of future directions are possible for expanding this work to improve our understanding of metagenomic and metabolomic perturbations in IBD. To better differentiate UC, for example, it is possible that the metabolomic methods employed here missed classes of molecules that specifically vary with UC status. Alternatively, the stool metabolome may be imperfect for capturing UC-specific signals. In such cases, profiles of serum metabolites might augment serum antibodies50 as diagnostic biomarkers for IBD/UC, while remaining less-invasive than biopsy but still associable with the microbiome. While this study employed cross-sectional sampling of a larger number of subjects, dense longitudinal sampling of a subset of individuals would further aid in the dissection of putative microbe-metabolite associations (by intrinsically controlling for within-specific properties), and would further illuminate whether the observed population substructure was stable over time, or correlated with changes in metabolomic or microbiome composition. This would, critically, also help disentangle causality—which metabolite shifts precede microbial or host phenotypes, and vice versa—as well as provide a potential predictive target for interception of disease activity. However, even without these additional studies, the multi’omic screens and associations uncovered here provide many actionable hypotheses regarding the role of specific known and yet-to-be characterized metabolites and their microbial partners in IBD pathogenesis. While many of these changes likely result from physiological changes on the host-side, the subset that can be confirmed to result from microbial activity will provide promising targets for microbiome-based IBD diagnostics and therapies.
Methods
We performed untargeted metabolomic and metagenomic profiling on two IBD cohorts containing subjects with Crohn’s disease (CD), ulcerative colitis (UC), and non-IBD controls. One cohort consisted of patients seen at the Massachusetts General Hospital (Boston, MA), and formed the basis of most analyses. A second (more heterogeneous) group of subjects from the Netherlands was used to validate findings. Microbial species, microbial enzymes, and >8K metabolites were tested for differential abundance (DA) in IBD. DA metabolites were clustered to identify groups of functionally related compounds that were similarly perturbed in IBD. DA metabolites and microbial features were compared via multi’omic correlation to identify putative mechanistic associations. Finally, all features were applied to build and validate multi’omic classifiers for IBD status and subtype.
PRISM cohort description and sample handling
The Prospective Registry in IBD Study at MGH (PRISM) is a referral center-based, prospective cohort of IBD patients. 161 adult patients (>18 y.o.) enrolled in PRISM and diagnosed with CD, UC, and non-IBD (control) conditions were selected for this study, with diagnoses based on standard endoscopic, radiographic, and histologic criteria. PRISM research protocols were reviewed and approved by the Partners Human Research Committee (#2004-P-001067), and all experiments adhered to the regulations of this review board. PRISM subject stool samples were collected at the MGH gastroenterology clinic and stored at −80°C prior to DNA extraction.
Approval for human subjects research
Human subjects research in the discovery (PRISM) cohort was reviewed and approved by the Partners Human Research Committee (#2004-P-001067), and all experiments adhered to the regulations of this review board. Human subjects research in the validation cohorts (LLDeep and NLIBD) was approved by the University Medical Center Groningen review board (ref. M12.113965 and IRB number 2008.338, respectively). All study procedures were performed in compliance with all relevant ethical regulations for the validation cohorts. Each participant signed an informed consent form prior to participation for PRISM and both validation cohorts.
Validation cohort description and sample handling
The validation cohort consisted of 65 subjects enrolled in two distinct studies from the Netherlands. 22 control subjects were enrolled in the LifeLines-DEEP general population study51. 43 subjects with IBD were enrolled in a study at the Department of Gastroenterology and Hepatology at University Medical Center Groningen (UMCG). Subjects enrolled in both studies collected stool via the same protocol: a single stool sample was collected at home and then frozen within 15 min in a conventional freezer. A research nurse visited all participants at home to collect home-frozen stool samples, which were then transported and stored at −80°C. The stool samples were kept frozen prior to DNA extraction or metabolomic profiling as described below.
DNA extraction and metagenomic sequencing
Metagenomic data generation and processing were performed at the Broad Institute (Cambridge, MA, USA). Stool DNA extractions were carried out using the QIAamp DNA Stool Mini Kit (QIAGEN, Inc.). Whole genome shotgun (WGS) libraries were prepared by quantifying metagenomic DNA samples by Quant-iT PicoGreen dsDNA Assay (Life Technologies) and normalized to a concentration of 50 pg/μL. Illumina sequencing libraries were prepared from 100–250 pg of DNA using the Nextera XT DNA Library Preparation kit (Illumina) according to the manufacturer’s recommended protocol, with reaction volumes scaled accordingly. Batches of 24, 48, or 96 libraries were pooled by transferring equal volumes of each library using a Labcyte Echo 550 liquid handler. Insert sizes and concentrations for each pooled library were determined using an Agilent Bioanalyzer DNA 1000 kit (Agilent Technologies). Metagenomic libraries were sequenced on the Illumina HiSeq 2500 platform, targeting ~2.5 Gb of sequence per sample with 101 bp, paired-end reads.
Read-level quality control and metagenomic profiling
Raw sequencing reads were quality-controlled using KneadData v0.5.1 (available via http://huttenhower.sph.harvard.edu/kneaddata). Briefly, this involved trimming low-quality bases from the 3’ end of reads with Trimmomatic52 and then discarding trimmed reads <60 nt in length. Host (human) reads were identified and removed by mapping against the human genome (hg19 build) with bowtie253.
Quality-filtered metagenomes were taxonomically profiled using MetaPhlAn2 v2.2.054 with default parameters. Only species-level relative abundance data were considered in this study. Species that failed to exceed 0.1% relative abundance in at least 5 samples were excluded. Functional profiling was performed using HUMAnN2 v0.9.4 in UniRef90 mode (available via http://huttenhower.sph.harvard.edu/humann2)55. HUMAnN2 initially maps metagenomic reads to the pangenomes of species identified during taxonomic profiling (using bowtie2). Coding sequences in these pangenomes have been pre-annotated to their respective UniRef90 families56. Reads that did not align to a pangenome are mapped to UniRef90 by translated search with DIAMOND57. Hits to UniRef90-annotated sequences are weighted by alignment quality, sequence length, and sequence coverage. Gene-level outputs are produced in reads per kilobase (RPK) units and stratified according to known/unclassified community contributions. Per-sample gene abundances are sum-normalized to parts (copies) per million units. Gene abundances can be regrouped to other functional annotation systems based on annotations from UniProt58. For this study, gene abundances were regrouped (summed) according to Enzyme Commission (EC) number.
Metabolite profiling from stool samples
Subjects’ gut metabolomic profiles were measured from stool samples using a combination of four liquid chromatography tandem mass spectrometry (LC-MS) methods that measure complementary metabolite classes. These range from polar metabolites (e.g. organic acids), lipids (e.g. triglycerides), free fatty acids, and bile acids. In each method, the MS data were acquired using sensitive, high-resolution mass spectrometers (Q Exactive, Thermo Scientific) that enabled non-targeted measurement of 1) metabolites of known identity and 2) heretofore unidentified metabolites (e.g. microbe-derived) in the same run.
Stool samples (weight range 50.5167.8 mg) were homogenized in 4 μL of water per milligram stool sample weight using a bead mill (TissueLyser II; Qiagen) and the aqueous homogenates were aliquoted for metabolite profiling analyses. Four separate liquid chromatography tandem mass spectrometry (LCMS) methods were used to measure polar metabolites and lipids in each sample. Methods 1, 2 and 3 below were conducted using two LCMS systems comprised of Nexera X2 UHPLC systems (Shimadzu Scientific Instruments; Marlborough, MA) and Q Exactive hybrid quadrupole orbitrap mass spectrometers (Thermo Fisher Scientific; Waltham, MA) and method 4 was conducted using a Nexera X2 UHPLC (Shimadzu Scientific Instruments; Marlborough, MA) coupled to an Exactive Plus orbitrap MS (Thermo Fisher Scientific; Waltham, MA).
Method 1: positive ion mode MS analyses of polar metabolites (feature prefix: HILIC-pos).
LCMS samples were prepared from stool homogenates (10 μL) via protein precipitation with the addition of nine volumes of 74.9:24.9:0.2 v/v/v acetonitrile/methanol/formic acid containing stable isotopelabeled internal standards (valined8, Isotec; and phenylalanined8, Cambridge Isotope Laboratories; Andover, MA). The samples are centrifuged (10 min, 9,000 x g, 4°C), and the supernatants were injected directly onto a 150 × 2 mm Atlantis HILIC column (Waters; Milford, MA). The column was eluted isocratically at a flow rate of 250 μL/min with 5% mobile phase A (10 mM ammonium formate and 0.1% formic acid in water) for 1 minute followed by a linear gradient to 40% mobile phase B (acetonitrile with 0.1% formic acid) over 10 minutes. MS analyses were carried out using electrospray ionization in the positive ion mode using full scan analysis over m/z 70800 at 70,000 resolution and 3 Hz data acquisition rate. Additional MS settings were: ion spray voltage, 3.5 kV; capillary temperature, 350°C; probe heater temperature, 300 °C; sheath gas, 40; auxiliary gas, 15; and Slens RF level 40.
Method 2: negative ion mode MS analysis of polar metabolites (feature prefix: HILIC-neg).
LCMS samples were prepared from stool homogenates (30 μL) via protein precipitation with the addition of four volumes of 80% methanol containing inosine15N4, thymined4 and glycocholated4 internal standards (Cambridge Isotope Laboratories; Andover, MA). The samples were centrifuged (10 min, 9,000 x g, 4°C) and the supernatants were injected directly onto a 150 × 2.0 mm Luna NH2 column (Phenomenex; Torrance, CA). The column was eluted at a flow rate of 400 μL/min with initial conditions of 10% mobile phase A (20 mM ammonium acetate and 20 mM ammonium hydroxide in water) and 90% mobile phase B (10 mM ammonium hydroxide in 75:25 v/v acetonitrile/methanol) followed by a 10 min linear gradient to 100% mobile phase A. MS analyses were carried out using electrospray ionization in the negative ion mode using full scan analysis over m/z 60750 at 70,000 resolution and 3 Hz data acquisition rate. Additional MS settings were: ion spray voltage, 3.0 kV; capillary temperature, 350°C; probe heater temperature, 325 °C; sheath gas, 55; auxiliary gas, 10; and Slens RF level 40.
Method 3: negative ion mode analysis of metabolites of intermediate polarity (e.g. bile acids and free fatty acids; feature prefix: C18-neg).
Stool homogenates (30 μL) were extracted using 90 μL of methanol containing PGE2d4 as an internal standard (Cayman Chemical Co.; Ann Arbor, MI) and centrifuged (10 min, 9,000 x g, 4°C). The supernatants (10 μL) were injected onto a 150 × 2 mm ACQUITY T3 column (Waters; Milford, MA). The column was eluted isocratically at a flow rate of 400 μL/min with 25% mobile phase A (0.1% formic acid in water) for 1 minute followed by a linear gradient to 100% mobile phase B (acetonitrile with 0.1% formic acid) over 11 minutes. MS analyses were carried out using electrospray ionization in the negative ion mode using full scan analysis over m/z 200550 at 70,000 resolution and 3 Hz data acquisition rate. Additional MS settings were: ion spray voltage, 3.5 kV; capillary temperature, 320°C; probe heater temperature, 300 °C; sheath gas, 45; auxiliary gas, 10; and Slens RF level 60.
Method 4: polar and nonpolar lipids (feature prefix: C8-pos).
Lipids were extracted from stool homogenates (10 μL) using 190 μL of isopropanol containing 1dodecanoyl2tridecanoylsnglycero3phosphocholine as an internal standard (Avanti Polar Lipids; Alabaster, AL). After centrifugation (10 min, 9,000 x g, ambient temperature), supernatants (10 μL) were injected directly onto a 100 × 2.1 mm ACQUITY BEH C8 column (1.7 μm; Waters; Milford, MA). The column was eluted at a flow rate of 450 μL/min isocratically for 1 minute at 80% mobile phase A (95:5:0.1 vol/vol/vol 10 mM ammonium acetate/methanol/acetic acid), followed by a linear gradient to 80% mobilephase B (99.9:0.1 vol/vol methanol/acetic acid) over 2 minutes, a linear gradient to 100% mobile phase B over 7 minutes, and then 3 minutes at 100% mobilephase B. MS analyses were carried out using electrospray ionization in the positive ion mode using full scan analysis over m/z 2001100 at 70,000 resolution and 3 Hz data acquisition rate. Additional MS settings were: ion spray voltage, 3.0 kV; capillary temperature, 300°C; probe heater temperature, 300 °C; sheath gas, 50; auxiliary gas, 15; and Slens RF level 60.
Post-processing.
We used Genedata Expressionist (program: “Refiner MS”; software version=“9.0”) to process raw LC-M data for chemical noise removal, to detect chromatographic peaks and isotope clusters, align retention times between samples, and assign putative metabolite identities via database look up. Detailed parameter settings are provided as Supplementary Dataset 8. Across samples, the combination of the four LC-MS methods generated 8,869 clustered features, characterized by chromatographic retention time and exact mass to <5 ppm accuracy. Note that these clustered features, referred to as “metabolites” or “metabolite features” elsewhere in the text, are presumed to represent a single molecular species. Broader clusters of metabolite features, presumed to represent families of related molecular species, were also constructed using the results of linear regression analysis (and are described later). Within each sample and LC-MS method, feature intensities were sum-normalized to parts per million (PPM) units.
A subset of 466 metabolites were identified more precisely using reference data generated from an in-house compound library. 3,829 metabolite features were linked to putative identifiers based on accurate m/z matching against the Human Metabolome Database (HMDB). Analyses of putatively matched features in the text focus on their molecular classes, rather than their identities. More specifically, we assigned HMDB subclasses to these features as a form of broad chemical classification. Subclasses assigned to >100 features (e.g. “fatty acyls”) were further broken down according HMDB’s “direct parent” annotations.
Profile-level quality control
Prior to downstream analysis, metagenomic and metabolomic samples were subjected to profile-level quality control. First, we isolated the set of subjects with complete profiles of both types. All 65 Netherlands subjects passed this filter, while 6 of 161 PRISM subjects were missing one of the two profiles (e.g. due to a failed sequencing run), and were excluded from subsequent analysis. Next, for both profile types, we considered the median Bray-Curtis distance of each PRISM sample to other samples in the PRISM cohort (the same distances form the basis of the ordinations in Fig. 1). If this distance was unusually large (defined as “above the upper inner fence of all values”), the sample was considered an outlier. All PRISM metagenomic and metabolomic profiles passed this filter. Repeating this procedure within-phenotype (CD, UC, Non-IBD control), we identified one potential control outlier among the PRISM metagenomic profiles, and a different control outlier plus one UC outlier among the PRISM metabolomic profiles. Because these profiles were representative of the human gut microbiome as a whole (if not their specific phenotype) they were retained for further analyses.
Statistical analyses
We carried out ordination analyses (Fig. 1; Supplementary Fig. 3 and Supplementary Fig. 4) using classical multidimensional scaling (CMDS) on matrices of between-sample diversity scores (Bray-Curtis distance). We used the Shannon index to quantify within-sample diversity. Metabolomic diversity scores considered all measured metabolites (sum-normalized first within method, and then within-sample), while taxonomic diversity scores focused on species-level relative abundances. To control for within-sample diversity when comparing ordination axes, we generated best-fit lines between axis values and dataset-specific diversity measures, saved the resulting residual values, and then compared dataset-specific residuals by Spearman correlation. Other comparisons involving between-sample diversity and sample metadata were made using permutational analysis of variance (PERMANOVA) as implemented in the adonis function from R’s vegan package (using 104 permutations). Specifically, we computed the influence of diagnosis (CD/UC/non-IBD) across all subjects’ metabolomic and metagenomic distances, and the influence of disease localization across CD subjects’ metabolomic and metagenomic profiles. These analyses did not consider additional covariates.
We used linear models implemented in Python’s statsmodels package to identify microbial species, enzymes, and metabolite features that were differentially abundant in IBD (http://www.statsmodels.org). Each data type was analyzed separately in each cohort. Relative abundance values were log-transformed to variance-stabilize the data. Zero values were additively smoothed by half the smallest non-zero measurement on a per-sample basis. For both cohorts, we modeled the transformed abundance of each feature as a function of IBD phenotype (modeled as a categorical variable with “non-IBD control” as the reference state), with age as a continuous covariate in both cohorts, and four medications (antibiotics, immunosuppressants, mesalamine, and steroids) as binary covariates in the PRISM cohort. Effect sizes take the form of model t-statistics (CD vs. non-IBD control and UC vs. non-IBD control) with associated two-tailed p-values. Nominal p-values were adjusted for multiple hypothesis testing with a target False Discovery Rate (FDR)59 of 0.05. A feature (metabolite, species, or enzyme) was considered “Differentially Abundant (DA)” in IBD if it passed this filter in either the CD- or UC-centered comparisons. Residual abundance values from the linear models were retained for use in subsequent analyses.
We identified molecular classes (as defined above) that were significantly enriched or depleted in IBD using rank-based enrichment analysis. Specifically, each metabolite was ranked according to its above-described t-statistics for CD- or UC-focused comparisons. For each class of molecule, we then evaluated if its members were enriched at the top or bottom of the list by performing a Wilcoxon rank-sum comparison of t-values in the class versus those outside the class. Only classes with at least 10 putative members were evaluated. Enrichment p-values were corrected for multiple hypothesis testing as described above.
Unsupervised clustering
We performed clustering of DA features using a custom approach. Features were clustered on their residual abundance values from the above-described linear modeling approach. This procedure enriches for covariation between features that is independent of mutual covariation with disease status (or other subject metadata, such as age or medication use). Features were ranked according to the significance of their association with IBD (the smaller of the two p-values from the CD- and UC-centered comparisons). The highest-ranked feature was seeded into an initial cluster. Each subsequent feature was then compared to each extant cluster: if the feature had a mean similarity to the cluster’s members exceeding a threshold, the feature was added to that cluster. (For all clustering analyses, we applied Spearman correlation as a similarity measure with a threshold of r=0.7.) If the feature was not added to a cluster in this way, it was used to seed a new cluster. After considering all features, clusters were renumbered according to their size (such that cluster 1 had the most members, etc.). Each cluster was characterized by a representative member. For metabolite clusters containing standards, this representative was the standard closest to the cluster centroid; the true centroid was used for clusters without standards. Similarly, characterized (vs. “unclassified”) species-level taxonomic features were preferred as representative features in microbial species clusters.
To evaluate “guilt-by-association” principles across the metabolite clusters, we compared pairs of metabolites present in the same cluster to all pairs of metabolites present in clusters with two or more members (i.e. ignoring singleton clusters). To compare retention times, we evaluated the median difference in retention time for co-clustered vs. all metabolite pairs (1.4 vs 3.8 min, a 2.7-fold reduction). The same procedure was used to compare mass/charge ratios (59 vs. 174 amu, a 3.0-fold reduction). To compare chemical class, we restricted the analysis to annotated metabolites present in clusters with at least two annotated members. 18.8% of co-clustered metabolites were annotated to the same class, compared with 1.2% of all metabolites: a 15-fold enrichment for similarity.
Random forest classification
We performed Random Forest (RF) classification using the implementation of this method in Python’s scikit-learn package (http://scikit-learn.org/). We considered separate RF classifiers for predicting 1) IBD/control status and 2) CD/UC/control status. We trained RF classifiers on the PRISM cohort using 1) five-fold cross-validation and 2) treating the entire cohort as a training set for independent validation against the Netherlands cohort. In each case, subject labels were randomly balanced prior to training and 100 trees were considered (other scikit-learn defaults were left unchanged). Features were not filtered in any way prior to RF training (i.e. the classifier could sample from any of the measured metabolites and/or species). Feature importance scores were retained for downstream analysis.
Growth effects of metabolites on Ruminococcus gnavus
We grew R. gnavus ATCC 29149 in BHI medium (37 g/L) containing: 5% sterile-filtered fetal bovine serum (Sigma-Aldrich), 1% vitamin K1-hemin solution (BD Biosciene), 1% trace mineral supplement (ATCC), 1% vitamin supplement (ATCC), 1 g/L D-(+)-cellobiose (Sigma-Aldrich), 1 g/L D-(+)-maltose (Sigma-Aldrich), 1 g/L D-(+)-fructose (Sigma-Aldrich) and 0.5 g/L L-cysteine (Sigma-Aldrich). Growth occurred under anaerobic conditions (atmosphere 5% H2, 20% CO2, 75% N2) in a soft-sided vinyl chamber (Coy Laboratory Products, Michigan, USA). We sterilized the media using a Corning filter unit (0.22 μm pore diameter). All metabolite standards (Sigma-Aldrich) were brought to 100 mM in DMSO (Sigma-Aldrich, D2438) prior to dilution for dose assays. Overnight bacterial cultures were diluted 100-fold in appropriate media and 40 μL were dispensed per well in 384-well plates (low evaporation lid, Costar 3680) containing metabolites or DMSO control. The plates were shaken to ensure homogeneity and bacterial growth was monitored anaerobically (absorbance at 600 nm) in a microplate reader (PowerWave HT Microplate Spectrophotometer, BioTek) for 24 hours at 37°C without shaking. Values recorded for DMSO controls and metabolite-treated triplicates were averaged.
Supplementary Material
Acknowledgements
The authors are grateful to the members of the PRISM, LLDeep, and NLIBD cohorts for participating in the study and providing sample material. We thank Tiffany Poon for project management and coordination of data generation, Theresa Reimels for editorial assistance and Ashley Garner for providing helpful feedback on the manuscript. The Dutch research team was funded by: IN-CONTROL CVON (CVON2012–03 to AZ and JF); the MLDS Dutch Digestive Foundation (D16–14 to RKW and AZ); the Netherlands Organization for Scientific Research (NWO-VIDI 864.13.013 to JF, NWO-VIDI 016.Vidi.178.056 to AZ, NWOOW-VIDI 016.136.308 to RKW); a Spinoza Prize (SPI 92–266 to CW); and the European Research Council (ERC-Starting #715772 to AZ and ERC-Advanced 2012–322698 to CW). The Boston Research team was funded by: the National Science Foundation (NSF CAREER DBI-1053486 and NSF EAGER MCB-1453942 to CH); The National Institutes of Health (R01HG00596 to CH, U54DK102557 to CH and RJX, R01DK92405 to RJX, R24DK110499 to CH), the Crohn’s and Colitis Foundation of America to RJX and CH, and the Center for Microbiome Informatics and Therapeutics (6933665 PO # 5710004058 to RJX). ABH is a Merck Fellow of the Helen Hay Whitney Foundation.
Footnotes
Competing interests
FI received a speaker’s fee from AbbVie.
Data availability
Metagenomic sequences for the PRISM, LLDeep, and NLIBD cohorts are available via SRA with BioProject number PRJNA400072. Metabolomics data (accession number PR000677) are available at the NIH Common Fund’s Metabolomics Data Repository and Coordinating Center (supported by NIH grant, U01-DK097430): Metabolomics Workbench (http://www.metabolomicsworkbench.org). Tables of processed metabolite, microbial species, and microbial enzyme abundance are available as Supplementary Datasets 2, 4, and 6.
Code availability
With the exception of Genedata Expressionist, the software packages used in this work are free and open source. The bioBakery methods (including kneadData, MetaPhlAn2, and HUMAnN2) are available via http://huttenhower.sph.harvard.edu/biobakery as source code and installable packages. The Python packages scipy, matplotlib (used for all data visualizations), statsmodels, and scikit-learn are available via http://pypi.python.org. The R package vegan is available via http://cran.r-project.org. Analysis scripts employing these packages (and associated usage notes) are available from the authors upon request.
Competing interests
References
- 1.Wlodarska M, Kostic AD & Xavier RJ An integrative view of microbiome-host interactions in inflammatory bowel diseases. Cell Host Microbe 17, 577–591 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imhann F et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huttenhower C, Kostic AD & Xavier RJ Inflammatory bowel disease as a model for translating the microbiome. Immunity 40, 843–854 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morgan XC et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13, R79 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gevers D et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haberman Y et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Invest. 124, 3617–3633 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lane ER, Zisman TL & Suskind DL The microbiota in inflammatory bowel disease: current and therapeutic insights. J. Inflamm. Res. 10, 63–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blander JM, Longman RS, Iliev ID, Sonnenberg GF & Artis D Regulation of inflammation by microbiota interactions with the host. Nat. Immunol. 18, 851–860 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorrestein PC, Mazmanian SK & Knight R Finding the missing links among metabolites, microbes, and the host. Immunity 40, 824–832 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McHardy IH et al. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome 1, 17 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu GD Diet, the gut microbiome and the metabolome in IBD. Nestle Nutr. Inst. Workshop Ser. 79, 73–82 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Kim S, Kim J-H, Park BO & Kwak YS Perspectives on the therapeutic potential of short-chain fatty acid receptors. BMB Rep. 47, 173–178 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith PM et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernando MR, Saxena A, Reyes J-L & McKay DM Butyrate enhances antibacterial effects while suppressing other features of alternative activation in IL-4-induced macrophages. Am. J. Physiol. Gastrointest. Liver Physiol. 310, G822–831 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Marchesi JR et al. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J. Proteome Res. 6, 546–551 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Wikoff WR et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. U. S. A. 106, 3698–3703 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams BB et al. Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe 16, 495–503 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zelante T et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39, 372–385 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Lamas B et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 22, 598–605 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Gall G et al. Metabolomics of fecal extracts detects altered metabolic activity of gut microbiota in ulcerative colitis and irritable bowel syndrome. J. Proteome Res. 10, 4208–4218 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Bjerrum JT et al. Metabonomics of human fecal extracts characterize ulcerative colitis, Crohn’s disease and healthy individuals. Metabolomics 11, 122–133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Preter V et al. Faecal metabolite profiling identifies medium-chain fatty acids as discriminating compounds in IBD. Gut 64, 447–458 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Jansson J et al. Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS One 4, e6386 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kolho K-L, Pessia A, Jaakkola T, de Vos WM & Velagapudi V Faecal and Serum Metabolomics in Paediatric Inflammatory Bowel Disease. J. Crohns. Colitis 11, 321–334 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Jacobs JP et al. A Disease-Associated Microbial and Metabolomics State in Relatives of Pediatric Inflammatory Bowel Disease Patients. Cell Mol Gastroenterol Hepatol 2, 750–766 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melnik AV et al. Coupling Targeted and Untargeted Mass Spectrometry for Metabolome-Microbiome-Wide Association Studies of Human Fecal Samples. Anal. Chem. 89, 7549–7559 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Sokol H & Seksik P The intestinal microbiota in inflammatory bowel diseases: time to connect with the host. Curr. Opin. Gastroenterol. 26, 327–331 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Joossens M et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 60, 631–637 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Sokol H et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis. 15, 1183–1189 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Wishart DS et al. HMDB: the Human Metabolome Database. Nucleic Acids Res. 35, D521–526 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mosli MH et al. C-Reactive Protein, Fecal Calprotectin, and Stool Lactoferrin for Detection of Endoscopic Activity in Symptomatic Inflammatory Bowel Disease Patients: A Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 110, 802–819; quiz 820 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Benjamini Y & Hochberg Y Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol, 289–300 (1995). [Google Scholar]
- 33.Duboc H et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 62, 531–539 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Abdel Hadi L, Di Vito C & Riboni L Fostering Inflammatory Bowel Disease: Sphingolipid Strategies to Join Forces. Mediators Inflamm. 2016, 3827684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.An D et al. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell 156, 123–133 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Braun A et al. Alterations of phospholipid concentration and species composition of the intestinal mucus barrier in ulcerative colitis: a clue to pathogenesis. Inflamm. Bowel Dis. 15, 1705–1720 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Qi Y et al. PPARα-dependent exacerbation of experimental colitis by the hypolipidemic drug fenofibrate. Am. J. Physiol. Gastrointest. Liver Physiol. 307, G564–573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fischbeck A et al. Sphingomyelin induces cathepsin D-mediated apoptosis in intestinal epithelial cells and increases inflammation in DSS colitis. Gut 60, 55–65 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Heimerl S et al. Alterations in intestinal fatty acid metabolism in inflammatory bowel disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1762, 341–350 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Hove H & Mortensen PB Influence of intestinal inflammation (IBD) and small and large bowel length on fecal short-chain fatty acids and lactate. Dig. Dis. Sci. 40, 1372–1380 (1995). [DOI] [PubMed] [Google Scholar]
- 41.Stuart JM, Segal E, Koller D & Kim SK A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Wolfe CJ, Kohane IS & Butte AJ Systematic survey reveals general applicability of” guilt-by-association” within gene coexpression networks. BMC Bioinformatics 6, 227 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frank DN et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. U. S. A. 104, 13780–13785 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lewis JD et al. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe 18, 489–500 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Desbois AP & Smith VJ Antibacterial free fatty acids: activities, mechanisms of action and biotechnological potential. Appl. Microbiol. Biotechnol. 85, 1629–1642 (2010). [DOI] [PubMed] [Google Scholar]
- 46.German JB & Dillard CJ Saturated fats: a perspective from lactation and milk composition. Lipids 45, 915–923 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galland L Magnesium and inflammatory bowel disease. Magnesium 7, 78–83 (1988). [PubMed] [Google Scholar]
- 48.Lih-Brody L et al. Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig. Dis. Sci. 41, 2078–2086 (1996). [DOI] [PubMed] [Google Scholar]
- 49.Yang JY et al. Molecular networking as a dereplication strategy. J Nat Prod 76, 1686–1699 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jaskowski TD, Litwin CM & Hill HR Analysis of serum antibodies in patients suspected of having inflammatory bowel disease. Clin. Vaccine Immunol. 13, 655–660 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tigchelaar EF et al. Cohort profile: LifeLines DEEP, a prospective, general population cohort study in the northern Netherlands: study design and baseline characteristics. BMJ Open 5, e006772 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Segata N et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 9, 811–814 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Franzosa E et al. Functionally profiling metagenomes and metatranscriptomes at species-level resolution. Nature Methods (In Proof). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzek BE et al. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buchfink B, Xie C & Huson DH Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Apweiler R et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 32, D115–119 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benjamini Y & Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300 (1995). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.