

Abstract
Methylmercury (MeHg), an environmental neurotoxicant primarily found in fish, produces neurobehavioral impairment when exposure occurs during gestation. Whether other developmental periods, such as adolescence, display enhanced vulnerability to the behavioral effects of MeHg exposure is only beginning to be explored. Further, little is known about the effects of repeated administration of lysine deacetylase inhibitors, such as sodium butyrate (NaB), on operant behavior. In Experiment 1, male C57BL6/n mice were exposed to 0, 0.3, and 3.0 ppm MeHg (n = 12 each) via drinking water from postnatal days 21 to 60 (murine adolescence). As adults, mice were trained to lever press under an ascending series of fixed-ratio schedules of milk reinforcement selected to enable the analysis of three important parameters of operant behavior using the framework provided by Mathematical Principles of Reinforcement. Adolescent MeHg exposure dose-dependently increased saturation rate, a measure of the retroactive reach of a reinforcer, and decreased minimum response time relative to controls. In Experiment 2, the behavioral effects of repeated NaB administration both alone and following adolescent MeHg exposure were examined. Male C57BL6/n mice were given either 0 or 3.0 ppm MeHg during adolescence and, before behavioral testing, two weeks of once daily i.p. injections of saline or 0.6 g/kg NaB (n = 12 in each cell). Adolescent MeHg exposure again increased saturation rate but did not significantly alter minimum response time. NaB also increased saturation rate in both MeHg exposure groups. These data suggest that the behavioral mechanisms of adolescent MeHg exposure and NaB may be related to the impact of reinforcement on prior responses. Specifically, MeHg and NaB concentrated the effects of reinforcers onto the most recent responses.
Keywords: adolescence, fixed ratio, lysine deacetylase inhibitor, Mathematical Principles of Reinforcement, methylmercury, sodium butyrate
Adolescence is a time of continued neurobehavioral development in both humans and nonhumans. The density of gray matter decreases in the human frontal cortex (Giedd et al., 1999), and dopamine-receptor pruning occurs in the rodent midbrain (Teicher et al., 1995). In rodents, adolescence ranges between postnatal days 21 and 60 (Spear, 2000) and marks a period of dysregulated choice and sensitivity to reinforcers. Adolescent rodents perform poorly on measures of executive function relative to adult conspecifics in that they prefer small-immediate reinforcers over larger-delayed ones (Doremus-Fitzwater et al., 2012; Pinkston and Lamb, 2011) and demonstrate impaired reversal learning and extradimensional shifting (Newman and McGaughy, 2011). Similar patterns of behavior related to executive-function dysregulation occur in human adolescents (Chambers and Potenza, 2003; Olson et al., 2007). The neurobehavioral profile of adolescence may escalate vulnerability to neurotoxicants and other chemicals that alter brain function. Thus, understanding how neurotoxic perturbations during adolescence affect behavior in adulthood is important to public health.
Methylmercury (MeHg) is an environmental neurotoxicant found in fish and is a significant public-health concern (National Research Council, 2000). In animal models, gestational exposure to doses of MeHg that encompass human exposures impairs reversal learning and choice in adulthood (Newland et al., 2008, 2004; Reed et al., 2006), suggesting deficiencies in executive functioning (Newland et al., 2015). However, the long-term behavioral effects of MeHg exposure in adolescence remain under explored. This is especially troubling since human adolescents both consume fish known to contain mercury and have higher blood-mercury concentrations compared to younger age groups (Nielsen et al., 2015; Sichert-Hellert et al., 2009). In mice, MeHg exposure during adolescence increases the number of trials required to transition through a spatial-discrimination reversal and extradimensional shift in adulthood (Boomhower and Newland, 2017). Adolescent MeHg exposure also dose-dependently reduces choice for a larger reinforcer relative to a smaller reinforcer in adult mice (Boomhower and Newland, 2016). These experiments suggest that MeHg exposure during adolescence can produce long-lasting behavioral effects related to choice in adulthood, but the behavioral mechanisms underlying these effects remain unclear. For example, adolescent MeHg exposure may alter reinforcer efficacy as with gestational exposures (Newland et al., 2015), or it may change some other aspect of reinforcement or behavior.
Mathematical Principles of Reinforcement (MPR) offers one way to characterize the behavioral mechanisms underlying chemical exposure (Killeen, 1994; Killeen and Sitomer, 2003). MPR is a theory-driven model of operant (voluntary) behavior and posits that three fundamental behavioral mechanisms are involved in reinforcement learning. Specifically, response rates under fixed-ratio (FR) schedules of reinforcement are a function of (a) minimum response time, which is a measure of the time (in seconds) required to complete a target response (e.g., a lever press), (b) specific activation, which is a measure of reinforcer value (in responses), and (c) saturation rate, which is a measure of how quickly response rates are maximized as a function of the number of target responses that precede reinforcer delivery. A high saturation rate indicates that relatively few target responses are strengthened by a reinforcer, whereas a low saturation rate indicates that relatively many target responses are strengthened by a reinforcer. Thus, saturation rate provides information about the effects of a reinforcer on target responding, and whether these effects are concentrated (high saturation rate) or dispersed (low saturation rate). MPR predicts response rate (b, responses/sec) as a function of fixed-ratio size (n) using Eq. 1 (Killeen and Sitomer, 2003):
| (1) |
where a is specific activation, δ is minimum response time, and λ is saturation rate. Figure 1 illustrates how changes in MPR’s parameters affect the shape of the function drawn by Eq. 1. MeHg-induced changes in MPR’s parameters would reveal information about the behavioral mechanisms targeted by adolescent MeHg exposure. Whether the lasting behavioral effects following adolescent MeHg exposure can be reversed or mitigated via drug therapy is unclear, however.
Figure 1.
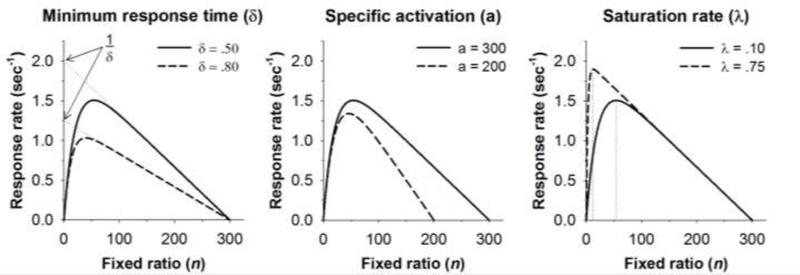
The effects of changes in minimum response time (δ), specific activation (a), and saturation rate (λ) on the shape of the MPR function drawn by Eq. 1. The base function (solid line) in each panel is drawn with δ = .50, a = 300, and λ = .10. An increase in minimum response time to δ = .80 (left panel) is evident in a reduction in the maximum response rate (1/δ), which is the extrapolated Y-intercept of the descending limb of the function (dotted lines, left panel). A reduction in specific activation to a = 200 (middle panel) is reflected as a decrease in the X-intercept of the descending limb of the function. Finally, an increase in saturation rate to λ = .75 shifts the peak of the function to the left. The vertical dotted lines (right panel) indicate that an FR 54 maximizes response rates when λ = .10 whereas an FR 12 maximizes response rates when λ = .75.
A growing literature suggests lysine deacetylase (LDAC) inhibitors ameliorate cognitive impairment in rodents (Day and Sweatt, 2012) as a result of normal or diseased aging (Fischer et al., 2007; Kilgore et al., 2010; Rane et al., 2012). LDAC inhibitors, such as sodium butyrate (NaB), target both histone deacetylases and DNA methyltransferases in the cell nucleus to increase histone acetylation and neuronal gene expression (Sarkar et al., 2011; Xu et al., 2007). Histone acetylation plays a crucial role in neuronal development, synaptic plasticity, learning, and memory (Day and Sweatt, 2012; Levenson et al., 2004; Miller et al., 2008), but the effects of repeated LDAC inhibition on operant behavior are underexplored. Some reports show that acute administration of NaB and other LDAC inhibitors do not affect responding under fixed- and progressive-ratio schedules of reinforcement (Romieu et al., 2008; Sun et al., 2008), but it is unclear whether NaB alters other aspects of reinforcement. It is important to characterize the effects of NaB on operant behavior both alone and after neurotoxicant exposure due to the growing use of NaB and other LDAC inhibitors for treating cognitive and behavioral dysfunction (Haggarty and Tsai, 2011). Whether repeated NaB administration mitigates the behavioral effects of adolescent MeHg exposure is unknown.
MeHg exposure has been linked to distorted gene expression and epigenetic dysregulation in some model systems, including cultured cells, nonhuman animals, and humans (Robinson et al., 2011). MeHg reduces Bdnf expression, a gene related to synaptic plasticity and learning, and increases Hdac4 expression in cultured rat cortical neurons (Guida et al., 2017, 2016). Further, the LDAC inhibitor trichostatin A prevents MeHg-induced changes in gene expression and histone deacetylation in vitro (Guida et al., 2016). In behaving animals, gestational MeHg exposure is correlated with reduced Bdnf expression in the adult rat hippocampus, with a concurrent decrease in histone H3 acetylation and increase in DNA methylation at the Bdnf promoter (Onishchenko, Karpova, Sabri, Castrén, & Ceccatelli, 2008). Thus, developmental MeHg exposure may produce its neurobehavioral effects in part by increasing neuroepigenetic marks that suppress gene expression. Treatment with a broad-acting LDAC inhibitor, such as NaB, following MeHg exposure may prevent behavioral impairment.
Experiment 1
Experiment 1 was designed to parse the behavioral mechanisms of adolescent MeHg exposure using Mathematical Principles of Reinforcement (Eq. 1; Killeen & Sitomer, 2003). The effects of adolescent MeHg exposure on MPR’s parameters—specific activation (a), minimum response time (δ), and saturation rate (λ)—were assessed using response rates derived from a multiple fixed-ratio procedure.
Method
Subjects and MeHg exposure
Thirty-six male C57BL/6n mice were purchased from a commercial vendor (Envigo, Indianapolis, IN). The mice were derived from 12 litters (3 littermates/litter). These mice have previously been reported on (Boomhower & Newland, 2016). Briefly, all mice were pair-housed and maintained on a 12-hr light/dark cycle (lights on at 6:00 AM) in a temperature- and humidity-controlled, AAALAC-accredited animal facility. Upon arrival, 21-day-old littermates were divided among three MeHg-exposure groups (via drinking water): 0 ppm (control), 0.3 ppm, and 3.0 ppm MeHg. Thus, a litter was equally represented across exposure groups. MeHg was delivered as methylmercuric chloride (MeHgCl2) dissolved in drinking water, and exposure occurred from postnatal day (PND) 21 through 59. All water was replaced with tap water on PND 60. Dosing was calculated by weighing water bottles, and sham water bottles were weighed to account for spillage. The low (0.3 ppm) and high (3.0 ppm) doses of MeHg corresponded to approximately 40 and 400 μg/kg/day of MeHg in mice, respectively (Boomhower and Newland, 2017, 2016). The dose range of MeHg used here has been associated with neurobehavioral impairment in past work (Boomhower and Newland, 2017, 2016). Mice were maintained at 25 (± 1) g body mass by restricting daily food intake to 2.4 (± .02) g chow. The Auburn University Institutional Animal Care and Use Committee approved all procedures.
Apparatus
Twelve standard operant chambers (Med Associates, St. Albans, VT) modified for mice were used for data collection. Each chamber was equipped with two retractable levers on a front wall panel. Situated between the two levers was an alcove where a dipper system delivered .01-cc presentations of a 3:1 water and sweetened-condensed milk solution (hereafter, milk). Two Sonalert® tone generators (high tone: 4500 Hz, low tone: 2700 Hz) were located at the top of the front wall. A sound-attenuating cubicle enclosed each chamber. All experimental contingencies were controlled within 0.01-sec resolution by a computer in an adjacent room. Each mouse was assigned a particular chamber for the duration of the study. Mice were divided into four squads that ran at approximately the same time every day (±15 min) Monday through Friday. The number of mice in each exposure group was counterbalanced across chambers and squads.
Procedure
All mice had prior experience lever pressing as described in Boomhower and Newland (2016). Beginning at 200 days of age, mice were trained under a multiple fixed-ratio (FR) schedule of reinforcement based on Reilly (2003) with some modifications. A session was divided into six components, each associated with a different FR schedule and signaled by a unique, high/low-alternating tone (Table 1). The FR schedule was increased across components in the following order: FR 1, 5, 15, 30, 60, and 120. At the beginning of a session, one lever was inserted into the chamber and the tones sounded. Upon completion of the FR requirement, the lever retracted, the tones were extinguished, and 3 sec of access to milk was made available. The lever was then re-inserted into the chamber. A component ended after the delivery of 12 reinforcers or 10 min, whichever came first. Components were separated by 30 sec during which the lever was retracted and tones were extinguished. The multiple FR schedule was in effect for 40 sessions to allow responding to stabilize.
Table 1.
Tone durations for the FR procedure.
| FR | Low/high tone durations (sec) |
|---|---|
| 1 | 0.15/1.19 |
| 5 | 0.74/0.60 |
| 15 | 0.92/0.42 |
| 30 | 1.04/0.30 |
| 60 | 1.13/0.21 |
| 120 | 1.19/0.15 |
FR = fixed-ratio schedule of reinforcement
Data analysis
Responding under the multiple FR schedule among exposure groups was assessed by averaging response rates, calculated as the number of responses divided by the duration of the component (in seconds), for each subject from the last 10 sessions. Eq. 1 was fit to individual-subject response-rate functions using nonlinear least-squares regression. Response rates were compared among groups using a repeated-measures ANOVA with exposure and FR as the within-subjects variables. Estimates of a, δ, and λ were compared across exposure groups using a repeated-measures ANOVA. Planned Tukey post-hoc comparisons were conducted between MeHg-exposed animals and controls.
Results and Discussion
Figure 3 shows mean response rates as a function of FR for the MeHg exposure groups along with best fits of Eq. 1 (solid lines). Individual-subject data for two mice from each exposure group also are shown to demonstrate the range of goodness of fit of Eq. 1: one was the best fit and one was the worst fit for each exposure group. One 3.0-ppm MeHg exposed mouse, was a statistical outlier due to his low rate of responding (Fig. 1, lower right panel) so his data were not included in the mean for the 3.0-ppm group. There were main effects of both FR [F(5, 197) = 37.95, p < .001] and exposure [F(2, 197) = 7.53, p < .001] in that response rates were a bitonic function of FR and MeHg-exposed mice responded more quickly on average. Eq. 1 provided a good fit of all individual-subject data with the mean (SEM) R2 values being 0.93 (0.02), 0.96 (0.01), and 0.94 (0.03) for the 0-, 0.3-, and 3.0-ppm MeHg groups, respectively. The quality of the fits was not significantly different among exposure groups.
Figure 3.
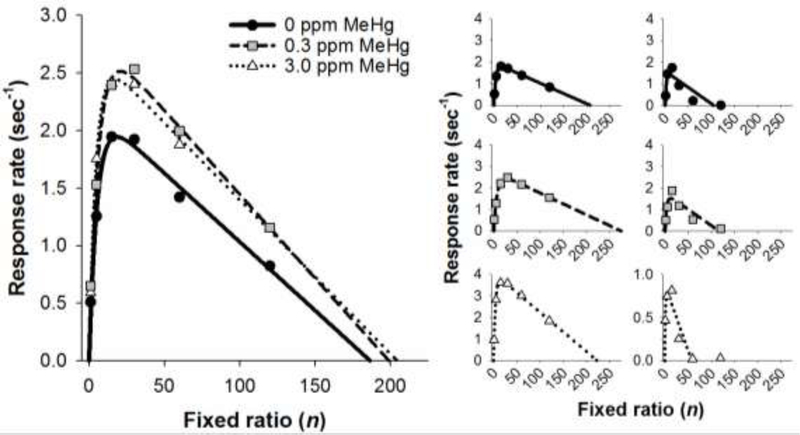
Left panel: Mean response rate as a function of fixed ratio for mice exposed to MeHg in adolescence. Lines represent mean predictions of Eq. 1. Right panel: Response rate (symbols) and predictions of Eq. 1 (lines) as a function of fixed ratio for individual mice. The left column shows data from mice with the best-fitting curves, and the right column shows data from mice with the worst-fitting curves. Note the Y-axis scaling in the lower-right panel. MeHg = methylmercury
Figure 4 shows mean parameter estimates of λ, δ, and a for the MeHg exposure groups. The outlier is denoted as an open circle. Estimates of λ were significantly different among the exposure groups [F(2, 33) = 4.16, p = .02], and post-hoc comparisons revealed that mice in the 3.0-ppm group had higher saturation rates compared to Controls (p < .05). Thus, fewer target responses maximized response rates in MeHg-exposed mice than Controls. Estimates of δ were significantly different among exposure groups [F(2, 33) = 9.15, p < .001]. Post-hoc comparisons revealed that the 0.3- and 3ppm groups had faster minimum response times than Controls (p <.02). This was evident in higher maximum response rates (1/δ) in exposed mice. It should be noted that the inclusion of the statistical outlier in the 3-ppm group rendered δ estimates for this group statistically similar to Controls’ δ estimates. Estimates of a were not significantly different among exposure groups [F(2, 33) = 0.89, p = .42], indicating that the value of a reinforcer was similar following adolescent MeHg exposure.
Figure 4.
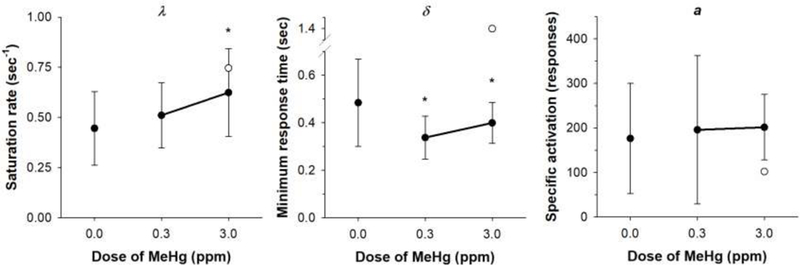
Mean (±SD) parameter estimates from Eq. 1 as a function of dose of MeHg in adolescence. An outlier in the 3.0 ppm group is denoted as an open circle. MeHg = methylmercury *p < .05 relative to control
As revealed by MPR (Killeen and Sitomer, 2003), adolescent exposure to MeHg manifested as alterations in saturation rate and minimum response time in adulthood. This suggests for the MeHg-exposed mice, the reinforcer’s effects are more concentrated on the responses that immediately preceded reinforcement than on the earlier responses. In other words, adolescent MeHg exposure reduced the number of responses being strengthened by reinforcement. Further, an enhancement in psychomotor responding following MeHg exposure is consistent with past work showing gestationally-exposed rats respond more quickly on fixed-(Paletz et al., 2006) and progressive-ratio schedules of reinforcement (Reed et al., 2008). These findings suggest that adolescent MeHg exposure may produce long-lasting distortions in both the ability of a reinforcer to strengthen past responses and psychomotor responding. Whether MeHg’s behavioral effects can be mitigated by repeated administration of sodium butyrate (NaB), a lysine deacetylase (LDAC) inhibitor, is unknown.
Experiment 2
Developmental MeHg exposure may produce its neurobehavioral effects in part by increasing neuroepigenetic marks that suppress gene expression (Guida et al., 2017; Onishchenko et al., 2008). Treatment with a broad-acting LDAC inhibitor, such as NaB, following MeHg exposure may prevent behavioral impairment. Due to the growing use of NaB and other LDAC inhibitors for treating cognitive and behavioral dysfunction (Haggarty and Tsai, 2011), it is also important to characterize the effects of NaB alone on operant behavior. Acute administration of NaB and other LDAC inhibitors does not affect responding under fixed- and progressive-ratio schedules of reinforcement in rodents (Romieu et al., 2008; Sun et al., 2008), but it is unclear whether NaB alters other aspects of reinforcement. The use of Mathematical Principles of Reinforcement (MPR) would parse the effects of NaB on three unique processes of operant behavior: saturation rate, minimum response time, and reinforcer value. Thus, Experiment 2 was designed to (a) replicate the effects of adolescent MeHg exposure on MPR’s parameters in a different cohort of mice, and (b) determine the effects of repeated NaB administration, both alone and after MeHg exposure, on MPR’s parameters. We chose to examine the highest dose (3 ppm) of MeHg because it produced significant changes in both saturation rate and minimum response time (Experiment 1) as well as behavioral impairment in other work (Boomhower and Newland, 2017) to determine the degree to which NaB prevented MeHg’s effects.
Method
Subjects and MeHg exposure
Forty-eight male C57Bl/6n mice derived from 24 litters (2 littermates/litter) were purchased from a commercial vendor (Envigo, Indianapolis, IN). Mice arrived at our facility on PND 23, and 12 litters (24 mice) were exposed to 3 ppm MeHg beginning on PND 24. MeHg was delivered as methylmercuric chloride dissolved in drinking water. The other 12 litters (24 mice) received control (tap) water. Dosing was calculated by weighing water bottles, and sham water bottles were used to control for spillage. On PND 63, MeHg bottles were removed and replaced with tap water. Thus, mice in Experiment 2 were exposed to MeHg for an identical number of days (39 days) as mice in Experiment 1 except MeHg exposure spanned PND 24–62 in Experiment 2 (a 3-day displacement). All mice were given ad libitum access to food until just prior to behavioral testing, which began on PND 100. Two weeks prior to behavioral testing, mice were maintained at a body mass of 25 (± 1) g by food restricting daily chow intake to 2.4 (± 0.2) g. All mice were individually-housed and maintained under a 12-hr light/dark cycle (lights on at 6:00 AM) in a temperature- and humidity-controlled, AAALAC-approved animal facility. The Auburn University Institutional Animal Care and Use Committee approved all procedures.
Sodium butyrate (NaB) administration
Two weeks before behavioral testing, half the mice in each MeHg-exposure group received daily acute i.p. injections of 0.9% saline or 0.6 g/kg NaB under a 2 (MeHg) X 2 (NaB) full-factorial design. Litters (2 littermates/litter) within a MeHg exposure group were divided equally between the saline and NaB conditions. Thus, a litter was equally represented across NaB conditions. Drug administration spanned PND 86–99, a regimen similar to past work (Kim et al., 2009; Rane et al., 2012). Sodium butyrate (Sigma) was dissolved in saline. Saline and NaB were injected at a .01 mL/g volume.
Apparatus
The same apparatuses were used as in Experiment 1.
Procedure
All mice had previous experience lever pressing under a spatial-discrimination-reversal and visual-discrimination procedure, similar to Boomhower and Newland (2017). Beginning at 200 days of age, mice were trained under the multiple fixed-ratio procedure described in Experiment 1. The number of mice from each group was counterbalanced across chambers and session time. One 0-ppm MeHg mouse (Control) and three 3-ppm MeHg mice (Control, n = 1; NaB, n = 2) were euthanized before behavioral testing for reasons unrelated to the present experiments.
Data analysis
Similar to Experiment 1, Eq. 1 was fit to individual-subject response-rate functions using nonlinear least-squares regression. Response rates were analyzed using a linear-mixed effects (LME) model with MeHg, NaB, and FR as fixed effects and litter as a random effect. LME was chosen because it is able to model incomplete repeated-measures data more effectively than does traditional repeated-measures analysis of variance. Estimates of a, δ, and λ were compared across groups using a LME model with MeHg and NaB as fixed effects and litter as a random effect. Planned Tukey post-hoc comparisons were conducted to assess significant differences between groups.
Results and Discussion
Figure 5 shows MeHg consumption across adolescence for each MeHg exposure group. The dose of MeHg was highest at the beginning of exposure and gradually decreased to about 400 μg/kg/day. This pattern of dosing was similar to that seen in Experiment 1 (see Boomhower & Newland, 2016) and Boomhower and Newland (2017) and is due to changes in fluid intake that occurs through early adolescence. Administration of NaB did not significantly alter MeHg consumption.
Figure 5.
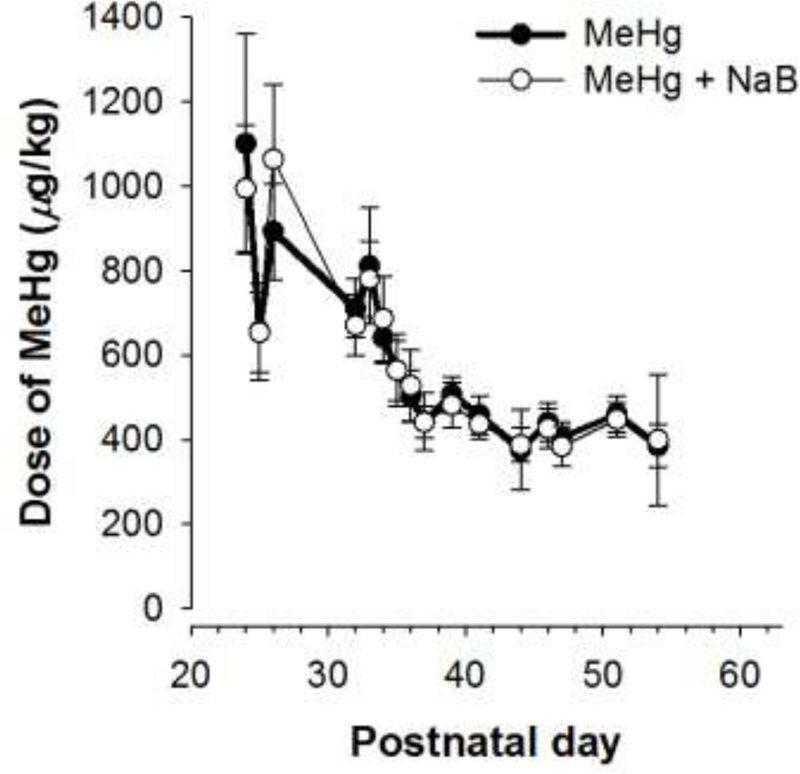
Mean (±SD) dose of MeHg as a function of postnatal day for each MeHg-exposure group in Experiment 2. Note the error bars may be obscured by the symbols in some cases. MeHg = methylmercury, NaB = sodium butyrate
Figure 6 shows mean response rates and best fits of Eq. 1 for each treatment group. There was a main effect of FR [F(5, 239) = 37.37, p < .001], which was reflected in a bitonic relation between response rates and FR size. There were no main effects of MeHg or NaB, and there were no significant interactions. Eq. 1 fit individual response-rate functions well with a mean pseudo R2 = 0.94 (SD: .06) across all groups.
Figure 6.
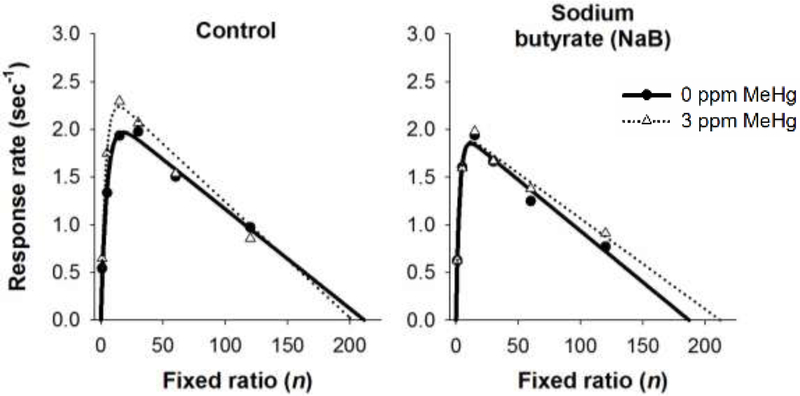
Mean response rate as a function of fixed ratio for mice exposed to 0 or 3 ppm MeHg in adolescence and treated with control (left panel), sodium butyrate (middle panel), or environmental enrichment (right panel) before behavioral testing. Lines represent mean predictions of Eq. 1. MeHg = methylmercury
Figure 7 shows mean parameter estimates from Eq. 1 for each exposure and treatment group. For saturation rate (λ), there was a main effect of NaB [F(1, 39) = 4.91, p = .03] with post-hoc comparisons revealing a significant increase in λ following MeHg alone, NaB alone, and the combination relative to the 0-ppm Control group (p’s < .05). Neither minimum response time (δ) nor specific activation (a) estimates were significantly altered relative to 0-ppm Control.
Figure 7.
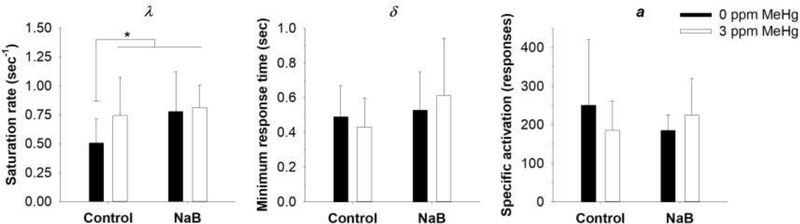
Mean (+SD) parameter estimates from Eq. 1 for each treatment group. MeHg = methylmercury, NaB = sodium butyrate. *p < .05
The finding that adolescent MeHg exposure increases saturation rate, or the degree to which prior responses are coupled to reinforcers, was replicated in Experiment 2. Further, MeHg-exposed mice tended to have higher maximum response rates (smaller δ’s) than unexposed mice similar to Experiment 2, but this was more modest than in Experiment 1 and was not reflected in a statistically significant decrease in minimum response time or maximum response rate. Differences in the exposure regimen might explain why the effects on minimum response time were not replicated in Experiment 2. Though mice in Experiment 2 were exposed to MeHg for 39 days, identical to Experiment 1, the exposure duration began 3 days later in development. Some work suggests the effects of MeHg on the postnatal rat brain, particularly dopamine signaling, are mitigated after 7 days of continued striatal development (from PND 14 to 21) (Dreiem et al., 2009). Of all three parameter estimates from MPR, the ability of reinforcers to strengthen past responding (λ) appears to be the one most consistently sensitive to adolescent MeHg exposure. The MeHg-induced increase in λ estimates persisted following treatment with NaB, suggesting no therapeutic benefits of NaB under these levels and durations. NaB treatment alone, however, produced a significant increase in saturation rate relative to 0-ppm Controls, an effect that was similar in magnitude to MeHg’s. These findings suggests repeated inhibition of lysine deacetylases prior to behavioral testing changes the extent to which reinforcers strengthen past responding.
General Discussion and Conclusions
The effects of adolescent methylmercury (MeHg) exposure on reinforcer value, motoric capacity, and the coupling of responses to reinforcement in mice were evaluated using Mathematical Principles of Reinforcement (Killeen and Sitomer, 2003) in two separate experiments. Further, the effects of sodium butyrate (NaB) both alone and after MeHg exposure were examined due to the growing use of NaB and other LDAC inhibitors for treating cognitive and behavioral dysfunction (Haggarty and Tsai, 2011). Overall, MPR described response-rate functions collected from individual mice quite well and captured alterations in responding following the various exposures and treatments.
In both Experiment 1 and 2, adolescent MeHg exposure increased saturation rate (λ), the rate at which response-reinforcer coupling decreased across time. Stated differently, adolescent MeHg exposure decreased the number of responses that were strengthened by reinforcers. Reduced coupling of all responses except the one that immediately preceded reinforcement following adolescent MeHg exposure could be related to past work reporting MeHg-induced impairments in choice. Adolescent MeHg dose-dependently reduced sensitivity to a larger reinforcer compared to a smaller reinforcer in these mice (Boomhower and Newland, 2016), and 3 ppm of MeHg increased the number of trials required to transition through repeated spatial-discrimination reversals and an extradimensional shift (Boomhower and Newland, 2017). These findings suggest a degradation in the ability of a reinforcer to strengthen prior responses and is consistent with past work showing that saturation rate (λ) is correlated with impaired performance on spatial discrimination reversals in mice (Pope et al., 2016).
The finding that adolescent MeHg exposure diminishes the impact of reinforcement on previous responses implicates neural substrates linked to motivation in adolescent MeHg toxicity. The nucleus accumbens, its projections and interaction with the prefrontal cortex, and dopamine signaling are important for reward processing (Everitt and Robbins, 2005; Fiorillo et al., 2003). Lesions of the rat nucleus accumbens core increase a parameter similar to saturation rate (λ) (Bezzina et al., 2008), whereas rat orbital prefrontal-cortex lesions are associated with reductions in specific activation (a) (Kheramin et al., 2005). Changes in the parameters of Mathematical Principles of Reinforcement are linked intimately with dopaminergic function, as both neuroleptics and amphetamines can alter saturation rate, minimum response time, and specific activation (Mobini et al., 2000; Reilly, 2003). Our data suggest adolescent MeHg exposure interferes with the coupling of behavior to reinforcement, which is linked to nucleus-accumbens and prefrontal-cortex function. The finding that the effects of reinforcement on behavior are impaired following adolescent MeHg exposure is consistent with Newland et al.’s (2015) hypothesis that developmental MeHg exposure impacts reinforcer processing, primarily through MeHg’s effects on dopamine. Though Newland et al.’s (2015) review relied on models of gestational MeHg exposure, the present study suggests that adolescent MeHg exposure also contributes to alterations in brain-behavior function.
Minimum response times (δ) were decreased by adolescent MeHg exposure in Experiment 1, particularly under the 0.3-ppm dose, but were not significantly altered in Experiment 2. Reduced minimum response time manifests as increased maximum response rate, which is correlated with 1/δ both theoretically (Killeen and Sitomer, 2003) and experimentally (Hutsell and Newland, 2013). Past work in gestationally-exposed rats showed that MeHg increases response rates under fixed-ratio (Paletz et al., 2006) and progressive-ratio schedules of reinforcement (Reed et al., 2008). In Experiment 1, both doses of MeHg reduced minimum response time. The greatest decrease in minimum response time occurred after 0.3 ppm MeHg, and the 3.0-ppm dose of MeHg also produced a significant decrease in δ estimates after a statistical outlier was removed. It is unclear why the same dose of MeHg in Experiment 2 did not produce a significant reduction in minimum response time. We have noted previously dose-specific behavioral effects of adolescent MeHg exposure in the cohort of animals used in Experiment 1. Specifically, delay sensitivity was reduced in the 0.3-ppm group whereas mice in the 3-ppm group displayed similar estimates of delay sensitivity as controls (Boomhower and Newland, 2016). Key differences in the exposure regimen between Experiment 1 and 2 could explain also why our findings related to minimum response time were only partially replicated. We shifted the 39-day exposure window three days later in Experiment 2 because we had shown previously MeHg consumption is dramatically elevated during the first few days (PND 21–23) of exposure (Boomhower and Newland, 2016). We were concerned this could be an artifact of elevated water consumption during shipment from commercial vendors (see Obernier & Baldwin, 2006). Some work suggests the effects of MeHg on the postnatal rat brain, particularly related to striatal dopamine signaling and brain mercury uptake, are mitigated with development (Dreiem et al., 2009; Sakamoto et al., 2018). Specifically, striatal synaptosomes isolated from 7-day-old rats are more susceptible to MeHg’s effects on dopamine release and dopamine uptake than striatal synaptosomes from 21-day-old rats (Dreiem et al., 2009). Brain mercury uptake also decreases from the neonatal (PND 1–7) to the mid-adolescent period (PND 35–41) in rats (Sakamoto et al., 2018). The striatum and striatal dopamine have been implicated as targets of MeHg (Faro et al., 2002, 2000; Tiernan et al., 2015) and most likely underlie MeHg’s effects on operant behavior in part (Newland et al., 2015). Because both the sensitivity of striatal dopamine to MeHg exposure and MeHg uptake may decrease early in life (Dreiem et al., 2009; Sakamoto et al., 2018), this could explain why mice in Experiment 2 did not show impairment in minimum response time following MeHg exposure and would implicate the early-adolescent period in susceptibility to MeHg. Interestingly, we still observed elevated MeHg consumption early in the exposure regimen in Experiment 2 even after delaying exposure three days.
Repeated administration of sodium butyrate (NaB), a lysine deacetylase inhibitor, increased saturate rate in adulthood and NaB did not interact with adolescent MeHg exposure to change saturation-rate estimates. Past work has shown that MeHg reduces Bdnf expression in vitro (Guida et al., 2017) and in vivo (Onishchenko et al., 2008) with concomitant changes in histone acetylation, suggesting MeHg’s behavioral impairment may result in part from alterations to neuroepigenetic mechanisms. Guida et al. (2016) found that trichostatin A, an LDAC inhibitor, prevented MeHg-induced changes in gene expression and histone H4 deacetylation in vitro. However, we saw no therapeutic benefit of NaB following MeHg exposure and found that NaB alone increased saturation rate to a comparable level as MeHg exposure alone in behaving mice. Studies using NaB as a treatment for both normal (Peleg et al., 2010; Reolon et al., 2011) and diseased aging (Fischer et al., 2007; Kilgore et al., 2010; Rane et al., 2012) as well as following neurotoxicant exposure (Guida et al., 2016; Sharma and Sharma, 2013; Song et al., 2010) have noted a range of therapeutic effects. One explanation for our findings related to NaB is that LDAC inhibitors often target a broad set of proteins, sometimes related to cell death or oxidative stress (Xu et al., 2007). The broad enzymatic targets of NaB relative to trichostatin A may explain not only why NaB was ineffective at prevent MeHg behavioral toxicity, but also why NaB alone increased saturation rate in our study. NaB induces differentiation in neurons (Balasubramaniyan et al., 2006; Hsieh et al., 2004) and apoptosis in neuroblastoma cells (Nuydens et al., 1995; Rozental et al., 2004). Further characterization of the neuroprotective and neurotoxic effects of NaB, and other lysine deacetylase inhibitors, on operant behavior are important for future therapeutic treatments of chemical exposure.
Across two studies, exposure to environmentally-relevant levels of MeHg in adolescent mice reduced the degree to which responses were coupled to reinforcement in adulthood. We also found some evidence that adolescent MeHg exposure reduces minimum response time, particularly following exposure to 0.3 ppm MeHg, which is consistent with past work on developmental MeHg exposure’s psychomotor effects. Repeated administration of sodium butyrate alone increased saturation rate to a similar extent as MeHg exposure but did not reverse MeHg’s effects. The present study suggests adolescence is a time of both behavioral and neurobiological vulnerability to MeHg exposure. The finding that both adolescent MeHg exposure and sodium butyrate administration concentrates the effects of reinforcers on recently-made responses provide insight into the behavioral mechanisms that permit neurotoxicity.
Figure 2.
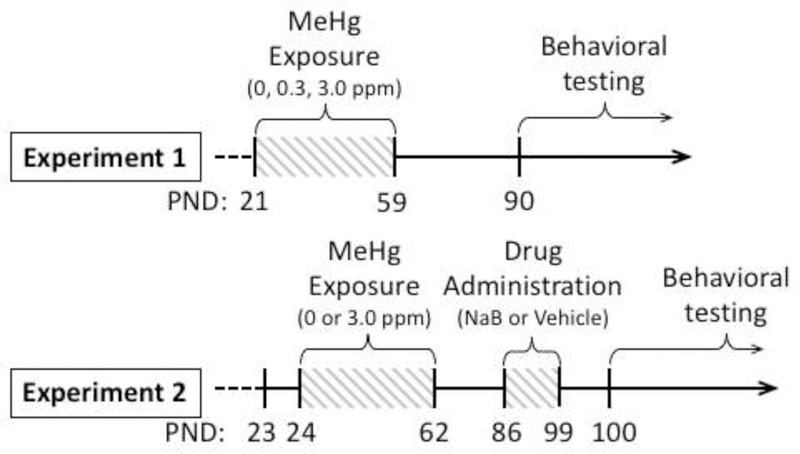
Timeline of events for Experiment 1 and Experiment 2. Mice arrived at PND 21 (Experiment 1) or PND 23 (Experiment 2) and were exposed to MeHg during adolescence for 39 days. In Experiment 2, sodium butyrate (NaB; 0.6 g/kg, i.p.) or saline vehicle were injected daily for two weeks prior to behavioral testing. Behavioral testing began in adulthood (PND 90 in Experiment 1, PND 100 in Experiment 2). MeHg = methylmercury, PND = postnatal day, NaB = sodium butyrate
Highlights.
Adolescent mice were exposed to 0, 0.3, and 3.0 ppm MeHg via drinking water.
NaB (0.6 g/kg) was administered for two weeks prior to behavioral testing.
MeHg concentrated the effects of reinforcers on responding in adulthood.
NaB concentrated the effects of reinforcers and did not prevent MeHg’s effects.
MeHg reduced minimum response time in one of two experiments.
Acknowledgements
We thank Megan Arnold, Douglas Glenn, Kevin Honeywell, Kate Johnson, Dalisa Kendricks, Sahar Moghadam, Madison Morlan, Alex Sauer, Savannah Simpson, and Kristyn Suelflow for help with data collection and animal care. Portions of this manuscript served as the first author’s dissertation and were presented as posters at Society of Toxicology.
Funding Information
This work was supported by graduate research grant awards from Psi Chi, Sigma Xi, the American Psychological Association, the American Psychological Foundation, Auburn University Graduate School, the National Science Foundation Graduate Research Fellowship Program [grant number DGE-1414475 to S.R.B.], and the National Institutes of Environmental Health Sciences [grant number ES024850 to M.C.N.].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
The authors declare no conflicts of interest.
References
- Balasubramaniyan V, Boddeke E, Bakels R, Kust B, Kooistra S, Veneman A, Copray S, 2006. Effects of histone deacetylation inhibition on neuronal differentiation of embryonic mouse neural stem cells. Neuroscience 143, 939–951. 10.1016/j.neuroscience.2006.08.082 [DOI] [PubMed] [Google Scholar]
- Bezzina G, Body S, Cheung THC, Hampson CL, Deakin JFW, Anderson IM, Szabadi E, Bradshaw CM, 2008. Effect of quinolinic acid-induced lesions of the nucleus accumbens core on performance on a progressive ratio schedule of reinforcement: Implications for inter-temporal choice. Psychopharmacology (Berl) 197, 339–350. 10.1007/s00213-007-1036-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boomhower SR, Newland MC, 2017. Effects of adolescent exposure to methylmercury and d-amphetamine on reversal learning and an extradimensional shift in male mice. Exp. Clin. Psychopharmacol 25, 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boomhower SR, Newland MC, 2016. Adolescent methylmercury exposure affects choice and delay discounting in mice. Neurotoxicology 57, 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers RA, Potenza MN, 2003. Neurodevelopment, impulsivity, and adolescent gambling. J. Gambl. Stud 19, 53–84. [DOI] [PubMed] [Google Scholar]
- Day JJ, Sweatt JD, 2012. Epigenetic treatments for cognitive impairments. Neuropsychopharmacology 37, 247–260. 10.1038/npp.2011.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doremus-Fitzwater TL, Barreto M, Spear LP, 2012. Age-related differences in impulsivity among adolescent and adult Sprague-Dawley rats. Behav. Neurosci 126, 735–741. 10.1037/a0029697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreiem A, Shan M, Okoniewski RJ, Sanchez-Morrissey S, Seegal RF, 2009. Methylmercury inhibits dopaminergic function in rat pup synaptosomes in an age-dependent manner. Neurotoxicol. Teratol 31, 312–317. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW, 2005. Neural systems of reinforcement for drug addiction: From actions to habits to compulsion. Nat. Neurosci 8, 1481–1489. 10.1038/nn1579 [DOI] [PubMed] [Google Scholar]
- Faro L, do Nascimento J, Alfonso M, Durán R, 2002. Mechanism of action of methylmercury on in vivo striatal dopamine release: Possible involvement of dopamine transporter. Neurochem. Int 40, 455–465. [DOI] [PubMed] [Google Scholar]
- Faro L, Do Nascimento JLM, San José JM, Alfonso M, Durán R, 2000. Intrastriatal administration of methylmercury increases in vivo dopamine release. Neurochem. Res 25, 225–229. [DOI] [PubMed] [Google Scholar]
- Fiorillo CD, Tobler PN, Schultz W, 2003. Discrete Coding of Reward Dopamine Neurons. Science (80) 299, 1898–1902. 10.1126/science.1077349 [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai L-H, 2007. Recovery of learning and memory is associated with chromatin remodelling. Nature 447, 178–182. 10.1038/nature05772 [DOI] [PubMed] [Google Scholar]
- Giedd JN, Blumenthal J, Jeffries NO, Castellanos FX, Liu H, Zijdenbos A, Paus T, Evans AC, Rapoport JL, 1999. Brain development during childhood and adolescence: a longitudinal MRI study. Nat. Neurosci 2, 861–3. [DOI] [PubMed] [Google Scholar]
- Guida N, Laudati G, Anzilotti S, Sirabella R, Cuomo O, Brancaccio P, Santopaolo M, Galgani M, Montuori P, Di Renzo G, Canzoniero LMT, Formisano L, 2016. Methylmercury upregulates RE-1 silencing transcription factor (REST) in SH-SY5Y cells and mouse cerebellum. Neurotoxicology 52, 89–97. 10.1016/j.neuro.2015.11.007 [DOI] [PubMed] [Google Scholar]
- Guida N, Laudati G, Mascolo L, Valsecchi V, Sirabella R, Selleri C, Di Renzo G, Canzoniero LMT, Formisano L, 2017. p38/Sp1/Sp4/HDAC4/BDNF Axis Is a Novel Molecular Pathway of the Neurotoxic Effect of the Methylmercury. Front. Neurosci 11, 1–10. 10.3389/fnins.2017.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty SJ, Tsai LH, 2011. Probing the role of HDACs and mechanisms of chromatin-mediated neuroplasticity. Neurobiol. Learn. Mem 96, 41–52. 10.1016/j.nlm.2011.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH, 2004. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. PNAS 101, 16659–16664. 10.1073/pnas.0407643101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutsell BA, Newland MC, 2013. A quantitative analysis of the effects of qualitatively different reinforcers on fixed ratio responding in inbred strains of mice. Neurobiol. Learn. Mem 101, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheramin S, Body S, Herrera FM, Bradshaw CM, Szabadi E, Deakin JFW, Anderson IM, 2005. The effect of orbital prefrontal cortex lesions on performance on a progressive ratio schedule: implications for models of inter-temporal choice. Behav. Brain Res 156, 145–52. 10.1016/j.bbr.2004.05.017 [DOI] [PubMed] [Google Scholar]
- Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G, 2010. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 35, 870–80. 10.1038/npp.2009.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killeen PR, 1994. Mathematical principles of reinforcement. Behav. Brain Sci 17, 105–172. [Google Scholar]
- Killeen PR, Sitomer MT, 2003. MPR. Behav. Processes 62, 49–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Leeds P, Chuang DM, 2009. The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J. Neurochem 110, 1226–1240. 10.1111/j.1471-4159.2009.06212.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, O’Riordan KJ, Brown KD, Trinh M. a., Molfese DL, Sweatt JD, 2004. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem 279, 40545–40559. 10.1074/jbc.M402229200 [DOI] [PubMed] [Google Scholar]
- Miller CA, Campbell SL, Sweatt JD, 2008. DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol. Learn. Mem 89, 599–603. 10.1016/j.nlm.2007.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobini S, Chiang T-J, Ho M-Y, Bradshaw CM, Szabadi E, 2000. Comparison of the effects of clozapine, haloperidol, chlorpromazine and d -amphetamine on performance on a time-constrained progressive ratio schedule and on locomotor behaviour in the rat. Psychopharmacology (Berl) 152, 47–54. 10.1007/s002130000486 [DOI] [PubMed] [Google Scholar]
- National Research Council, 2000. Toxicological effects of methylmercury National Academy Press, Washington, D.C. 10.17226/9899 [DOI] [Google Scholar]
- Newland MC, Paletz EM, Reed MN, 2008. Methylmercury and nutrition: Adult effects of fetal exposure in experimental models. Neurotoxicology 29, 783–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newland MC, Reed MN, Rasmussen E, 2015. A hypothesis about how early developmental methylmercury exposure disrupts behavior in adulthood. Behav. Processes 114, 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newland MC, Reile PA, Langston JL, 2004. Gestational exposure to methylmercury retards choice in transition in aging rats. Neurotoxicol. Teratol 26, 179–194. [DOI] [PubMed] [Google Scholar]
- Newman L, McGaughy J, 2011. Adolescent rats show cognitive rigidity in a test of attentional set shifting. Dev. Psychobiol 53, 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen SJ, Aoki Y, Kit BK, Ogden CL, 2015. More than half of US youth consume seafood and most have blood mercury concentrations below the EPA reference level, 2009–2012. J. Nutr 145, 322–327. 10.3945/jn.114.203786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuydens R, Heers C, Chadarevian A, De Jong M, Nuyens R, Cornelissen F, Geerts H, 1995. Sodium butyrate induces aberrant tau phosphorylation and programmed cell death in human neuroblastoma cells. Brain Res 688, 86–94. 10.1016/0006-8993(95)00514-Q [DOI] [PubMed] [Google Scholar]
- Obernier JA, Baldwin RL, 2006. Establishing an appropriate period of acclimatization following transportation of laboratory animals. ILAR J 47, 364–369. 10.1093/ilar.47.4.364 [DOI] [PubMed] [Google Scholar]
- Olson EA, Hooper CJ, Collins P, Luciana M, 2007. Adolescents’ performance on delay and probability discounting tasks: contributions of age, intelligence, executive functioning, and self-reported externalizing behavior. Pers. Individ. Dif 43, 1886–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishchenko N, Karpova N, Sabri F, Castrén E, Ceccatelli S, 2008. Long-lasting depression-like behavior and epigenetic changes of BDNF gene expression induced by perinatal exposure to methylmercury. J. Neurochem 106, 1378–1387. 10.1111/j.1471-4159.2008.05484.x [DOI] [PubMed] [Google Scholar]
- Paletz EM, Craig-Schmidt MC, Newland MC, 2006. Gestational exposure to methylmercury and n-3 fatty acids: Effects on high- and low-rate operant behavior in adulthood. Neurotoxicol. Teratol 28, 59–73. [DOI] [PubMed] [Google Scholar]
- Peleg S, Sananbenesi F, Zovoilis A, Burkhardt S, Bahari-Javan S, Agis-Balboa RC, Cota P, Wittnam JL, Gogol-Doering A, Opitz L, Salinas-Riester G, Dettenhofer M, Kang H, Farinelli L, Chen W, Fischer A, 2010. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science (80) 328, 753–756. https://doi.org/328/5979/753 [pii]\r10.1126/science.1186088 [DOI] [PubMed] [Google Scholar]
- Pinkston JW, Lamb RJ, 2011. Delay discounting in C57BL/6J and DBA/2J mice: Adolescent-limited and life-persistent patterns of impulsivity. Behav. Neurosci 125, 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope DA, Boomhower SR, Hutsell BA, Teixeira K, Newland MC, 2016. Chronic cocaine exposure in adolescence: Effects on spatial discrimination reversal, delay discounting, and performance on fixed-ratio schedules in mice. Neurobiol. Learn. Mem 130, 93–104. 10.1016/j.nlm.2016.01.017 [DOI] [PubMed] [Google Scholar]
- Rane P, Shields J, Heffernan M, Guo Y, Akbarian S, King JA, 2012. The histone deacetylase inhibitor, sodium butyrate, alleviates cognitive deficits in pre-motor stage PD. Neuropharmacology 62, 2408–2411. 10.1016/j.neuropharm.2012.01.026 [DOI] [PubMed] [Google Scholar]
- Reed MN, Banna KM, Donlin WD, Newland MC, 2008. Effects of gestational exposure to methylmercury and dietary selenium on reinforcement efficacy in adulthood. Neurotoxicol. Teratol 30, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed MN, Paletz EM, Newland MC, 2006. Gestational exposure to methylmercury and selenium: Effects on a spatial discrimination reversal in adulthood. Neurotoxicology 27, 721–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly MP, 2003. Extending mathematical principles of reinforcement into the domain of behavioral pharmacology. Behav. Processes 62, 75–88. [DOI] [PubMed] [Google Scholar]
- Reolon GK, Maurmann N, Werenicz A, Garcia VA, Schröder N, Wood MA, Roesler Rafael R, 2011. Posttraining systemic administration of the histone deacetylase inhibitor sodium butyrate ameliorates aging-related memory decline in rats. Behav. Brain Res 221, 329–332. 10.1016/j.bbr.2011.03.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JF, Theunissen PT, van Dartel DAM, Pennings JL, Faustman EM, Piersma AH, 2011. Comparison of MeHg-induced toxicogenomic responses across in vivo and in vitro models used in developmental toxicology. Reprod. Toxicol 32, 180–188. 10.1016/j.reprotox.2011.05.011 [DOI] [PubMed] [Google Scholar]
- Romieu P, Host L, Gobaille S, Sandner G, Aunis D, Zwiller J, 2008. Histone Deacetylase Inhibitors Decrease Cocaine But Not Sucrose Self-Administration in Rats. J. Neurosci 28, 9342–9348. 10.1523/JNEUROSCI.0379-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozental R, Faharani R, Yu Y, Johnson JM, Chan SO, Chiu FC, 2004. Sodium butyrate induces apoptosis in MSN neuroblastoma cells in a calcium independent pathway. Neurochem. Res 29, 2125–2134. 10.1007/s11064-004-6886-9 [DOI] [PubMed] [Google Scholar]
- Sakamoto M, Tatsuta N, Chan HM, Domingo JL, Murata K, Nakai K, 2018. Brain methylmercury uptake in fetal, neonate, weanling, and adult rats. Environ. Res 167, 15–20. 10.1016/j.envres.2018.06.038 [DOI] [PubMed] [Google Scholar]
- Sarkar S, Abujamra AL, Loew JE, Forman LW, Perrine SP, Faller DV, 2011. Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res 31, 2723–2732. [PubMed] [Google Scholar]
- Sharma B, Sharma PM, 2013. Arsenic toxicity induced endothelial dysfunction and dementia: Pharmacological interdiction by histone deacetylase and inducible nitric oxide synthase inhibitors. Toxicol. Appl. Pharmacol 273, 180–188. 10.1016/j.taap.2013.07.017 [DOI] [PubMed] [Google Scholar]
- Sichert-Hellert W, Wicher M, Kersting M, 2009. Age and time trends in fish consumption pattern of children and adolescents, and consequences for the intake of long-chain n-3 polyunsaturated fatty acids. Eur. J. Clin. Nutr 63, 1071–1075. [DOI] [PubMed] [Google Scholar]
- Song C, Kanthasamy A, Anantharam V, Sun F, Kanthasamy a G., 2010. Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: relevance to epigenetic mechanisms of neurodegeneration. Mol. Pharmacol 77, 621–632. 10.1124/mol.109.062174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP, 2000. The adolescent brain and age-related behavioral manifestations. Neurosci. Biobehav. Rev 24, 417–463. [DOI] [PubMed] [Google Scholar]
- Sun J, Wang L, Jiang B, Hui B, Lv Z, Ma L, 2008. The effects of sodium butyrate, an inhibitor of histone deacetylase, on the cocaine- and sucrose-maintained self-administration in rats. Neurosci. Lett 441, 72–76. 10.1016/j.neulet.2008.05.010 [DOI] [PubMed] [Google Scholar]
- Teicher MH, Andersen SL, Hostetter JC, 1995. Evidence for dopamine receptor pruning between adolescence and adulthood in striatum but not nucleus accumbens. Dev. Brain Res 89, 167–172. [DOI] [PubMed] [Google Scholar]
- Tiernan CT, Edwin EA, Hawong HY, Rios-Cabanillas M, Goudreau JL, Atchison WD, Lookingland KJ, 2015. Methylmercury impairs canonical dopamine metabolism in rat undifferentiated pheochromocytoma (pc12) cells by indirect inhibition of aldehyde dehydrogenase. Toxicol. Sci 144, 347–356. 10.1093/toxsci/kfv001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu WS, Parmigiani RB, Marks P, 2007. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 26, 5541–5552. 10.1038/sj.onc.1210620 [DOI] [PubMed] [Google Scholar]