

Abstract
Affiliative social motivation and behavior, i.e. sociability that includes attachment, prosocial behavior (sharing, caring, and helping), and empathy (the ability to understand and share the feelings of others), has high variability in the human population, with a portion of people outside of the normal range. While psychiatric disorders and autism spectrum disorders are typically associated with a deficit in social behavior, the opposite trait of hypersociability and indiscriminate friendliness are exhibited by individual with specific neurodevelopmental disorders and following early adverse care. Here we discuss both genetic and environmental factors that cause or increase the risk for developing pathological hypersociability from human to rodent models.
Introduction
As shown in Fig. 1, sociability is variable in a population1–3. Similar to many continuously variable behavioral phenotypes, sociability fits a normal (Gaussian) distribution, with most individuals falling in the middle of the range and some individuals, exhibiting pathological/abnormal phenotypes, at the left and right sides of the distribution2,4–6. Deficits in social interactions and socialization are often diagnosed in anxiety, antisocial and psychopathic personality disorders, and autism spectrum disorders7. The opposite traits of excessive need for social contact, emotional dependence on continuous social company and attention have also been recognized in adolescents and adults8. Hypersociability and indiscriminate sociability are associated with some neurodevelopmental disorders and child maltreatment. In contrast to social behavioral deficits, abnormal hypersociability in human and animals models has not been comprehensively reviewed.
Figure 1.
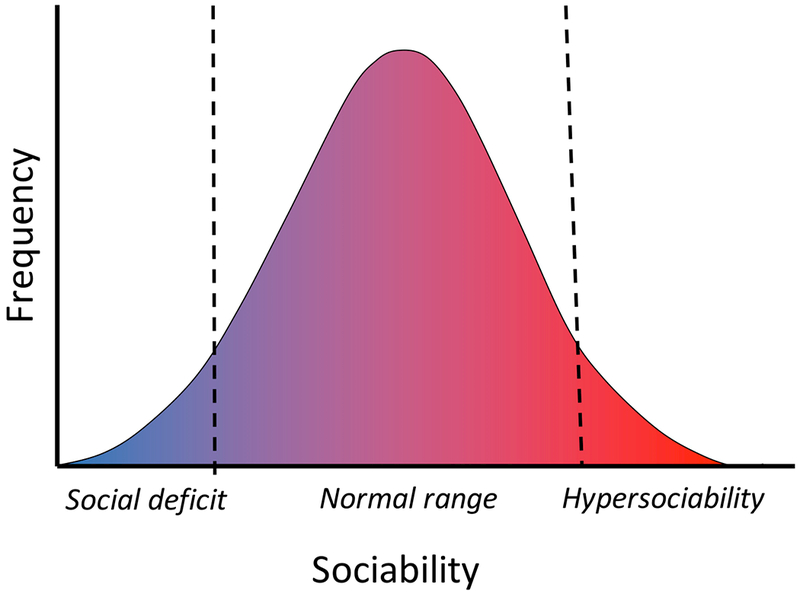
Variability of sociability in a population.
1. Genetic variability in sociability
Twin and family studies in human, and genetic mapping in rodent genetic reference panels showed that individual levels of sociability, both high and low on the behavioral scale, are heritable, at least partly3. Furthermore, genetic association studies (GWAS) between measures of sociability and single nucleotide polymorphisms (SNPs) identified chromosomal regions/genes that, via gene expression, may explain some of the differences in sociability between individuals in a population. Genes involved in sociability/hypersociability are listed in Table 1.
Table 1.
Sociability/hypersociability associated genes
| Genes | Condition | Species | Function | haploinsifficient | reference | |
|---|---|---|---|---|---|---|
| OXTR | rs53576GG | human | gain of | receptor | N | 17,18, 19 |
| CD38 | rs12644506 | human | coregulator of OTX | 20 | ||
| Gtf2I and Gtf2ird1 | Williams syndrome | human | loss of | transcription factor | Y | 44 |
| GTF2I | rs13227433 | human | transcription factor | 46 | ||
| Gtf2I and Gtf2ird1 | structural variants | dog | transcription factor | 21 | ||
| from Gtf2i to Fkbp6 | targeted deletion | mouse | loss of | Y | 69 | |
| from Gtf2i to Limk1 | targeted deletion | mouse | loss of | Y | 70, 71 | |
| Gtf2ird1 | targeted deletion | mouse | loss of | transcription factor | Y | 73, 74 |
| UBE3A | Angelman syndrome | human | loss of | ubiquitin-protein ligase | 52 | |
| Ube3a | targeted deletion | mouse | loss of | 77 | ||
| KANSL1 | Koolen-De Vries syndrome | human | loss of | member of HAT complex | Y | 64–67 |
| DLG4 | targeted deletion | mouse | loss of | synaptic protein | Y | 64 |
| DLG2 | targeted deletion | mouse | loss of | synaptic protein | N | 64 |
| nNOS | targeted deletion | mouse | loss of | synaptic regulator | 79 | |
| Nrg1 | targeted deletion | mouse | loss of | signaling molecule | Y | 103 |
| Erbb3 | targeted deletion | mouse | loss of | tyrosine kinase receptor | 103 | |
1.1. Genetic variability in sociability in human: Twin studies
In behavioral genetics, twin studies are fundamental to establish trait heritability. A classic twin study design compares trait similarity in same sex monozygotic (MZ) twins raised in the same family to that in same sex dizygotic (DZ) twins. MZ twins share virtually all, while DZ twins approximately 50% of their genes. Although twin studies have significant limitations and confounds (e.g. reciprocal influences between twins)9, difference in correlations between MZ and DZ twins reflects heritability. Heritability between 56%–72% was reported in twin studies for self-reported measures of altruism, empathy, and nurturance10. In another study, genetic influences accounted for 43% and 34% of the variance in observed self-initiated and compliant pro-social behavior in children, respectively11. Further, genetic influences, although not apparent at 14 and 20 months, explained 34%–47% of the variance in empathy at 24 and 36 months of age12. Although twin studies indicate a surprisingly high heritability for social behavior, they provide no information on trait-associated chromosomal regions and genes.
1.2. Genetic polymorphism in oxytocin pathway genes is associated with variability in social attachment and empathy in human
Sociability is a polygenic trait determined by the interaction of many genes, each with a relatively small contribution to the behavioral phenotype. GWAS identified SNPs in oxytocin pathway genes that influence the individual level of sociability. Oxytocin (OXT) is a neuromodulator that affects a range of social behaviors in humans and animals, including social attachment and higher order human social functions such as empathy13,14. It is believed that OTX increases the salience and reinforcing value of social stimuli15. OTX is produced in the hypothalamus and is released via axons projecting to the pituitary, and via axon collateral to the nucleus accumbens (NAc) and other brain regions16. Beside of OTX, the genes for the OTX receptor (OTXR) and CD38, a protein that regulates OTX release (CD38), have been studied to explain variability in human social behavior. Evidence links more efficient signaling variants of OTX pathway genes to increased affiliative behavior. For example, OXTR rs53576GG homozygotes in one study self-report higher levels of sociality17, while in another study are rated as more prosocial than A carriers (AA, AG genotypes)18. Also, the OXTR rs53576GG genotype was associate with a higher level empathy in a population, as well as in a comprehensive meta-analysis of 13 independent studies19. Similarly, SNP rs12644506 in the CD38 gene was associated with social integration and social connectedness20. Finally, the rs53576A allele that predisposes for low sociability was linked to morphometric alterations of the hypothalamus, specifically to a decrease in gray matter volume and to an increase in functional coupling between hypothalamus and amygdala17.
1.3. Sociability traits identified by GWAS in domestic dogs.
Dogs exhibit a heightened propensity to initiate social contact that is extended to members of other species, when compared with human-socialized gray wolves21. This behavior is likely driven by behavioral neoteny, which is the retention of juvenile features in the adult. A genome-wide association scan of more than 48,000 SNP genotypes from 701 dogs from 85 breeds and 92 gray wolves identified a top ranking outlier site located within the polymorphic Wbsrc17 gene, which is within the region typically deleted in Williams syndrome22. Williams syndrome patient exhibit hypersociability as discussed in section 2.1. A follow up study found that a 5-Mb genomic region around the Williams critical region was under positive selection in domestic dog breeds and that hypersociability is a core element of domestication that distinguishes dogs from wolves21. Structural variants in two genes in the Williams syndrome critical regions, Gtf2I and Gtf2ird1 (encoding two related transcription factors of the TFII-I family, likely contribute to the hypersociability of domestic dogs. These data indicate that human directed evolution of dogs through selective breeding retained specific structural variants of a unique set of linked genes with a large effect on sociability (i.e. survival of the friendliest23).
1.4. Mapping of “sociability” traits in mice by using recombinant inbred strains.
Over the past decades, murine recombinant inbred (RI) strain families from naturally divergent strains have been developed via chromosome substitution to dissect complex genetic traits, including sociability24,25. The BXD family, currently the largest and best characterized mouse RI collection, is composed of ~160 lines that descend from crosses between C57BL/6J and DBA/2J26. Statistical genetic techniques in BXDs allow the identification of quantitative trait loci (QTL) that define regions of the genome (intervals or loci) and their modulating effects on the phenotype. The BXD family has been exploited to study the genetics of immune function, infectious disease, and behavior27. More recently, BXDs were used to identify QTLs associated with sociability. Although the parental C57BL/6 and DBA/2 inbred strains exhibit low and intermediate levels of sociability, Knoll and coworkers reported a wide variability in social approach and vocal communication in BXDs, extending well above the average sociability of DBA mice28. The quantitative trait locus for vocal communication, and associated candidate genes, were mapped to chromosome 18. However, the large size of the QTL does not allow specifying a single gene or set of genes that may directly contribute to the trait.
2. Pathological hypersociability in genetic conditions
Affiliative behavior and empathy that exceed the normal range of sociability can be a part of neurodevelopmental disorders such as Williams syndrome29.
2.1. Williams syndrome
Williams syndrome arises from a genetic deletion of 7q11.23, in 1 out of approximately 20,000 births29. Williams syndrome is characterized by cognitive deficits in the IQ range of 50-60, with relative strengths in face and object recognition and in some aspects of auditory and verbal abilities, but with severe weaknesses in spatial, motor and visual-motor abilities30–32. However, Williams syndrome is most associated with exuberant gregariousness, lack of social inhibition, and enjoyment of conversations, a distinct phenotype from most neurodevelopmental disorders that are typically associated with social anxiety. Williams syndrome affects males and females in equal numbers29, in contrast to a male bias in autism spectrum disorders (characterized by social anxiety). However, hypersociability in Williams syndrome is accompanied by a poor understanding of social dynamics, facial expression, and body language and Williams patients have high rates of symptoms of generalized and anticipatory anxiety33. Toddlers with Williams syndrome have an intense, penetrating gaze, that is the opposite of the aversion of eye contacts in fragile X syndrome and autism spectrum disorders. Although individuals with Williams syndrome can hold simple jobs, they require assistance managing their lives. Distinctive cardiovascular problems limit the average lifespan of a person with Williams to around fifty.
Because of the well-defined, albeit multigene deletion, Williams syndrome provides an opportunity to study how hemizygosity in a relatively small set of genes shape complex social behavior such as an innate drive to connect with others. Williams syndrome usually occurs sporadically, though familial cases have been reported34,35. Due to repetitive sequences flanking the commonly deleted regions, 98% of Williams individuals studied have the same deletion breakpoints. The commonly deleted region spans 1.5 Mb and include GTF2l and NCF1 on one (the telomeric) end, and their pseudogenes (P) on the other end30,36–38 (https://www.jci.org/articles/view/35309/figure/2). Duplication of the identical region also occurs, albeit with lower frequency, causing an opposite behavioral phenotype that falls within the autism spectrum39,40. This indicates that the dosage of at least some of the genes within the Williams critical region is essential for the development of normal social behavior, and that lower and higher gene dosage results in behavioral abnormalities in the opposite direction.
The commonly deleted region contains approximately 25 genes, encoding transcriptional regulators (GTF2I, GTF2IRD1, BAZ1B, MLXIPL), signaling molecules (FZD9, TBL2, LIMK1) and molecules with functions in various cellular processes (STX1A, CYLN2, FKBP6, EIF4H, CLDN3, CLDN4, VPS39D and RFC2)41. Although most Williams patients have the full deletion, there are individuals with partial deletions that prompted studies to associate individual genes with specific phenotypes/behaviors. Deletion of only ELN (encoding elastin) was linked to supravalvar aortic stenosis and cutis laxa with no cognitive and behavioral abnormalities, while deletion of LIMK1 was associated with visual-spatial constructive cognition, but not with most of the Williams symptoms29,42. However, another study found that visual-spatial construction was associated with GTF2IRD143. Still another study that analyzed partial 7q11.23 deletions implicated GTF2IRD1, as well as its close relative GTF2I, as the main genes responsible for the Williams syndrome neurocognitive profile44. The discrepancy between these studies could be due to the difficulty to establish specific gene–phenotype correlations in Williams syndrome because of phenotypic variability, presumably due to modifier loci. Nevertheless, a report describing a deletion of all genes but GTF2I in the critical region is noteworthy because the affected individual, unlike most Williams patients, did not show increased gaze and attention to strangers43,45. This suggests that loss of GTF2I, at least partly, is involved in the social behavioral phenotype of Williams syndrome. However, it is important to point out that some individuals with the full deletion (i.e. including GTF2I) also have fairly normal social behavior. Nonetheless, genetic variability (instead of a hemizygous deletion) in GTF2I in healthy individuals supports a possible role for this gene in social behavior. Specifically, rs13227433AA, an SNP in GTF2I showed association with low social anxiety, combined with reduced social-communication abilities46, a phenotype reminiscent to that seen in Williams syndrome.
Although none of the genes deleted in Williams syndrome have been functionally connected to the behavioral phenotype, brain structural47 and functional48 abnormalities provide a possible link between Williams syndrome genes and social disinhibition. A large number of brain abnormalities have been described in Williams syndrome, but probably the most prominent are reduced amygdala response to fearful facial expressions48–50 and heightened amygdala reactivity to positive social stimuli49. Reduced amygdala response to fearful facial expressions may underlie social disinhibition (i.e. approach of strangers), while increased amygdala response to happy facial expressions could explain the high motivation of Williams patients to interact with others. Consistent with the notion that loss of function in GTF2I is a major contributor to the clinical phenotype of Williams43,45, the GTF2I rs13227433 AA genotype, previously associated with lower social anxiety in the population46 was also associated with reduced amygdala response to threat51, linking this gene, via amygdala function, to social behavior.
2.2. Other neurodevelopmental disorders with hypersocial behavior
Angelman syndrome.
Deletion of chromosome 15q11-q13 in Angelman syndrome causes developmental delay (which becomes apparent by 6–12 months of age), lack of vocal speech, muscular hypotonia, epilepsy, and a typical behavioral profile that includes a happy demeanor, hypermotor behavior, and low attention span52. Frequent smiling and laughing are considered to be part of repetitive and stereotyped behavior in Angelman patients53–55. However, some studies showed that laughing and smiling increase during social situations and occur at low levels during non-social situations56, raising the possibility of increased motivation to interact with others.
Angelman syndrome is typically associated with the deletion of the maternal UBE3A gene at 15q12-q13, encoding the ubiquitin-protein ligase E3A57. Since the paternal allele is imprinted and inactivated in the developing brain, Angelman patients lack UBE3A during development, one of the enzymes required for proper ubiquitination of proteins destined for proteolysis. Typical deletion in Angelman also includes genes with biallelic expression; three gamma-aminobutyric acid (GABA) receptor subunit genes and the P gene which, when mutated in both alleles, is responsible for oculocutaneous albinism type II.
17q21.31 microdeletion syndrome (Koolen-De Vries syndrome).
17q21.31 microdeletion syndrome, characterized by developmental delay, moderate intellectual disability, facial dysmorphisms, and anomalies of the brain and multiple organ systems, is a relatively recently discovered condition58–60. Interestingly, clinical data indicate an amiable, friendly disposition, less social fear and more approach behavior61–63. The 17q21.31 microdeletion includes the microtubule associated protein tau (MAPT ) gene58–60 that normally promotes microtubule assembly and stability in neurites, corticotropin releasing hormone receptor 1 (CRHR1), C17orf69 (long intergenic non-protein coding RNA 2210) and KANSL1 (member of histone acetyltransferase complex). Koolen-De Vries syndrome, including intellectual disability and happy disposition, can be caused by heterozygous truncating mutations in KANSL164,65, demonstrating that inactivation of this gene is central to the 17q21.31 microdeletion syndrome. Indeed, recent comparison of the phenotype of microdeletion and KANSL1 SNV (single nucleotide variant) patients did not show differences of clinical importance, stressing that haploinsufficiency of KANSL1 is sufficient to cause the full Koolen-De Vries syndrome phenotype66,67.
2.3. Mouse models of neurodevelopmental disorders exhibiting increased social behavior
Neurodevelopmenal conditions have been generated in mice by the genetic inactivation of Williams, Angelman, and 17q21.31 microdeletion syndrome candidate genes. These mouse models can reproduce the hypersociability phenotype of the human conditions.
Williams syndrome models.
The entire Williams syndrome critical region is conserved in the mouse68 and its genetic manipulation provided further insight into the possible contribution of individual genes to the phenotypes of Williams syndrome. Heterozygous deletion of the mouse Williams critical region between Gtf2i and Fkbp6 (that includes Gtf2ird1) resulted in increased social interactions and less habituation in a repeated social approach task69. Mutants with a partial deletion of the Williams critical region have also been generated70. Proximal deletion (PD) mice lack Gtf2i to Limk1 (that includes Gtf2ird1), distal deletion (DD) mice lack Limk1 to Fkbp6, and double heterozygotes (PD/DD) model the complete human deletion. In the three chamber social test71, male PD and PD/DD mice spent significantly more time interacting with s stranger mouse, while in the tube test72 that assess social dominance, PD and PD/DD animals were less competitive than WT and DD mice against a control C57BL/6 mouse. This suggests that haploinsufficiency in the proximal part of the Williams critical region, that includes Limk1, Eif4h, Rfc2, Cyln2, Gtf2ird1, and Gtf2i, is associated with increased sociability and subordination. However, only PD but not PD/DD mice exhibited increased activity in direct social interaction suggesting a more complex gene-phenotype association in mouse models of Williams syndrome. Nevertheless, knockout mice heterozygous for Gtf2ird1 (a close relative of Gtf2i) was shown to exhibit an increase in the number and length of social interactions and a decrease in the number of aggressive interactions in the resident intruder test73. Similarly, a more recent study reported that mice with a heterozygous deletion of Gtf2ird1 have increased social interaction with unfamiliar mice and do not show typical social habituation74. Other genes within the proximal region of the Williams syndrome critical region may contribute to phenotypes other than hypersociability. Cyln2 heterozygote knockout mice exhibit mild growth retardation, mild brain abnormality, attenuated contextual fear, and reduced synaptic plasticity and motor function75. No apparent behavioral phenotype was found in LimK1 heterozygote knockouts76. Taken together, haploinsufficiency in GTF2I (human studies, see 2.1.) and/or in the related GTF2IRD1 (human and mouse studies) is probably critical in the development of the hypersociable phenotype of Williams patients.
Angelman syndrome model.
Consistent with the central role of UBE3A deletion on the maternal allele in the development of the Angelman syndrome behavioral phenotype, including typical happy demeanor, genetic deletion of Ube3a in mice was reported to increase preference for social stimuli in the three chamber social approach task and physical contacts of both males and females paired with unfamiliar genotype-matched females77.
17q21.31 microdeletion syndrome model.
Human studies indicated that haploinsufficiency in KANSL1 causes 17q21.31 microdeletion syndrome. However, heterozygous ablation of Kansl1 caused no apparent change in social behavior in the three-chamber sociability test, albeit exhibited other behaviors typically seen in 17q21.31 microdeletion syndrome (i.e. impaired recognition memory and associative learning and memory and brain malformations)78. A larger deletion that included Mapt and Crhr1 and mimicked the 17q21.31 microdeletion however resulted in increased sociability, similar to the human disease phenotype. The authors speculate that the discrepancy between the human and mouse phenotypes is due to mice requiring more reduction in Kansl1 gene dosage to exhibit increased sociability and that haploinsufficiency in another gene(s) from the larger deletion could contribute to lower KANSL1 levels in heterozygotes.
2.4. Genetic inactivation of synaptic genes in mice exhibiting increased social behavior
Behavioral testing of genetically engineered mice revealed genes whose deletion causes hypersociability. Interestingly, all of these genes are associated, directly or indirectly, with synaptic functions.
Hypersociability of PSD95 null mice.
PSD-95 (postsynaptic density-95, DLG4), a member of the membrane-associated guanylate kinase family of synaptic molecules, is located in the postsynaptic density79 and is involved in anchoring synaptic proteins at excitatory synapses, including neuroligins, NMDA receptors, AMPA receptors, and potassium channels80. PSD-93 (DLG2) is a similar protein, forming heterotrimers with PSD9581.
Dlg4−/− mice were reported to interact with unfamiliar conspecifics more than control wild-type mice in the three chamber social approach and social novelty tests82. Null mice were indistinguishable from controls in investigating nonsocial objects, indicating that increased sociability was not due to a general preference for novelty.
While Dlg4−/− mice, alongside of an increased social interaction phenotype, exhibit impaired motor coordination, increased stress reactivity, and anxiety-related responses82, heterozygote Dlg4+/− mice have only hypersociability. Heterozygote mice of both sexes exhibited increased interest towards an unfamiliar conspecific in dyadic social interactions in neutral testing cages83, and males had enhanced aggression as measured in the resident-intruder paradigm. This suggests that Dlg4, similar to some of the Williams genes, is haploinsufficient. Interestingly, Dlg2 null mice (PSD93) displayed a similar, increased sociability phenotype in dyadic social interaction tests, while heterozygote mice were not different from controls, indicating that Dlg2 is not a haploinsufficient gene83. These data suggest that PSD95 and PSD93 are both involved in controlling social behavior, presumably in a redundant manner. The authors speculate that the increased social phenotype of PSD95 and PSD93 mutant mice could be the consequence of ‘social blindness’, i.e. the inability to adequately process and respond to social signals83. Indeed Dlg4+/− mice exhibited impaired social recognition while nonsocial cognitive functioning was retained84.
It is surprising that genetic deletion of PSD95 interacting proteins, including the transynaptic proteins of neuroligins, neurexins and shanks, results in reduced sociability, and that their loss of function has been implicated in monogenetic forms of autism85–88. However, PSDs have a much wider function in organizing postsynaptic complexes than the individual transynaptic proteins.
Hypersociability in nitric oxide synthase (NOS) null mice.
NO, a highly diffusible gas with function in neurotransmission, synaptic plasticity, gene expression and neurotoxicity, is produced by neuronal (n) NOS from L-arginine in the brain89,90. nNOS is expressed in the hippocampus, cortex, striatum, cerebellum, olfactory bulb, and brain stem91,92. Unlike traditional neurotransmitters, NO levels are not regulated by release but rather via production by the phosphorylation dependent catalytic activity of nNOS93. nNOS is part of a macromolecular signaling complex at excitatory synapses that consists of PSD95 and the NMDA receptor, rendering nNOS activity dependent on Ca2+94. In turn, NO acts via retrograde signaling to activate soluble guanylyl cyclase in the presynaptic terminal and regulate neurotransmitter release95,96. At higher concentrations, NO is covalently attached to a cysteine residue to form an S-nitrosothiol in various proteins that include NMDA subunits97.
Mice with targeted disruption in nNOS98 (partially backcrossed to the C57BL/6 background), was reported to exhibit increased social interaction in their home cage under familiar conditions, but less sociability (i.e. time spent around a stranger mouse) in the three chamber social test, suggesting a different response with a familiar and unfamiliar conspecific99. In another report, genetic and pharmacological inhibition of nNOS in mice on mixed genetic background was reported to have reduced social investigation and increased aggression toward an unfamiliar mouse in a modified resident–intruder partition test100. These data indicate that the social behavior of nNOS null mice is altered, with higher and lower sociability depending on familiarity with a conspecific.
Neuregulin (NRG1) and its receptors ERBB3 and ERBB4 are linked to sociability in mice.
NRG1 is an EGF-like signaling molecule, which, by interacting with transmembrane tyrosine kinase receptors of the ErbB family, is involved in cell-cell communication, both during development and in the adult. In the brain, it regulates neuron migration and progenitor cell proliferation, but also glutamate release, NMDA and GABA receptor subunit expression and synaptic plasticity101,102. Moy and coworkers reported that Nrg1+/− mice spent significantly more time in the side containing an unfamiliar stranger mouse, in comparison to the wild-type mice, in a three chamber social test103. A similar pattern was found with CNS specific Erbb3 mutants (Erbb3tm2Dwt/tm2Dwt, Nestin-Cre) and a milder phenotype was apparent in CNS specific Erbb4 mutants (Erbb4lox/-,hGFAP-Cre). NRG1 is a schizophrenia candidate gene104,105, and several studies reported increased expression of NRG1 mRNA isoforms in frontal cortex and hippocampus of schizophrenia patients106,107 and in neurons derived from induced pluripotent stem cells of patients108. Given the presumably higher NRG1-EERB signaling in schizophrenia, and the association of schizophrenia with impaired social functioning, the higher sociability of Nrg1, Erbb3, and Erbb4 deficient mice indicates a bidirectional modulation of social behavior by the NRG1-EERB pathway.
3. Effect of the environment on social behavior
3.1. Hypersociability (indiscriminate friendliness) in adopted children with early adverse care experiences
In addition to genetic factors, the environment has a significant effect on shaping social behavior. After the overthrow of the Ceausescu regime in December 1989, the world discovered that 170,000 children were being raised in Romania’s impoverished institutions. It was noted that adopted children, who have had early adverse care experiences during the first two years of life, approached and interacted with unfamiliar adults in a way expected and typical with familiar individuals109. The prominent social abnormality of children adopted following institutional rearing has been variably called indiscriminate friendliness109, disinhibited social behavior110, disinhibited attachment behavior111, or disinhibited social engagement disorder (DSM-5 313.89). No sex difference was found in indiscriminate friendliness109. Beside social disinhibition, children adopted following institutional rearing tend to have difficulty with executive functions such as cognitive flexibility, inhibitory control and working memory. They struggle to regulate their emotions and suffer from anxiety. These children can be exposed to safety risks112, have later difficulties in peer relationships113,114 and have impaired attention and impulse control115,116.
A similar clinical phenotype is also apparent in maltreated foster children117. Moreover, disinhibited social engagement disorder can develop in noninstitutionalized children due to parental adjustment problems that include poverty, teen parenting, substance abuse and mental health problems. Children, once removed from abusive or neglectful environments tend to recover physically but can still present the symptoms of disinhibited social engagement disorder118.
Although the overall behavioral phenotype of individuals with prior childhood maltreatment and Williams syndrome are different, they share increased and indiscriminate sociability suggesting a common neurobiological basis of hypersociability. Indeed, similar to Williams syndrome, amygdala activity in previously institutionalized youths is altered. It was reported that, while controls showed an amygdala response that clearly discriminated mother versus stranger stimuli, previously institutionalized individuals exhibited reduced amygdala discrimination between mothers and strangers119. Reduced amygdala differentiation correlated with greater reports of indiscriminate friendliness that in turn correlated with age-at-adoption. Lack of discrimination was the result of atypically high amygdala response to strangers in previously institutionalized youth, whereas responses to mother stimuli were equivalent across groups.
An animal model of early life neglect or early adverse care would be an invaluable tool for understanding the potential mechanisms underlying increased sociability of post-institutionalized children. Although daily separation of pups from their mothers during the early postnatal period has been extensively used to study persistent alterations in emotional and anxiety-related behaviors, less is known about the impact of maternal separation on social behavior. The limited literature suggests that current maternal separation models and even maternal separation combined with early weaning (to mimic early life neglect) may not be able to reproduce the increased sociability phenotype seen in post-institutionalized children120–122. Development of animal models of early life neglect that recapitulate indiscriminate sociability would help to understand the underlying neurobiological mechanism.
3.3. Parental genotype as an environmental effects in programming hypersociability
Genetic variants in parents can affect the behavior of their offspring, even if the child does not carry the allele. For example, the maternal genotype can influence the in utero environment that in turn, could alter prenatal development and the long-term health of the offspring123. The parental genotype may also alter the postnatal development of the offspring, a phenomenon recently named genetic nurturing124. Although no study, to the best of our knowledge, has linked a specific parental genotype (i.e. SNP) to measures of offspring social behavior in human, parental serotonin and dopamine transporter and brain-derived neurotrophic factor (BDNF) polymorphisms have been reported to influence parenting behaviors125–127 that in turn, could modulate offspring social behavior.
The animal literature regarding parental genotype effects on offspring social behavior is also unexplored, but we reported that heterozygous deletion of the Fmr1 gene (encoding the fragile X mental retardation protein FMRP) in dams programs higher sociability, specifically reduced avoidance and increased approach in both a three-chamber sociability assay71 and a one-chamber social interaction procedure128, in genetically wild-type male FVB offspring129. In contrast to this maternal Fmr1 genotype effect, absence of FMRP in male offspring had no significant effect on their social interaction (i.e. no offspring genotype effect)129. This was surprising because lack of FMRP in fragile X syndrome is typically associated with a high degree of gaze aversion, interpreted as social anxiety130. Nevertheless, similar to our results, Spencer and coworkers found no effect of deleting Fmr1 in C57BL/6 males either on social behavior using a “partitioned” social interaction test apparatus 131,132. However, more in line with human studies, others reported social anxiety in FMRP deficient mice in the presence of a stranger in various tests133–136, suggesting that offspring Fmr1 genotype has no or variable effect on social behavior, depending on testing conditions and the genetic background. In summary, FMRP seems to have a dual, genetic and non-genetic role in social behavior; inactivating Fmr1 in mothers increases sociability of their genetically normal male offspring, in contrast to increased social anxiety in fragile X syndrome and in some mouse models of the disease.
Because increased social approach could be elicited by the FMRP-deficient maternal environment both pre- and postnatally, mechanisms that may mediate this maternal effect include maternal bioactive molecules that cross the placenta or are transferred to the offspring via milk, or behavioral interactions between the mother and offspring137. The relevance of the maternal FMRP genotype effect, if present in human, is potentially broad because it includes the genetically unaffected children of mothers with full mutation, as well as mothers with the far more prevalent premutation (which is also associated with deficits in FMRP138).
Brain-wide imaging of neuronal activity (via the expression of the immediate early gene c-fos) upon social interaction with a stranger showed that, while control male offspring (of WT mothers) exhibit activation in the ventral tegmental area (VTA) and its connected region, the NAc shell, the hypersocial male offspring (of FMRP deficient mothers) exhibit suppression of activity in these regions129. This result is consistent with the model that medium spiny neurons in the NAc shell encode, via activation and suppression, aversion and reward, respectively, first proposed by Carlezon and Thomas139 and also shown by others140. Therefore, our interpretation is that programmed hypersociability is associated with a failure to recognize social cues and activate NAc, resulting in a failure to avoid the unfamiliar mouse.
Discussion/Synthesis
Both genetic (Table 1) and environmental effects (early life neglect) contribute to the development of hypersociability. While genetic effects can be directly linked to specific genes and, via gene expression, to neurobiological processes, this association is more difficult to establish with environmental effects. However, early life insults produce persistent changes in DNA methylation and chromatic modifications141–144, and epigenetic changes, similar to genetic polymorphisms and mutations, can alter gene expression145–147.
Although the precise functions of sociability-related genetic and epigenetic effects are not understood, they seem to converge on two neuronal functions that regulate social behavior (Fig. 2). The first is the ability of the amygdala to discriminate between fear-inducing and friendly social clues and to produce an appropriate behavioral response. The second is the dopaminergic reward/aversion system that assesses the salience of social situations and initiates approach (social reward) or avoidance (social aversion) behavior.
Figure 2.
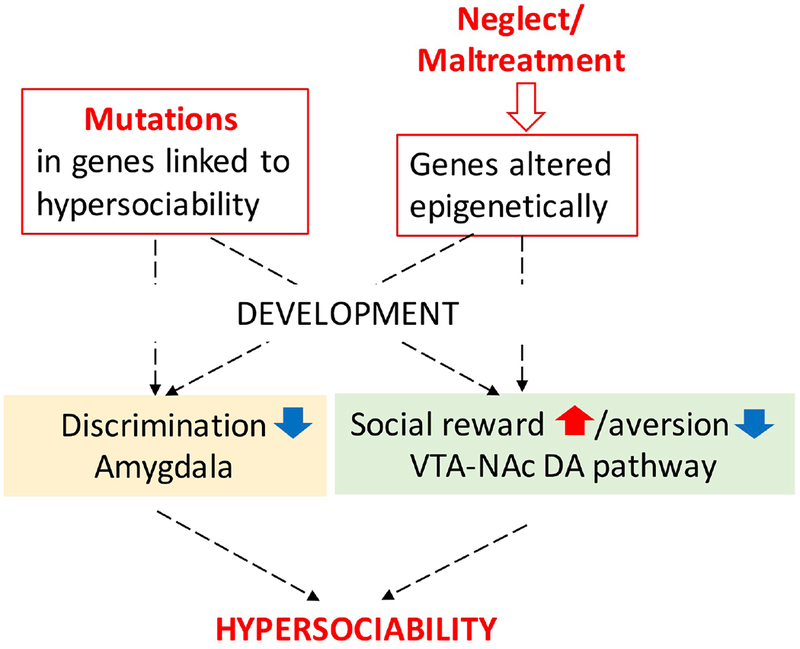
Convergence of genes and early adverse care experience on neuronal networks to produce hypersocial behavior.
Hypersociability can be conceptualized as a developmental abnormality leading to a failure in discrimination between familiar and stranger in the amygdala and/or increased reward or reduced aversion in the VTA-NAc pathway. For example, Williams patients were reported to exhibit reduced amygdala response to fearful facial expressions that may explain their social disinhibition 48–50. In addition, these patients tend to show heightened amygdala reactivity to positive social stimuli that could underlie their high motivation to interact with others49. Similarly, previously institutionalized individuals were reported to exhibit atypically high amygdala response to strangers119. Regarding the reward pathway, its possible role in social behavior, specifically in pair bonding, was first suggested by higher OTXR densities (i.e. the receptor for the prosocial OTX) in the NAc of monogamous, compared to nonmonogamous, prairie voles148. The involvement of VTA-NAc system in social reward was also demonstrated in mice149, and a more efficient signaling variants of OTXR has been linked to prosocial behavior17,18. However, the VTA-NAc pathway also codes for aversion139,140 that seems to play a role in the hypersocial behavior of male offspring of FMRP deficient mothers129.
To better understand the neurobiological basis of hypersociability, it will be important to link hypersociability-related genes and environment to specific neuronal functions. Because of the diversity of conditions that lead to hypersociability, multiple neuronal mechanisms may be involved. Nevertheless, abnormalities at the neuronal level can facilitate the understanding of neuronal network abnormalities that ultimately may explain how hypersocial behavior is generated in these circuits.
Acknowledgment:
This work was supported by US National Institute of Health grants 5R01 MH086883, R01 MH100894, and R01 MH103102 to MT.
References
- 1.Reeb-Sutherland BC, Levitt P, Fox NA. The predictive nature of individual differences in early associative learning and emerging social behavior. PLoS One. 2012;7(1):e30511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robinson EB, St Pourcain B, Anttila V, et al. Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nat Genet. 2016;48(5):552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ebstein RP, Israel S, Chew SH, Zhong S, Knafo A. Genetics of human social behavior. Neuron. 2010;65(6):831–844. [DOI] [PubMed] [Google Scholar]
- 4.Constantino JN, Todd RD. Autistic traits in the general population: a twin study. Arch Gen Psychiatry. 2003;60(5):524–530. [DOI] [PubMed] [Google Scholar]
- 5.Lundström S, Chang Z, Råstam M, et al. Autism spectrum disorders and autistic like traits: similar etiology in the extreme end and the normal variation. Arch Gen Psychiatry. 2012;69(1):46–52. [DOI] [PubMed] [Google Scholar]
- 6.St Pourcain B, Whitehouse AJ, Ang WQ, et al. Common variation contributes to the genetic architecture of social communication traits. Mol Autism. 2013;4(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trull TJ, Widiger TA. Dimensional models of personality: the five-factor model and the DSM-5. Dialogues Clin Neurosci. 2013;15(2):135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martens WH. The suffering of hypersocial patients. Psychopathology. 2001;34(6):328. [DOI] [PubMed] [Google Scholar]
- 9.Constantino JN, Todd RD. Genetic structure of reciprocal social behavior. Am J Psychiatry. 2000;157(12):2043–2045. [DOI] [PubMed] [Google Scholar]
- 10.Rushton JP. Genetic and environmental contributions to pro-social attitudes: a twin study of social responsibility. Proc Biol Sci. 2004;271(1557):2583–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knafo A, Israel S, Ebstein RP. Heritability of children’s prosocial behavior and differential susceptibility to parenting by variation in the dopamine receptor D4 gene. Dev Psychopathol 2011;23(1):53–67. [DOI] [PubMed] [Google Scholar]
- 12.Knafo A, Zahn-Waxler C, Van Hulle C, Robinson JL, Rhee SH. The developmental origins of a disposition toward empathy: Genetic and environmental contributions. Emotion 2008;8(6):737–752. [DOI] [PubMed] [Google Scholar]
- 13.Donaldson ZR, Young LJ. Oxytocin, vasopressin, and the neurogenetics of sociality. Science. 2008;322(5903):900–904. [DOI] [PubMed] [Google Scholar]
- 14.Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiol Rev. 2001;81(2):629–683. [DOI] [PubMed] [Google Scholar]
- 15.Young LJ. Oxytocin, social cognition and psychiatry. Neuropsychopharmacology. 2015;40(1):243–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landgraf R, Neumann ID. Vasopressin and oxytocin release within the brain: a dynamic concept of multiple and variable modes of neuropeptide communication. Front Neuroendocrinol. 2004;25(3–4):150–176. [DOI] [PubMed] [Google Scholar]
- 17.Tost H, Kolachana B, Hakimi S, et al. A common allele in the oxytocin receptor gene (OXTR) impacts prosocial temperament and human hypothalamic-limbic structure and function. Proc Natl Acad Sci U S A. 2010;107(31):13936–13941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kogan A, Saslow LR, Impett EA, Oveis C, Keltner D, Rodrigues Saturn S. Thin-slicing study of the oxytocin receptor (OXTR) gene and the evaluation and expression of the prosocial disposition. Proc Natl Acad Sci U S A. 2011;108(48):19189–19192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gong P, Fan H, Liu J, Yang X, Zhang K, Zhou X. Revisiting the impact of OXTR rs53576 on empathy: A population-based study and a meta-analysis. Psychoneuroendocrinology. 2017;80:131–136. [DOI] [PubMed] [Google Scholar]
- 20.Chang SC, Glymour MM, Rewak M, et al. Are genetic variations in OXTR, AVPR1A, and CD38 genes important to social integration? Results from two large U.S. cohorts. Psychoneuroendocrinology. 2014;39:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.vonHoldt BM, Shuldiner E, Koch IJ, et al. Structural variants in genes associated with human Williams-Beuren syndrome underlie stereotypical hypersociability in domestic dogs. Sci Adv. 2017;3(7):e1700398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vonholdt BM, Pollinger JP, Lohmueller KE, et al. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature. 2010;464(7290):898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hare B, Brown M, Williamson C, Tomasello M. The domestication of social cognition in dogs. Science. 2002;298(5598):1634–1636. [DOI] [PubMed] [Google Scholar]
- 24.Singer JB, Hill AE, Burrage LC, et al. Genetic dissection of complex traits with chromosome substitution strains of mice. Science. 2004;304(5669):445–448. [DOI] [PubMed] [Google Scholar]
- 25.Williams RW, Gu J, Qi S, Lu L. The genetic structure of recombinant inbred mice: high-resolution consensus maps for complex trait analysis. Genome Biol. 2001;2(11):RESEARCH0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peirce JL, Lu L, Gu J, Silver LM, Williams RW. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet. 2004;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Philip VM, Duvvuru S, Gomero B, et al. High-throughput behavioral phenotyping in the expanded panel of BXD recombinant inbred strains. Genes Brain Behav. 2010;9(2):129–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knoll AT, Jiang K, Levitt P. Quantitative trait locus mapping and analysis of heritable variation in affiliative social behavior and co-occurring traits. Genes Brain Behav. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris CA, Mervis CB. Williams syndrome and related disorders. Annu Rev Genomics Hum Genet. 2000;1:461–484. [DOI] [PubMed] [Google Scholar]
- 30.Korenberg JR, Chen XN, Hirota H, et al. VI. Genome structure and cognitive map of Williams syndrome. J Cogn Neurosci. 2000;12 Suppl 1:89–107. [DOI] [PubMed] [Google Scholar]
- 31.Schultz RT, Grelotti DJ, Pober B. Genetics of childhood disorders: XXVI. Williams syndrome and brain-behavior relationships. J Am Acad Child Adolesc Psychiatry. 2001;40(5):606–609. [DOI] [PubMed] [Google Scholar]
- 32.Bellugi U, Bihrle A, Jernigan T, Trauner D, Doherty S. Neuropsychological, neurological, and neuroanatomical profile of Williams syndrome. Am J Med Genet Suppl. 1990;6:115–125. [DOI] [PubMed] [Google Scholar]
- 33.Dykens EM. Anxiety, fears, and phobias in persons with Williams syndrome. Dev Neuropsychol. 2003;23(1–2):291–316. [DOI] [PubMed] [Google Scholar]
- 34.Morris CA, Thomas IT, Greenberg F. Williams syndrome: autosomal dominant inheritance. Am J Med Genet. 1993;47(4):478–481. [DOI] [PubMed] [Google Scholar]
- 35.Sadler LS, Robinson LK, Verdaasdonk KR, Gingell R. The Williams syndrome: evidence for possible autosomal dominant inheritance. Am J Med Genet. 1993;47(4):468–470. [DOI] [PubMed] [Google Scholar]
- 36.Meng X, Lu X, Li Z, et al. Complete physical map of the common deletion region in Williams syndrome and identification and characterization of three novel genes. Hum Genet. 1998;103(5):590–599. [DOI] [PubMed] [Google Scholar]
- 37.Peoples R, Franke Y, Wang YK, et al. A physical map, including a BAC/PAC clone contig, of the Williams-Beuren syndrome--deletion region at 7q11.23. Am J Hum Genet. 2000;66(1):47–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osborne LR. Williams-Beuren syndrome: unraveling the mysteries of a microdeletion disorder. Mol Genet Metab. 1999;67(1):1–10. [DOI] [PubMed] [Google Scholar]
- 39.Turner DJ, Miretti M, Rajan D, et al. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40(1):90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berg JS, Brunetti-Pierri N, Peters SU, et al. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams-Beuren syndrome region. Genet Med. 2007;9(7):427–441. [DOI] [PubMed] [Google Scholar]
- 41.Francke U Williams-Beuren syndrome: genes and mechanisms. Hum Mol Genet. 1999;8(10):1947–1954. [DOI] [PubMed] [Google Scholar]
- 42.Ewart AK, Morris CA, Atkinson D, et al. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet. 1993;5(1):11–16. [DOI] [PubMed] [Google Scholar]
- 43.Dai L, Bellugi U, Chen XN, et al. Is it Williams syndrome? GTF2IRD1 implicated in visual-spatial construction and GTF2I in sociability revealed by high resolution arrays. Am J Med Genet A. 2009;149A(3):302–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Antonell A, Del Campo M, Magano LF, et al. Partial 7q11.23 deletions further implicate GTF2I and GTF2IRD1 as the main genes responsible for the Williams-Beuren syndrome neurocognitive profile. J Med Genet. 2010;47(5):312–320. [DOI] [PubMed] [Google Scholar]
- 45.Doyle TF, Bellugi U, Korenberg JR, Graham J. “Everybody in the world is my friend” hypersociability in young children with Williams syndrome. Am J Med Genet A. 2004;124A(3):263–273. [DOI] [PubMed] [Google Scholar]
- 46.Crespi BJ, Hurd PL. Cognitive-behavioral phenotypes of Williams syndrome are associated with genetic variation in the GTF2I gene, in a healthy population. BMC Neurosci 2014;15:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jackowski AP, Rando K, Maria de Araújo C, Del Cole CG, Silva I, Tavares de Lacerda AL. Brain abnormalities in Williams syndrome: a review of structural and functional magnetic resonance imaging findings. Eur J Paediatr Neurol. 2009;13(4):305–316. [DOI] [PubMed] [Google Scholar]
- 48.Meyer-Lindenberg A, Hariri AR, Munoz KE, et al. Neural correlates of genetically abnormal social cognition in Williams syndrome. Nat Neurosci. 2005;8(8):991–993. [DOI] [PubMed] [Google Scholar]
- 49.Haas BW, Mills D, Yam A, Hoeft F, Bellugi U, Reiss A. Genetic influences on sociability: heightened amygdala reactivity and event-related responses to positive social stimuli in Williams syndrome. J Neurosci. 2009;29(4):1132–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haas BW, Hoeft F, Searcy YM, Mills D, Bellugi U, Reiss A. Individual differences in social behavior predict amygdala response to fearful facial expressions in Williams syndrome. Neuropsychologia 2010;48(5):1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swartz JR, Waller R, Bogdan R, et al. A Common Polymorphism in a Williams Syndrome Gene Predicts Amygdala Reactivity and Extraversion in Healthy Adults. Biol Psychiatry. 2017;81(3):203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bower BD, Jeavons PM. The “happy puppet” syndrome. Arch Dis Child. 1967;42(223):298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buntinx IM, Hennekam RC, Brouwer OF, et al. Clinical profile of Angelman syndrome at different ages. Am J Med Genet. 1995;56(2):176–183. [DOI] [PubMed] [Google Scholar]
- 54.Elian M Fourteen happy puppets. Clin Pediatr (Phila). 1975;14(10):902–908. [DOI] [PubMed] [Google Scholar]
- 55.Richman DM, Gernat E, Teichman H. Effects of social stimuli on laughing and smiling in young children with Angelman syndrome. Am J Ment Retard. 2006;111(6):442–446. [DOI] [PubMed] [Google Scholar]
- 56.Oliver C, Demetriades L, Hall S. Effects of environmental events on smiling and laughing behavior in Angelman syndrome. Am J Ment Retard. 2002;107(3):194–200. [DOI] [PubMed] [Google Scholar]
- 57.Jiang Y, Lev-Lehman E, Bressler J, Tsai TF, Beaudet AL. Genetics of Angelman syndrome. Am J Hum Genet. 1999;65(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koolen DA, Vissers LE, Pfundt R, et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet. 2006;38(9):999–1001. [DOI] [PubMed] [Google Scholar]
- 59.Sharp AJ, Hansen S, Selzer RR, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38(9):1038–1042. [DOI] [PubMed] [Google Scholar]
- 60.Shaw-Smith C, Pittman AM, Willatt L, et al. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat Genet. 2006;38(9):1032–1037. [DOI] [PubMed] [Google Scholar]
- 61.Koolen DA, Sharp AJ, Hurst JA, et al. Clinical and molecular delineation of the 17q21.31 microdeletion syndrome. J Med Genet. 2008;45(11):710–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tan TY, Aftimos S, Worgan L, et al. Phenotypic expansion and further characterisation of the 17q21.31 microdeletion syndrome. J Med Genet. 2009;46(7):480–489. [DOI] [PubMed] [Google Scholar]
- 63.Egger JI, Wingbermühle E, Verhoeven WM, et al. Hypersociability in the behavioral phenotype of 17q21.31 microdeletion syndrome. Am J Med Genet A. 2013;161A(1):21–26. [DOI] [PubMed] [Google Scholar]
- 64.Zollino M, Orteschi D, Murdolo M, et al. Mutations in KANSL1 cause the 17q21.31 microdeletion syndrome phenotype. Nat Genet. 2012;44(6):636–638. [DOI] [PubMed] [Google Scholar]
- 65.Koolen DA, Kramer JM, Neveling K, et al. Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nat Genet. 2012;44(6):639–641. [DOI] [PubMed] [Google Scholar]
- 66.Koolen DA, Pfundt R, Linda K, et al. The Koolen-de Vries syndrome: a phenotypic comparison of patients with a 17q21.31 microdeletion versus a KANSL1 sequence variant. Eur J Hum Genet. 2016;24(5):652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moreno-Igoa M, Hernández-Charro B, Bengoa-Alonso A, et al. KANSL1 gene disruption associated with the full clinical spectrum of 17q21.31 microdeletion syndrome. BMC Med Genet. 2015;16:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.DeSilva U, Elnitski L, Idol JR, et al. Generation and comparative analysis of approximately 3.3 Mb of mouse genomic sequence orthologous to the region of human chromosome 7q11.23 implicated in Williams syndrome. Genome Res. 2002;12(1):3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Segura-Puimedon M, Sahún I, Velot E, et al. Heterozygous deletion of the Williams-Beuren syndrome critical interval in mice recapitulates most features of the human disorder. Hum Mol Genet. 2014;23(24):6481–6494. [DOI] [PubMed] [Google Scholar]
- 70.Li HH, Roy M, Kuscuoglu U, et al. Induced chromosome deletions cause hypersociability and other features of Williams-Beuren syndrome in mice. EMBO Mol Med. 2009;1(1):50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moy SS, Nadler JJ, Perez A, et al. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes, brain, and behavior. 2004;3(5):287–302. [DOI] [PubMed] [Google Scholar]
- 72.LINDZEY G, WINSTON H, MANOSEVITZ M Social dominance in inbred mouse strains. Nature. 1961;191:474–476. [DOI] [PubMed] [Google Scholar]
- 73.Young EJ, Lipina T, Tam E, et al. Reduced fear and aggression and altered serotonin metabolism in Gtf2ird1-targeted mice. Genes Brain Behav. 2008;7(2):224–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sakurai T, Dorr NP, Takahashi N, McInnes LA, Elder GA, Buxbaum JD. Haploinsufficiency of Gtf2i, a gene deleted in Williams Syndrome, leads to increases in social interactions. Autism Res 2011;4(1):28–39. [DOI] [PubMed] [Google Scholar]
- 75.Hoogenraad CC, Koekkoek B, Akhmanova A, et al. Targeted mutation of Cyln2 in the Williams syndrome critical region links CLIP-115 haploinsufficiency to neurodevelopmental abnormalities in mice. Nat Genet. 2002;32(1):116–127. [DOI] [PubMed] [Google Scholar]
- 76.Meng Y, Zhang Y, Tregoubov V, et al. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35(1):121–133. [DOI] [PubMed] [Google Scholar]
- 77.Stoppel DC, Anderson MP. Hypersociability in the Angelman syndrome mouse model. Exp Neurol. 2017;293:137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arbogast T, Iacono G, Chevalier C, et al. Mouse models of 17q21.31 microdeletion and microduplication syndromes highlight the importance of Kansl1 for cognition. PLoS Genet. 2017;13(7):e1006886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hunt CA, Schenker LJ, Kennedy MB. PSD-95 is associated with the postsynaptic density and not with the presynaptic membrane at forebrain synapses. J Neurosci. 1996;16(4):1380–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sheng M, Sala C. PDZ domains and the organization of supramolecular complexes. Annu Rev Neurosci. 2001;24:1–29. [DOI] [PubMed] [Google Scholar]
- 81.Kim E, Cho KO, Rothschild A, Sheng M. Heteromultimerization and NMDA receptor-clustering activity of Chapsyn-110, a member of the PSD-95 family of proteins. Neuron. 1996;17(1):103–113. [DOI] [PubMed] [Google Scholar]
- 82.Feyder M, Karlsson RM, Mathur P, et al. Association of mouse Dlg4 (PSD-95) gene deletion and human DLG4 gene variation with phenotypes relevant to autism spectrum disorders and Williams’ syndrome. Am J Psychiatry. 2010;167(12):1508–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Winkler D, Daher F, Wüstefeld L, et al. Hypersocial behavior and biological redundancy in mice with reduced expression of PSD95 or PSD93. Behav Brain Res. 2017. [DOI] [PubMed] [Google Scholar]
- 84.Krueger-Burg D, Winkler D, Mitkovski M, et al. The SocioBox: A Novel Paradigm to Assess Complex Social Recognition in Male Mice. Front Behav Neurosci 2016;10:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ju A, Hammerschmidt K, Tantra M, Krueger D, Brose N, Ehrenreich H. Juvenile manifestation of ultrasound communication deficits in the neuroligin-4 null mutant mouse model of autism. Behav Brain Res. 2014;270:159–164. [DOI] [PubMed] [Google Scholar]
- 86.Jamain S, Radyushkin K, Hammerschmidt K, et al. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc Natl Acad Sci U S A. 2008;105(5):1710–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bey AL, Jiang YH. Overview of mouse models of autism spectrum disorders. Curr Protoc Pharmacol. 2014;66:5.6661–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang X, Xu Q, Bey AL, Lee Y, Jiang YH. Transcriptional and functional complexity of Shank3 provides a molecular framework to understand the phenotypic heterogeneity of SHANK3 causing autism and Shank3 mutant mice. Mol Autism 2014;5:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhuo M, Small SA, Kandel ER, Hawkins RD. Nitric oxide and carbon monoxide produce activity-dependent long-term synaptic enhancement in hippocampus. Science. 1993;260(5116):1946–1950. [DOI] [PubMed] [Google Scholar]
- 90.Dawson VL, Dawson TM. Nitric oxide in neurodegeneration. Prog Brain Res. 1998;118:215–229. [DOI] [PubMed] [Google Scholar]
- 91.Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci U S A. 1990;87(2):682–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bredt DS, Glatt CE, Hwang PM, Fotuhi M, Dawson TM, Snyder SH. Nitric oxide synthase protein and mRNA are discretely localized in neuronal populations of the mammalian CNS together with NADPH diaphorase. Neuron. 1991;7(4):615–624. [DOI] [PubMed] [Google Scholar]
- 93.Bredt DS, Ferris CD, Snyder SH. Nitric oxide synthase regulatory sites. Phosphorylation by cyclic AMP-dependent protein kinase, protein kinase C, and calcium/calmodulin protein kinase; identification of flavin and calmodulin binding sites. J Biol Chem. 1992;267(16):10976–10981. [PubMed] [Google Scholar]
- 94.Schuman EM, Madison DV. Nitric oxide and synaptic function. Annu Rev Neurosci. 1994;17:153–183. [DOI] [PubMed] [Google Scholar]
- 95.Roy B, Halvey EJ, Garthwaite J. An enzyme-linked receptor mechanism for nitric oxide-activated guanylyl cyclase. J Biol Chem. 2008;283(27):18841–18851. [DOI] [PubMed] [Google Scholar]
- 96.Neitz A, Mergia E, Eysel UT, Koesling D, Mittmann T. Presynaptic nitric oxide/cGMP facilitates glutamate release via hyperpolarization-activated cyclic nucleotide-gated channels in the hippocampus. Eur J Neurosci. 2011;33(9):1611–1621. [DOI] [PubMed] [Google Scholar]
- 97.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3(2):193–197. [DOI] [PubMed] [Google Scholar]
- 98.Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993;75(7):1273–1286. [DOI] [PubMed] [Google Scholar]
- 99.Tanda K, Nishi A, Matsuo N, et al. Abnormal social behavior, hyperactivity, impaired remote spatial memory, and increased D1-mediated dopaminergic signaling in neuronal nitric oxide synthase knockout mice. Mol Brain. 2009;2:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Trainor BC, Workman JL, Jessen R, Nelson RJ. Impaired nitric oxide synthase signaling dissociates social investigation and aggression. Behav Neurosci. 2007;121(2):362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Britsch S The neuregulin-I/ErbB signaling system in development and disease. Adv Anat Embryol Cell Biol. 2007;190:1–65. [PubMed] [Google Scholar]
- 102.Mei L, Nave KA. Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron. 2014;83(1):27–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moy SS, Ghashghaei HT, Nonneman RJ, et al. Deficient NRG1-ERBB signaling alters social approach: relevance to genetic mouse models of schizophrenia. J Neurodev Disord. 2009;1(4):302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang JZ, Si TM, Ruan Y, et al. Association study of neuregulin 1 gene with schizophrenia. Mol Psychiatry. 2003;8(7):706–709. [DOI] [PubMed] [Google Scholar]
- 105.Stefánsson H, Thorgeirsson TE, Gulcher JR, Stefánsson K. Neuregulin 1 in schizophrenia: out of Iceland. Mol Psychiatry. 2003;8(7):639–640. [DOI] [PubMed] [Google Scholar]
- 106.Hashimoto R, Straub RE, Weickert CS, Hyde TM, Kleinman JE, Weinberger DR. Expression analysis of neuregulin-1 in the dorsolateral prefrontal cortex in schizophrenia. Mol Psychiatry. 2004;9(3):299–307. [DOI] [PubMed] [Google Scholar]
- 107.Law AJ, Lipska BK, Weickert CS, et al. Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5’ SNPs associated with the disease. Proc Natl Acad Sci U S A. 2006;103(17):6747–6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brennand KJ, Gage FH. Modeling psychiatric disorders through reprogramming. Dis Model Mech. 2012;5(1):26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chisholm K A three year follow-up of attachment and indiscriminate friendliness in children adopted from Romanian orphanages. Child Dev. 1998;69(4):1092–1106. [PubMed] [Google Scholar]
- 110.Bruce J, Tarullo AR, Gunnar MR. Disinhibited social behavior among internationally adopted children. Dev Psychopathol. 2009;21(1):157–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.O’Connor TG, Marvin RS, Rutter M, Olrick JT, Britner PA, Team EaRAS. Child-parent attachment following early institutional deprivation. Dev Psychopathol 2003;15(1):19–38. [DOI] [PubMed] [Google Scholar]
- 112.O’Connor TG, Rutter M. Attachment disorder behavior following early severe deprivation: extension and longitudinal follow-up. English and Romanian Adoptees Study Team. J Am Acad Child Adolesc Psychiatry 2000;39(6):703–712. [DOI] [PubMed] [Google Scholar]
- 113.Hodges J, Tizard B. Social and family relationships of ex-institutional adolescents. J Child Psychol Psychiatry. 1989;30(1):77–97. [DOI] [PubMed] [Google Scholar]
- 114.Roy P, Rutter M, Pickles A. Institutional care: associations between overactivity and lack of selectivity in social relationships. J Child Psychol Psychiatry. 2004;45(4):866–873. [DOI] [PubMed] [Google Scholar]
- 115.Kreppner JM, O’Connor TG, Rutter M, Team EaRAS. Can inattention/overactivity be an institutional deprivation syndrome? J Abnorm Child Psychol. 2001;29(6):513–528. [DOI] [PubMed] [Google Scholar]
- 116.Rutter ML, Kreppner JM, O’Connor TG, team EaRAEs. Specificity and heterogeneity in children’s responses to profound institutional privation. Br J Psychiatry. 2001;179:97–103. [DOI] [PubMed] [Google Scholar]
- 117.Pears KC, Bruce J, Fisher PA, Kim HK. Indiscriminate friendliness in maltreated foster children. Child Maltreat. 2010;15(1):64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bakermans-Kranenburg MJ, Steele H, Zeanah CH, et al. Attachment and Emotional Development in Institutional Care: Characteristics and Catch-Up. Monogr Soc Res Child Dev. 2011;76(4):62–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Olsavsky AK, Telzer EH, Shapiro M, et al. Indiscriminate amygdala response to mothers and strangers after early maternal deprivation. Biol Psychiatry. 2013;74(11):853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bouet V, Lecrux B, Tran G, Freret T. Effect of pre- versus post-weaning environmental disturbances on social behaviour in mice. Neurosci Lett. 2011;488(2):221–224. [DOI] [PubMed] [Google Scholar]
- 121.Tsuda MC, Ogawa S. Long-lasting consequences of neonatal maternal separation on social behaviors in ovariectomized female mice. PLoS One. 2012;7(3):e33028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Carlyle BC, Duque A, Kitchen RR, et al. Maternal separation with early weaning: a rodent model providing novel insights into neglect associated developmental deficits. Dev Psychopathol. 2012;24(4):1401–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Halmoy A, Johansson S, Winge I, McKinney JA, Knappskog PM, Haavik J. Attention-deficit/hyperactivity disorder symptoms in offspring of mothers with impaired serotonin production. Arch Gen Psychiatry. 2011;67(10):1033–1043. [DOI] [PubMed] [Google Scholar]
- 124.Kong A, Thorleifsson G, Frigge ML, et al. The nature of nurture: Effects of parental genotypes. Science. 2018;359(6374):424–428. [DOI] [PubMed] [Google Scholar]
- 125.Morgan JE, Hammen C, Lee SS. Parental Serotonin Transporter Polymorphism (5-HTTLPR) Moderates Associations of Stress and Child Behavior With Parenting Behavior. J Clin Child Adolesc Psychol 2016:1–12. [DOI] [PubMed] [Google Scholar]
- 126.Lee SS, Chronis-Tuscano A, Keenan K, et al. Association of maternal dopamine transporter genotype with negative parenting: evidence for gene x environment interaction with child disruptive behavior. Mol Psychiatry. 2010;15(5):548–558. [DOI] [PubMed] [Google Scholar]
- 127.Avinun R, Knafo-Noam A. Parental brain-derived neurotrophic factor genotype, child prosociality, and their interaction as predictors of parents’ warmth. Brain Behav. 2017;7(5):e00685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Berton O, McClung CA, Dileone RJ, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science (New York, NY). 2006;311(5762):864–868. [DOI] [PubMed] [Google Scholar]
- 129.Zupan B, Sharma A, Frazier A, Klein S, Toth M. Programming social behavior by the maternal fragile X protein. Genes Brain and Behavior. 2016;15(6):578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cohen IL, Vietze PM, Sudhalter V, Jenkins EC, Brown WT. Effects of age and communication level on eye contact in fragile X males and non-fragile X autistic males. Am J Med Genet. 1991;38(2–3):498–502. [DOI] [PubMed] [Google Scholar]
- 131.Spencer CM, Alekseyenko O, Serysheva E, Yuva-Paylor LA, Paylor R. Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes, brain, and behavior. 2005;4(7):420–430. [DOI] [PubMed] [Google Scholar]
- 132.Spencer CM, Graham DF, Yuva-Paylor LA, Nelson DL, Paylor R. Social behavior in Fmr1 knockout mice carrying a human FMR1 transgene. Behavioral neuroscience. 2008;122(3):710–715. [DOI] [PubMed] [Google Scholar]
- 133.McNaughton CH, Moon J, Strawderman MS, Maclean KN, Evans J, Strupp BJ. Evidence for social anxiety and impaired social cognition in a mouse model of fragile X syndrome. Behavioral neuroscience. 2008;122(2):293–300. [DOI] [PubMed] [Google Scholar]
- 134.Liu ZH, Smith CB. Dissociation of social and nonsocial anxiety in a mouse model of fragile X syndrome. Neurosci Lett. 2009;454(1):62–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mines MA, Yuskaitis CJ, King MK, Beurel E, Jope RS. GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS One. 2010;5(3):e9706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Moy SS, Nadler JJ, Young NB, et al. Social approach in genetically engineered mouse lines relevant to autism. Genes Brain Behav. 2009;8(2):129–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Toth M Mechanisms of non-genetic inheritance and psychiatric disorders. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2015;40(1):129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hessl D, Wang JM, Schneider A, et al. Decreased fragile X mental retardation protein expression underlies amygdala dysfunction in carriers of the fragile X premutation. Biological psychiatry. 2011;70(9):859–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Carlezon WA, Jr., Thomas MJ. Biological substrates of reward and aversion: a nucleus accumbens activity hypothesis. Neuropharmacology. 2009;56 Suppl 1:122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Al-Hasani R, McCall JG, Shin G, et al. Distinct Subpopulations of Nucleus Accumbens Dynorphin Neurons Drive Aversion and Reward. Neuron. 2015;87(5):1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cecil CA, Smith RG, Walton E, Mill J, McCrory EJ, Viding E. Epigenetic signatures of childhood abuse and neglect: Implications for psychiatric vulnerability. J Psychiatr Res. 2016;83:184–194. [DOI] [PubMed] [Google Scholar]
- 142.Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13(2):97–109. [DOI] [PubMed] [Google Scholar]
- 143.Roth TL, Lubin FD, Funk AJ, Sweatt JD. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry. 2009;65(9):760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Murgatroyd C, Patchev AV, Wu Y, et al. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci. 2009;12(12):1559–1566. [DOI] [PubMed] [Google Scholar]
- 145.Gutierrez-Arcelus M, Lappalainen T, Montgomery SB, et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. Elife. 2013;2:e00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Heller EA, Cates HM, Peña CJ, et al. Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nat Neurosci. 2014;17(12):1720–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Insel TR, Shapiro LE. Oxytocin receptor distribution reflects social organization in monogamous and polygamous voles. Proc Natl Acad Sci U S A. 1992;89(13):5981–5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hung LW, Neuner S, Polepalli JS, et al. Gating of social reward by oxytocin in the ventral tegmental area. Science. 2017;357(6358):1406–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]