

Abstract
Introduction:
We sought to determine which therapeutically targetable immune checkpoints, costimulatory signals, and other tumor microenvironment (TME) factors are independently associated with immune cytolytic activity (CYT), a gene expression signature of activated effector T cells, in human glioblastoma (GBM).
Methods:
GlioVis was accessed for RNA-seq data from The Cancer Genome Atlas (TCGA). For subjects with treatment-naïve, primary GBM, we quantified mRNA expression of 28 therapeutically targetable TME factors. CYT (geometric mean of GZMA and PRF1 expression) was calculated for each tumor. Multiple linear regression was performed to determine the relationship between the dependent variable (CYT) and mRNA expression of each of the 28 factors. Variables associated with CYT in multivariate analysis were subsequently evaluated for this association in an independent cohort of newly diagnosed GBMs from the Chinese Glioma Cooperative Group (CGCG).
Results:
109 TCGA tumors were analyzed. The final multiple linear regression model included the following variables, each positively associated with CYT except VEGF-A (negative association): CSF-1 (p=0.003), CD137 (p=0.042), VEGF-A (p<0.001), CTLA4 (p=0.028), CD40 (p=0.023), GITR (p=0.020), IL6 (p=0.02), and OX40 (p<0.001). In CGCG (n=52), each of these variables remained significantly associated with CYT in univariate analysis except for VEGF-A. In multivariate analysis, only CTLA4 and CD40 remained statistically significant.
Conclusions:
Using multivariate modeling of RNA-seq gene expression data, we identified therapeutically targetable TME factors that are independently associated with intratumoral cytolytic T-cell activity in human GBM. As a myriad of systemic immunotherapies are now available for investigation, our results could inform rational combinations for evaluation in GBM.
Keywords: Glioblastoma, T cells, RNA-seq, Immunotherapy
Introduction
Immunotherapy, led by programmed cell death-1 / programmed death-ligand 1 (PD-1/PD-L1) immune checkpoint blockade, has dramatically altered the landscape of cancer treatment in multiple tumor types [1]. However, despite promise in preclinical and early phase studies [2,3], single agent programmed death 1 (PD-1) inhibition is ineffective for recurrent GBM in the absence of a rare, markedly hypermutated tumor [4–6]. This is due, in part, to the relative paucity of intratumoral T cells in GBM compared to cancers with carcinogen-induced mutational signatures and high tumor mutational burden (TMB) [7–11]. Even in GBMs with higher than average TMB, there does not appear to be a resultant influx of CD8+ T cells or increase in PD-1/PD-L1 expression [7]. Furthermore, approaches for increasing intratumoral T cells in GBM, such as vaccines, oncolytic viruses, or chimeric antigen receptor (CAR) T cells, are hindered by a severely immunosuppressive tumor microenvironment [12–14]. An improved understanding of the factors that influence both the infiltration and killing activity of CD8+ T cells in GBM may allow for rational immunotherapeutic targeting in this disease.
The intratumoral cytolytic T cell activity index [henceforth referred to as immune cytolytic activity (CYT)] is a validated gene expression signature of granzyme A (GZMA) and perforin-1 (PRF1) [10]. GZMA is a tryptase that induces caspase-independent programmed cell death, and PRF1 is a pore-forming enzyme that mediates entry of granzymes into target cells [15]. Both enzymes are produced by activated cytolytic CD8+ T cells and are upregulated during productive clinical responses to immune checkpoint inhibitors [16,17]. Using in silico RNAseq analysis from previously untreated tumor samples, we aimed to determine which therapeutically targetable inhibitory and stimulatory immune microenvironment factors are associated with CYT in newly diagnosed, primary GBM. Because many of these factors are correlated with one another and may not truly be driving CYT, we used multiple linear regression modeling to determine those that are independently associated with CYT.
Methods
GBM Samples and RNAseq
Using GlioVis (http://gliovis.bioinfo.cnio.es/), a web application for data visualization and analysis to explore previously published brain tumor gene expression datasets [18], we downloaded clinical and RNAseq data from The Cancer Genome Atlas (TCGA) Project [19]. We included only patients with newly diagnosed, treatment-naïve, IDH-wild type GBM whose tumors had RNAseq expression data for our genes of interest. In addition, we downloaded clinical and RNAseq data for 52 patients with these same characteristics from the Chinese Glioma Cooperative Group (CGCG) cohort [20]. RNAseq data is processed in GlioVis through normalization of count reads from the pre-processed data (sequence alignment and transcript abundance estimation), followed by addition of a 0.5 pseudocount (to avoid infinite value upon log transformation) and subsequent log2 transformation. CYT for patients in the TCGA cohort was downloaded directly from the original Cell publication through PubMed Central (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4856474) [10], and CYT for patients in the CGCG cohort [20] was calculated as the geometric mean of GZMA and PRF1 mRNA expression according to the method of Rooney et al [10].
We pre-specified 28 immune checkpoints, immune co-stimulatory receptors, and other immunomodulatory factors for analysis, each of which is currently therapeutically targetable with approved or investigational agents and hypothesized to be associated with intratumoral T cell number and/or effector function [21–24]. The 28 factors are displayed in Table 1 and categorized as an immune checkpoint, immune co-stimulatory receptor, or other immunomodulatory factor.
Table 1.
List of 28 therapeutically targetable immune checkpoints, immune co-stimulatory receptors, and other immunomodulatory cytokines and receptors evaluated for an association with immune cytolytic activity (CYT) in newly diagnosed, untreated glioblastoma (GBM) (alphabetical order)
| Category |
|---|
| Candidate variable |
| Immune checkpoints |
| A2aR (ADORA2A) |
| CD73 (NT5E) |
| CEACAM1 |
| CTLA-4 |
| ICOS |
| LAG3 |
| PDL1 |
| PDL2 |
| PVRIG |
| TIGIT |
| TIM3 (HAVCR2) |
| Immune co-stimulatory receptors |
| CD40 (TNFRSF5) |
| CD137 (TNFRSF9) |
| GITR (TNFRSF18) |
| OX40 (TNFRSF9) |
| Other Immunomodulatory Factors |
| ARG1 |
| CSF-1 |
| GLUT1 (SLC2A1) |
| IDO1 |
| IL-6 |
| IL10 |
| MCT1 |
| MCT2 |
| MTOR |
| STAT3 |
| TDO |
| TGFB1 |
| VEGF-A |
Statistical Analysis
Patient and tumor characteristics were examined using descriptive statistics. The dependent variable CYT in the TCGA dataset was heavily right skewed (Figure 1) and therefore log10-transformed for the linear regression analysis. In the TCGA cohort, simple linear regression was used to screen mRNA expression of each of the 28 variables, as well as age, sex, O(6)-methylguanine-DNA methyltransferase (MGMT) promoter methylation status, TCGA molecular subtype (classical, proneural, mesenchymal, or neural) and TMB [calculated as the number of non-synonymous mutations per megabase (Mb)], for its association with log-transformed CYT. Each variable was also assessed visually for its association with CYT using scatterplots. Significance of each of the regression coefficients (i.e., p-value) was adjusted using the Benjamini-Hochberg false discovery rate (FDR) to account for multiple testing. All variables with a raw p-value smaller than the associated Benjamini-Hochberg critical value [critical value (i/m)Q, where i = rank of raw p-value from smallest to largest, m = number of tests (28), and Q false discovery rate (0.05)] were entered into a multiple linear regression model. Correlation between the variables entered into the multiple linear regression model was examined to determine whether collinearity was a concern for the multivariate model; no pair of variables had a correlation coefficient greater than 0.75. Backward selection was performed manually in the multivariate analysis with an exit criterion of p>0.05. Once the final multivariate model was reached, we examined model diagnostics using residual plots versus predicted plots and quantile-quantile plots for the residuals. All model assumptions were met adequately. Six patients were identified as possible outliers based on Cook’s distance values greater than 0.04 (4/n = 4/109). Results were not substantially different when these outliers were excluded, and results reported are therefore based on the full dataset. Adjusted R-squared was calculated for the final model. Next, each of the variables included in the final model derived from the TCGA cohort was assessed in univariate analysis in the CGCG cohort (n=52) for its correlation with CYT (untransformed due to normal distribution) using the Spearman correlation coefficient. In addition, the final multivariate model from the TCGA cohort was applied to the CGCG cohort. Statistical tests were two-sided, with a P value of 0.05 or lower considered to indicate statistical significance. All statistical tests were performed with the use of Stata software, version 14 (StataCorp, College Station, TX).
Fig. 1.
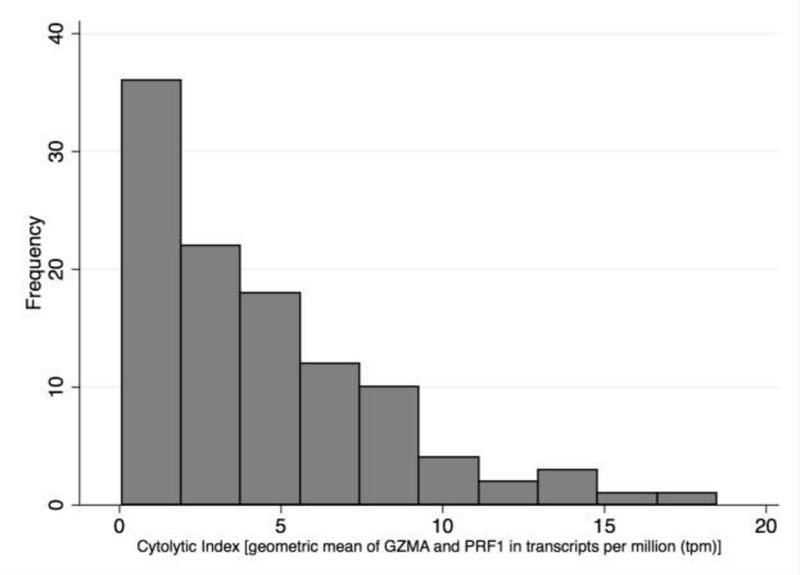
Cytolytic activity (CYT), defined as the geometric mean of GZMA and PRF1 expression in transcripts per million (TPM), in The Cancer Genome Atlas (TCGA) cohort of previously untreated glioblastoma (GBM) specimens [isocitrate dehydrogenase (IDH) wild-type only, N=109]
Results
Patient Characteristics
109 patients were included in the TCGA derivation cohort. Median age was 65 (range, 24–89; IQR, 55–73), and 72 (66%) were male. MGMT promoter methylation status was available for 83 patients; 49 (59%) were unmethylated, and 34 (41%) were methylated. TCGA molecular subtype was available for 106 patients; 31 (29%) were classical, 31 (29%) mesenchymal, 21 (20%) neural, and 23 (22%) proneural. Median TMB was 1.4 mutations/Mb (range, 0.1–3.8; IQR, 1.1–1.6). Median CYT (log-average expression of GZMA and PRF1, transcripts per million) was 3.57 (range, 0.08–18.5; IQR, 1.36–6.34; Figure 1).
Fifty-two patients were included in the CGCG validation cohort. Median age was 54 (range, 25–81; IQR, 43–59), and 36 (70%) were male. Twenty-two (42%) were classical TCGA subtype, 17 (33%) mesenchymal, 5 (10%) neural, and 8 (15%) proneural. MGMT methylation status and TMB were not available. Median CYT was 5.7 (range, 2.4–9.3; IQR, 4.9–6.5).
TCGA Cohort
The results of univariate linear regression analyses for each of the 28 immune microenvironment candidate variables, as well as associated p values and critical values according to the Benjamini Hochberg FDR, are displayed in Table 2. Sex (p=0.63), age (p=0.99), MGMT promoter methylation status (p=0.56), TCGA molecular subtype (p=0.34), and TMB (p=0.30) were not associated with CYT. The final multivariate model is displayed in Table 3. Cytotoxic T-lymphocyte antigen-4 (CTLA-4), CD40, CD137, glucocorticoid-induced TNFR-related protein (GITR), OX40, colony-stimulating factor 1 (CSF1), interleukin-6 (IL6), and vascular endothelial growth factor A (VEGF-A) were statistically significantly and independently associated with log-transformed CYT in multivariate analysis (p<0.05 for each variable, adjusted R2 = 0.65, F=26.48 on 100 df). Scatter plots demonstrating the association between each of these variables and CYT (untransformed), accompanied by associated Spearman correlation coefficients, are displayed in Figure 2A.
Table 2.
Results of univariate linear regression for the association between 28 molecules in the glioblastoma (GBM) immune microenvironment and intratumoral immune cytolytic activity (CYT) in The Cancer Genome Atlas (TCGA) cohort (bolded variables are statistically significant after controlling the false discovery rate using the Benjamini-Hochberg method)
| Variable | Coefficient | Standard Error |
t value | p value | Benjamini- Hochberg critical value |
|---|---|---|---|---|---|
| Immune checkpoints | |||||
| A2aR (ADORA2A) | 0.11 | 0.07 | 1.7 | 0.10 | 0.038 |
| CD73 (NT5E) | 0.05 | 0.05 | 1.2 | 0.25 | 0.043 |
| CEACAM1 | 0.04 | 0.03 | 1.6 | 0.12 | 0.039 |
| CTLA-4 | 0.16 | 0.03 | 6.0 | <0.001 | 0.007 |
| ICOS | 0.12 | 0.02 | 5.1 | <0.001 | 0.014 |
| LAG3 | 0.07 | 0.04 | 1.9 | 0.065 | 0.036 |
| PDL1 | 0.11 | 0.03 | 4.3 | <0.001 | 0.027 |
| PDL2 | 0.15 | 0.03 | 5.1 | <0.001 | 0.016 |
| PVRIG | −0.03 | 0.04 | −0.7 | 0.47 | 0.048 |
| TIGIT | 0.09 | 0.03 | 3.5 | 0.001 | 0.029 |
| TIM3 (HAVCR2) | 0.24 | 0.04 | 6.5 | <0.001 | 0.002 |
| Immune co-stimulatory receptors | |||||
| CD40 (TNFRSF5) | 0.25 | 0.04 | 6.0 | <0.001 | 0.009 |
| CD137 (TNFRSF9) | 0.16 | 0.02 | 6.5 | <0.001 | 0.004 |
| GITR (TNFRSF18) | 0.15 | 0.02 | 5.9 | <0.001 | 0.012 |
| OX40 (TNFRSF9) | 0.17 | 0.04 | 4.6 | <0.001 | 0.023 |
| Other immunomodulatory factors | |||||
| ARG1 | 0.05 | 0.03 | 2.0 | 0.046 | 0.034 |
| CSF-1 | 0.19 | 0.04 | 4.4 | <0.001 | 0.025 |
| GLUT1 (SLC2A1) | −0.05 | 0.05 | −1.1 | 0.30 | 0.045 |
| IDO1 | 0.08 | 0.02 | 4.9 | <0.001 | 0.018 |
| IL-6 | 0.11 | 0.02 | 5.9 | <0.001 | 0.013 |
| IL10 | 0.10 | 0.02 | 4.6 | <0.001 | 0.021 |
| MCT1 | 0.11 | 0.09 | 1.3 | 0.21 | 0.041 |
| MCT2 | 0.02 | 0.03 | 0.6 | 0.55 | 0.050 |
| MTOR | 0.08 | 0.09 | 0.9 | 0.38 | 0.046 |
| STAT3 | 0.23 | 0.10 | 2.3 | 0.025 | 0.032 |
| TDO | 0.09 | 0.02 | 4.9 | <0.001 | 0.020 |
| TGFB1 | 0.31 | 0.05 | 6.1 | <0.001 | 0.005 |
| VEGF-A | −0.07 | 0.03 | −2.8 | 0.007 | 0.030 |
Table 3.
Final multiple linear regression model for the association of intratumoral immune cytolytic activity (CYT) with factors present in the glioblastoma (GBM) immune microenvironment
| Variable | Standardized beta coefficient |
Standard error |
t value | p value |
|---|---|---|---|---|
| Immune checkpoints | ||||
| CTLA-4 | 0.15 | 0.02 | 2.2 | 0.028 |
| Immune co-stimulatory receptors | ||||
| CD40 (TNFRSF5) | 0.16 | 0.03 | 2.3 | 0.023 |
| CD137 (TNFRSF9) | 0.15 | 0.02 | 2.1 | 0.042 |
| GITR (TNFRSF18) | 0.15 | 0.02 | 2.4 | 0.020 |
| OX40 (TNFRSF9) | 0.27 | 0.03 | 4.3 | <0.001 |
| Other immunomodulatory factors | ||||
| CSF-1 | 0.21 | 0.03 | 3.1 | 0.003 |
| IL-6 | 0.20 | 0.02 | 2.4 | 0.020 |
| VEGF-A | −0.32 | 0.02 | −4.6 | <0.001 |
Fig 2.
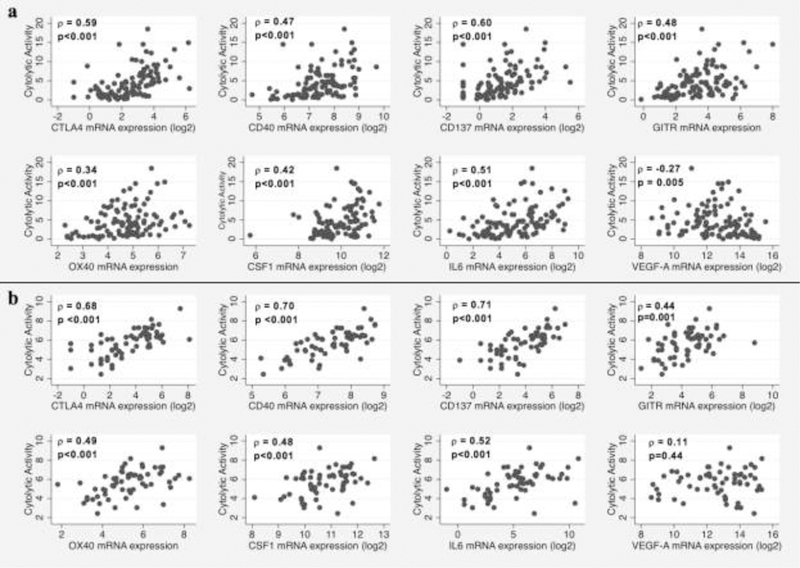
Scatterplots demonstrating a the relationship between the tumor immune microenvironment variables included in the final multivariate linear regression model and cytolytic activity (CYT) in the The Cancer Genome Atlas (TCGA) glioblastoma (GBM) cohort and b the relationship between these variables and CYT in the Chinese Glioma Cooperative Group (CGCG) validation cohort
Independent CGCG Cohort
Each of the immune checkpoints (CTLA-4), immune co-stimulatory receptors (CD40, CD137, GITR, OX40), and other immunomodulatory cytokines (CSF1, IL-6, VEGF-A) included in the final multivariate model from the TCGA cohort were evaluated in univariate analysis in the CGCG cohort for their relationship with CYT. Each of these factors was statistically significantly associated with CYT (untransformed) in univariate analysis with the exception of VEGF-A (Figure 2B). When the final multivariate model derived from the TCGA cohort was applied to the CGCG cohort, the adjusted R2 was similar (0.63). However, only CTLA4 (standardized beta coefficient = 0.41, p=0.002) and CD40 (standardized beta coefficient 0.36, p=0.009) remained statistically significantly associated with CYT (p<0.05). With the exception of IL-6, all other variables (CD137, GITR, OX40, CSF-1, and VEGF-A) maintained the same direction of association with CYT.
Discussion
Immune checkpoint inhibitors have demonstrated little efficacy as monotherapy in GBM, and studies of other immunotherapeutic approaches, including oncolytic viral therapy and dendritic cell vaccination, have generally failed to produce rates of response or stable disease above 20% [4,12,13,25,26]. Immunologically, GBM is characterized by a highly suppressive tumor microenvironment [12], and for most patients, there is scant intratumoral infiltration of effector T cells [27,28]. Well-described barriers to infiltration and/or activation of effector T cells in the GBM tumor microenvironment include immunosuppressive microglial cells and tumor-associated (M2) macrophages [29,30], upregulation of immune checkpoints on functionally exhausted cytotoxic T lymphocytes and other immune cells [31], the presence of regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) [32,33], and increased expression and secretion of immune inhibitory molecules by tumor and other cells [2,14]. With each of these immunosuppressive mechanisms involving modulation of different signaling pathways and gene expression levels, as well as the unprecedented number of targeted and immunomodulatory therapies in development or already approved across oncology [34], prioritization of individual targets and combinations of targets for preclinical and clinical evaluation in GBM is a significant challenge.
In this study utilizing in silico RNAseq analysis in untreated, de novo (IDH-wild type) GBM specimens from the TCGA database, we identified a set of immune checkpoints, immune co-stimulatory receptors, and other immunomodulatory cytokines whose mRNA expression levels are independently associated with CYT, a validated gene expression signature of immune cytolytic activity [10]. In addition, all factors that were evaluated for their relationship with CYT are currently therapeutically targetable by either approved or investigational systemic agents. Identification of markers in the tumor cells and tumor microenvironment that are associated with higher CYT in GBM leads to two hypotheses that may have therapeutic implications for patients: 1) for certain markers, increased expression occurs as a compensatory response by the tumor to evade a cytolytic attack mounted by tumor-specific T cells; 2) for other markers, increased expression that had initially driven a cytolytic attack by tumor-specific T cells is now being overmatched by tumor-mediated immunosuppression. In the former scenario, therapeutic inhibition of these factors may result in increased T cell cytolytic activity. In the latter scenario, therapeutic agonism of these factors may result in increased T cell cytolytic activity [10]. However, because up- or down-regulation of one checkpoint or cytokine in the tumor immune microenvironment is typically accompanied by altered expression of many others, it is difficult to know which are pathways are truly driving or suppressing immune activation against the tumor (as opposed to being turned on or off as a “bystander” to another more important mechanistic pathway) [24]. Through the use of multiple linear regression modeling, we were able to identify factors in the GBM immune microenvironment that are independently associated with CYT (i.e., factors that remained associated with CYT even after adjusting for the impact of other relevant immune checkpoints, co-stimulatory receptors, and cytokines). Further studies will be needed to determine whether therapeutic drug combinations to manipulate these factors will, in turn, directly impact intratumoral immune cytolytic activity and lead to an immune-mediated anti-tumor effect. Consistent with other studies [7,27,35], we found no association between the non-synonymous TMB and the degree of intratumoral immune cytolytic activity in GBM.
Each of the immune microenvironment factors that we identified as being independently associated with CYT have previously been shown to play a role in glioma-associated immune suppression or immune activation.[36] CTLA-4, an immune checkpoint molecule, is expressed on both “exhausted” CD8+ T cells and regulatory T cells (Tregs) and reduces early stages of T-cell expansion, primarily in tumor draining lymph nodes [37]. Consistent with its clinical activity in other solid tumors, CTLA-4 blockade in preclinical glioma models has shown efficacy only when used in combination with other immune checkpoint inhibitors [3,38]. Clinical data for CTLA-4 blockade in GBM is limited thus far to phase I data safety data [39]. CD40 is a cell-surface member of the tumor necrosis factor (TNF) receptor superfamily that is most prominently expressed on dendritic cells, B cells, and myeloid cells and leads to dendritic cell activation, thus playing a critical role in regulating T cell priming in tumors [40]. In GBM, CD40/CD40L mRNA expression has been associated with improved survival [41], and antitumor effects have been demonstrated with an anti-CD40 agonistic monoclonal antibody in mouse glioma models [42]. CD137, GITR, and OX40 are also members of the TNF receptor superfamily, and each are immune co-stimulatory receptors expressed on CD4+ and CD8+ T cells, as well as Tregs [43]. Preclinical models have demonstrated efficacy for agonist monoclonal antibodies against CD137 [44], GITR [45], and OX40 [46] in murine glioma, and a clinical trial is currently underway for an anti-CD137 antibody in recurrent GBM (NCT02658981). Lastly, CSF-1 and IL-6 are cytokines whose predominant immunosuppressive role in GBM has recently been linked to promoting tumor-associated macrophages and microglia [47,48]. Despite evidence of efficacy in preclinical glioma models [49], CSF1 receptor inhibition has not demonstrated efficacy in human glioma to date [50]. Trials of anti-IL-6 monoclonal antibodies have not yet commenced in GBM.
The primary limitation of our study is that it is currently unknown whether increased expression of GZMA and PRF1 in tumor-infiltrating lymphocytes, which comprise the quantitative measure of immune cytolytic activity (CYT) used as the dependent variable in our study, is associated with clinical benefit from immunotherapy in GBM. This is contrast to multiple other solid tumors with higher TMB and likelihood of response to immunotherapy, where productive clinical responses to immune checkpoint inhibitors are correlated with dramatically upregulated transcript levels of GZMA and PRF [16,17]. Thus, we cannot prove from our study that therapeutic manipulation of these factors would result in improved immune control of GBM. However, since increased T cell cytolytic activity is prerequisite to any effective anti-tumor immune response, our identification of factors that are associated with CYT can be used to stimulate hypotheses for rational immunotherapy combinations. Combinations of systemic therapies targeted against the markers we identified should be studied and proven efficacious in animal glioma models prior to being instituted in clinical trials for GBM.
Another limitation of this study was that our findings from the TCGA cohort could only be replicated in the CGCG cohort in univariate analysis (i.e., not all variables in the final multivariate model from the TCGA cohort maintained a statistically significant, independent association with CYT when the model was applied to the CGCG). We suspect this is primarily due to the small sample size available for analysis in the CGCG cohort. In addition, our findings may not be generalizable to recurrent GBM (post-radiation and temozolomide). Lastly, while we attempted to comprehensively evaluate as many immune microenvironment markers as possible for their relationship with CYT, there are likely other unidentified or understudied molecules that play important roles in determining T cell cytolytic activity in GBM. However, our list of 28 candidate variables was based on extensive literature review of relevant immune microenvironment factors [21–24] and was intentionally limited to targets for which there are currently drugs in preclinical and/or clinical development.
In summary, through in silico analysis of RNASeq gene expression data, we demonstrated that increased expression of CTLA-4, CD40, CD137, GITR, OX40, CSF-1, and IL-6 are independently associated with intratumoral immune cytolytic activity in newly diagnosed, previously untreated human GBM. CTLA-4 and CD40 were the only two molecules that remain independently associated with CYT in two separate datasets, implying a particularly strong relationship with T cell cytolytic activity in GBM. These results are hypothesis-generating and may inform rational choices of immunotherapeutic combinations for future evaluation in this disease. Additional studies are needed to determine whether therapeutic alteration of these targets (inhibition of CTLA-4, CSF-1 and IL-6 or stimulation of CD40, CD137, GITR, and OX40) alone or in combination with other immunotherapeutic strategies increases intratumoral cytolytic T-cell activity in GBM, and whether this translates into antitumor efficacy. Similarly, future studies are needed to determine whether CYT can be used as a predictive biomarker of response to immunotherapy in GBM. However, such studies would require large sample sizes of patients who have responded to immunotherapy, which are currently difficult to obtain in GBM.
Acknowledgments
The results published here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/
Footnotes
Conflict of interest: The authors declare that they have no conflict of interest.
Human participants: This article does not contain any studies with human participants or animals performed by any of the authors
References
- 1.Gnjatic S, Bronte V, Brunet LR, et al. Identifying baseline immune-related biomarkers to predict clinical outcome of immunotherapy. Journal for immunotherapy of cancer 2017;5:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ladomersky E, Zhai L, Lenzen A, et al. IDO1 inhibition synergizes with radiation and PD-1 blockade to durably increase survival against advanced glioblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2018. [DOI] [PMC free article] [PubMed]
- 3.Reardon DA, Gokhale PC, Klein SR, et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer immunology research 2016;4(2):124–135. [DOI] [PubMed] [Google Scholar]
- 4.Reardon DA. Randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro Oncology 2017;19(suppl_3). [Google Scholar]
- 5.Bouffet E, Larouche V, Campbell BB, et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2016;34(19):2206–2211. [DOI] [PubMed] [Google Scholar]
- 6.Johanns TM, Miller CA, Dorward IG, et al. Immunogenomics of Hypermutated Glioblastoma: A Patient with Germline POLE Deficiency Treated with Checkpoint Blockade Immunotherapy. Cancer discovery 2016;6(11):1230–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodges TR, Ott M, Xiu J, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro-oncology 2017. [DOI] [PMC free article] [PubMed]
- 8.Sims JS, Grinshpun B, Feng Y, et al. Diversity and divergence of the glioma-infiltrating T-cell receptor repertoire. Proceedings of the National Academy of Sciences of the United States of America 2016;113(25):E3529–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome medicine 2017;9(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160(1–2):48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thorsson V, Gibbs DL, Brown SD, et al. The Immune Landscape of Cancer. Immunity 2018;48(4):812–830.e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nature reviews Clinical oncology 2018;15(7):422–442. [DOI] [PubMed] [Google Scholar]
- 13.Lang FF, Conrad C, Gomez-Manzano C, et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2018;36(14):1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Science translational medicine 2017;9(399). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annual review of immunology 2008;26:389–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ji RR, Chasalow SD, Wang L, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother 2012;61(7):1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515(7528):563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowman RL, Wang Q, Carro A, Verhaak RG, Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-oncology 2017;19(1):139–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell 2013;155(2):462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bao ZS, Chen HM, Yang MY, et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res 2014;24(11):1765–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blank CU, Haanen JB, Ribas A, Schumacher TN. CANCER IMMUNOLOGY. The “cancer immunogram”. Science (New York, NY) 2016;352(6286):658–660. [DOI] [PubMed] [Google Scholar]
- 22.Patel SA, Minn AJ. Combination Cancer Therapy with Immune Checkpoint Blockade: Mechanisms and Strategies. Immunity 2018;48(3):417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mirzaei R, Sarkar S, Yong VW. T Cell Exhaustion in Glioblastoma: Intricacies of Immune Checkpoints. Trends in immunology 2017;38(2):104–115. [DOI] [PubMed] [Google Scholar]
- 24.Zappasodi R, Merghoub T, Wolchok JD. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer cell 2018;33(4):581–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desjardins A, Gromeier M, Herndon JE 2nd, et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. The New England journal of medicine 2018;379(2):150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liau LM, Ashkan K, Tran DD, et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. Journal of translational medicine 2018;16(1):142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garg AD, Vandenberk L, Van Woensel M, et al. Preclinical efficacy of immune-checkpoint monotherapy does not recapitulate corresponding biomarkers-based clinical predictions in glioblastoma. Oncoimmunology 2017;6(4):e1295903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berghoff AS, Kiesel B, Widhalm G, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-oncology 2015;17(8):1064–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Q, Hu B, Hu X, et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer cell 2017;32(1):42–56.e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antonios JP, Soto H, Everson RG, et al. Immunosuppressive tumor-infiltrating myeloid cells mediate adaptive immune resistance via a PD-1/PD-L1 mechanism in glioblastoma. Neuro-oncology 2017;19(6):796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nduom EK, Wei J, Yaghi NK, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-oncology 2016;18(2):195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fecci PE, Mitchell DA, Whitesides JF, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer research 2006;66(6):3294–3302. [DOI] [PubMed] [Google Scholar]
- 33.Jacobs JF, Idema AJ, Bol KF, et al. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro-oncology 2009;11(4):394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (New York, NY) 2018;359(6382):1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen YP, Zhang Y, Lv JW, et al. Genomic Analysis of Tumor Microenvironment Immune Types across 14 Solid Cancer Types: Immunotherapeutic Implications. Theranostics 2017;7(14):3585–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mangani D, Weller M, Roth P. The network of immunosuppressive pathways in glioblastoma. Biochemical pharmacology 2017;130:1–9. [DOI] [PubMed] [Google Scholar]
- 37.Maes W, Van Gool SW. Experimental immunotherapy for malignant glioma: lessons from two decades of research in the GL261 model. Cancer immunology, immunotherapy : CII 2011;60(2):153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wainwright DA, Chang AL, Dey M, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 2014;20(20):5290–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Omuro A, Vlahovic G, Lim M, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro-oncology 2018;20(5):674–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vonderheide RH. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer cell 2018;33(4):563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chonan M, Saito R, Shoji T, et al. CD40/CD40L expression correlates with the survival of patients with glioblastomas and an augmentation in CD40 signaling enhances the efficacy of vaccinations against glioma models. Neuro-oncology 2015;17(11):1453–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shoji T, Saito R, Chonan M, et al. Local convection-enhanced delivery of an anti-CD40 agonistic monoclonal antibody induces antitumor effects in mouse glioma models. Neuro-oncology 2016;18(8):1120–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cabo M, Offringa R, Zitvogel L, Kroemer G, Muntasell A, Galluzzi L. Trial Watch: Immunostimulatory monoclonal antibodies for oncological indications. Oncoimmunology 2017;6(12):e1371896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Belcaid Z, Phallen JA, Zeng J, et al. Focal radiation therapy combined with 4–1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PloS one 2014;9(7):e101764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patel MA, Kim JE, Theodros D, et al. Agonist anti-GITR monoclonal antibody and stereotactic radiation induce immune-mediated survival advantage in murine intracranial glioma. Journal for immunotherapy of cancer 2016;4:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jahan N, Talat H, Curry WT. Agonist OX40 immunotherapy improves survival in glioma-bearing mice and is complementary with vaccination with irradiated GM-CSF-expressing tumor cells. Neuro-oncology 2018;20(1):44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quail DF, Bowman RL, Akkari L, et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science (New York, NY) 2016;352(6288):aad3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Q, He Z, Huang M, et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2alpha. Nature communications 2018;9(1):559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pyonteck SM, Akkari L, Schuhmacher AJ, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nature medicine 2013;19(10):1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Butowski N, Colman H, De Groot JF, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-oncology 2016;18(4):557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]