

Abstract
Cancer cell resistance to taxanes is a complex, multifactorial process, which results from the combination of several molecular and cellular changes. In breast cancer cells adapted to long-term paclitaxel treatment, we previously identified a new adaptive mechanism that contributes to resistance and involves high levels of tubulin tyrosination and long-chain polyglutamylation coupled with high levels of septin expression, especially that of SEPT9_i1. This in turn led to higher CLIP-170 and MCAK recruitment to microtubules to enhance microtubule dynamics and therefore counteract the stabilizing effects of taxanes. Here, we explored to which extent this new mechanism alone could trigger taxane resistance. We show that coupling septins (including SEPT9_i1) overexpression together with long-chain tubulin polyglutamylation induce significant paclitaxel resistance in several naive (taxane-sensitive) cell lines and accordingly stimulate the binding of CLIP-170 and MCAK to microtubules. Strikingly, such resistance was paralleled by a systematic relocalization of septin filaments from actin fibers to microtubules. We further show that this relocalization resulted from the overexpression of septins in a context of enhanced tubulin polyglutamylation and reveal that it could also be promoted by an acute treatment with paclitaxel of sensitve cell displaying a high basal level of SEPT9_i1. These findings point out the functional importance and the complex cellular dynamics of septins in the onset of cell resistance to death caused by microtubule-targeting antimitotic drugs of the taxane family.
Introduction
Paclitaxel induces cell death, making it a successful drug for anticancer chemotherapy. However, several superimposed mechanisms of resistance limit the extent of paclitaxel use in therapeutics1. A new mechanism contributing to such chemoresistance was uncovered in the laboratory, involving the overexpression of septins coupled to tubulin modifications2,3.
Septins are filamentous GTPases involved in a vast array of cellular functions in which they mainly behave as diffusion barriers or as scaffolds4,5. In mammals, there are 13 septin genes grouped in four families6. Septins arrange into palindromic octamers: SEPT9-SEPT7-SEPT6-SEPT2-SEPT2-SEPT6-SEPT7-SEPT9, which then can assemble into higher structures like filaments, gauzes or rings7,8. Each of the septin gene loci can generate several transcripts. The sept9 locus engenders at least 15 isoforms9 and the overexpression of SEPT9_i1, one of the largest isoforms, has already been involved in ovarian cancer tumorigenesis10, head and neck cancers11, and breast cancer progression12,13. In interphase cells, septins can be found on membranes14,15, on actin stress fibers7,16 and/or on microtubules (MTs) in a few cell types17 where they were proposed to play a role in the regulation of MT guidance and organization18.
MT dynamics can be modulated by post-translational modifications (PTMs) of tubulin19. The detyrosination/retyrosination cycle20–22 was involved in breast cancer cell resistance to paclitaxel23. Tubulin polyglutamylation (polyE), which was shown to modulate protein interactions with MTs24,25 and thus to control MT dynamics, consists in the branching and in the elongation of (Glu)n side chains on both α- and/or β-tubulin. It is catalyzed by tubulin tyrosine ligase like (TTLL) glutamylases. TTLL4, 5, or 7 start the branching by adding a single glutamate whereas the elongation is catalyzed by TTLL1, 6, 11, or 1326,27.
Previous studies have shown that cell resistance to paclitaxel is a multifactorial process1,28. In addition, we have shown previously2,3 that long-term paclitaxel adaptation of MDA-MB 231 breast cancer cells (paclitaxel-resistant; Tr) resulted in additional changes: (i) in the occurrence of high levels of long-chain polyE and in TTL (Tubulin Tyrosine Ligase)-mediated tubulin retyrosination, (ii) in global septin overexpression together with a partial replacement of SEPT9_i3 (the main isoform of paclitaxel-sensitive cells; Ts) by SEPT9_i1, and (iii) in a higher recruitment to MTs of plus end-tracking proteins (+TIPs) that control catastrophes (MCAK) and rescues (CLIP-170). Knocking-down each of these actors led to the reversion of chemoresistance, allowing us to propose this new resistance mechanism. Strikingly, it was paralleled by a dramatic relocalization of septins from actin filaments to MTs in resistant cells3.
Here, to determine to which extent this mechanism alone could trigger taxane resistance, we studied the respective contributions of tubulin modifications and of septin overexpression to the chemoresistant phenotype in MDA-MB 231 taxane-sensitive (Ts) and in a variety of naive cells. We found that the most effective set of modifications consisted in the simultaneous overexpressions of TTLL5, TTLL11, SEPT2, SEPT6, SEPT7 and SEPT9_i1, and that this combination caused the relocalization of septin filaments from actin to MTs, consistent with the phenotype observed in long-term paclitaxel-adapted cells. We also show for the first time that septin relocalization occurred early in response to acute paclitaxel treatment, and that cell lines that constitutively express a high level of SEPT9_i1 were more prone to undergo such a phenotype. Together, these results indicate that septin overexpression and relocalization to MTs is a key event to allow paclitaxel resistance to take place.
Results
Paclitaxel resistance is promoted by the overexpression of octamer-forming septins, and is further enhanced by MT polyglutamylation
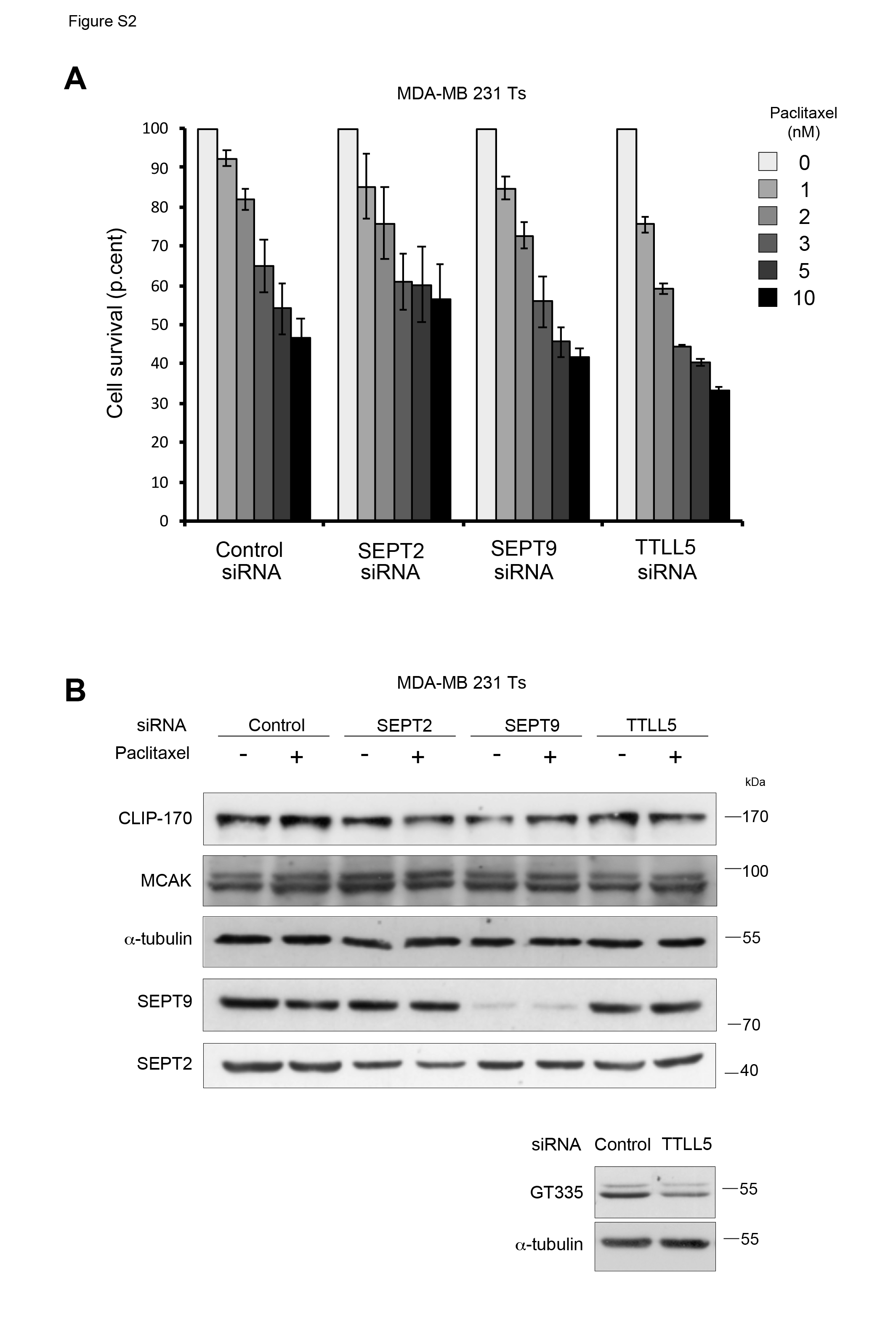
By RNAi depletion, we previously identified four new factors involved in paclitaxel resistance: TTL, TTLLs, septins and +TIPs3. Here, in a reverse approach, we investigated which of these actors, alone or in combination, are sufficient to induce a significant level of paclitaxel resistance. As we showed that +TIPs function downstream of tubulin modifications and septins to confer chemoresistance3, we focused on the three other factors to determine their role in taxane-resistance acquisition. To this end, we first overexpressed combinations of TTLLs and septins in taxane-senstive (Ts) MDA-MB 231 cells, the cell line from which the long-term paclitaxel-adapted (Tr) cells originated2. To determine whether resistance was restricted to breast cancer cells, we also studied cancer cells from other origins (HeLa and HuH7). The study was further extended to immortalized cells (RPE-1) and even to non-human fibroblasts (CHO), to evaluate the universality of the MT-dependent resistance mechanism we uncovered3. First of all, transfection efficacy and specificity were verified (Figs. S1 and S2). Drug resistance was determined from experiments that consisted in 72 h-exposure to increasing paclitaxel concentrations followed by measurement of cell survival in MTT assays relative to cells expressing eGFP as a control. Figure 1a.1 shows that the overexpression of TTLL5 (branching) and TTLL11 (elongating) is not sufficient to cause Ts cells to resist paclitaxel. A low level of resistance was induced when each of the SEPT9 isoforms identified in MDA-MB 231 cells (either SEPT9_i3 or the bad prognosis SEPT9_i1 isoform) was overexpressed alone (Fig. 1a.1). When the whole set of filament-forming septins (SEPT2,6,7,9) was overexpressed, especially when it included SEPT9_i1 (Fig. 1a.3) or when MT polyglutamylation was enhanced in the case of the presence of SEPT9_i3 (Fig. 1a.2), a proportion of Ts cells resisted paclitaxel up to ~10 nM. The overexpression of the SEPT9_i1 isoform together with that of other septins enhanced cell resistance to paclitaxel to an intermediate level, and resistance was even higher when septin overexpression was combined with that of polyglutamylation enzymes (Fig. 1a.3 and 1a.4). To reinforce the significance of the above MTT assays, we further verified that the overexpression of septins in association or not with polyglutamylases limits the release of DNA fragments in the cytoplasm, reflecting the reduction in apoptotic cell death (Fig. 1b).
Fig. 1. The overexpression of a SEPT9_i1-containing set of filament-forming septins is necessary and sufficient to confer paclitaxel resistance, and is further enhanced upon microtubule polyglutamylation.

a MTT assays were conducted in paclitaxel-sensitive (Ts) MDA-MB 231 cells in conditions that enhanced either tubulin long-chain polyglutamylation (overexpression of the branching TTLL5 and elongating TTLL11 enzymes) or overexpression of a septin pattern including SEPT9_i1 or_i3, or both. After a 72h-exposure to the paclitaxel concentrations indicated, cell survival was measured. Each point is the mean ± s.e.m of at least 10 independent experiments. Statistical analysis is based on the overall comparison of the curves *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Statistical significance of the comparisons with the negative control is color-coded as indicated. The colors also correspond to the additional percentages of cell survival compared to those of control cells at the IC50: absence of resistance (blue), low level of resistance (light orange, ≤10%), intermediate level (orange, between 10 and 20%) and high level of resistance (red, >20%). The table summarizes the effect (color-coded) of each effector alone or in combination, and indicates the panel(s) displaying the data. b MDA-MB 231 Ts cells were subjected to the overexpression of the indicated plasmids and cell death was quantified 24 h after exposure to paclitaxel by measuring the cytoplasmic release of histone-associated DNA fragments relative to the values without paclitaxel. Each point is the mean ± s.e.m. of at least three independent experiments. *p ≤ 0.05, **p ≤ 0.01
Overexpressing TTL alone also caused cells to resist paclitaxel to an intermediate level (Fig. 2.1). Note that combining TTL overexpression with that of septins alone, whatever the SEPT9 isoform (Fig. 2.1), or with that of the polyglutamylases TTLL5 and 11 did not further enhance Ts cells resistance (Fig. 2.2 and 2.3). When SEPT9_i1 was included in the combination, overexpressing TTL on top of TTLLs even had a negative effect on resistance (Fig. 2.3). In our model of taxane resistance, TTL was an important upstream actor to allow effective long-chain polyglutamyation of tubulin and septin recruitment to MTs3. The fact that TTL overexpression failed to increase the resistance conveyed by TTLL and septin overexpression revealed that overexpressed TTLLs bypassed the need for TTL to stimulate long-chain polyE. It also indicated that TTL conferred some level of taxane resistance independently from the polyE/septins pathway, in keeping with the notion that tubulin tyrosination is required for the recruitment of CLIP-170 and MCAK on MTs29,30.
Fig. 2. TTL overexpression does not enhance paclitaxel resistance induced by the overexpression of septins and polyglutamylases.
MTT assays were conducted in paclitaxel-sensitive (Ts) MDA-MB 231cells in conditions that enhanced or not tubulin retyrosination (overexpression of the Tubulin Tyrosine Ligase TTL) combined with increased tubulin long-chain polyglutamylation (overexpression of TTLL5 and TTLL11 enzymes) or overexpression of a septin set including SEPT9_i1 or_i3, or both. Each bar is the mean ± s.e.m of at least 10 independent experiments. Statistical analysis is based on the overall comparison of the curves *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Statistical significance of the comparisons with the negative control is color-coded as indicated. The colors also correspond to the additional percentages of cell survival compared to those of control cells at the IC50: absence of resistance (blue), low level of resistance (light orange, ≤10%), intermediate level (orange, between 10 and 20%) and high level of resistance (red, >20%). The table summarizes the effect (color-coded) of each effector alone or in combination, and indicates the panel(s) displaying the data
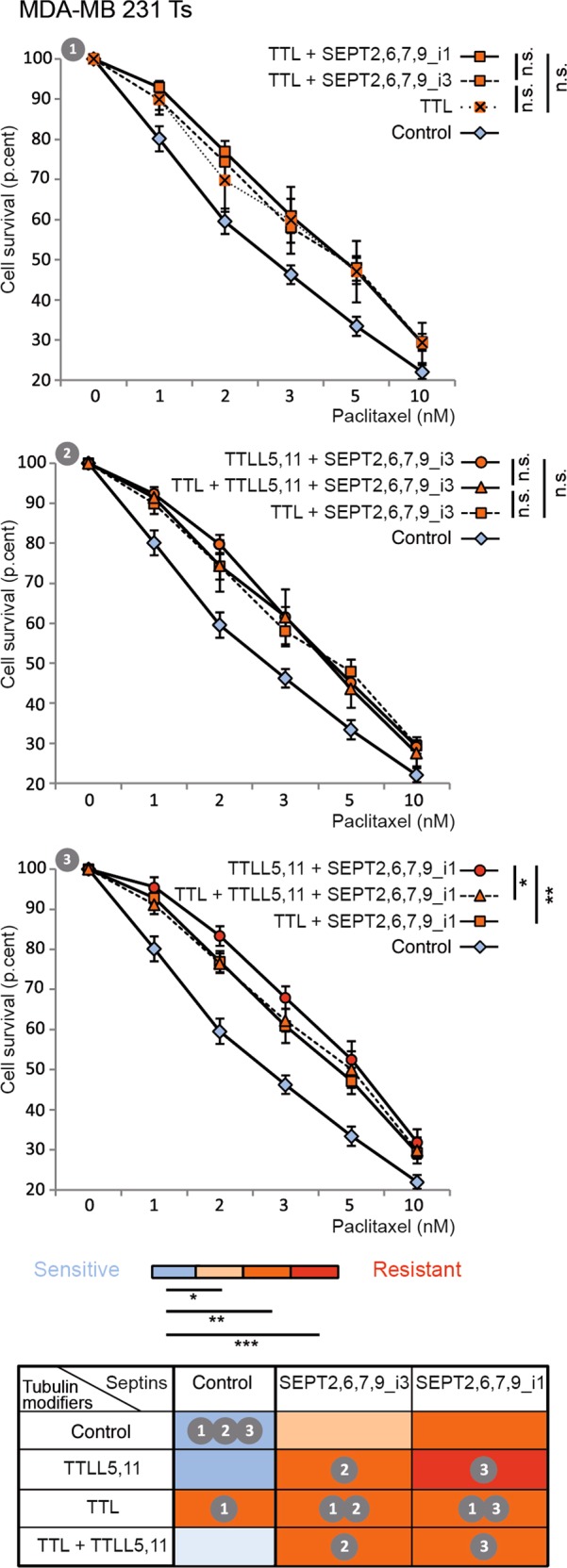
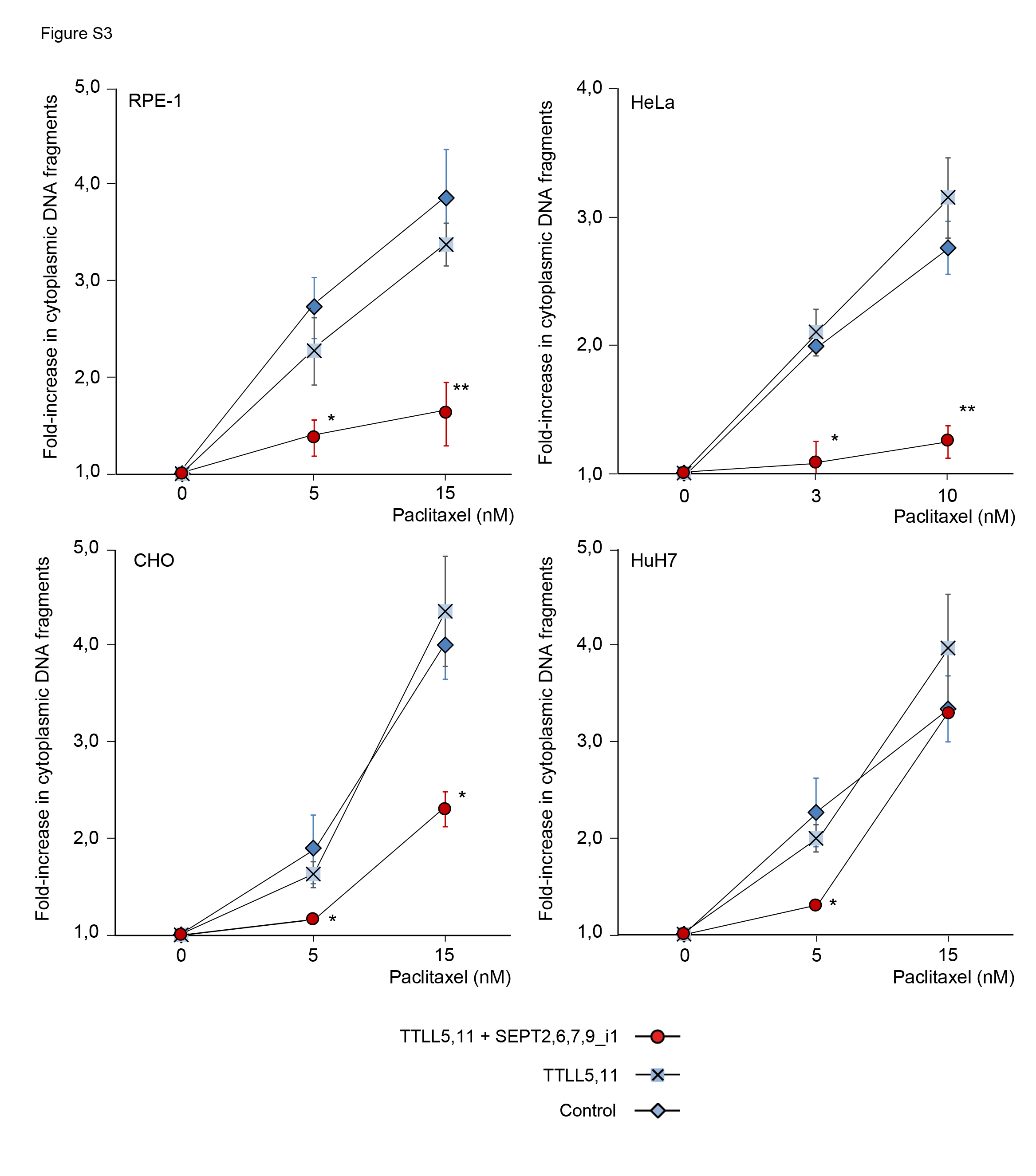
The same viability assays were repeated in RPE-1, HeLa, CHO and HuH7 cells, which are all sensitive to paclitaxel as evidenced by their respective IC50 (Fig. 3a, top panel). MTT cell survival assays were conducted in four different conditions according to the levels of septin expression and of tubulin polyglutamylation: Control, TTLL5,11, SEPT2,6,7,9_i1 and TTLL5,11 + SEPT2,6,7,9_i1. Except for HuH7 hepatoma cells, which displayed low to intermediate resistance only at very low concentrations of paclitaxel (1 nM and 2 nM, respectively), each of the other cell lines behaved like MDA-MB 231 Ts cells (Figs. 3b, c and S3). They resisted paclitaxel when the whole set of polyglutamylation enzymes and septins was overexpressed. The overexpression of septins alone caused a milder resistant phenotype. Regarding polyglutamylase overexpression, it consistently failed to cause resistance, except perhaps in CHO and HuH7 cells at 2 nM paclitaxel (Fig. 3c). Figure 3a (bottom panel) shows the relative increases in IC50 for paclitaxel, as determined from the responses to MTT cell survival assays (Fig. 3b, c).
Fig. 3. Long-chain polyglutamylation of tubulin and septin overexpression are necessary and sufficient to confer paclitaxel resistance to multiple cell lines.

a The IC50 for paclitaxel of untreated paclitaxel-sensitive (Ts) MDA-MB-231cells, RPE-1, HeLa, CHO and HuH7 cells or in conditions that enhanced either tubulin long-chain polyglutamylation or septin pattern overexpression including SEPT9_i1 or both, were measured using MTT assays with drug concentrations ranging from 0 to 100 nM paclitaxel. The values shown are the mean ± s.e.m. of at least four independent experiments. b, c MTT assays were conducted in paclitaxel-sensitive (Ts) MDA-MB-231cells, RPE-1, HeLa, CHO, and HuH7 cells in conditions that enhanced either tubulin long-chain polyglutamylation or septin pattern overexpression including SEPT9_i1, or both. After a 72h-exposure to the paclitaxel concentrations indicated, cell survival was measured. Each bar is the mean ± s.e.m of at least four independent experiments. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001
Septins and tubulin polyglutamylation contribute to CLIP-170 and MCAK recruitment to MTs
We previously showed in our Tr model3 that the increase of CLIP-170 and MCAK on MTs contributed to chemoresistance, downstream of tubulin modifications and septin recruitement to MTs. As tubulin tyrosination is a key factor to allow the binding of these +TIPs to MTs29,30, we next wondered whether tubulin polyglutamylation and/or septins also contributed to this recruitment. In a way that paralleled what we observed above for the induction of paclitaxel resistance, Ts cells overexpressing the combination of TTLL5,11 alone did not display a higher recruitment of CLIP-170 and MCAK in MT fractions (Fig. 4a). In contrast, overexpressing a combination of septins (SEPT2,6,7,9_i1) increased CLIP-170 and MCAK levels in the MT fractions. This effect was even higher when long-chain tubulin polyglutamylation was stimulated in parallel (Fig. 4a). Note that these phenotypes occurred while TTL was not overexpressed, suggesting that an increased recruitment of these +TIPs is also stimulated by septin recruitment on MTs. Increased levels of CLIP-170 and MCAK in the MT fractions were also observed in the RPE-1 cell line (Fig. 4b), confirming that the overexpression of septins combined with that of polyglutamylases may cause an enhanced recruitment of these proteins not only in cancer cells.
Fig. 4. Overexpression of a SEPT9_i1-containing set of septins stimulates the recruitment of CLIP-170 and MCAK to microtubules, which is amplified by long-chain tubulin polyglutamylation.

MT fractions from Ts (a) or RPE-1 (b) cells subjected to TTLL and/or septin overexpression as indicated were analyzed for the recruitment of various +TIPs (EB1, CLIP-170, and MCAK). The polyE antibody recognizes glutamate chains composed of at least three glutamic acids. The polyE signal is therefore a reflection of elongating TTLL activity. Tubulin was used as a loading control. The quantitative data are the ratio of the intensity of the protein of interest to that of α-tubulin for the blot shown
The combined overexpression of septins and of polyglutamylation enzymes that optimizes paclitaxel resistance causes the relocalization of septin filaments to MTs, independently of paclitaxel stimulation
Another striking phenotype that distinguished Ts from Tr cells is the subcellular localization of septin filaments that shifted from actin fibers to MTs, respectively (ref. 3 and Fig. 5a). Furthermore, SEPT9_i1 expression was increased in Tr cells while SEPT9_i3 was the main SEPT9 isoform in Ts cells. Also, SEPT9_i1-enriched septin filaments are associated with MTs that bear long polyglutamate side chains in resistant Tr cells3. We wanted to determine to which extent this change of localization resulted from the induction of tubulin polyglutamylation and/or from the expression of a specific SEPT9 isotype.
Fig. 5. Paclitaxel-independent septin relocalization from actin fibers to microtubules is complete upon the overexpression of SEPT9_i1-containing set of septins combined with long-chain polyglutamylation in the MDA-MB 231 breast cancer cell line.

a Subcellular localization of septin filaments in the paclitaxel-sensitive (Ts) and -resistant (Tr) MDA-MB 231 cell lines. Septin filaments associated with actin fibers in Ts cells mainly comprise SEPT9_i3, while SEPT9_i1 is the main long SEPT9 isoform recovered in septin filaments, which are associated with long-chain polyglutamylated MTs in paclitaxel-adapted Tr cells3. Endogenous SEPT2 was used to reveal the localization of septin filaments in Ts and Tr cells. b Subcellular septin localization in Ts cells overexpressing various combinations of septins and TTLL enzymes alone or in association. SEPT2 labelling was used to detect endogenous septin filaments. With the exception of SEPT9_i1, which bears the V5 tag, all septins are GFP-tagged. TTLL enzymes are coexpressed with CFP. The bottom-left panels show examples of differential septin localization in transfected (t) versus non-transfected (nt) cells in the same field. The images shown are representative of at least three independent experiments. Scale bars = 10 µm
We thus tested which of tubulin long-chain polyglutamylation and/or septin overexpression (including SEPT9 isoform variation) was required to make the endogenous septin filaments coalign with MTs. As shown in Fig. 5b, the stimulation of tubulin polyglutamylation alone was not sufficient to displace septin filaments from actin fibers to MTs. Next, all the conditions in which SEPT9 alone (either the _i1 or the _i3 isoform) was overexpressed, in combination or not with the polyglutamylation enzymes, also failed to relocalize endogenous septin filaments, indicating that SEPT9 (even the _i1 isoform which can physically interact with tubulin31) does not have a role by itself in this change of septin subcellular localization. Moreover, as the overexpression of an individual septin may form artifactual homo-oligomers32 and mimic physiological filament coalignment with MTs, we also overexpressed SEPT9 isoforms together with other filament-forming septins and followed total SEPT2 (endogenous and overexpressed) by immunofluorescence to actually reflect the behavior of endogenous septin filaments. In these conditions, when filament-forming septins included SEPT9_i1, a partial relocalization of septin filaments to MTs could be observed, mostly on perinuclear MTs forming bundles (Fig. 5b). This relocalization was complete when septin overexpression was combined with effective long-chain polyglutamylation that resulted from the overexpression of both TTLL5 and TTLL11 (Fig. 5b). The latter situations contrasted with the conditions that comprised SEPT9_i3. In these cases, overexpressing septins alone did not allow septin filaments to relocalize from actin fibers to MTs (Fig. 5b), and combining the overexpression of this septin profile with long-chain polyglutamylation only moderately caused septin filaments to coalign with MTs, which is also in accordance with the lower level of resistance shown by cell viability data (see Fig. 1, right panels).
The functional importance of combining long-chain tubulin polyglutamylation and SEPT9_i1 expression to trigger septin coalignment with MTs was further verified in other cell types. As shown in Fig. 6, the relocalization of endogenous septin filaments was also observed in RPE-1, HeLa and CHO cells, the same 3 cell lines in which paclitaxel resistance could be significantly induced (see Fig. 3c). Consistent with this parallel between paclitaxel resistance and septin relocalization, HuH7 cells, which only resisted very low concentrations of paclitaxel (see Fig. 3c), did not exhibit a displacement of their septin filaments to MTs, as visualized by the labelling of SEPT2 (Fig. 6, bottom line).
Fig. 6. Paclitaxel-independent subcellular relocalization of endogenous septin filaments to microtubules occurs following the combined overexpression of polyglutamylases (TTLL5,11) and septins (2,6,7,9_i1) in diverse cell lines.

a Immunofluorescent images of endogenous SEPT2 (green) and actin (phalloidin; red) in cells. The overlay images show that septin fibers colocalize with actin stress fibers in each cell line. b Immunofluorescent images show endogenous SEPT2 (green), overexpressed SEPT9_i1 and alpha-tubulin (red). Septin filaments relocalize to MTs when polyglutamylases and septins (including SEPT9_i1) are overexpressed in CHO, RPE-1, and HeLa cells and only partially in HuH7 cell line. The images shown are representative of at least three independent experiments. Scale bars = 10 µm
Cells that survive acute paclitaxel treatment exhibit MT-bound septins
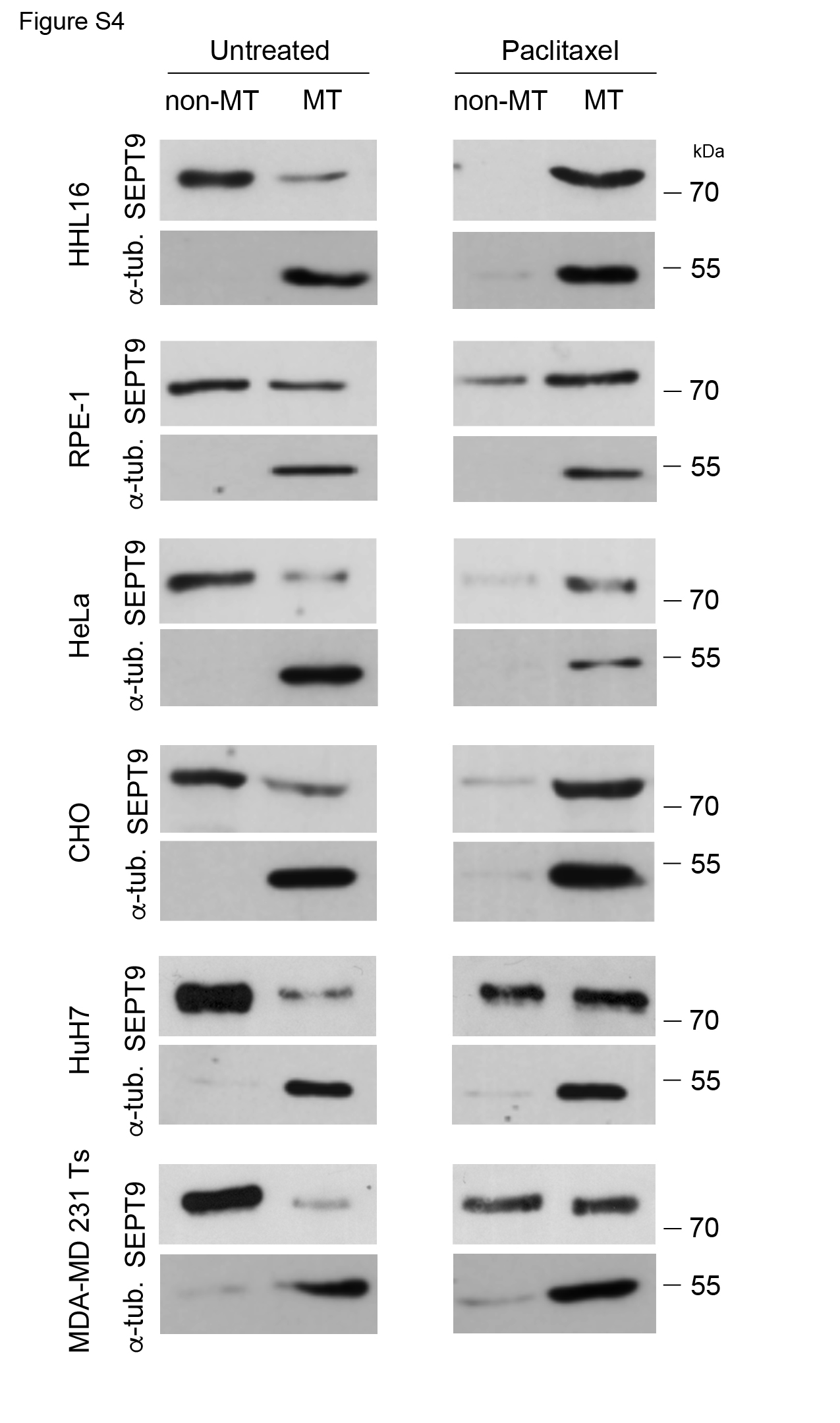
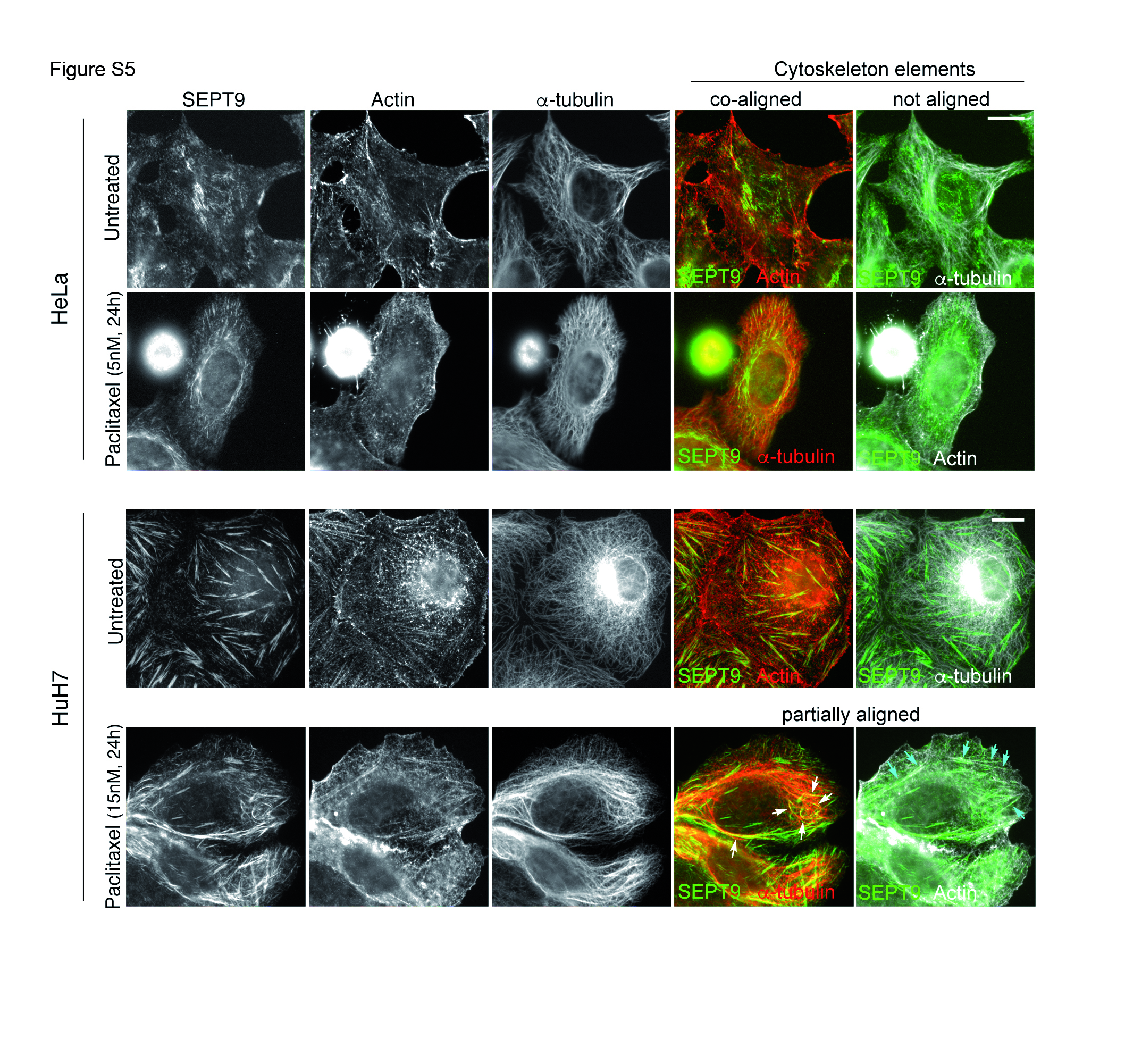
Our above results show that septin relocalization triggered by septin overexpression (including SEPT9_i1) in a context of long-chain tubulin polyglutamylation was well correlated with cell survival to a 72 h exposure to paclitaxel. To confirm that this change in septin subcellular location could be actually causative of the onset of a cell resistance, we treated different untransfected cell lines using 24 h acute treatment with paclitaxel at a concentration three-times higher than their respective IC50. Figures 7a, d, S4 and S5 show that, except for MDA-MB 231 Ts, a partial or complete relocalization of septin filaments occurred from the actin cytoskeleton to the MT network. As this relocalization seems to be a general feature of cancer and non-cancer cell response to acute paclitaxel treatment, we further analyzed this cytoskeleton reorganization in the two RPE-1 and HHL16 immortalized cell lines that were both good responders to paclitaxel and easy to image. We next eliminated the hypothesis that the change in septin location could originate from enhanced septin expression following paclitaxel treatment (Fig. 7b). It is worth noting, especially in RPE1 (Fig. 7a) or HeLa (Fig. S5) cells, that the relocalization of septins to MTs is accompanied by a loss of thick actin stress fibers. This is in agreement with the role of septins in stabilizing actin fibers33. We also noticed that the higher the concentration of paclitaxel we used, the faster septins relocalized to MTs (Data not shown). Measuring the time-course of cell survival in both RPE-1 and HHL16 cell lines showed that strikingly, all the cells that survived the two highest concentrations of paclitaxel, exhibited septin filaments coaligned with their MTs (Fig. 7c), further indicating that when it actually occurs, this septin relocalization plays a key role in cell survival/resistance to paclitaxel treatment. As long-term adapted MDA-MB 231 Tr cells exhibit an elevated level of SEPT9_i1 compared to their sensitive counterpart Ts3, the differential behavior between MDA-MB 231 Ts, HuH7 and the other cell lines is likely to originate from differences in the basal level of SEPT9_i1. Indeed, all the cell lines that displayed strong septin relocalization after paclitaxel treatment actually exhibited a high basal level of SEPT9_i1 expression (Fig. 7e, f). The low basal level of SEPT9_i1 expression in HuH7 and its absence in MDA-MB 231 Ts cells correlates well with their respective partial or barely detectable relocalization of septin filaments to MTs (Fig. 7f). In addition, short-term paclitaxel treatment failed to stimulate SEPT9_i1 expression in MDA-MB 231 Ts and HuH7 cells, pointing out that a high basal level is determinant to induce such a quick relocalization, whether the cells are cancerous or not. This result also suggests that cell lines with a low basal level of SEPT9_i1 expression might need a longer adaptation to resist the drug.
Fig. 7. Cells that survived acute paclitaxel treatment exhibit subcellular relocalization of endogenous septin filaments to microtubules.

a Immunofluorescent images of endogenous SEPT2 (green), actin (phalloidin; red or white) and α-tubulin in HHL16 and RPE-1 cells following 24 h paclitaxel treatment. The overlay images show that septin coalign with actin fibers in untreated cells, and with tubulin in paclitaxel treated cells. The paclitaxel concentration used corresponds to three times the IC50 for the drug. b Septin cellular amount does not change upon treatment with increasing concentrations of paclitaxel. Note that tubulin polyglutamylation starts increasing only after 24 h of 10 nM paclitaxel treatment. c Quantification of cells with MT-bound septins among cells that have survived 24 h-paclitaxel treatment. d Immunofluorescent images of breast cancer MDA-MB 231 cells after 15 nM paclitaxel treatment for 24 h. Note that septin filaments are still mainly associated with actin fibers in these cells. e Immunoblot showing the basal levels of SEPT9 long isoforms (SEPT9_i1 and _i3) in total cell extracts of untreated sensitive MDA-MB 231 Ts, HHL16, and RPE-1 cells. f Immunoblot showing the basal levels of SEPT9_i1 and _i3 in total cell lysates of untreated or paclitaxel-treated MDA-MB 231 Ts, HeLa, CHO, and HuH7 cells. The images, immunoblots shown and cell quantifications are representative of at least three independent experiments. Scale bars = 10 µm
Discussion
Cancer cell resistance to taxanes is a multifactorial process that involves a great variety of cellular adaptation means1,28. We previously uncovered a new septin- and tubulin polyglutamylation-dependent mechanism that contributes to long-term cell adaptation to taxanes3. While the inhibition of each of these actors partially restored paclitaxel sensitivity3, here we sought to determine to which extent they were sufficient to trigger resistance in various paclitaxel-naive cells. We show that modifying tubulin alone failed to cause resistance. In contrast, overexpressing a set of filament-forming septins (SEPT2,6,7,9), especially when it comprised SEPT9_i1, was sufficient to cause significant paclitaxel resistance that was further enhanced upon the induction of MT polyglutamylation. Strikingly, this resistance appeared to be tightly linked to the early relocalization of septin filaments from actin fibers to MTs, as shown upon acute paclitaxel treatment.
Tubulin polyglutamylation is a complex modification34, which plays an important role in neuropathology as shown in mice defective for a glutamate chain-reducing enzyme (CCP1), which display Purkinje cell degeneration35,36. A link between tubulin polyglutamylation and cancer cell resistance to MT-targeting agents has already been proposed in estramustine-resistant prostatic cancer cells37, but no mechanism was proposed to explain it.
Septin filaments, which have been identified as a new cytoskeleton element38, may display a great variety of subcellular organizations according to the organelle or structure they associate with. Apart from their assembly on membranes of specific curvature and lipid composition39 or their ability to form free rings in the cytoplasm7, they also associate with cytoskeleton components, mostly with actin filaments16,40,41 and with MTs in a few cell types7,17,42,43. The control of septin subcellular localization may thus regulate key cellular functions44,45. When septins are associated with actin microfilaments, they may impact on cell morphology, actin cytoskeleton organization, and cell migration46–48. They would function by crosslinking actin microfilaments in stress fibers, protecting them against depolymerization33. In agreement with this role, we observed that upon acute paclitaxel treatment, thick actin stress fibers disappear when septin filaments relocalized to MTs (see Fig. 7a). When coaligned with MTs, septins contribute to promote cell asymmetry49. Indeed, in polarized epithelial cells, post-Golgi trafficking operates along polyglutamylated MT tracks that bear septins50. Septins can also enhance MT dynamics by preventing the binding of the stabilizing protein MAP4 to MTs51 and/or by enhancing the recruitment of the rescue-promoting and catastrophe-promoting factors CLIP-170 and MCAK, respectively (ref3. and this study). We further show now that septin relocalization to MTs is linked to the early development of paclitaxel resistance.
Septin expression is modulated in pathology including in cancer4,6,52. Some septins like SEPT2, SEPT8, SEPT9, and SEPT11 are often overexpressed while SEPT4 and SEPT10 are frequently downregulated5,53. Many cancers also display differences in the level of SEPT9 isoform expression54. SEPT9_i2, SEPT9_i3, and SEPT9_i4 are often downregulated13,55 while SEPT9_i1 and SEPT9_i5 are overexpressed in various human tumors10–13,56. In our experiments, we observed a perfect correlation between the efficacy of early relocalization of septin filaments to MTs and the basal level of SEPT9_i1 expression (Fig. 7a–f and Fig. S5). SEPT9_i1 has already been proposed to be a marker of bad prognosis11,57 and to correlate with cell resistance to MT-disrupting agents58. The mechanisms proposed thus far include a role for SEPT9_i1 in the control of HIF-1α nuclear translocation and transcriptional effects59,60 or in a putative stimulation of MT dynamics through a delay in JNK degradation61,62. The latter effect could also contribute to cell adaptation to the MT-stabilizing action of taxanes. In addition, the SEPT9_i1-facilitated relocalization of septin filaments to MTs (this manuscript) could represent another means of modulating MT dynamics as recently shown in vitro63 and as observed in our long-term adapted cells3. Even though all cell types may adapt to taxanes following long-term exposure to paclitaxel as exemplified in our Tr cells2,3, the basal level of SEPT9_i1 could thus be predictive of the easier emergence of cell resistance to taxanes, especially upon acute treatments. Moreover, the fact that a broad range of cells express a sufficient level of SEPT9_i1 suggests that this mechanism could be relevant not only in cancer cells but also in any other tumor-associated cells.
Some molecular determinants of septin association with cytoskeleton elements have been identified, but the reasons for which septins associate with either cytoskeleton element are still far from clear. It has been shown that septins may bind directly to actin filaments64 or to actin-associated proteins like non-muscular myosin II41 or anillin7. SEPT9 can directly interact with F-actin33 via its N-terminal region. The same region is also involved in the interaction with tubulin through basic motifs present in the long isoforms of SEPT9 (SEPT9_i1, _i2 and _i3)31. SEPT9_i1 but not SEPT9_i3, when overexpressed with other octamer-forming septins, can specifically contribute to endogenous septin filament binding to MTs, and especially when the latter are polyglutamylated (these data). In spite of the minor sequence differences between the three long SEPT9 isoforms, SEPT9_i2 was recently shown to limit tumorogenicity contrary to SEPT9_i155. SEPT9_i1 only differs from SEPT9_i3 by the presence of an additional 17-aminoacid sequence at its N-terminus and by its next eight aminoacids, which makes SEPT9_i1 to bear five more positive charges than SEPT9_i3. Such difference would favor SEPT9_i1 over the other long isoforms for interacting with the acidic tail of tubulin, and even more when tubulin bears long polyglutamylate chains. Such interaction could be a cause of septin filament retention on the MTs of a variety of cells, making them develop a resistance to low concentrations of paclitaxel.
However, it is likely that other proteins are involved in stimulating septin interaction with MTs. Indeed, SEPT9_i1 is necessary but not sufficient to relocalize septin filaments to MTs, as high basal levels of SEPT9_i1 alone do not cause septins to coalign with MTs, as illustrated in RPE-1 or HHL16 cells (Fig. 7). Cell exposure to paclitaxel is also needed to trigger this relocalization. The molecular determinants of septin compartmentalization may involve a complex interplay between MTs, septin filaments including SEPT9_i1, and actin filaments, which will deserve further exploration, with a special emphasis on Borg proteins, which were evidenced to reinforce the binding between septin and actin filaments in non-dividing cells65. Some motors may also be involved as the EB1-binding kinesin KIF17 has been shown to bind SEPT966,67. Targeting this re-localization process would be a valuable approach to limit cell adaptation and resistance to cancer chemotherapies using MT poisons in the future.
Materials and methods
Cell culture and treatments
Experiments were performed with human cancer cells: MDA-MB 231 (breast), HeLa (cervix), HuH7 (hepatoma), human immortalized cells: RPE-1 (retinal epithelium), HHL16 (hepatocytes), or Chinese Hamster Ovary fibroblasts (CHO). All cell lines were cultured with 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM, Gibco™, Fisher Scientific, Illkirch, France) containing an antibiotic–antifungal mixture and supplemented with 10% fetal calf serum (GE Healthcare HyClone™, Fisher Scientific), 1% sodium pyruvate and 1% L-Glutamine for all the cell lines except MDA-MB 231 (2%). The paclitaxel-resistant (Tr) and -sensitive (Ts) sublines of the human breast carcinoma cell line MDA-MB 231 were continuously cultured without (Ts) or with 25 nM paclitaxel (Tr) as previously described2. When appropriate, cells were treated with different concentrations of paclitaxel (Sigma, Darmstadt, Germany) for 24, 48 or 72 h.
Cell transfections
Transfections of RPE-1 and HuH7 cells were performed 48 h or 72 h prior to analysis using X-tremeGENE while those of Ts, HeLa, and CHO cells were done with Turbofect (ThermoFischer, Illkirch, France), according to the manufacturer’s instructions. Note that the HHL16 cell line was not used in these experiments because they are hard to transfect. Plasmids were used alone or up to a combination of six but the maximal amount of total DNA used per well (6-well plate, 50% confluence) never exceeded 1.5 µg. The amounts of plasmids were determined from the measurements of the efficacy of transfection of the combination involving the six plasmids of interest (SEPT2, SEPT6, SEPT7, SEPT9_i1 or SEPT9_i3, TTLL5, and TTLL11; Fig. S1). Importantly, the same individual amounts were used in every transfection experiment even when the plasmids were used alone. The pEGFP plasmid (Addgene) was used as a negative control. GFP-TTL, CFP-TTLL5, and CFP-TTLL11 expression vectors were kind gifts of Dr. C. Janke (Institut Curie, Orsay, France). GFP- and His-tagged septins (SEPT2, 6, 7 or 9_i3) and V5-SEPT9_i1 were kindly provided by Pr W. Trimble (Hospital for Sick Children, Toronto, Canada) and Dr. A. Gassama (CHB Paul Brousse, Villejuif, France), respectively.
Cell viability assays
Twenty-four hours after cell transfection or not, sensitivity to paclitaxel was measured using either 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma) cytotoxicity assay after 72 h exposition to increasing concentrations of paclitaxel, or the Cell Death Detection ELISAPLUS assay, according to the manufacturer’s instructions (Roche, Sigma) after 24 h paclitaxel treatment. The IC50 was determined with Prism4 software (GraphPad software). Each value represents the mean ± s.e.m. of at least four independent experiments.
Immunoblotting
Total cell lysates and MT fractions were prepared as previously described2. Constant protein amounts (10 μg) were loaded onto gels for total cell lysate analysis, while constant α-tubulin amounts were used for MT fraction analysis. Blots were probed with selected primary antibodies against: α-tubulin (clone DM1-A, Sigma), SEPT2 (Atlas, Ozyme, St Quentin en Yvelines, France), SEPT9 (Proteintech, Manchester, United Kingdom), EB1 (H70, Santa Cruz Biotechnology, Clinisciences, Nanterre, france), CLIP-170 (H300, Santa Cruz Biotechnology), MCAK (Abnova, VWR, Fontenay sous bois, France), GFP (Cell Signaling, Ozyme), V5 (Invitrogen, ThermoFischer). The anti-long chain-polyglutamylated-tubulin (polyE) antibody was prepared and characterized by Drs C. Janke (Institut Curie, Orsay, France) and T. Surrey (London Research Institute, London, England). Protein bands were visualized with respective HRP-conjugated secondary antibodies and the ECL detection kit (Pierce, ThermoFischer). Western-blot quantification was performed after film digitization using the ImageJ software (National Institutes of Health, Bethesda, MD; imagej.nih.gov/ij/).
Immunofluorescence and microscopy
Cells grown on glass coverslips were immunostained with primary α-tubulin (clone DM1-A, Sigma), SEPT2 (Atlas), GFP (Cell Signaling) or V5 (Invitrogen) antibodies, after fixation with 3.75% paraformaldehyde/PBS, permeabilization with 0.1% Triton X-100/PBS and blocking with 0.5% BSA/PBS. Secondary antibodies were Alexa Fluor 488- or 555-conjugated antibodies against rabbit or mouse IgGs (Molecular Probes, Invitrogen) and Cyanine 5 antibody against mouse IgGs (Abcam, Paris, France). Actin microfilaments were visualized by fluorescent phalloidin (Sigma).
Images were acquired using either a Zeiss LSM 510 confocal microscope (63 × 1.4 NA objective) or a Leica DMLB microscope (40 or 100 × 1.3 NA objective). In the latter case, image acquisitions were performed using a Scion CFW1312M CCD camera driven from an Apple Macintosh G4 computer and homemade software. Data were quantified using the ImageJ software.
Statistical analysis
Quantitative data are the means ± s.e.m. of at least four independent experiments. Paclitaxel cytotoxicity measurements were compared using multiple covariance analysis on StatView 5 software (SAS institute). The following symbols were used: *p ≤ 0.05, **p ≤ 0.01 and *** p ≤ 0.001.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank V. Nicolas (Platform MIPSIT, Institut Paris-Sud d’Innovation Thérapeutique, Châtenay- Malabry, France) for valuable help in cell imaging. The authors are grateful to Pr W. Trimble, Drs C. Janke and A. Gassama for sharing materials. This work was supported by the Ministère de l’Enseignement Supérieur et de la Recherche, the Comité des Hauts de Seine of the Ligue contre le Cancer and by grants to B.T., L.K., and J.S. from the Ministère de la Recherche.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by A. Stephanou
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Laurence Klipfel, Isabelle Cantaloube
Contributor Information
Christian Poüs, Email: christian.pous@u-psud.fr.
Anita Baillet, Email: anita.baillet@u-psud.fr.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41419-019-1318-6).
References
- 1.Murray S, Briasoulis E, Linardou H, Bafaloukos D, Papadimitriou C. Taxane resistance in breast cancer: mechanisms, predictive biomarkers and circumvention strategies. Cancer Treat. Rev. 2012;38:890–903. doi: 10.1016/j.ctrv.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 2.Froidevaux-Klipfel L, et al. Modulation of septin and molecular motor recruitment in the microtubule environment of the Paclitaxel-resistant human breast cancer cell line MDA-MB-231. Proteomics. 2011;11:3877–3886. doi: 10.1002/pmic.201000789. [DOI] [PubMed] [Google Scholar]
- 3.Froidevaux-Klipfel L, et al. Septin cooperation with tubulin polyglutamylation contributes to cancer cell adaptation to taxanes. Oncotarget. 2015;6:36063–36080. doi: 10.18632/oncotarget.5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fung KYY, Dai L, Trimble WS. Cell and molecular biology of septins. Int. Rev. Cell. Mol. Biol. 2014;310:289–339. doi: 10.1016/B978-0-12-800180-6.00007-4. [DOI] [PubMed] [Google Scholar]
- 5.Montagna C, Bejerano-Sagie M, Zechmeister JR. Mammalian septins in health and disease. Res. Rep. Biochem. 2015;5:59–72. doi: 10.2147/RRBC.S59060. [DOI] [Google Scholar]
- 6.Peterson EA, Petty EM. Conquering the complex world of human septins: implications for health and disease. Clin. Genet. 2010;77:511–524. doi: 10.1111/j.1399-0004.2010.01392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kinoshita M, Field CM, Coughlin ML, Straight AF, Mitchison TJ. Self- and actin-templated assembly of Mammalian septins. Dev. Cell. 2002;3:791–802. doi: 10.1016/S1534-5807(02)00366-0. [DOI] [PubMed] [Google Scholar]
- 8.Sellin ME, Sandblad L, Stenmark S, Gullberg M. Deciphering the rules governing assembly order of mammalian septin complexes. Mol. Biol. Cell. 2011;22:3152–3164. doi: 10.1091/mbc.e11-03-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalikin LM, Sims HL, Petty EM. Genomic and expression analyses of alternatively spliced transcripts of the MLL septin-like fusion gene (MSF) that map to a 17q25 region of loss in breast and ovarian tumors. Genomics. 2000;63:165–172. doi: 10.1006/geno.1999.6077. [DOI] [PubMed] [Google Scholar]
- 10.Scott M, McCluggage WG, Hillan KJ, Hall PA, Russell SE. Altered patterns of transcription of the septin gene, SEPT9, in ovarian tumorigenesis. Int. J. Cancer. 2006;118:1325–1329. doi: 10.1002/ijc.21486. [DOI] [PubMed] [Google Scholar]
- 11.Stanbery L, et al. High SEPT9_v1 expression is associated with poor clinical outcomes in head and neck squamous cell carcinoma. Transl. Oncol. 2010;3:239–245. doi: 10.1593/tlo.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez ME, et al. High SEPT9_v1 expression in human breast cancer cells is associated with oncogenic phenotypes. Cancer Res. 2007;67:8554–8564. doi: 10.1158/0008-5472.CAN-07-1474. [DOI] [PubMed] [Google Scholar]
- 13.Connolly D, et al. Septin 9 isoform expression, localization and epigenetic changes during human and mouse breast cancer progression. Breast Cancer Res. 2011;13:R76. doi: 10.1186/bcr2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chacko AD, et al. Expression of the SEPT9_i4 isoform confers resistance to microtubule-interacting drugs. Cell. Oncol. 2012;35:85–93. doi: 10.1007/s13402-011-0066-0. [DOI] [PubMed] [Google Scholar]
- 15.Bertin A, et al. Phosphatidylinositol-4,5-bisphosphate promotes budding yeast septin filament assembly and organization. J. Mol. Biol. 2010;404:711–731. doi: 10.1016/j.jmb.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinoshita M, et al. Nedd5, a mammalian septin, is a novel cytoskeletal component interacting with actin-based structures. Genes Dev. 1997;11:1535–1547. doi: 10.1101/gad.11.12.1535. [DOI] [PubMed] [Google Scholar]
- 17.Silverman-Gavrila RV, Silverman-Gavrila LB. Septins: new microtubule interacting partners. Sci. World J. 2008;8:611–620. doi: 10.1100/tsw.2008.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spiliotis ET. Regulation of microtubule organization and functions by septin GTPases. Cytoskeleton. 2010;67:339–345. doi: 10.1002/cm.20448. [DOI] [PubMed] [Google Scholar]
- 19.Wloga D, Gaertig J. Post-translational modifications of microtubules. J. Cell Sci. 2010;123:3447–3455. doi: 10.1242/jcs.063727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hallak ME, Rodriguez JA, Barra HS, Caputto R. Release of tyrosine from tyrosinated tubulin. Some common factors that affect this process and the assembly of tubulin. FEBS Lett. 1977;73:147–150. doi: 10.1016/0014-5793(77)80968-X. [DOI] [PubMed] [Google Scholar]
- 21.Aillaud C, et al. Vasohibins/SVBP are tubulin carboxypeptidases (TCPs) that regulate neuron differentiation. Science. 2017;358:1448–1453. doi: 10.1126/science.aao4165. [DOI] [PubMed] [Google Scholar]
- 22.Ersfeld K, et al. Characterization of the tubulin-tyrosine ligase. J. Cell Biol. 1993;120:725–732. doi: 10.1083/jcb.120.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banerjee A. Increased levels of tyrosinated alpha-, beta(III)-, and beta(IV)-tubulin isotypes in paclitaxel-resistant MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun. 2002;293:598–601. doi: 10.1016/S0006-291X(02)00269-3. [DOI] [PubMed] [Google Scholar]
- 24.Bonnet C, et al. Differential binding regulation of microtubule-associated proteins MAP1A, MAP1B, and MAP2 by tubulin polyglutamylation. J. Biol. Chem. 2001;276:12839–12848. doi: 10.1074/jbc.M011380200. [DOI] [PubMed] [Google Scholar]
- 25.Lacroix B, et al. Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. J. Cell. Biol. 2010;189:945–954. doi: 10.1083/jcb.201001024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magiera MM, Janke C. Posttranslational modifications of tubulin. Curr. Biol. 2014;24:R351–R354. doi: 10.1016/j.cub.2014.03.032. [DOI] [PubMed] [Google Scholar]
- 27.Song Y, Brady ST. Post-translational modifications of tubulin: pathways to functional diversity of microtubules. Trends Cell Biol. 2015;25:125–136. doi: 10.1016/j.tcb.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGrogan, B. T., Gilmartin, B., Carney, D. N. & McCann, A. Taxanes, microtubules and chemoresistant breast cancer. Biochim. Biophys. Acta1785, 96–132 (2008). [DOI] [PubMed]
- 29.Peris L, et al. Tubulin tyrosination is a major factor affecting the recruitment of CAP-Gly proteins at microtubule plus ends. J. Cell Biol. 2006;174:839–849. doi: 10.1083/jcb.200512058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peris L, et al. Motor-dependent microtubule disassembly driven by tubulin tyrosination. J. Cell Biol. 2009;185:1159–1166. doi: 10.1083/jcb.200902142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bai X, et al. Novel septin 9 repeat motifs altered in neuralgic amyotrophy bind and bundle microtubules. J. Cell Biol. 2013;203:895–905. doi: 10.1083/jcb.201308068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sellin ME, Stenmark S, Gullberg M. Cell type-specific expression of SEPT3-homology subgroup members controls the subunit number of heteromeric septin complexes. Mol. Biol. Cell. 2014;25:1594–1607. doi: 10.1091/mbc.e13-09-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith C, et al. Septin 9 exhibits polymorphic binding to F-actin and inhibits myosin and cofilin activity. J. Mol. Biol. 2015;427:3273–3284. doi: 10.1016/j.jmb.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Janke C, Rogowski K, van Dijk J. Polyglutamylation: a fine-regulator of protein function? ‘Protein Modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:636–641. doi: 10.1038/embor.2008.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rogowski K, et al. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell. 2010;143:564–578. doi: 10.1016/j.cell.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 36.Berezniuk I, et al. Cytosolic carboxypeptidase 1 is involved in processing α- and β-tubulin. J. Biol. Chem. 2012;287:6503–6517. doi: 10.1074/jbc.M111.309138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sangrajrang S, et al. Association of estramustine resistance in human prostatic carcinoma cells with modified patterns of tubulin expression. Biochem. Pharmacol. 1998;55:325–331. doi: 10.1016/S0006-2952(97)00472-3. [DOI] [PubMed] [Google Scholar]
- 38.Mostowy S, Cossart P. Septins: the fourth component of the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2012;13:183–194. doi: 10.1038/nrm3284. [DOI] [PubMed] [Google Scholar]
- 39.Bridges AA, Gladfelter AS. Septin form and function at the cell cortex. J. Biol. Chem. 2015;290:17173–17180. doi: 10.1074/jbc.R114.634444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oegema K, Savoian MS, Mitchison TJ, Field CM. Functional analysis of a human homologue of the Drosophila actin binding protein anillin suggests a role in cytokinesis. J. Cell Biol. 2000;150:539–552. doi: 10.1083/jcb.150.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joo E, Surka MC, Trimble WS. Mammalian SEPT2 is required for scaffolding nonmuscle myosin II and its kinases. Dev. Cell. 2007;13:677–690. doi: 10.1016/j.devcel.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 42.Surka MC, Tsang CW, Trimble WS. The mammalian septin MSF localizes with microtubules and is required for completion of cytokinesis. Mol. Biol. Cell. 2002;13:3532–3545. doi: 10.1091/mbc.e02-01-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagata K, et al. Filament formation of MSF-A, a mammalian septin, in human mammary epithelial cells depends on interactions with microtubules. J. Biol. Chem. 2003;278:18538–18543. doi: 10.1074/jbc.M205246200. [DOI] [PubMed] [Google Scholar]
- 44.Poüs C, Klipfel L, Baillet A. Cancer-related functions and subcellular localizations of septins. Front. Cell Dev. Biol. 2016;4:126–133. doi: 10.3389/fcell.2016.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spiliotis ET. Spatial effects - site-specific regulation of actin and microtubule organization by septin GTPases. J. Cell Sci. 2018;131:207555–207564. doi: 10.1242/jcs.207555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chacko AD, et al. SEPT9_v4 expression induces morphological change, increased motility and disturbed polarity. J. Pathol. 2005;206:458–465. doi: 10.1002/path.1794. [DOI] [PubMed] [Google Scholar]
- 47.Dolat L, et al. Septins promote stress fiber-mediated maturation of focal adhesions and renal epithelial motility. J. Cell Biol. 2014;207:225–235. doi: 10.1083/jcb.201405050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kremer BE, Adang LA, Macara IG. Septins regulate actin organization and cell-cycle arrest through nuclear accumulation of NCK mediated by SOCS7. Cell. 2007;130:837–850. doi: 10.1016/j.cell.2007.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spiliotis ET, Gladfelter AS. Spatial guidance of cell asymmetry: septin GTPases show the way. Traffic. 2012;13:195–203. doi: 10.1111/j.1600-0854.2011.01268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spiliotis ET, Hunt SJ, Hu Q, Kinoshita M, Nelson WJ. Epithelial polarity requires septin coupling of vesicle transport to polyglutamylated microtubules. J. Cell Biol. 2008;180:295–303. doi: 10.1083/jcb.200710039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kremer BE, Haystead T, Macara IG. Mammalian septins regulate microtubule stability through interaction with the microtubule-binding protein MAP4. Mol. Biol. Cell. 2005;16:4648–4659. doi: 10.1091/mbc.e05-03-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beise N, Trimble W. Septins at a glance. J. Cell Sci. 2011;124:4141–4146. doi: 10.1242/jcs.087007. [DOI] [PubMed] [Google Scholar]
- 53.Liu M, Shen S, Chen F, Yu W, Yu L. Linking the septin expression with carcinogenesis. Mol. Biol. Rep. 2010;37:3601–3608. doi: 10.1007/s11033-010-0009-2. [DOI] [PubMed] [Google Scholar]
- 54.Stanbery L, Petty EM. Steps solidifying a role for SEPT9 in breast cancer suggest that greater strides are needed. Breast Cancer Res. 2012;14:101–103. doi: 10.1186/bcr3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Verdier-Pinard P, et al. Septin9_i2 is downregulated in tumors, impairs cancer cell migration and alters subnuclear actin filaments. Sci. Rep. 2017;7:44976–44993. doi: 10.1038/srep44976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Amir S, Golan M, Mabjeesh NJ. Targeted knockdown of SEPT9_v1 inhibits tumor growth and angiogenesis of human prostate cancer cells concomitant with disruption of hypoxia-inducible factor-1 pathway. Mol. Cancer Res. 2010;8:643–652. doi: 10.1158/1541-7786.MCR-09-0497. [DOI] [PubMed] [Google Scholar]
- 57.Gilad R, et al. High SEPT9_i1 Protein expression is associated with high-grade prostate cancers. PLoS ONE. 2015;10:e0124251. doi: 10.1371/journal.pone.0124251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amir S, Mabjeesh NJ. SEPT9_V1 protein expression is associated with human cancer cell resistance to microtubule disrupting agents. Cancer Biol. Ther. 2007;6:1926–1931. doi: 10.4161/cbt.6.12.4971. [DOI] [PubMed] [Google Scholar]
- 59.Golan M, Mabjeesh NJ. SEPT9_i1 is required for the association between HIF-1α and importin-α to promote efficient nuclear translocation. Cell Cycle. 2013;12:2297–2308. doi: 10.4161/cc.25379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tazat, K., Schindler, S., Deppeing, R. & Mabjeesh, N. J. Septin 9 isoform 1 (SEPT9_i1) specifically interacts with importin-a1 to drive hypoxia-inducible factor (HIF)-1a nuclear translocation. Cytoskeleton10.1002/cm.21450 (2018). [DOI] [PubMed]
- 61.Gonzalez ME, Makarova O, Peterson EA, Privette LM, Petty EM. Up-regulation of SEPT9_v1 stabilizes c-Jun-N-terminal kinase and contributes to itspro-proliferative activity in mammary epithelial cells. Cell Signal. 2009;21:477–487. doi: 10.1016/j.cellsig.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Daire V, et al. Kinesin-1 regulates microtubule dynamics via a c-Jun N-terminal kinase-dependent mechanism. J. Biol. Chem. 2009;284:31992–32001. doi: 10.1074/jbc.M109.007906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakos K, Rosenberg M, Spiliotis ET. Regulation of microtubule plus end dynamics by septin 9. Cytoskeleton. 2018 doi: 10.1002/cm.21488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mavrakis M, et al. Septins promote F-actin ring formation by crosslinking actin filaments into curved bundles. Nat. Cell Biol. 2014;16:322–334. doi: 10.1038/ncb2921. [DOI] [PubMed] [Google Scholar]
- 65.Farrugia AJ, Calvo F. The Borg family of Cdc42 effector proteins Cdc42EP1-5. Biochem. Soc. Trans. 2016;44:1709–1716. doi: 10.1042/BST20160219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bai X, Karasmanis EP, Spiliotis ET. Septin 9 interacts with kinesin KIF17 and interferes with the mechanism of NMDA receptor cargo binding and transport. Mol. Biol. Cell. 2016;27:897–906. doi: 10.1091/mbc.E15-07-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nölke T, et al. Septins guide microtubule protrusions induced by actin-depolymerizing toxins like Clostridium difficile transferase (CDT) Proc. Natl Acad. Sci. USA. 2016;113:7870–7875. doi: 10.1073/pnas.1522717113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.