

Abstract
Studies of Plasmodium vivax gene expression are complicated by the lack of in vitro culture system and the difficulties associated with studying clinical infections that often contain multiple clones and a mixture of parasite stages. Here, we characterize the transcriptomes of P. vivax parasites from 26 malaria patients. We show that most parasite mRNAs derive from trophozoites and that the asynchronicity of P. vivax infections is therefore unlikely to confound gene expression studies. Analyses of gametocyte genes reveal two distinct clusters of co-regulated genes, suggesting that male and female gametocytes are independently regulated. Finally, we analyze gene expression changes induced by chloroquine and show that this antimalarial drug efficiently eliminates most P. vivax parasite stages but, in contrast to P. falciparum, does not affect trophozoites.
Plasmodium vivax biology is not well understood, due to a lack of in vitro culture systems and difficulties associated with studying clinical blood samples. Here, Kim et al. use gene expression profiles from P. vivax infected patient blood and show stage-specific chloroquine response and differential regulation of male and female gametocytes.
Introduction
Plasmodium vivax is the most widespread human malaria parasite, responsible for more than 8.5 million clinical malaria cases worldwide in 2016 and threatening more than 2 billion people in 90 countries1,2. P. vivax diverged more than 100 million years ago from Plasmodium falciparum3 and displays unique biological features complicating malaria treatment and elimination, including a lower parasitemia and pyrogenic threshold4, an elusive dormant liver-stage5, and a rapid gametocytogenesis6 often leading to the apparition of transmittable parasites before the onset of malaria symptoms. Unfortunately, our understanding of the biology of this important human pathogen remains limited and lags behind that of P. falciparum, primarily due to our inability to continuously propagate P. vivax parasites in vitro4. Most studies of P. vivax have to rely on infected blood samples, which complicates biological and molecular investigations due to the polyclonality of many P. vivax infections7–14, the concurrent presence of different parasite stages (caused by the lack of sequestration4,15 typical in P. falciparum16) with their specific regulatory programs and responses, and the abundance of host molecules that hamper genomic studies. As a result, studies of P. vivax transcriptomes, which could provide unique insights on the biology of this parasite and its differences with P. falciparum and rodent parasites, have been few and far between, and limited to parasites grown in short-term ex vivo culture17–19 (which might alter the gene expression profiles20). We therefore still have a very limited understanding of the patterns of P. vivax gene expression during clinical infections or of their changes upon antimalarial drug treatment.
We previously analyzed blood samples from three P. vivax infected patients and showed that, despite extensive differences in stage composition, the parasite gene expression profiles were very similar across infections21. Here, we expand these analyses and characterize the in vivo parasite gene expression profiles from 26 vivax malaria patients enrolled in a chloroquine efficacy study22,23. First, we use gene expression deconvolution to estimate the stage of origin of the transcripts characterized and rigorously address composition heterogeneity. Second, we analyze variations in the expression of gametocyte genes among infected patients to better understand the mechanisms underlying successful parasite transmission. Finally, we compare the gene expression profiles of parasites before and after chloroquine administration to gain insights on the drug mode of action and examine how P. vivax parasites respond to this therapeutic stress. Overall, our study provides a global perspective on the diversity of expression profiles of P. vivax parasites in vivo and of their regulation.
Results
Variations in P. vivax gene expression among infections
We extracted RNA from ~50 µL of whole blood collected from 26 Cambodian individuals seeking treatment for vivax malaria. All patients were tested positive for P. vivax by RDT and blood smears, and PCR confirmed that they were solely infected with P. vivax22,23. We prepared and sequenced a stranded RNA-seq library from each blood sample after globin and rRNA reduction21. After removing reads originating from human transcripts, we aligned all remaining reads to the most recent P. vivax genome sequence24. The percentage of reads originating from P. vivax transcripts varied greatly among samples, from 1.09% to 41.63% (median: 11.97%), with only a moderate correlation with the samples’ parasitemia (Pearson’s R = 0.37, p = 0.06, Supplementary Figure 1). After stringent quality filters, 253,914–36,491,854 reads aligned to the P. vivax genome and 20 of the 26 samples yielded more than one million P. vivax reads (Supplementary Data 1). Out of the 6823 annotated P. vivax genes, 4999 were deemed expressed in at least ten patients and were further analyzed. Principal component analysis of the gene expression profiles showed no clear separation between samples (Supplementary Figure 2), nor according to the parasitemia, gametocytemia, or the stage composition determined by microscopy.
P. vivax infections are typically asynchronous and, at enrollment, most patient blood samples displayed multiple parasite stages in variable proportions (Fig. 1a). To statistically determine the contribution of the different developmental stages present in each infection to the overall P. vivax expression patterns, we performed gene expression deconvolution using single-cell RNA-seq data from P. falciparum synchronized cultures25 (see Methods). We first used Cibersort26 to determine the signature gene expression profile of each developmental stage using reference datasets (in our case from single-cell P. falciparum transcriptome data) and used these signatures to infer the relative contributions of these different cell types to bulk RNA-seq data. To validate this approach, we first analyzed RNA-seq data from synchronized P. falciparum cultures27 as well as artificial bulk RNA-seq data generated by mixing, in silico, known proportions of single-cell RNA-seq from Plasmodium berghei28. Overall, the deconvolution results from these control datasets were concordant with the expected composition of these samples (Supplementary Figure 3), indicating that gene expression deconvolution analysis is able to infer the stage of origin of the transcripts characterized from an asynchronous infection. We then used this approach to determine the stage composition of each P. vivax clinical infection based on its parasite gene expression profile. Consistent with our previous analyses21, we observed that, in each patient infection, the parasite transcripts derived almost exclusively from trophozoites, regardless of the stage composition determined by microscopy (Fig. 1b).
Fig. 1.

P. vivax stage composition of all clinical infections. Each vertical bar represents one clinical infection and is colored according to the proportion of schizonts (purple), trophozoites (green and blue), and rings (red). The top panel displays the stage composition determined by microscopy (a) while the bottom panel shows the stage composition inferred from gene expression deconvolution of the same infections (b). (The gene expression deconvolution for the M infection was not successful and is not shown.)
Regulation of the expression of gametocyte genes
The reference gene expression datasets used for gene expression deconvolution did not include gametocytes and the contribution of sexual parasites to the overall profile of each sample could therefore not be estimated using this approach. Examination of the transcript levels of the gametocyte genes Pvs47 and Pvs48/4529 revealed highly correlated expression among patients (Fig. 2a, Pearson’s R = 0.93, p < 0.01). To systematically investigate this pattern, we first compared the gene expression levels of 21 P. vivax genes thought to be expressed in gametocytes (Table 1). The gene expression of these 21 predicted gametocyte genes was highly correlated among samples but, interestingly, clustered into two distinct subsets poorly correlated with each other (Fig. 2b). Thus, Pvs47, Pvs48/45, Hap2, the gamete egress and sporozoite traversal protein, s16, and three CPW-WPC proteins were all positively correlated with each other (Cluster A in Fig. 2b, Pearson’s R = 0.15–0.96, p < 0.01), while Pvs25, ULG8, the gametocyte associated protein, gametocyte developmental protein 1, guanylate kinase, HMGB1, and five CPW-WPC proteins all clustered into a second group (Cluster B, Pearson’s R = 0.05–0.86, p < 0.01). Only genes belonging to this second group (Cluster B) had their expression correlated with the gametocytemia determined by microscopy (Pearson’s R > 0.5, p < 0.01, Table 1). To expand our knowledge of P. vivax genes possibly expressed in gametocytes, we searched for additional genes whose expression correlated with any of these gametocyte genes. Overall, the expression of 1613 genes was correlated with that of at least one of these known gametocyte genes (Pearson’s R > 0.8, p < 0.01) and could represent putative novel gametocyte genes. These included 460 genes correlated with members of Cluster A and 1153 correlated with genes in Cluster B (Supplementary Data 2). Gene ontology analyses showed that genes whose expression correlated with those of gametocyte genes from Cluster A were enriched in biological processes such as microtubule-related genes, including dynein, kinesin, and tubulin. By contrast, genes associated with intracellular trafficking and histone remodeling were overrepresented in Cluster B.
Fig. 2.

Expression of gametocyte genes. a Correlation between the expression level of two gametocyte genes. Each dot on the figure represents a single P. vivax patient infection and is displayed according to the normalized gene expression values of Pvs48/45 (x-axis) and Pvs47 (y-axis). b Heatmap showing the extent of gene expression correlation (Pearson’s R, in red scale) among 21 gametocyte candidate genes. The bordering tree shows the results of unsupervised clustering of these genes according to their gene expression pattern. Genes whose expression levels are highly correlated with the gametocytemia determined by microscopy are also indicated
Table 1.
Clustering of known gametocyte genes
| Cluster A | Cluster B | ||
|---|---|---|---|
| PVP01_0814300 | Male gamete fusion factor HAP2 | PVP01_0616100 | Ookinete surface protein Pvs25a |
| PVP01_0305600 | Sexual stage antigen s16 | PVP01_0820000 | CPW-WPC family proteina |
| PVP01_1258000 | Gamete egress sporozoite traversal protein | PVP01_1403000 | Gametocyte associated proteina |
| PVP01_1208100 | 6-Cysteine protein | PVP01_1125200 | CPW-WPC family proteina |
| PVP01_1208000 | 6-Cysteine protein | PVP01_0415800 | 6-Cysteine protein |
| PVP01_1215900 | CPW-WPC family protein | PVP01_0904300 | CPW-WPC family protein |
| PVP01_1119500 | CPW-WPC family protein | PVP01_0727400 | Guanylate kinase |
| PVP01_1320100 | CPW-WPC family protein | PVP01_1452800 | Upregulated in late gametocytes ULG8 |
| PVP01_0734100 | Gametocyte development protein 1 | ||
| PVP01_1003000 | CPW-WPC family protein | ||
| PVP01_1223200 | CPW-WPC family protein | ||
| PVP01_1302200 | High mobility group protein B1 | ||
| PVP01_1128100 | Inner membrane complex protein 1j | ||
aIndicates genes whose expression levels are correlated with the gametocytemia measured by microscopy
Effect of chloroquine exposure on P. vivax transcriptome
To assess how P. vivax parasites respond to chloroquine exposure, we compared the expression profiles of the parasites collected, from the same 20 infections, before and 8 h after the first dose of chloroquine. Consistent with the drug-induced clearance observed by microscopy, we observed that the proportion of RNA-seq reads aligned to the P. vivax genome decreased after chloroquine treatment (Fig. 3a). However, while the parasitemia measured by microscopy decreased by 57% (median) in the 8 h following the administration of the first dose of chloroquine, the proportion of P. vivax reads decreased by more than 81% (with no clear correlation with the change in parasitemia determined by microscopy, Supplementary Figure 4). Gene expression deconvolution analyses indicated that, 8 h after treatment, most parasite RNAs still primarily derived from trophozoites (Supplementary Figure 5).
Fig. 3.
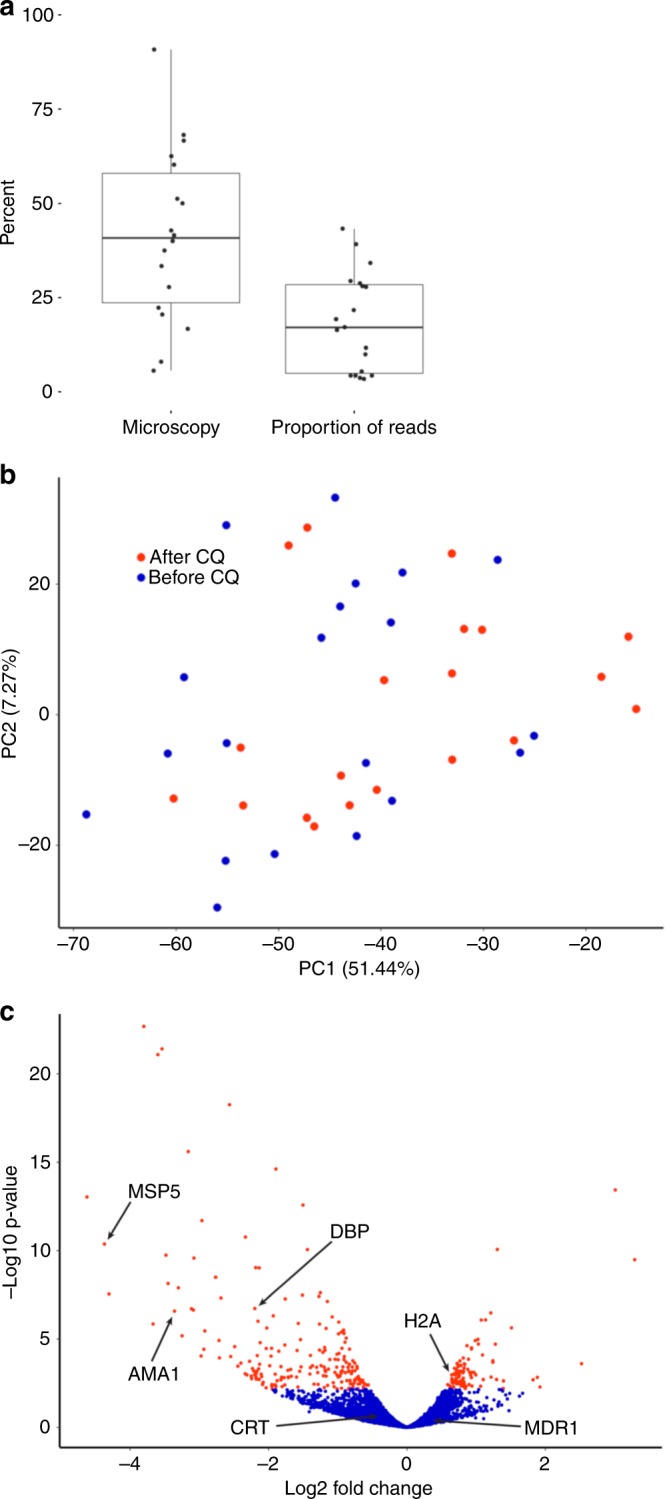
Effect of chloroquine on parasite gene expression. a Decrease in the number of parasites 8 h after chloroquine administration (represented by the proportion of remaining parasites, in %) measured by microscopy and by the proportion of parasite reads. Each dot represents one infection, the centerline shows the median, the box limits the quartiles, and the whiskers indicate the range (with outliers removed). b Principal component analysis of the parasite gene expression profiles from each sample before (blue dots) and 8 h after chloroquine administration (red dots). c Volcano plot of the gene expression changes after chloroquine treatment. Each dot represents one annotated P. vivax gene and is displayed according to the fold-change in expression (x-axis, in log2) and statistical significance (y-axis, in negative logarithm to the base 10 of the p-value). Red indicates significantly affected genes (FDR < 0.1)
To account for the extensive decrease in P. vivax reads post-treatment, that can lead to statistical artefacts, we randomly subsampled the datasets generated prior to chloroquine administration to the same number of reads as in the post-treatment datasets (see Methods for details). Principal component analysis showed that the parasite RNA profiles did not separate samples pre- and post-treatment (Fig. 3b), suggesting that, despite the large decrease in total parasite RNA induced by chloroquine, the overall gene expression patterns were not dramatically affected qualitatively. Indeed, permutation analyses showed that the differences in gene expression between paired samples (i.e., before and after chloroquine from the same patient) were significantly lower than those between randomly paired samples (i.e., comparing before and after chloroquine samples from different patients) (p = 4.2 × 10−4). This result indicated that the combination of parasite genetic diversity and host response to the infection had a greater effect on the parasite gene expression than the exposure to chloroquine.
While it did not separate the samples collected before and after chloroquine, PC1 was statistically associated with chloroquine treatment in paired analyses (p = 0.002, Supplementary Figure 6) suggesting that chloroquine did influence parasite gene expression. We therefore tested the effect of chloroquine exposure on each annotated P. vivax gene. Out of 4280 genes tested, 293 genes (195 downregulated and 98 upregulated) showed a statistically significant change in expression following chloroquine treatment (FDR < 0.1, Fig. 3c). Interestingly, a large number of exported protein genes (including 23 PHIST genes) and PIR genes were significantly downregulated after treatment, as were many genes involved in erythrocyte invasion (e.g., AMA1, DBP, MSP5, RBP2a, RBP2e, or RBP3) (Supplementary Data 3). Genes upregulated included ribosomal RNAs and histones. None of the candidate genes suspected to be associated with chloroquine susceptibility in P. vivax or P. falciparum showed significant changes in expression: for example, the chloroquine resistance transporter gene (CRT) showed only a modest, non-significant decrease in expression (log2 fold change = −0.47, FDR = 0.42) as did the multidrug resistance gene (MDR1) (log2 fold change = −1.48, FDR = 0.27). (Note that previous clinical and genetic analyses suggested that none of these P. vivax isolates were resistant to chloroquine23.) Since we previously showed that both CRT and MDR1 could be spliced into multiple isoforms in P. vivax21, we also tested whether the transcription of different isoforms was associated with chloroquine susceptibility. In samples pre-treatment, we did not observe any association between the isoforms expressed and the subsequent response to chloroquine: retention of the CRT intron 9, that leads to a predicted early stop codon, was highly variable among infections (Supplementary Figure 7A) but was not associated with the decrease in parasitemia (p = 0.58) nor with the proportion of P. vivax reads post-treatment (p = 0.95). Similarly, the extent of splicing of the 3′ UTR of MDR1 varied greatly among infections (Supplementary Figure 7B) but was not associated with the response to chloroquine.
Discussion
RNA-sequencing has provided invaluable insights on some of the fundamental characteristics of Plasmodium gene expression and, for example, highlighted the complexity of these parasites’ transcriptomes, with extensive noncoding transcripts, long 5′ and 3′ untranslated regions, and, often, multiple isoforms per gene17,21,30–32. Additionally, gene expression studies have been extremely useful to identify transcriptional differences between parasite stages18,19,27,33,34, in response to antimalarial drugs35–38 or to culture conditions20,39–41. However, most of these studies have been conducted using rodent malaria parasites or in vitro cultures of P. falciparum, and our understanding of gene regulation in other human malaria parasites remain very incomplete (but see also ref. 34). This is notably true for P. vivax for which the lack of in vitro culture system restricts studies to patient samples, with all the associated problems due to overwhelming host RNA and asynchronous parasites.
In this study, we showed that parasite gene expression profiles could be consistently generated from patients presenting with clinical vivax malaria. In contrast with the variable stage composition observed by microscopy among patient infections, gene expression deconvolution analyses revealed that the vast majority of RNA molecules in a P. vivax blood infection derived from a single developmental stage, the trophozoites. This observation is consistent with our previous analyses of three P. vivax-infected patients21, and with nuclear run-on experiments31,42, RNA polymerase II profiling31 and single-cell RNA-seq25 that showed that Plasmodium trophozoites are much more transcriptionally active than the other asexual parasite stages. In addition, the relatively low sensitivity of microscopy likely contributes to the discrepancy between the microscopy and gene expression deconvolution data: given the low parasitemia typical of P. vivax infections, only a small number of parasites are typically observed by microscopy, which might miss lowly abundant stages (e.g., the schizonts in Fig. 1) and limit the accuracy. For example, in an infection with a parasitemia of 0.1%, analysis of 200 fields by microscopy only provides stage information based on ~50 observed parasites, while RNA-seq data generated from 50 µL of blood would summarize information from ~250,000 parasites.
The overwhelming transcriptional signal of trophozoite parasites circumvents the limitation of studying asynchronous infections and indicates that analyses of parasite RNA-seq profiles generated directly from infected patient blood are unlikely to be confounded by stage differences (and could be further controlled by using the gene expression deconvolution results as covariates in the analyses). However, these results also imply that differences in gene regulation occurring in the less transcriptionally-active stages (e.g., rings) will be more difficult to detect using whole blood RNA-seq since fewer transcripts from these stages will be represented in the dataset (though our analysis of gametocyte genes show that this might not be impossible). Thus, analysis of a mechanism specific to ring-stage parasites (e.g., dormancy) might be difficult using whole-blood RNA-seq and may require other approaches, such as profiling after short-term cultures or single-cell RNA-seq25,28, to specifically investigate gene expression regulation at this stage.
We believe that the gene expression deconvolution analyses described here could be very useful in many Plasmodium gene expression studies to identify, and correct for (if necessary), the stages from which the mRNA molecules are derived. However, it is important to emphasize the limitations of these analyses that rely on a number of assumptions. First, the accuracy of the deconvolution depends on the reference datasets used for determining the signature of each stage (in our case scRNA-seq generated from synchronized P. falciparum cultures25). Using these profiles to deconvolute datasets from other synchronized P. falciparum cultures27 revealed some stage heterogeneity suggesting that (i) stage deconvolution is not entirely accurate and/or (ii) that some of these cultures might not have been perfectly synchronized and contained different parasite stages (Supplementary Figure 3). In addition, we used here profiles from P. falciparum to analyze P. vivax infections which may introduce some errors if the transcriptional regulation of the different stages differs between Plasmodium species. However, the analysis of mock P. berghei mixtures suggests that species differences are unlikely to dramatically influence deconvolution results, probably because the signature profile of each stage is a composite of hundreds of genes expressed at this stage (Supplementary Data 4) and is therefore robust to limited species-specific gene expression differences34. Finally, it is possible that the reference profiles of in vitro parasites may not truly recapitulate in vivo gene expression patterns20. Note that all these potential issues could be alleviated in the near future by using, for the reference stage profiles, scRNA-seq data generated from P. vivax in vivo infections. We also used here gene expression deconvolution to determine the proportion of mRNA derived from each stage, assuming that these developmental stages are discrete entities, when in fact parasite gene expression varies continuously along the intraerythrocytic developmental stages25,28,34,35,43 (but see also ref. 44). However, we chose to classify parasites into discrete developmental categories to facilitate the comparison with microscopy data and the data interpretation (see e.g., the discussion of the effect of chloroquine below). Along the same idea, the discrete parasite stages used to categorize the origin of each transcript are crudely defined and likely encompass a range of more subtle developmental changes. This low resolution might fail to capture the exact biological mechanisms at play and, for example, the overall conservation of the gene expression profiles observed in broadly-defined trophozoites upon chloroquine exposure might mask differences among parasites at different points of this extended developmental period. Again, these limitations could be alleviated in future studies by grouping reference datasets from scRNA-seq into more bins, representing intermediary and transitional stages, and thus providing a better temporal resolution.
The dataset used for gene expression deconvolution did not include profiles from gametocytes and these were therefore not considered in these analyses. However, we observed that the expression levels of putative gametocyte markers were highly correlated with each other across infections but, surprisingly, clustered in two distinct groups. Our co-expression analyses also revealed novel putative gametocyte genes and indicated that microtubule-associated proteins were overrepresented among the genes from one cluster, while intracellular trafficking and histone remodeling genes were overrepresented in the second cluster. One explanation for these observations is that the Cluster A (that includes hap2) represented genes expressed in male gametocytes while Cluster B (that includes Pvs25) corresponded to female gametocyte genes (Table 1). This hypothesis is consistent with the observation that gametocytemia (determined by microscopy) was only correlated with the expression of genes from Cluster B since P. vivax female gametocytes are much more abundant and easier to detect by microscopy45,46. These data indicate that, not only do the proportions of gametocytes vary across infections but, more importantly, that the ratio of male to female gametocytes differs among infections. This observation is consistent with previous studies47,48 and implies that the male and female terminal gametocytogeneses are independently-regulated, which could provide a mechanism for the parasites to reduce the probability of self-fertilization in mosquito by isolating, perhaps temporally, the generation of gametes of different sexes by a single clone. Note that by identifying genes that are potentially highly expressed in male and female P. vivax gametocytes (Supplementary Data 2), our findings will also facilitate further investigations of this hypothesis by enabling the monitoring, over time, of changes in the relative proportions of male and female gametocytes in P. vivax infections by qRT-PCR. In addition, these findings, if confirmed, would suggest that estimates of transmission potential need to account for both sexes, rather than just the presence of gametocytes, since some infections might, at a given time, only contain gametocytes from one sex (or a non-optimal proportion of both sexes).
We also explored how exposure to chloroquine affects the regulation of P. vivax gene expression in vivo by sequencing RNA from parasites collected 8 h after the first dose of chloroquine treatment. As expected, we observed a large decrease in the proportion of reads aligned to the P. vivax genome when compared to samples pre-treatment. However, the magnitude of this decrease was significantly greater than the decrease in parasitemia measured by microscopy. We hypothesize that the discrepancy between the microscopy data and the number of RNA-seq reads could be caused by counting dead or metabolically-inactive parasites, resulting in an overestimation of the parasitemia after chloroquine treatment. This hypothesis, that will need to be validated in future analyses, would suggest that clearance rates, typically determined for antimalarial drugs by microscopy, could be underestimated due to the confounding rate at which dead parasites are cleared from the circulation. In striking contrast with this important decrease in the proportion of parasite mRNAs after treatment, we noted few qualitative changes in parasite gene expression upon treatment. In fact, only a handful of genes were differentially expressed after drug treatment and the overall profiles of gene expression remained unchanged between infections. One possible explanation for this observation is that the trophozoites, who contribute the vast majority of the parasite transcripts, are unaffected by chloroquine while the other blood stages are rapidly eliminated (the decrease in the expression level of invasion genes and exported proteins after treatment could thus reflect a lower proportion of schizonts after treatment). This hypothesis would explain the similar pattern of gene expression before and after treatment, while (1) the lack of new trophozoites 8 h after treatment and (2) the continued development of trophozoites into chloroquine-susceptible schizonts would explain the observed decrease in total parasite RNAs (Fig. 4). (All P. vivax infections included in this study were efficiently cleared by chloroquine and we did not observe any evidence of drug resistance in these parasites23.) This hypothesis is consistent with the results of ex vivo studies that showed that P. vivax trophozoites are insensitive to chloroquine49–51 in contrast to P. falciparum trophozoites. This difference in the stage-susceptibility to chloroquine could indicate that the drug acts through a different mechanism in P. vivax than it does in P. falciparum (see e.g., ref. 49). For example, we could speculate that inhibition of haemozoin synthesis by chloroquine might have a strong detrimental effect on P. falciparum trophozoites that have a single large food vacuole but a lesser effect on P. vivax trophozoites that have many small vacuoles. These findings emphasize the marked differences between P. falciparum and P. vivax and the need to specifically study P. vivax as the observations from one Plasmodium species might translate poorly to another.
Fig. 4.
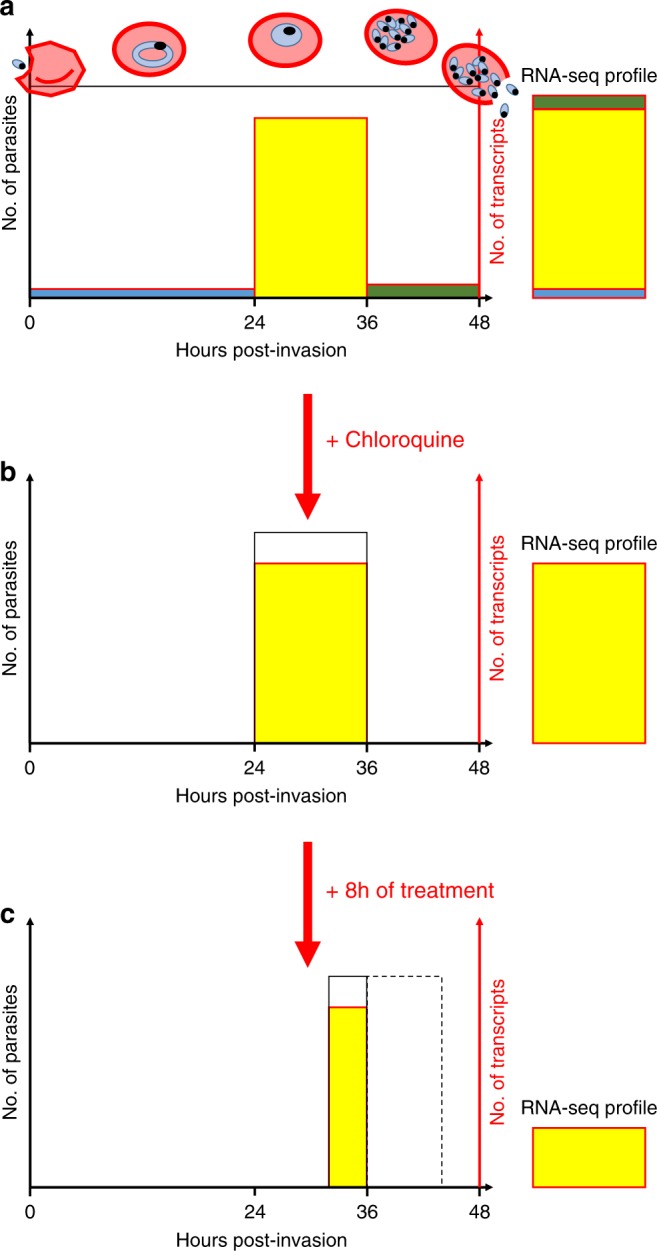
Possible effect of chloroquine on P. vivax blood-stage parasites. a The figure shows a simplified P. vivax infection containing uniform amount of parasites (black line) along their intraerythrocytic cycle (x-axis, in hours). The amount of mRNA produced by the different stages is represented in color (blue-rings, yellow-trophozoites, and green-schizonts) and well as the resulting RNA-seq profile that is dominated by the transcriptionally active trophozoites (right). b Upon treatment with chloroquine, all rings and schizonts are killed while the trophozoites are not affected. c Eight hours after chloroquine treatment, there are no new trophozoites, since the rings have been eliminated, and the trophozoites developing into schizonts die (dashed box), producing much fewer P. vivax reads by RNA-seq, but identical expression profiles
Overall, our study confirmed previous analyses21 that robust gene expression profiles could be generated directly from blood samples of vivax malaria patients and that stage composition differences between infections were unlikely to confound gene expression studies. We expanded the gene expression analysis to all patients participating in a drug efficacy study in Cambodia22,23 and, using a statistical approach to infer the stage of origin of the transcripts, showed that parasite mRNAs overwhelmingly derived from trophozoites. Our analyses also revealed that the production of male and female gametocytes was not correlated with each other, and that the gametocyte sex ratio varied significantly among infections, providing a possible mechanism for the parasite to reduce self-fertilization. Finally and consistently with our clinical observations23, we showed that chloroquine efficiently cleared most blood-stage parasites but had little effect on P. vivax trophozoites, indicating that chloroquine may act according to different mode of action in P. vivax than it does in P. falciparum. In summary, our study highlighted the biological knowledge that can be obtained from studying in vivo the gene expression profiles of P. vivax clinical infections and, combined with in-depth analysis of genetic diversity22,23, showed that genomic approaches provide a promising framework to better understand this important human pathogen.
Methods
Patients
We analyzed blood samples collected from 26 vivax malaria patients enrolled in a chloroquine efficacy study23. All patients originated from villages within 50 km of BanLung city (Ratanakiri Province, Cambodia), presented with fever (or history of fever within 48 h) and were positive for P. vivax DNA and no other Plasmodium DNA (see refs. 22,23 for details). After providing written informed consent, all patients were treated with a supervised standard 3-day course of chloroquine (30 mg/kg, Nivaquine) and monitored for 60 days. The study was approved by the Cambodian National Ethics Committee for Health Research (038 NECHR 24/02/14) and registered at ClinicalTrials.gov (NTC02118090).
Sample collection and stranded RNA-seq library preparation
Upon enrollment, and prior to the first administration of chloroquine, we collected ~50 µL of blood by finger prick from all patients (n = 26) and immediately stored it in 500 µL of Trizol at −80 °C. Additional blood samples were collected similarly from 20 of the patients 8 h after the initial chloroquine administration.
We extracted RNA from all blood samples stored in Trizol using the Zymo Direct-zol kit with an in-column DNAse step and eluted RNA into 20 µL of water. We then prepared Illumina stranded RNA-seq libraries after ribosomal RNA and globin mRNA reduction21 and sequenced them on a HiSeq 2500 to generate a total of 1.2 billion paired-end reads of 50 bp with a median of 24 million reads per sample (Supplementary Data 1).
Read alignment
We aligned all reads, first to the human genome (Hg38), then to the P01 P. vivax genome (PlasmoDB P01 3424) using Tophat252 with the following parameters: -g 1 (to assign each read to a single location), -I 5000 (maximum intron length), -library-type fr-firststrand (for stranded libraries), resulting in a median of 5.5 million reads per sample aligned to P. vivax before chloroquine and 0.5 million reads after chloroquine (Supplementary Data 1). We removed potential PCR duplicates using the samtools rmdup function to end up with a median of 3.5 million reads per sample before chloroquine and 0.27 million reads after chloroquine. For each annotated P. vivax gene and each sample, we determined the number of reads overlapping any exon using custom perl scripts21. We then transformed the raw counts into normalized transcripts per million (tpm) by dividing each gene count by the gene length (in kb) and by the sum of these values (in millions) in each sample. For further analyses, we only considered annotated P. vivax genes with more than 20 transcripts per million (tpm) in at least 10 samples, resulting in 4999 genes analyzed.
Gene expression deconvolution
To determine the proportion of mRNA molecules derived from parasites at each developmental stage in each sample, we performed gene expression deconvolution using Cibersort26. Briefly, by comparing the profiles of “pure” cells, Cibersort first determines the gene expression signature that characterizes each specific cell type. For our analysis, we used single-cell RNA-seq data from different stages of the P. falciparum intraerythrocytic developmental cycle25. This dataset was generated using synchronized P. falciparum in vitro cultures collected at different time points to represent the following stages: ring, early trophozoites, late trophozoites, and schizonts. For each stage, we selected the transcriptomes of 100 cells and normalized the data into transcripts per million before removing all genes without reads in more than 90% of the cells. We then retrieved the orthologous P. vivax gene names using PlasmoDB, and any genes with no orthologs or multiple orthologs were removed, resulting in a total of 2143 genes. We used this dataset to determine signature gene expression profiles of each stage (provided as Supplementary Data 4) and estimated the proportion of the different stages in different mixed samples.
Gene expression deconvolution has been extensively used to investigate cell-type heterogeneity but we verified its performance for differentiating Plasmodium developmental stages using several datasets. First, we analyzed RNA-seq data generated from another study of synchronized in vitro cultures of P. falciparum parasites27. Second, we generated mock bulk RNA-seq data by mixing, in different proportions, 20 parasites at different stages from a third dataset consisting of single-cell RNA-seq generated from P. berghei28. All datasets were converted to orthologous P. vivax genes in order to use the same signature gene set for all analyses. Finally, we used Cibersort to analyze the P. vivax RNA-seq profiles generated from all patient infections. For all analyses, we ran Cibersort with 100 permutations and quantile normalization disabled.
Gene co-expression analysis
To examine the gene expression of gametocytes, we first retrieved putative gametocyte genes from the literature and determined the Pearson correlation between the gene expression level of each pair of gametocyte genes across all samples. We then grouped these genes using unsupervised hierarchical clustering and assessed the significance of the clusters using pvclust53. We also identified putative novel gametocyte genes by retrieving genes whose expression was statistically correlated with one of the known gametocyte genes (with Pearson’s R > 0.8 and p < 0.01).
Determination of gene expression profile similarity
To assess the overall effect of the first dose of chloroquine on the parasite gene expression profiles, we compared the mean difference in gene expression across all genes between paired samples, to the mean difference observed between randomly paired samples. We first calculated the pair-wise normalized gene expression differences for each pair of samples (i.e., before and after chloroquine) across all expressed genes using the following formula:
where Xi,0 stands for the normalized gene expression (in tpm) of the sample i at time 0 for gene X. We then summed all pair-wise differences across all pairs of samples and compared this number to the sum of the pair-wise differences obtained by randomly pairing samples (n = 100) (i.e., Xi,0 − Xj,1 where i and j are different patients).
Identification of genes differentially expressed
We also determined which P. vivax genes were significantly affected by chloroquine by testing for differential gene expression using EdgeR and paired analyses54. To account for the large difference in the number of reads obtained from P. vivax mRNAs before and after chloroquine treatment, we randomly subsampled all pre-treatment datasets to the same number of reads as in their corresponding post-treatment datasets. Due to the lower read counts in samples after treatment and the subsampling, only 4280 genes remained expressed above the threshold described above and were included in the analysis of the effect of chloroquine on parasite gene expression. All analyses were corrected for multiple testing using false discovery rates55,56.
Code availability
All the programs, and the specific versions and parameters, used in this study are described in Methods. Custom code available at https://github.com/atomadam2/PvCQ_RNAseq.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
The authors would like to thank all patients and healthcare workers involved in this study and the staff of the Malaria Molecular Epidemiology Unit at the Institut Pasteur in Cambodia and of the National Center for Parasitology, Entomology and Malaria Control in Cambodia for their collaboration and sample collection. The authors would also like to thank Dr. Kafsack and Dr. Lawniczak for facilitating access to their scRNA-seq data. This work was funded by a National Institutes of Health—NIAID award to D.S. (R01 AI103228).
Author contributions
A.K. performed all the molecular experiments and analyses described in this study. J.P. collected the blood samples and conducted the microscopy analyses. D.M. supervised the field study. A.K. and D.S. analyzed the data and wrote the manuscript. J.P. contributed to the manuscript preparation. The manuscript was reviewed by all authors.
Data availability
The sequence data are freely available in NCBI SRA under the BioProject PRJNA378759.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information accompanies this paper at 10.1038/s41467-019-08312-z.
References
- 1.WHO. World Malaria Report 2017 (2017).
- 2.Howes, R. E. et al. Global epidemiology of Plasmodium vivax. Am. J. Trop. Med. Hyg.10.4269/ajtmh.16-0141 (2016). [DOI] [PMC free article] [PubMed]
- 3.Loy, D. E. et al. Out of Africa: origins and evolution of the human malaria parasites Plasmodium falciparum and Plasmodium vivax. Int. J. Parasitol. 47, 87–97 (2017). [DOI] [PMC free article] [PubMed]
- 4.Mueller I, et al. Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect. Dis. 2009;9:555–566. doi: 10.1016/S1473-3099(09)70177-X. [DOI] [PubMed] [Google Scholar]
- 5.White NJ, Imwong M. Relapse. Adv. Parasitol. 2012;80:113–150. doi: 10.1016/B978-0-12-397900-1.00002-5. [DOI] [PubMed] [Google Scholar]
- 6.Baker DA. Malaria gametocytogenesis. Mol. Biochem. Parasitol. 2010;172:57–65. doi: 10.1016/j.molbiopara.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fola AA, et al. Higher complexity of infection and genetic diversity of Plasmodium vivax than Plasmodium falciparum across all malaria transmission zones of Papua New Guinea. Am. J. Trop. Med. Hyg. 2017;96:630–641. doi: 10.4269/ajtmh.16-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedrich LR, et al. Complexity of infection and genetic diversity in Cambodian Plasmodium vivax. PLoS Negl. Trop. Dis. 2016;10:e0004526. doi: 10.1371/journal.pntd.0004526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan ER, et al. Whole genome sequencing of field isolates provides robust characterization of genetic diversity in Plasmodium vivax. PLoS Negl. Trop. Dis. 2012;6:e1811. doi: 10.1371/journal.pntd.0001811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parobek CM, et al. Selective sweep suggests transcriptional regulation may underlie Plasmodium vivax resilience to malaria control measures in Cambodia. Proc. Natl Acad. Sci. USA. 2016;113:E8096–E8105. doi: 10.1073/pnas.1608828113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nair S, et al. Single-cell genomics for dissection of complex malaria infections. Genome Res. 2014;24:1028–1038. doi: 10.1101/gr.168286.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cui L, et al. Genetic diversity and multiple infections of Plasmodium vivax malaria in Western Thailand. Am. J. Trop. Med. Hyg. 2003;68:613–619. doi: 10.4269/ajtmh.2003.68.613. [DOI] [PubMed] [Google Scholar]
- 13.Karunaweera ND, et al. Extensive microsatellite diversity in the human malaria parasite Plasmodium vivax. Gene. 2008;410:105–112. doi: 10.1016/j.gene.2007.11.022. [DOI] [PubMed] [Google Scholar]
- 14.Lin JT, et al. Individual Plasmodium vivax msp1 variants within polyclonal P. vivax infections display different propensities for relapse. J. Clin. Microbiol. 2012;50:1449–1451. doi: 10.1128/JCM.06212-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Price RN, et al. Vivax malaria: neglected and not benign. Am. J. Trop. Med. Hyg. 2007;77:79–87. doi: 10.4269/ajtmh.2007.77.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller LH, Good MF, Milon G. Malaria pathogenesis. Science. 1994;264:1878–1883. doi: 10.1126/science.8009217. [DOI] [PubMed] [Google Scholar]
- 17.Zhu L, et al. New insights into the Plasmodium vivax transcriptome using RNA-Seq. Sci. Rep. 2016;6:20498. doi: 10.1038/srep20498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bozdech Z, et al. The transcriptome of Plasmodium vivax reveals divergence and diversity of transcriptional regulation in malaria parasites. Proc. Natl Acad. Sci. USA. 2008;105:16290–16295. doi: 10.1073/pnas.0807404105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Westenberger SJ, et al. A systems-based analysis of Plasmodium vivax lifecycle transcription from human to mosquito. PLoS Negl. Trop. Dis. 2010;4:e653. doi: 10.1371/journal.pntd.0000653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lapp SA, et al. Plasmodium knowlesi gene expression differs in ex vivo compared to in vitro blood-stage cultures. Malar. J. 2015;14:110. doi: 10.1186/s12936-015-0612-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim A, et al. Characterization of P. vivax blood stage transcriptomes from field isolates reveals similarities among infections and complex gene isoforms. Sci. Rep. 2017;7:7761. doi: 10.1038/s41598-017-07275-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Popovici, J. et al. Genomic analyses reveal the common occurrence and complexity of Plasmodium vivax relapses in Cambodia. mBio9, 10.1128/mBio.01888-17 (2018). [DOI] [PMC free article] [PubMed]
- 23.Popovici, J. et al. Recrudescence, reinfection or relapse? A more rigorous framework to assess chloroquine efficacy for vivax malaria. J. Infect. Dis.10.1093/infdis/jiy484 (2018). [DOI] [PMC free article] [PubMed]
- 24.Auburn S, et al. A new Plasmodium vivax reference sequence with improved assembly of the subtelomeres reveals an abundance of pir genes. Wellcome Open Res. 2016;1:4. doi: 10.12688/wellcomeopenres.9876.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poran A, et al. Single-cell RNA sequencing reveals a signature of sexual commitment in malaria parasites. Nature. 2017;551:95–99. doi: 10.1038/nature24280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newman AM, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otto TD, et al. New insights into the blood-stage transcriptome of Plasmodium falciparum using RNA-Seq. Mol. Microbiol. 2010;76:12–24. doi: 10.1111/j.1365-2958.2009.07026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reid, A. J. et al. Single-cell RNA-seq reveals hidden transcriptional variation in malaria parasites. eLife7, 10.7554/eLife.33105 (2018). [DOI] [PMC free article] [PubMed]
- 29.Tachibana M, et al. Plasmodium vivax gametocyte proteins, Pvs48/45 and Pvs47, induce transmission-reducing antibodies by DNA immunization. Vaccine. 2015;33:1901–1908. doi: 10.1016/j.vaccine.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 30.Broadbent KM, et al. Strand-specific RNA sequencing in Plasmodium falciparum malaria identifies developmentally regulated long non-coding RNA and circular RNA. BMC Genomics. 2015;16:454. doi: 10.1186/s12864-015-1603-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu XM, et al. Nascent RNA sequencing reveals mechanisms of gene regulation in the human malaria parasite Plasmodium falciparum. Nucleic Acids Res. 2017;45:7825–7840. doi: 10.1093/nar/gkx464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Painter HJ, Carrasquilla M, Llinas M. Capturing in vivo RNA transcriptional dynamics from the malaria parasite Plasmodium falciparum. Genome Res. 2017;27:1074–1086. doi: 10.1101/gr.217356.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otto TD, et al. A comprehensive evaluation of rodent malaria parasite genomes and gene expression. BMC Biol. 2014;12:86. doi: 10.1186/s12915-014-0086-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoo R, et al. Integrated analysis of the Plasmodium species transcriptome. EBioMedicine. 2016;7:255–266. doi: 10.1016/j.ebiom.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mok S, et al. Drug resistance. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance. Science. 2015;347:431–435. doi: 10.1126/science.1260403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gunasekera AM, Patankar S, Schug J, Eisen G, Wirth DF. Drug-induced alterations in gene expression of the asexual blood forms of Plasmodium falciparum. Mol. Microbiol. 2003;50:1229–1239. doi: 10.1046/j.1365-2958.2003.03787.x. [DOI] [PubMed] [Google Scholar]
- 37.Natalang O, et al. Dynamic RNA profiling in Plasmodium falciparum synchronized blood stages exposed to lethal doses of artesunate. BMC Genomics. 2008;9:388. doi: 10.1186/1471-2164-9-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ganesan K, et al. A genetically hard-wired metabolic transcriptome in Plasmodium falciparum fails to mount protective responses to lethal antifolates. PLoS Pathog. 2008;4:e1000214. doi: 10.1371/journal.ppat.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peters JM, Fowler EV, Krause DR, Cheng Q, Gatton ML. Differential changes in Plasmodium falciparum var transcription during adaptation to culture. J. Infect. Dis. 2007;195:748–755. doi: 10.1086/511436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Q, et al. From in vivo to in vitro: dynamic analysis of Plasmodium falciparum var gene expression patterns of patient isolates during adaptation to culture. PLoS ONE. 2011;6:e20591. doi: 10.1371/journal.pone.0020591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mackinnon MJ, et al. Comparative transcriptional and genomic analysis of Plasmodium falciparum field isolates. PLoS Pathog. 2009;5:e1000644. doi: 10.1371/journal.ppat.1000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sims JS, et al. Patterns of gene-specific and total transcriptional activity during the Plasmodium falciparum intraerythrocytic developmental cycle. Eukaryot. Cell. 2009;8:327–338. doi: 10.1128/EC.00340-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lemieux JE, et al. Statistical estimation of cell-cycle progression and lineage commitment in Plasmodium falciparum reveals a homogeneous pattern of transcription in ex vivo culture. Proc. Natl Acad. Sci. USA. 2009;106:7559–7564. doi: 10.1073/pnas.0811829106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adjalley SH, Scanfeld D, Kozlowski E, Llinas M, Fidock DA. Genome-wide transcriptome profiling reveals functional networks involving the Plasmodium falciparum drug resistance transporters PfCRT and PfMDR1. BMC Genomics. 2015;16:1090. doi: 10.1186/s12864-015-2320-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Annecke S. The relative numbers of male and female gametocytes in human malaria (Plasmodium vivax, Plasmodium falciparum and Plasmodium malariae) and Hoemoproteus in birds. Proc. R. Soc. Med. 1927;20:267–271. [PMC free article] [PubMed] [Google Scholar]
- 46.Furuya T, et al. Disruption of a Plasmodium falciparum gene linked to male sexual development causes early arrest in gametocytogenesis. Proc. Natl Acad. Sci. USA. 2005;102:16813–16818. doi: 10.1073/pnas.0501858102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reece SE, Read AF. Malaria sex ratios. Trends Ecol. Evol. 2000;15:259–260. doi: 10.1016/S0169-5347(00)01893-0. [DOI] [PubMed] [Google Scholar]
- 48.Reece SE, Drew DR, Gardner A. Sex ratio adjustment and kin discrimination in malaria parasites. Nature. 2008;453:609–614. doi: 10.1038/nature06954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharrock WW, et al. Plasmodium vivax trophozoites insensitive to chloroquine. Malar. J. 2008;7:94. doi: 10.1186/1475-2875-7-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Powell RD, Berglund EM. Effects of chloroquine upon the maturation of asexual erythrocytic forms of Plasmodium vivax in vitro. Am. J. Trop. Med. Hyg. 1974;23:1007–1014. doi: 10.4269/ajtmh.1974.23.1007. [DOI] [PubMed] [Google Scholar]
- 51.Suwanarusk R, et al. Chloroquine resistant Plasmodium vivax: in vitro characterisation and association with molecular polymorphisms. PLoS ONE. 2007;2:e1089. doi: 10.1371/journal.pone.0001089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22:1540–1542. doi: 10.1093/bioinformatics/btl117. [DOI] [PubMed] [Google Scholar]
- 54.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- 56.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The sequence data are freely available in NCBI SRA under the BioProject PRJNA378759.