

Abstract
Myotonic dystrophy type 1 (DM1) is an autosomal dominant, CTG•CAG microsatellite expansion disease. Expanded CUG repeat RNA sequester the muscleblind-like (MBNL) family of RNA-binding proteins, thereby disrupting their normal cellular function leading to global mis-regulation of RNA processing. Previously, the small molecule furamidine was shown to reduce CUG foci and rescue mis-splicing in a DM1 HeLa cell model and rescue mis-splicing in the HSALR DM1 mouse model, but furamidine’s mechanism of action was not explored. Here we use a combination of biochemical, cell toxicity and genomic studies in DM1 patient-derived myotubes and the HSALR DM1 mouse model to investigate furamidine’s mechanism of action. Mis-splicing rescue was observed in DM1 myotubes and the HSALR DM1 mouse with furamidine treatment. Interestingly, while furamidine was found to bind CTG•CAG repeat DNA with nanomolar affinity, a reduction in expanded CUG repeat transcript levels was observed in the HSALR DM1 mouse, but not DM1 patient-derived myotubes. Further investigation in these cells revealed that furamidine treatment at nanomolar concentrations led to up-regulation of MBNL1 and MBNL2 protein levels and a reduction of ribonuclear foci. Additionally, furamidine was shown to bind CUG RNA with nanomolar affinity and disrupted the MBNL1–CUG RNA complex in vitro at micromolar concentrations. Furamidine’s likely promiscuous interactions in vitro and in vivo appear to affect multiple pathways in the DM1 mechanism to rescue mis-splicing, yet surprisingly furamidine was shown globally to rescue many mis-splicing events with only modest off-target effects on gene expression in the HSALR DM1 mouse model. Importantly, over 20% of the differentially expressed genes were shown to be returned, to varying degrees, to wild type expression levels.
INTRODUCTION
Myotonic dystrophy type 1 (DM1) is a neuromuscular disorder caused by an unstable CTG•CAG expansion in the 3’ untranslated region (UTR) of the dystrophia myotonica protein kinase (DMPK) gene1. DM1 is an RNA gain-of-function disease, in which transcription of the expanded CTG repeats produces long tracts of CUG RNA that sequester the muscleblind-like (MBNL) family of RNA-binding proteins into nuclear foci, thereby disrupting their normal cellular function2-4. The DM1 repeats have also been shown to undergo bidirectional transcription and repeat-associated non-ATG (RAN) translation, which may have a role in disease pathogenesis5, 6. Members of the MBNL family regulate the alternative splicing of hundreds of tissue-specific transcripts and have been implicated in RNA localization and other RNA processing events7-9. DM1 patients exhibit a wide range of symptoms including myotonia, muscle weakness and wasting, cataracts, gastrointestinal issues and cognitive disabilities10. Many events that are misspliced in DM1 correspond directly with, or are linked to, disease symptoms. For example, mis-splicing of the insulin receptor (INSR), cardiac troponin T (TNNT2), and muscle-specific chloride channel (CLCN1) correspond to insulin insensitivity, cardiac defects, and myotonia, respectively11-13.
There are many potential molecular targets to consider in the development of therapeutics for DM1. These targets include genome editing to eliminate the expanded CTG repeats14, 15, inhibiting transcription from the CTG repeats16-18, degrading the CUG repeat RNA19-22, disrupting the MBNL–CUG RNA interaction23-26, increasing MBNL levels27, 28, and targeting downstream events such as single mis-spliced events29, among others. Much of the field has focused on targeting the toxic expanded CUG RNA. This can be achieved by degrading the RNA through the use of antisense oligonucleotides (ASOs), siRNAs and small molecules or by disrupting the MBNL–CUG RNA interaction with small molecules, peptides, or ASOs 19-23, 26. 30. We have focused on the strategy of reducing or eliminating transcription from the expanded CTG repeats, rather than targeting the RNA itself, due to the potential of ameliorating all downstream effects of the toxic RNA.
We previously found that the small molecule pentamidine, and its analog, heptamidine (Figure 1a), were able to rescue mis-spliced events and reduce CUG RNA levels in both cell and mouse models of DM1, albeit at relatively toxic concentrations16. To identify small molecules with increased specificity and decreased toxicity, we previously synthesized analogues of pentamidine that contained various substitutions to alter size, hydrophobicity, and the number of hydrogen bond donors31. From this study, we identified the analog furamidine (Figure 1b), which was shown to rescue mis-splicing of the INSR and TNNT2 minigene events, reduce ribonuclear foci in a DM1 HeLa cell model, as well as rescue the Atp2a1 exon22 and Clcn1 exon7a mis-splicing events in the HSALR DM1 mouse model with reduced toxicity compared to both pentamidine and heptamidine31. These previous findings motivated us to determine the mechanism of action by which furamidine rescues DM1 associated mis-splicing.
Figure 1. Structures of pentamidine analogs.
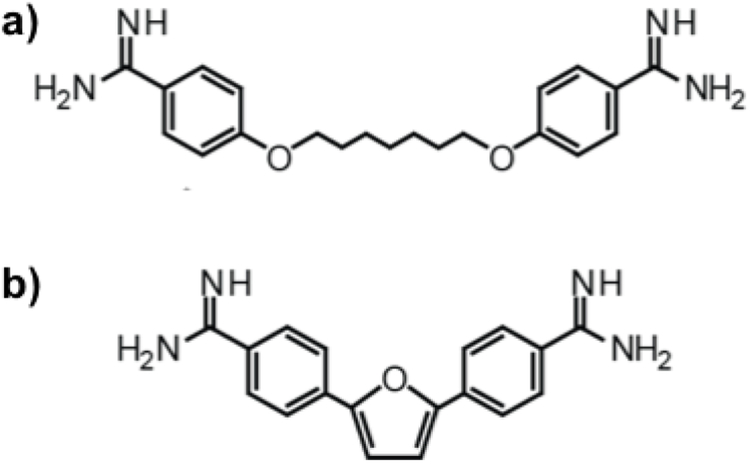
a) Heptamidine and b) furamidine have the characteristic structural features of diamidines that bind the minor groove of DNA: the ability to adopt a semi-curved shape, two terminal amidine groups that are positively charged at physiological pH, and a relatively flat conformation.
Furamidine is an analog of pentamidine with activity against Trypanosoma sp., Pneumocystis carinii, and Cryptosporidium parvum infections32-34. The mechanism of antimicrobial action of furamidine has not been elucidated; however, studies have shown that the primary mode of DNA binding for furamidine is at AT-rich sites of the minor groove35 and that it localizes to the nucleus when it enters cells36, 37. Therefore, it is proposed that furamidine and similar compounds work by binding the minor groove of DNA and inhibit DNA-dependent enzymes/regulatory factors or inhibit transcription or replication directly38, 39. Furamidine is also hypothesized to form non-canonical intercalation interactions at GpC dinucleotides at higher concentrations40. Further, it has been shown to bind A-form polyA·polyU dsRNA41, as well as displace the HIV-1 protein Rev from the Rev Response Element (RRE) RNA structure in the HIV-1 genome by binding the RRE in a structure dependent manner42. The promiscuous binding of furamidine to various nucleic acids suggests it could function to rescue DM1-associated mis-splicing through multiple mechanisms.
To determine the mechanism(s) of action of furamidine and its level of specificity in rescuing mis-splicing, furamidine was studied in two different DM1 models. In the HSALR DM1 mouse model treated with furamidine, RNA-seq and RT-qPCR were used to assess furamidine’s activity on gene expression and splicing compared to that of HSALR mice treated with heptamidine. Furamidine and heptamidine rescued many mis-splicing events, but furamidine caused markedly fewer off-target splicing and gene expression changes. In DM1 patient-derived myotubes, furamidine rescued mis-splicing in the nanomolar (nM) concentration range with no toxicity compared to heptamidine. Isothermal calorimetry (ITC) showed that furamidine binds CTG•CAG DNA repeats at nM concentrations, consistent with furamidine inhibiting transcription of expanded CUG repeats. In the HSALR mice, CUG repeat levels were reduced as expected, but the expanded CUG repeats were not significantly reduced in the DM1 myotubes with furamidine treatment. This surprising result motivated additional studies on furamidine’s mechanism of action in this cell line. We found furamidine treatment in DM1 myotubes reduced CUG ribonuclear foci and up-regulated MBNL1 and MBNL2 protein levels. In an in vitro gel mobility assay, furamidine displaced MBNL proteins from CUG RNA in the micromolar (μM) range and was shown to bind CUG RNA in the nM range via ITC. These results indicate that furamidine can affect multiple pathways of DM1 pathogenesis, suggesting that furamidine works through multiple mechanisms to rescue DM1-associated mis-splicing events.
RESULTS AND DISCUSSION
Furamidine reduced HSA transgene levels in the HSALR DM1 mouse model.
Previously, pentamidine and heptamidine were shown to rescue a few select mis-spliced events in the HSALR DM1 mouse model expressing approximately 220 CUG repeats in the 3’ UTR of human skeletal actin (HSA) trangene16, 43. This rescue of mis-splicing was likely due to the ability of pentamidine and heptamidine to reduce the transcript levels of the HSA transgene containing the CUG repeat RNA16. Furamidine was shown to rescue two mis-spliced events in the HSALR DM1 mouse, Atp2a1 and Clcn131, at 20 mg kg−1 daily for 7 days. Here, we treated HSALR mice with either 5% glucose in PBS (control) or 30 mg kg−1 furamidine daily for 7 days with the goal of achieving increased mis-splicing rescue with the higher dose. HSALR mice were also treated with 30 mg kg−1 heptamidine. We performed RT-PCR analysis on RNA samples from quadriceps muscle to confirm mis-splicing rescue of Atp2a1 exon22 and Clcn1 exon7a with furamidine treatment. In agreement with our previous data, furamidine rescued the mis-splicing of Atp2a1 and Clcn1, yielding percent rescues of 69 ± 11% and 84 ± 13%, respectively (p<0.01, purple points, Figure 2a and 2b). We previously observed percent rescues of 76 ± 6% and 82 ± 9% for Atp2a1 and Clcn1, respectively, with 20 mg kg−1 furamidine31, indicating that the 30 mg kg−1 treatment did not yield increased mis-splicing rescue as intended. With 30 mg kg−1 heptamidine treatment, we observed percent rescues of 63 10% and 106 1% for Atp2a1 and Clcn1, respectively, (p<0.01, green points, Figure 2a and 2b) consistent with our previous data43.
Figure 2. In HSALR mice, furamidine recued Atp2a1 and Clcn1 mis-splicing and reduced HSA transgene levels.

RT-PCR confirmed that both a) Atp2a1 exon22 and b) Clcn1 exon7a mis-splicing events were partially rescued by furamidine (purple) and heptamidine (green) treatment (p<0.01). RT-qPCR data showed that furamidine (purple) reduced c) HSA transgene levels (p<0.01) and did not affect d) endogenous Dmpk levels, while heptamidine (green) treatment drastically reduced both (p<0.001). e) A representative isothermal titration calorimetry (ITC) binding curve using a d(CTGCTGCAGCAG) palindrome sequence (inset lower right) shows that furamidine binds CTG repeat DNA with a K = (2.1 ± 0.3) ×106 M–1 when fit with a single binding model, where N = 7.2 ± 0.6, ∆H = −4104 ± 436 cal/mol, ∆S = 15 ± 1 cal/mol/deg. Raw heat of reaction versus time is inset in the upper left corner.
We performed RT-qPCR analysis to assess HSA transgene levels with furamidine treatment and found that furamidine reduced transgene levels to 62 ± 11% of HSALR control (p<0.01, purple bar, Figure 2c), but did not reduce endogenous Dmpk transcripts (purple bar, Figure 2d). Heptamidine treatment dramatically lowered both HSA transgene and Dmpk levels to 12 ± 11% and 24 ± 5% of control, respectively (p<0.001, green bar, Figure 2c and 2d). These results imply that, similar to other diamidines16, a primary mode of action of furamidine is inhibiting transcription from the CTG•CAG-containing transgene in the HSALR mouse model. Further, furamidine appears to have a higher specificity for reducing HSA transgene levels compared to heptamidine.
As furamidine is thought to be primarily an AT-rich DNA minor groove binder44, we performed ITC experiments to determine if furamidine could bind CTG•CAG repeat DNA. Furamidine did bind the CTG palindromic sequence d(CTGCTGCAGCAG) (inset bottom right, Figure 2e) with a KD of 485 ± 73 nM. A representative ITC binding curve is shown in Figure 2e with the raw heats of reaction versus time inset in the upper left corner. We compared the binding of furamidine to the CTG palindrome with that of an AT-rich DNA palindromic sequence, d(CGAAAATTTTCG) (inset bottom right, Figure S1). We observed curves indicative of two site binding for ITC experiments with furamidine and the AT-rich palindrome (Figure S1), consistent with previously published data using a similar sequence45. Interestingly, the KD for furamidine binding of the CTG palindrome was similar to that of furamidine binding to the lower affinity site of the AT-rich palindrome at 543 ± 16 nM. These data are consistent with the model that furamidine binds the expanded CTG•CAG repeats and inhibits transcription of toxic CUG repeat RNA in the HSALR mouse model.
Furamidine treatment partially rescued mis-splicing with little to no toxicity in DM1 patient-derived myotubes.
We next determined if furamidine rescued mis-splicing in DM1 patient-derived cell lines. We used two previously characterized myoblast lines, DM-04, a line derived from a non-DM individual, and DM-05, a DM1 patient-derived line expressing approximately 2900 CUG repeats46. The myoblasts were differentiated to myotubes over a 7-day period. A concentration range of 0.1 – 40 μM furamidine treatment was started on day three of differentiation and continued through day seven for a total of four days of treatment. Concentrations of 8 μM and above caused cell death and resulted in mis-splicing exacerbation and knockdown of MBNL1 and MBNL2 transcripts and proteins (supplemental information shows this data and is described below). The concentration range was narrowed to 0.1 – 4 μM to assess the mechanism of action of furamidine where mis-splicing rescue was observed. Toxicity data associated with the full 0.1 – 40 μM concentration range can be found in the supplementary information (Figure S2). To assess the effect of furamidine treatment on endogenous splicing events, RT-PCR analysis was performed for the exon-skipping (ES) events MBNL1 exon5, MBNL2 exon5, NUMA1 exon2, and SYNE1 exon137, as these events showed consistent differential splicing between the non-DM1 control and DM1 lines.
The splicing analyses for the concentration range of 0.1 – 4 μM furamidine treatments are shown in Figure 3. The percent rescue was calculated for each treatment, where percent rescue is the difference in exon inclusion levels, or percent spliced in (PSI), between the untreated and treated DM1 myotubes divided by the difference between the non-DM myotubes and untreated DM1 myotubes multiplied by 100. For MBNL1 and NUMA1 events, maximum rescue was observed at 1 μM, with percent rescues of 30 ± 3% and 22 ± 6%, respectively (p<0.001, Figure 3a and 3c), while MBNL2 and SYNE1 showed maximum rescue at 0.5 μM, with percent rescues of 47 ± 4% and 63 ± 9%, respectively (p<0.001, Figure 3b and 3d). The same general trend of rescue was observed for all events between 0.1 and 4 μM (p<0.01 or better, except NUMA1 was not statistically significant (NS) at 4 μM). Interestingly, heptamidine treatment in the DM1 patient-derived myotubes did not display mis-splicing rescue until 8 μM or above (Figure S3), which shows that furamidine has better activity for mis-splicing rescue in the DM1 myotubes. Further, furamidine treatment did not affect the inclusion levels of MBNL1 exon5, MBNL2 exon5, NUMA1 exon2, and SYNE1 exon137 in non-DM myotubes (Figure S4). We also determined if furamidine treatment affected the alternative splicing of four other endogenous pre-mRNAs that had previously been shown to be un-changed by expression of CUG repeats25. None of the four alternatively spliced exons tested were affected at any concentration of furamidine (Figure S5). These results suggest that furamidine does not globally affect alternative splicing in DM1 myotubes, but only a subset of pre-mRNAs that are mis-regulated in DM1.
Figure 3. Furamidine partially rescued mis-splicing in DM1 patient-derived myotubes.

a) MBNL1, b) MBNL2, c) NUMA1 and d) SYNE1 events displayed maximum percent rescue of 30 ± 3%, 47 ± 4%, 22 ± 6%, and 63 ± 9%, respectively, after 4 days of furamidine treatment (p<0.001). Maximum rescue occurred between 0.5 and 1 μM furamidine for all splicing events shown.
Next, we used an absorbance-based assay that measures the reducing power of living cells to measure cell toxicity. Importantly, between 0.1 and 4 μM treatment, furamidine displayed no cell toxicity in DM1 myotubes (Figure 4, purple bars). The same trends in toxicity held true with furamidine treatment in non-DM1 myotubes (Figure S6, purple bars). Heptamidine treatment caused cell toxicity by 0.5 μM in DM1 myotubes (p<0.001, Figure 4, green bars) and in non-DM myotubes (p<0.001, Figure S6, green bars). These results indicate that the presence of the expanded CUG repeats does not alter the toxicity of either of these compounds.
Figure 4. Furamidine displayed no toxicity in DM1 patient-derived myotubes.

Furamidine (purple) did not affect cell viability in the concentration range where mis-spicing rescue was observed (0.1 – 4 μM). Heptamidine (green) started to display significant cell toxicity after 4 days of treatment at 0.5 μM (p<0.001).
Furamidine modestly affected CUG RNA levels in DM1 patient-derived myotubes.
Both heptamidine and pentamidine have previously been shown to reduce the levels of CUG repeat RNA in DM1 cell and mouse models16. We performed northern blot analysis in the DM1 myotubes to determine if furamidine, like other diamidines, would reduce CUG repeat RNA levels. Surprisingly, while there does appear to be a modest decrease in CUG RNA levels between 0.1 and 0.75 μM furamidine treatment, the change was not statistically significant compared to that of CUG RNA levels of untreated DM1 myotubes (Figure 5). The same general trend was observed with RT-qPCR to evaluate DMPK expression levels (Figure S7). Therefore, in the concentration range where mis-splicing rescue was observed, furamidine did not significantly reduce the levels of the expanded CUG repeats.
Figure 5. CUG RNA levels were not significantly reduced with furamidine treatment in DM1 myotubes.

Northern blot quantification of CUG repeat RNA levels relative to GAPDH with the untreated CUG levels set to 1. There were no significant changes in CUG RNA levels at any concentration of furamidine treatment.
Furamidine treatment clearly reduced the expression of the CUG repeats in the HSALR mouse; however, it is puzzling that it did not reduce CUG RNA levels in the DM1 myotubes. We were concerned that this could be a transgene specific effect, meaning furamidine specifically knocks down the human ACTA1 gene. To investigate this possibility, we checked the homology of the mouse Acta1 gene versus the human ACTA1 gene and determined if its expression changed in the HSALR mice treated with furamidine. The mouse Acta1 gene has 84% sequence identity with the human ACTA1. Acta1 showed no significant expression changes with furamidine treatment based on our RNA-seq data in the HSALR mice (purple bar, Figure S8), however Acta1 levels were significantly reduced with heptamidine treatment to 44 ± 3% of control (p<0.001, green bar, Figure S8). Further, we used RT-qPCR to assess ACTA1 levels in the DM1 myotubes treated with furamidine (Figure S9). In the concentration range where mis-splicing rescue was observed in the DM1 myotubes, ACTA1 gene expression was slightly elevated (p<0.5 or better for all concentrations except NS for 2 and 4 μM). Although indirect, these data suggest that furamidine did not specifically reduce the human actin gene, but rather shows a degree of specificity for the CTG•CAG-containing HSA transgene.
Furamidine significantly reduced ribonuclear foci abundance in DM1 patient-derived myotubes and disrupted MBNL1 binding to CUG repeat RNA.
As furamidine treatment did not reduce CUG RNA levels in DM1 myotubes, we looked at other potential mechanisms of action to explain the mis-splicing rescue observed. We had previously shown that furamidine reduced ribonuclear foci in a DM1 HeLa cell model at 80 μM31. Therefore, we performed fluorescent in situ hybridization (FISH) against the CUG repeat RNA to assess activity of furamidine on ribonuclear foci formation in DM1 myotubes. Furamidine reduced the number of ribonuclear foci per nucleus relative to untreated DM1 myotubes in the 0.25 to 4 μM concentration range tested (Figure 6a). Foci number in at least 100 nuclei were counted per concentration per experiment (blinded) and then normalized by setting the untreated ratio to 1. Representative FISH images of DM1 myotubes with furamidine treatment are shown in Figure S10. At 1 μM furamidine treatment, the foci abundance was reduced to 0.72 ± 0.02 of untreated levels, which corresponds to the highest level of mis-splicing rescue.
Figure 6. Furamidine treatment reduced ribonuclear foci abundance in DM1 myotubes and disrupted the MBNL1-CUG complex in vitro.

a) Quantified FISH data showing the number of ribonuclear foci per nucleus with foci abundance of untreated DM1 cells set to 1. A reduction in ribonuclear foci per nucleus was observed for all furamidine concentrations tested (p<0.01 or better). b) Using a CUG8 hairpin construct (inset upper right) in an electrophoretic mobility shift assay (EMSA), furamidine displaced MBNL1 from CUG RNA with an IC50 of 40 ± 3 μM. c) A representative isothermal titration calorimetry (ITC) binding curve using a r(CUG)4 sequence that forms a duplex (inset) shows that furamidine binds CUG repeat RNA with a K = (1.06 ± 0.30) ×107 M−1 when fit with a single binding model, where N = 6.6 ± 0.2, ∆H = −6400 ± 238 cal/mol, ∆S = 10.2 ± 0.2 cal/mol/deg. Raw heat of reaction versus time is inset.
Based upon these findings, we hypothesized that furamidine may bind the expanded CUG RNA and disrupt the MBNL–CUG RNA complex. To test this hypothesis, an MBNL1–CUG repeat electrophoretic mobility shift assay (EMSA) was used to determine if furamidine disrupted the protein-RNA complex. The CUG RNA construct used contains eight CUG repeats stabilized into a stem-loop structure using the stable UUCG loop (inset upper right in Figure 6b). We found that furamidine was able to disrupt the MBNL1–CUG complex with an IC50 of 40 ± 3 μM (Figure 6b). This corresponds with our previous finding that 80 μM furamidine treatment reduced ribonuclear foci in HeLa cells transfected with a CUG960 plasmid31 and is consistent with displacing MBNL proteins from CUG repeats. Further, to determine if furamidine binds CUG repeat RNA, we performed ITC experiments using the CUG RNA sequence r(CUG)4 that forms a duplex47 (inset, Figure 6c). Furamidine was found to bind the CUG RNA with a KD of 99 ± 25 nM. A representative ITC binding curve is shown in Figure 6c with the raw heats of reaction versus time inset. The highest levels of mis-splicing rescue and reduction of ribonuclear foci all occurred between 0.5 to 1 μM furamidine. These results paired with furamidine’s ability to bind CUG RNA suggest that furamidine may disrupt the MBNL–CUG complex formation in cellulo leading to the release of MBNL.
Furamidine treatment increased MBNL1 and MBNL2 protein levels in DM1 patient-derived myotubes.
The exacerbation of mis-splicing (Figure S11 and S12) coupled with the significant reduction of foci (Figure S13) at higher furamidine concentrations suggested that furamidine treatment could alter MBNL levels. Loss of MBNL function can recapitulate aspects of DM1 pathogenesis48, 49, such as mis-splicing, and MBNL1 knockdown has been shown to decrease RNA foci accumulation50. Therefore, we wanted to determine if furamidine treatment impacted the expression of MBNLs. Interestingly, we found that MBNL1 and MBNL2 transcript levels increased concurrently in a dose-dependent manor up to 4 μM furamidine, reaching 1.5-fold and 2-fold, respectively (p<0.01 or better, Figure 7a). Notably, the increased levels of MBNL1 and MBNL2 transcripts caused by furamidine treatment did not appear to be dependent on the DM1 disease background. We observed similar transcript increases in non-DM myotubes (Figure S14). Further, the modulation of MBNL1 and MBNL2 expression does not appear to be a characteristic of diamidines in general as heptamidine treatment in the DM1 myotubes did not have a significant impact on transcript levels within the concentration range tested (Figure S15).
Figure 7. MBNL transcript and protein levels increased with furamidine treatment in DM1 myotubes.

a) RT-qPCR data showing MBNL1 (sold bars) and MBNL2 (patterned bars) expression levels and western blot data for b) MBNL1 and c) MBNL2 protein levels in DM1 patient-derived myotubes treated with furamidine. Furamidine treatment caused increased levels of both MBNL1 and MBNL2 transcripts (p<0.01 or better for all). Also, MBNL1 protein levels increased between 0.25 and 0.75 μM furamidine (p<0.05 or better), while MBNL2 protein levels increased between 0.1 and 2 μM furamidine (p<0.05 or better).
Protein levels of MBNL1 and MBNL2 were measured at the same concentration range of furamidine (0.1 μM to 4 μM) in DM1 myotubes and both proteins were increased anywhere from 109% to 127% of untreated levels between 0.25 and 1 μM furamidine (Figure 7b and 7c). MBNL1 levels peaked at 116 ± 5% with 0.5 μM treatment (p<0.02) and then steadily decreased back to untreated levels by 2 μM (Figure 7b). At 4 μM treatment, MBNL1 levels dropped to 86 ± 11% of untreated levels (p<0.01). Similarly, MBNL2 levels peaked at 127 ± 8% with 0.5 μM furamidine and remained elevated to 2 μM (p<0.01), then dropped back to untreated levels at 4 μM (Figure 7c). We observed that MBNL1 and MBNL2 protein levels increased in non-DM myotubes, as well (Figure S16), however the increases were not as dramatic for MBNL2. MBNL1 levels peaked with 0.75 μM furamidine treatment at 113 ± 6% (p<0.05) and MBNL2 levels peaked with 1 μM furamidine at 112 ± 5% (p<0.02) in the non-DM myotubes.
The modulation of MBNL transcript and protein levels by furamidine is intriguing and multiple mechanisms are likely involved over the 100-fold concentration range studied. Possibilities include furamidine up-regulated transcription through interactions with DNA in the MBNL1/2 genes or altered transcription factor binding as has been shown previously for furamidine with the PU.1 transcription factor39. Alternatively, furamidine could interact directly with the MBNL1/2 transcripts and stabilized the mRNAs. Although of interest, determining the mechanism(s) through which furamidine modulates MBNL transcript and protein levels is beyond the scope of this work.
Furamidine rescued more mis-splicing events and had fewer off-target exon skipping events compared to heptamidine in the HSALR DM1 mouse model.
To assess the degree of global mis-splicing rescue achieved by furamidine in the HSALR mouse, RNA-seq analysis was used to measure the level of inclusion, or PSI, of alternatively spliced cassette exons in the WT, control HSALR, furamidine and heptamidine treated HSALR mice. RNA-seq libraries were prepared from the same RNA samples used for the splicing and transgene expression experiments. Consistent with our RT-PCR analysis, the RNA-seq data supported rescue of the mis-splicing of Atp2a1 exon22 event in the HSALR mouse (Figure S17a); however, read coverage was too low to confidently predict the PSI values for the Clcn1 exon7a event to determine rescue. Many additional events showing mis-splicing rescue were identified in the mice treated with furamidine and heptamidine. The events that showed mis-splicing rescue with furamidine included Rapgef1, Tnnt3 and Rilpl1 having percent rescues of 38 ± 9%, 24 ± 14% and 94 ± 29% rescue (p<0.05 or better), respectively (purple points, Figure S17b–d). Heptamidine treatment also showed rescue of these events at 48 ± 8%, 40 ± 23% and 88 ± 0% rescue (p<0.05 or better), respectively (green points, Figure S17b–d).
When comparing the WT and control HSALR mice, a total of 666 ES events showed evidence of mis-regulation greater than a 10% change in PSI (p<0.01, FDR<0.01), however these events were not validated specifically as DM1-associated mis-splicing events. Of these events, 74 showed at least a 10% rescue with 30 mg kg−1 furamidine treatment and 62 displayed at least a 10% rescue with 30 mg kg−1 heptamidine treatment (p<0.01, FDR<0.01, Figure 8a and 8b). A distribution of events shown by percent rescue by furamidine and heptamidine is shown in Figure S17e. On average, heptamidine displayed a higher degree of mis-splicing rescue compared to furamidine for the 58 ES events that were rescued by both, with furamidine having an average percent rescue of 52 ± 31% and heptamidine an average of 65 ± 39%.
Figure 8. Furamidine rescued more mis-splicing events, had fewer gene expression changes and rescued more gene expression changes compared to heptamidine treatment in HSALR mice.

When splicing was analyzed globally using RNA-seq, a) furamidine had a higher number of rescued events (purple) and fewer over-rescued (blue), mis-rescued (yellow), and off-target exon skipping (ES) events (grey) compared to b) heptamidine (green) treatment (p<0.01, FDR<0.01). Expression analysis of the RNA-seq data showed that c) furamidine had fewer transcripts with significantly altered expression (red dots) compared to d) heptamidine (p<0.1). Gray dots represent genes that were not significantly differentially expressed. When gene expression was analyzed for rescue back to wild type levels, the RNA-seq data showed that e) furamidine had a higher number and percentage of rescued gene expression changes (purple) and fewer over-rescued (blue), mis-rescued (yellow), and off-target gene expression changes (grey) compared to f) heptamidine (green) treatment (p<0.1).
Interestingly, both drugs caused some of the 666 ES events to be over-rescued or even mis-rescued, meaning that the mis-splicing was shifted further from WT. Furamidine caused 6 events to be over-rescued and 11 events to be mis-rescued, and heptamidine caused the over-rescue of 16 events and the mis-rescue of 18 events (p<0.01, FDR<0.01, Figure 8a and 8b). Along with over-rescue or mis-rescue of splicing, we wanted to know if the treatments caused any ‘off-target’ alternative splicing changes, which we identified by looking at changes in the PSI of ES events outside of the 666 mis-regulated events found between the WT and control HSALR mice. With heptamidine treatment, there were 331 ES events that showed greater than a 10% change in PSI versus control, while furamidine had less than half that number at 146 events (p<0.01, FDR<0.01, Figure 8a and 8b). These data show that furamidine rescued more mis-splicing events and had reduced off-target effects on ES events compared to heptamidine treatment in HSALR mice.
Furamidine had low off-target effects on global gene expression in the HSALR DM1 mouse model.
One challenge for small molecules as therapeutics is to identify concentration windows in which target engagement is maximized and off-target effects are minimized. For DM1, we have focused on small molecules that reduce levels of expanded CUG repeats with the goal of minimizing or eliminating changes to other transcripts. ActD was the first transcription inhibitor used in DM1 for which RNA-seq was performed to determine the off-target effects17. In that study, we observed that 0.25 mg kg−1 ActD changed the expression of 5.1% of genes in HSALR mice. Therefore, determining furamidine’s effect on global gene expression was an important next step. Analysis of the RNA-seq data showed that furamidine only modestly affected global gene expression patterns with dramatically fewer changes in gene expression (2.9% of genes, p<0.1, Figure 8c) compared to heptamidine (42% of genes, p<0.1, Figure 8d). However, these gene expression changes are those of the untreated versus treated HSALR transcriptome. Expression of CUG repeats has been shown to induce transcriptional changes that are linked to loss of Mbnl1 function in HSALR mice51. Similar to mis-splicing rescue, we wanted to determine if some of the gene expression changes in the untreated versus treated HSALR mice were actually the “rescue” of expression back to wild type levels.
To determine gene expression rescue, we identified genes that were differentially expressed between wild type FVB mice and the control HSALR mice and compared the expression of those to ones differentially expressed between control mice and furamidine or heptamidine treated HSALR mice. An explanation of how percent rescue of gene expression was calculated can be found in the methods section. Interestingly, the expression of 21% (153 genes) of the 708 genes differentially expressed with furamidine treatment were rescued by more than 10% back to wild type levels (Figure 8e). We also identified genes that were over-rescued and mis-rescued by more than 10% with furamidine treatment, corresponding to 15% (104 genes) and 14% (98 gene) of the 708 differentially expressed genes, respectively (Figure 8e). Heptamidine treatment resulted in differential expression of 10,435 genes and only 6% (587 genes) were rescued, while 10% were over-rescued (1094 genes) and 21% (2139 genes) were mis-rescued (Figure 8f). These data show that furamidine has fewer total gene expression changes compared to heptamidine treatment in the HSALR mice and that it rescued a larger percentage of those differentially expressed genes back toward wild type levels.
Because furamidine reduced HSA transgene levels, we wanted to determine if there was an enrichment of CTG motifs in the genes that were significantly differentially expressed with furamidine treatment. Of the 264 genes that were differentially expressed (p<0.1) with a log2 fold change of more than ±1, 123 were down regulated and 141 were up regulated. There was not a significant enrichment of CTG motifs in either the up or down regulated genes versus a randomly generated set of 150 genes that were not significantly affected by furamidine treatment. The 123 down regulated genes had an average of 25 ± 6 CTGs/kb and the 141 were up regulated had an average of 25 ± 5 CTGs/kb, where the randomly generated 150 genes had an average of 25 ± 9 CTGs/kb. There was also not a significant enrichment of AT-rich genes in the differentially expressed genes with furamidine treatment versus the randomly generated genes, at 54 ± 6% AT for both the up and down regulated genes and 55 ± 6% for the randomly generated genes. Our working model for furamidine’s mis-splicing rescue in the HSALR mouse is that it binds the expanded CTG•CAG repeats to reduce their transcription, but that its promiscuous binding to nucleic acids and proteins causes up and down regulation of the other transcripts through multiple mechanisms.
While furamidine’s primary mode of action appears to be inhibiting transcription from the CTG•CAG-containing transgene in the HSALR mouse, we cannot dismiss that furamidine may work through disruption of the MBNL–CUG complex to release MBNL proteins consistent with the ITC and EMSA studies. It is unlikely to be working through the up-regulation of MBNL proteins as the transcript levels of Mbnl1 and Mbnl2 are relatively unchanged with furamidine treatment (Figure S18); however, protein levels were not assessed to rule out this mechanism in the HSALR mouse model.
Conclusions.
Taken together, the results of mis-splicing rescue, changing MBNL protein levels, lack of significant change in expanded CUG levels, and reduction in ribonuclear foci suggest that increased levels of MBNL proteins and furamidine disruption of the MBNL–CUG complex are the primary drivers of mis-splicing rescue in the DM1 patient-derived myotubes. Interestingly, furamidine’s primary mode of action may be inhibition of transcription from the CTG•CAG-containing transgene in the HSALR mouse model, although disruption of MBNL–CUG repeats by furamidine is also possible. Our results, from this and previous studies, show that small molecules can have a broad range of off-target effects. These studies revealed that furamidine has the lowest number of off-target gene expression changes compared to other small molecules that have been studied globally for DM. Further, this is the first example showing the global gene expression rescue of small molecules for DM. The reduction of off-target effects and reduced toxicity from heptamidine to furamidine suggests that modifications to furamidine and screening of furamidine analogs could lead to new molecules with improved activity and selectivity. Moreover, our findings highlight the importance of assessing the activity of lead molecules for DM1 across a broad concentration range and in multiple systems (data associated with upregulation of DMPK transcript levels and knockdown of MBNL proteins at high furamidine treatment are shown in Figures S19 to S22). In the future, it will be exciting to determine if furamidine and analogs have the same multi-mechanism activity in related microsatellite diseases such as myotonic dystrophy type 2, spinocerebellar ataxia-8, and Fuchs’ Corneal Dystrophy52.
METHODS
A description of all chemicals, reagents, instrumentation, and procedures is available in the Supporting Information.
Supplementary Material
ACKNOWLEDGEMENTS
Special thanks to K. Dannehower, other members of the Berglund lab, J. Cleary, and T. Reid for helpful discussions, experimental advice, and comments on the manuscript. Thank you to the UF Center for NeuroGenetics, especially the Ranum, Swanson, and Wang labs, for general support and guidance. Also, thank you to J. Sydes for the custom PDR deduplication script, J. Richardson for help with bioinformatics, to the Xie lab for help with northern blotting, and to the McKenna lab, namely C. Lomelino and J. Andring, for assistance with ITC. These studies were supported by funding from the MDA (516314) and NIH (AR059833) to JAB, JSPS KAKENHI (15K15339 and 16H05321) to MN, MDF postdoctoral fellowship to LAC, and NSF pre-doctoral fellowship (2016204076) to JRJ. The authors declare the following competing financial interest(s): J. Andrew Berglund and the University of Oregon have patented diamidines for treating myotonic dystrophy (U.S. Patents 8463049 and 20130281462)
REFERENCES
- [1].Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O’Hoy K, and et al. (1992) Myotonic dystrophy mutation: an unstable CTG repeat in the 3’ untranslated region of the gene, Science (New York, N.Y.) 255, 1253–1255. [DOI] [PubMed] [Google Scholar]
- [2].Davis BM, McCurrach ME, Taneja KL, Singer RH, and Housman DE (1997) Expansion of a CUG trinucleotide repeat in the 3’ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts, Proceedings of the National Academy of Sciences of the United States of America 94, 7388–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, and Brook JD (2002) Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells, Human molecular genetics 11, 805–814. [DOI] [PubMed] [Google Scholar]
- [4].Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, and Swanson MS (2000) Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy, Embo j 19, 4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cho DH, Thienes CP, Mahoney SE, Analau E, Filippova GN, and Tapscott SJ (2005) Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF, Molecular cell 20, 483–489. [DOI] [PubMed] [Google Scholar]
- [6].Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, and Ranum LP (2011) Non-ATG-initiated translation directed by microsatellite expansions, Proceedings of the National Academy of Sciences of the United States of America 108, 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, Hall MP, Shiue L, Swanson MS, Thornton CA, and Ares M Jr. (2010) Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy, Nature structural & molecular biology 17, 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Masuda A, Andersen HS, Doktor TK, Okamoto T, Ito M, Andresen BS, and Ohno K (2012) CUGBP1 and MBNL1 preferentially bind to 3’ UTRs and facilitate mRNA decay, Scientific reports 2, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, Lecuyer E, and Burge CB (2012) Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins, Cell 150, 710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ranum LP, and Cooper TA (2006) RNA-mediated neuromuscular disorders, Annual review of neuroscience 29, 259–277. [DOI] [PubMed] [Google Scholar]
- [11].Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, Cannon SC, and Thornton CA (2002) Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy, Molecular cell 10, 35–44. [DOI] [PubMed] [Google Scholar]
- [12].Philips AV, Timchenko LT, and Cooper TA (1998) Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy, Science (New York, N.Y.) 280, 737–741. [DOI] [PubMed] [Google Scholar]
- [13].Savkur RS, Philips AV, and Cooper TA (2001) Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy, Nature genetics 29, 40–47. [DOI] [PubMed] [Google Scholar]
- [14].van Agtmaal EL, Andre LM, Willemse M, Cumming SA, van Kessel IDG, van den Broek W, Gourdon G, Furling D, Mouly V, Monckton DG, Wansink DG, and Wieringa B (2017) CRISPR/Cas9-Induced (CTGCAG)n Repeat Instability in the Myotonic Dystrophy Type 1 Locus: Implications for Therapeutic Genome Editing, Molecular therapy : the journal of the American Society of Gene Therapy 25, 24–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gao Y, Guo X, Santostefano K, Wang Y, Reid T, Zeng D, Terada N, Ashizawa T, and Xia G (2016) Genome Therapy of Myotonic Dystrophy Type 1 iPS Cells for Development of Autologous Stem Cell Therapy, Molecular therapy : the journal of the American Society of Gene Therapy 24, 1378–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Coonrod LA, Nakamori M, Wang W, Carrell S, Hilton CL, Bodner MJ, Siboni RB, Docter AG, Haley MM, Thornton CA, and Berglund JA (2013) Reducing levels of toxic RNA with small molecules, ACS chemical biology 8, 2528–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Siboni RB, Nakamori M, Wagner SD, Struck AJ, Coonrod LA, Harriott SA, Cass DM, Tanner MK, and Berglund JA (2015) Actinomycin D Specifically Reduces Expanded CUG Repeat RNA in Myotonic Dystrophy Models, Cell reports 13, 2386–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pinto BS, Saxena T, Oliveira R, Méndez-Gómez HR, Cleary JD, Denes LT, McConnell O, Arboleda J, Xia G, Swanson MS, and Wang ET (2017) Impeding Transcription of Expanded Microsatellite Repeats by Deactivated Cas9, Molecular cell 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee JE, Bennett CF, and Cooper TA (2012) RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1, Proceedings of the National Academy of Sciences of the United States of America 109, 4221–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, Cheng SH, Wentworth BM, Bennett CF, and Thornton CA (2012) Targeting nuclear RNA for in vivo correction of myotonic dystrophy, Nature 488, 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Guan L, and Disney MD (2013) Small Molecule-Mediated Cleavage of RNA in Living Cells, Angew Chem Int Ed Engl 52, 1462–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nguyen L, Luu LM, Peng S, Serrano JF, Chan HY, and Zimmerman SC (2015) Rationally designed small molecules that target both the DNA and RNA causing myotonic dystrophy type 1, Journal of the American Chemical Society 137, 14180–14189. [DOI] [PubMed] [Google Scholar]
- [23].Nakamori M, Taylor K, Mochizuki H, Sobczak K, and Takahashi MP (2016) Oral administration of erythromycin decreases RNA toxicity in myotonic dystrophy, Ann Clin Transl Neurol 3, 42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rzuczek SG, Colgan LA, Nakai Y, Cameron MD, Furling D, Yasuda R, and Disney MD (2017) Precise small-molecule recognition of a toxic CUG RNA repeat expansion, Nature chemical biology 13, 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Warf MB, Nakamori M, Matthys CM, Thornton CA, and Berglund JA (2009) Pentamidine reverses the splicing defects associated with myotonic dystrophy, Proceedings of the National Academy of Sciences of the United States of America 106, 18551–18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wong CH, Nguyen L, Peh J, Luu LM, Sanchez JS, Richardson SL, Tuccinardi T, Tsoi H, Chan WY, Chan HY, Baranger AM, Hergenrother PJ, and Zimmerman SC(2014) Targeting toxic RNAs that cause myotonic dystrophy type 1 (DM1) with a bisamidinium inhibitor, Journal of the American Chemical Society 136, 6355–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA, and Swanson MS (2006) Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy, Proceedings of the National Academy of Sciences of the United States of America 103, 11748–11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang F, Bodycombe NE, Haskell KM, Sun YL, Wang ET, Morris CA, Jones LH, Wood LD, and Pletcher MT (2017) A flow cytometry-based screen identifies MBNL1 modulators that rescue splicing defects in myotonic dystrophy type I, Human molecular genetics 26, 3056–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wheeler TM, Lueck JD, Swanson MS, Dirksen RT, and Thornton CA (2007) Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy, The Journal of clinical investigation 117, 3952–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, and Thornton CA (2009) Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA, Science (New York, N.Y.) 325, 336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Siboni RB, Bodner MJ, Khalifa MM, Docter AG, Choi JY, Nakamori M, Haley MM, and Berglund JA (2015) Biological Efficacy and Toxicity of Diamidines in Myotonic Dystrophy Type 1 Models, Journal of medicinal chemistry 58, 5770–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bell CA, Hall JE, Kyle DE, Grogl M, Ohemeng KA, Allen MA, and Tidwell RR (1990) Structure-activity relationships of analogs of pentamidine against Plasmodium falciparum and Leishmania mexicana amazonensis, Antimicrobial agents and chemotherapy 34, 1381–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Boykin DW, Kumar A, Spychala J, Zhou M, Lombardy RJ, Wilson WD, Dykstra CC, Jones SK, Hall JE, Tidwell RR, and et al. (1995) Dicationic diarylfurans as anti-Pneumocystis carinii agents, Journal of medicinal chemistry 38, 912–916. [DOI] [PubMed] [Google Scholar]
- [34].Yang S, Wenzler T, Miller PN, Wu H, Boykin DW, Brun R, and Wang MZ (2014) Pharmacokinetic comparison to determine the mechanisms underlying the differential efficacies of cationic diamidines against first- and second-stage human African trypanosomiasis, Antimicrobial agents and chemotherapy 58, 4064–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu Y, Collar CJ, Kumar A, Stephens CE, Boykin DW, and Wilson WD (2008) Heterocyclic diamidine interactions at AT base pairs in the DNA minor groove: effects of heterocycle differences, DNA AT sequence and length, J Phys Chem B 112, 11809–11818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lansiaux A, Tanious F, Mishal Z, Dassonneville L, Kumar A, Stephens CE, Hu Q, Wilson WD, Boykin DW, and Bailly C (2002) Distribution of furamidine analogues in tumor cells: targeting of the nucleus or mitochondria depending on the amidine substitution, Cancer Res 62, 7219–7229. [PubMed] [Google Scholar]
- [37].Purfield AE, Tidwell RR, and Meshnick SR (2009) The diamidine DB75 targets the nucleus of Plasmodium falciparum, Malaria journal 8, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bailly C, Dassonneville L, Carrasco C, Lucas D, Kumar A, Boykin DW, and Wilson WD(1999) Relationships between topoisomerase II inhibition, sequence-specificity and DNA binding mode of dicationic diphenylfuran derivatives, Anti-cancer drug design 14, 47–60. [PubMed] [Google Scholar]
- [39].Munde M, Wang S, Kumar A, Stephens CE, Farahat AA, Boykin DW, Wilson WD, and Poon GM (2014) Structure-dependent inhibition of the ETS-family transcription factor PU.1 by novel heterocyclic diamidines, Nucleic acids research 42, 1379–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Laughton CA, Tanious F, Nunn CM, Boykin DW, Wilson WD, and Neidle S (1996) A crystallographic and spectroscopic study of the complex between d(CGCGAATTCGCG)2 and 2,5-bis(4-guanylphenyl)furan, an analogue of berenil. Structural origins of enhanced DNA-binding affinity, Biochemistry 35, 5655–5661. [DOI] [PubMed] [Google Scholar]
- [41].Zhao M, Ratmeyer L, Peloquin RG, Yao S, Kumar A, Spychala J, Boykin DW, and Wilson WD (1995) Small changes in cationic substituents of diphenylfuran derivatives have major effects on the binding affinity and the binding mode with RNA helical duplexes, Bioorg Med Chem 3, 785–794. [DOI] [PubMed] [Google Scholar]
- [42].Ratmeyer L, Zapp ML, Green MR, Vinayak R, Kumar A, Boykin DW, and Wilson WD(1996) Inhibition of HIV-1 Rev-RRE interaction by diphenylfuran derivatives, Biochemistry 35, 13689–13696. [DOI] [PubMed] [Google Scholar]
- [43].Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, and Thornton CA (2000) Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat, Science (New York, N.Y.) 289, 1769–1773. [DOI] [PubMed] [Google Scholar]
- [44].Wilson WD, Nguyen B, Tanious FA, Mathis A, Hall JE, Stephens CE, and Boykin DW (2005) Dications that target the DNA minor groove: compound design and preparation, DNA interactions, cellular distribution and biological activity, Current medicinal chemistry. Anti-cancer agents 5, 389–408. [DOI] [PubMed] [Google Scholar]
- [45].Liu Y, Kumar A, Boykin DW, and Wilson WD (2007) Sequence and Length Dependent Thermodynamic Differences in Heterocyclic Diamidine Interactions at AT Base Pairs in the DNA Minor Groove, Biophysical chemistry 131, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Xia G, Santostefano KE, Goodwin M, Liu J, Subramony SH, Swanson MS, Terada N, and Ashizawa T (2013) Generation of neural cells from DM1 induced pluripotent stem cells as cellular model for the study of central nervous system neuropathogenesis, Cellular reprogramming 15, 166–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mooers BHM, Logue JS, and Berglund JA (2005) The structural basis of myotonic dystrophy from the crystal structure of CUG repeats, Proceedings of the National Academy of Sciences of the United States of America 102, 16626–16631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, and Swanson MS (2003) A muscleblind knockout model for myotonic dystrophy, Science (New York, N.Y.) 302, 1978–1980. [DOI] [PubMed] [Google Scholar]
- [49].Thomas JD, Sznajder LJ, Bardhi O, Aslam FN, Anastasiadis ZP, Scotti MM, Nishino I, Nakamori M, Wang ET, and Swanson MS (2017) Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy, Genes Dev 31, 1122–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Querido E, Gallardo F, Beaudoin M, Menard C, and Chartrand P (2011) Stochastic and reversible aggregation of mRNA with expanded CUG-triplet repeats, Journal of cell science 124, 1703–1714. [DOI] [PubMed] [Google Scholar]
- [51].Osborne RJ, Lin X, Welle S, Sobczak K, O’Rourke JR, Swanson MS, and Thornton CA (2009) Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy, Human molecular genetics 18, 1471–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhang N, and Ashizawa T (2017) RNA toxicity and foci formation in microsatellite expansion diseases, Current opinion in genetics & development 44, 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wu TD, and Nacu S (2010) Fast and SNP-tolerant detection of complex variants and splicing in short reads, Bioinformatics (Oxford, England) 26, 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Love MI, Huber W, and Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads, EMBnet.journal 17. [Google Scholar]
- [56].Bolger AM, Lohse M, and Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data, Bioinformatics (Oxford, England) 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014) rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data, Proceedings of the National Academy of Sciences of the United States of America 111, E5593–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zygmunt DA, Singhal N, Kim ML, Cramer ML, Crowe KE, Xu R, Jia Y, Adair J, Martinez-Pena y Valenzuela I, Akaaboune M, White P, Janssen PM, and Martin PT(2017) Deletion of Pofut1 in Mouse Skeletal Myofibers Induces Muscle Aging-Related Phenotypes in cis and in trans, Molecular and Cellular Biology 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.