

Summary
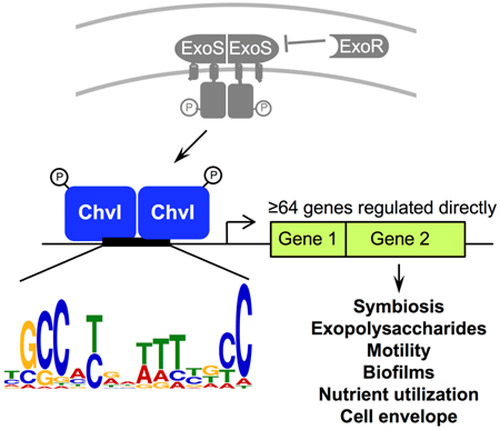
ExoS/ChvI two-component signaling in the nitrogen-fixing α-proteobacterium Sinorhizobium meliloti is required for symbiosis and regulates exopolysaccharide production, motility, cell envelope integrity, and nutrient utilization in free-living bacteria. However, identification of many ExoS/ChvI direct transcriptional target genes has remained elusive. Here, we performed chromatin immunoprecipitation followed by microarray analysis (chIP-chip) to globally identify DNA regions bound by ChvI protein in S. meliloti. We then performed qRT-PCR with chvI mutant strains to test ChvI-dependent expression of genes downstream of the ChvI-bound DNA regions. We identified 64 direct target genes of ChvI, including exoY, rem, and chvI itself. We also identified ChvI direct target candidates, like exoR, that are likely controlled by additional regulators. Analysis of upstream sequences from the 64 ChvI direct target genes identified a 15 bp-long consensus sequence. Using electrophoretic mobility shift assays and transcriptional fusions with exoY, SMb21440, SMc00084, SMc01580, chvI, and ropB1, we demonstrated this consensus sequence is important for ChvI binding to DNA and transcription of ChvI direct target genes. Thus, we have comprehensively identified ChvI regulon genes and a “ChvI box” bound by ChvI. Many ChvI direct target genes may influence the cell envelope, consistent with the critical role of ExoS/ChvI in growth and microbe-host interactions.
Keywords: Sinorhizobium meliloti, Symbiosis, Gene expression regulation, Regulon, Chromatin immunoprecipitation, Microarray analysis
Graphical Abstract
Abbreviated Summary
The Sinorhizobium meliloti ExoS/ChvI two-component signaling pathway is critical for establishing a nitrogen-fixing symbiosis with plant hosts and regulates many free-living bacterial phenotypes. We report the comprehensive identification of genes comprising the ChvI regulon in S. meliloti and demonstrate the importance of a consensus sequence for ChvI-DNA binding and regulation of ChvI direct target genes. These findings represent a major advance in understanding the role of this conserved signaling pathway involved in successful microbe-host interactions.
Introduction
Symbioses between nitrogen-fixing bacteria and legumes are a major source of fixed nitrogen in the biosphere, converting molecular dinitrogen into a form that is accessible to plants for growth (Gibson et al., 2008). Establishment of the symbiosis begins with communication via chemical signals between the legume plants and bacteria (Long, 2016). The plant roots secrete flavonoids (Liu and Murray, 2016), which are perceived by bacteria in the rhizosphere, and induce expression of rhizobial nod genes for synthesizing and secreting Nod factor, a lipochitooligosaccharide with various chemical substituents. Nod factor binds to Nod factor receptors in the plant, inducing root hair curling, root cortical cell division, and expression of receptors for bacterial exopolysaccharides (EPS) (Kawaharada et al., 2015). The bacteria colonize curled root hairs and infect the plant via infection threads. Eventually, bacteria are released from the ends of the infection threads to form symbiosomes in nodule cells. In symbiosomes, the bacteria differentiate into nitrogen-fixing bacteroids, which provide the plant with fixed nitrogen in exchange for dicarboxylic acids (Gibson et al., 2008). A microaerobic nodule environment and host plant nodule-specific cysteine-rich (NCR) peptides control bacteroid differentiation (Gibson et al., 2008; Maroti and Kondorosi, 2014).
In addition to helping rhizobia tolerate varied environmental stresses (Arnold et al., 2017; Barnett et al., 2012; Davies and Walker, 2007; Geddes et al., 2014; Lehman and Long, 2013; Miller-Williams et al., 2006; Morris and Gonzalez, 2009; Vriezen et al., 2007), EPS are important signaling molecules that contribute to the specificity of the host-microbe interaction and promote infection (Kawaharada et al., 2015; Kawaharada et al., 2017). In the Rm1021 strain of Sinorhizobium meliloti, a symbiont of Medicago spp., EPS-I or succinoglycan is the only symbiotically active EPS produced. Succinoglycan is required for infection (Pellock et al., 2002; Sharypova et al., 2006), appears to suppress host plant defense responses (Jones et al., 2008), and protects the bacteria from NCR peptides (Arnold et al., 2018). Mutants that lack succinoglycan, or that produce succinoglycan with an altered structure, have defects in initiation and elongation of infection threads, whereas some, but not all mutants that overproduce succinoglycan have symbiotic defects (Barnett and Long, 2018; Cheng and Walker, 1998; Jones, 2012). The structure and amount of succinoglycan produced by rhizobia thus influence the success of symbiosis.
A key regulator of succinoglycan synthesis in S. meliloti is the ExoS/ChvI two-component pathway. ExoS is a membrane-bound histidine kinase with a periplasmic sensing domain and ChvI is a response regulator of the OmpR/PhoB family of response regulators (Cheng and Walker, 1998). ExoS/ChvI has orthologs in related α-proteobacteria (Heavner et al., 2015; Vanderlinde and Yost, 2012), and orthologs such as BvrS/BvrR in the mammalian pathogen Brucella abortus and ChvG/ChvI in the plant pathogen Agrobacterium tumefaciens are required for virulence (Charles and Nester, 1993; Heavner et al., 2015; Sola-Landa et al., 1998). ChvG/ChvI-dependent transcription is activated by acidic pH and BvrS/BvrR-dependent transcription is activated by acidic pH and nutrient limitation (Altamirano-Silva et al., 2018; Yuan et al., 2008).
A third member of the ExoS/ChvI pathway in S. meliloti is ExoR, a periplasmic regulator protein that physically associates with ExoS to inhibit ExoS/ChvI signaling (Chen et al., 2008). Binding to ExoS stabilizes the ExoR protein (Chen et al., 2008), and ExoR activity may be regulated via proteolysis in the periplasm (Lu et al., 2012). The exoR and exoS genes were originally identified because transposon insertions in these genes (exoR95::Tn5 and exoS96::Tn5) resulted in overproduction of succinoglycan by increasing transcription of exo genes encoding succinoglycan biosynthesis enzymes (Doherty et al., 1988).
Characterization of exoS and chvI mutants revealed that ExoS/ChvI influences functions beyond succinoglycan production and symbiosis. ExoS/ChvI also regulates motility, biofilm formation, cell envelope integrity, galactoglucan production, and nutrient utilization in the free-living form of the bacteria (Bélanger et al., 2009; Wang et al., 2010; Wells et al., 2007; Yao et al., 2004). Initial attempts to construct null mutations in exoS or chvI were unsuccessful, suggesting that each was an essential gene (Cheng and Walker, 1998; Østerås et al., 1995). Two groups later reported construction of chvI null mutants, which could grow on minimal but not rich media (Bélanger et al., 2009; Wang et al., 2010). The Bélanger et al. chvI null mutant (chvI261), constructed by insertion of a neomycin resistance cassette into the chvI ORF, showed severe growth defects: the chvI261 null mutant failed to grow in liquid media and grew poorly on solid minimal media supplemented with a wide variety of carbon sources (Bélanger et al., 2009). In stark contrast, the Wang et al. chvI null mutant showed wild-type growth in liquid media and slight growth defects only in acidic media or media with acetoacetate or pyruvate (Wang et al., 2010). Because the Wang chvI null mutant strain had an unmarked chvI deletion allele, precluding transduction of the allele into a clean genetic background, the presence of additional mutations could account for the striking discrepancy in the chvI null growth phenotypes reported by both groups.
Initial transcriptional profiling experiments with exoR95::Tn5 and exoS96::Tn5 mutants showed that the expression of hundreds of genes was altered in these mutants (Wells et al., 2007; Yao et al., 2004). With so many potential regulatory targets, it was important to distinguish genes directly regulated by ChvI, namely, genes whose upstream region was bound by the ChvI protein to control their transcription, from genes whose expression changed indirectly as a consequence of other ExoS/ChvI mutant phenotypes.
The first attempt to identify direct target genes of ChvI involved two different methods: transcriptional profiling to measure ChvI-dependent transcription and electrophoretic mobility shift assays (EMSA) to detect ChvI-DNA binding in vitro (Chen et al., 2009). In the transcriptional profiling experiment, gene expression was compared in a chvI partial loss of function mutant strain (LOF; chvI (K214T)); a chvI gain of function mutant strain (GOF; chvI (D52E)/chvI+), and wild-type. The chvI (K214T) mutation is in the DNA-binding effector domain, whereas the chvI (D52E) mutation is at the likely phosphorylation site in the receiver domain and appears to be lethal in the absence of wild-type chvI (Chen et al., 2009; Wells et al., 2007). 59 genes showed at least a 1.4-fold reciprocal change in expression in both chvI mutants compared to wild-type and were designated ChvI-regulated genes. Many of these ChvI-regulated genes showed altered expression in other reported transcriptome data sets (Barnett and Long, 2015; Calatrava-Morales et al., 2017; Hellweg et al., 2009; Krol and Becker, 2004; Krol et al., 2016; Penterman et al., 2014; Santos et al., 2014).
In that first attempt to identify ChvI direct target genes, 19 of the upstream DNA regions of these ChvI-regulated genes were tested by EMSA for binding to purified ChvI protein (Chen et al., 2009). ChvI binding was detected with only 3 of these DNA regions (upstream of SMc01580-SMc01581, ropB1, and SMb21440), suggesting that it may be difficult to detect ChvI binding in vitro for most target genes (Chen et al., 2009). A more recent attempt to identify direct targets of ChvI used large-scale EMSA with genomic DNA (GD.EMSA), followed by cloning and sequencing DNA in slower migrating bands (Bélanger and Charles, 2013). 27 DNA fragments were identified using the GD.EMSA approach, but only 1 operon (SMb21188-msbA2) overlapped with the set of 59 ChvI-regulated genes previously identified (Chen et al., 2009). Together, these studies demonstrated a need for a more comprehensive approach to identify ChvI direct target genes.
Here, we have performed “chromatin” immunoprecipitation followed by hybridization to Affymetrix Symbiosis GeneChips (chIP-chip) to identify DNA regions in the entire genome of S. meliloti that are directly bound by ChvI protein. Because this method involves cross-linking protein-DNA complexes in vivo prior to immunoprecipitation, followed by hybridization to Affymetrix Symbiosis GeneChips to identify the DNA regions enriched by the immunoprecipitation, we expected that this method would yield more ChvI direct target genes than previous methods that depended on ChvI-DNA binding in vitro. After identifying the DNA regions enriched by chIP-chip, we used quantitative reverse transcription PCR (qRT-PCR) to test candidate genes for ChvI-dependent expression in a chvI partial LOF mutant, chvI GOF mutant, and wild-type strain. We identified 64 genes that are direct transcriptional targets of ChvI, greatly expanding the number of genes known to be directly regulated by ChvI. Further, we discovered a 15-bp consensus sequence upstream of these genes and demonstrated that this sequence is important for ChvI-dependent binding and transcriptional regulation.
Results
Identification of direct transcriptional target genes of ChvI
Previous transcriptome studies identified numerous genes that appeared to be transcriptionally regulated by ExoS/ChvI (Barnett and Long, 2015; Chen et al., 2009; Wells et al., 2007; Yao et al., 2004). However, of the ChvI-regulated genes identified in those previous studies, only ropB1, SMb21440, and the SMc01580-SMc01581 operon (Chen et al., 2009) and the SMb21188-msbA2 operon (Bélanger and Charles, 2013) were shown to be directly regulated by ChvI, using electrophoretic mobility shift assays (EMSA) to detect binding between purified ChvI protein and the upstream regions of these genes. Since mutation of ExoS/ChvI affects many genes and phenotypes, we hypothesized that many more ExoS/ChvI direct transcriptional target genes existed, but that the in vitro conditions in EMSA did not allow their detection. We decided to use “chromatin” immunoprecipitation followed by microarray analysis (chIP-chip) to identify DNA regions bound by ChvI protein in vivo.
To perform chIP, we replaced the chvI gene in the S. meliloti genome with an epitope-tagged chvI gene. We empirically found that ChvI with a C-terminal HA tag was functional. In contrast to the chvI (K214T) partial LOF mutant which fails to grow on TY medium, the strain with HA-tagged ChvI grew well on TY, like the wild-type strain with untagged ChvI (Figure 1). We also compared succinoglycan production in the ChvI-HA and ChvI-untagged strains. The ChvI-HA and ChvI-untagged strains had similar fluorescence under UV light when grown on media containing calcofluor, indicating that succinoglycan production is similar in both strains (Figure 1). These results confirmed the validity of using the ChvI-HA strain for the chIP-chip experiment.
Figure 1. HA-tagged ChvI is functional.

S. meliloti strains [ChvI-HA (ES147), ChvI untagged (Rm1021), chvI (K214T) (EC412), and chvI(D52E)/chvI+ (EC409) were spotted onto LB, TY, or LB + 0.02% calcofluor plates, incubated at 30°C for 2 days, and photographed with white light or UV.
chIP-chip was performed as described in Experimental Procedures using chIP with anti-HA antibody and Affymetrix Symbiosis GeneChip microarrays (Figure 2). The Symbiosis GeneChips include even tiling of both strands of intergenic regions >150 bp long, thus long intergenic regions are represented by multiple probe sets (Barnett et al., 2004). We categorized probe sets, or DNA regions, as “enriched by chIP” if they were enriched two-fold or more (≥0.96 signal-log ratio) in the ChvI-HA sample subjected to chIP with anti-HA antibody compared to (1) DNA regions in the ChvI-untagged sample subjected to chIP with anti-HA antibody, and (2) total DNA from the ChvI-HA strain. These comparisons in the chIP-chip experiment involved only strains with wild-type ChvI (tagged or untagged), not mutant ChvI. The comparison of chromatin-immunoprecipitated DNA to total DNA was included since previous chIP-chip studies employed this control (Laub et al., 2002; Molle et al., 2003a; Molle et al., 2003b). Using these criteria, we found 489 Affymetrix probe sets that were enriched by chIP (Supporting Information Table S1). 72% of these probe sets corresponded to S. meliloti intergenic regions, a proportion highly unlikely to be due to chance (hypergeometric probability P-value < 9.2×10−29). Binding of ChvI primarily to noncoding DNA regions is consistent with its role as a transcription regulator.
Figure 2. Summary of the procedure to globally identify genes directly regulated by ChvI.

We manually inspected the genomic context of each of the 489 DNA regions enriched by chIP and compared with operon prediction data from RhizoRegNet (Krol et al., 2011) to compile the list of 276 genes “identified by chIP” (Figure 2; Supporting Information Table S2). Each such gene overlapped with, or was within 300 bp downstream of, one or more DNA regions enriched by chIP. Only the first gene in each operon was included in Table S2, since operon promoters typically lie upstream of the first gene. These 276 genes corresponded to 462 of the 489 DNA regions enriched by chIP. Notably, (1) many of the 276 genes, particularly those with long upstream intergenic regions, were represented by multiple contiguous DNA regions enriched by chIP and (2) some DNA regions enriched by chIP, such as an intergenic region flanked by divergently transcribed genes, corresponded to two or more genes.
We then compared the list of 276 genes identified by chIP with previous transcriptome data (Chen et al., 2009) and found 115 protein-coding genes that were candidates for direct regulation by ChvI (Supporting Information Table S3; Experimental Procedures). We decided to test the ChvI-regulated expression of all 115 genes using qRT-PCR because qRT-PCR is more sensitive than Affymetrix GeneChip analysis for measuring transcription (Barnett et al., 2004). For consistency, for qRT-PCR we used the same chvI mutant and wild-type RNA samples that had been used in the transcriptome study (Chen et al., 2009).
49 of the 115 candidates were suspected to have ChvI-regulated expression since each of the 49 genes had shown an increase or decrease in expression of ≥1.4-fold in at least one chvI mutant (chvI partial LOF or chvI GOF) compared to wild-type in the previous transcriptome study (Chen et al., 2009). This criterion for suspecting ChvI-regulated expression in the present study was chosen to be less stringent than the criterion in the previous study (≥1.4-fold reciprocal change in both chvI partial LOF and chvI GOF mutants compared to wild-type), to yield a more inclusive set of genes to test by qRT-PCR. An example for our rationale is the rem (SMc03046) gene (Rotter et al., 2006). In the previous transcriptome study, rem showed a 15-fold decrease in expression in the chvI GOF mutant but no detectable change in expression in the chvI partial LOF mutant compared to wild type, and thus had not previously been categorized as ChvI-regulated (Chen et al., 2009). In the present study, using qRT-PCR, we observed a 15-fold decrease in rem expression in the chvI GOF mutant and a 1.8-fold increase in expression in the chvI partial LOF mutant compared to wild type (Table 1). Thus, in contrast to the transcriptome data, the qRT-PCR data showed that rem has a reciprocal change in expression in both chvI mutants and is indeed transcriptionally regulated by ChvI.
Table 1.
Direct transcriptional target genes of ChvI
| Gene | Description | Average Fold Change (SEM) | Other genes in operon | |||
|---|---|---|---|---|---|---|
| chvI(D52E) | chvI(K214T) | |||||
| Group 1. Genes identified by chIP with ≥1.4-fold reciprocal change in expression for both chvI mutants by qRT-PCR | ||||||
| SMa5027† | Hypothetical protein | 3.75 | (0.55) | −1.42 | (0.11) | |
| SMb20809 kpsF1‡ | Arabinose-5-phosphate isomerase | 2.09 | (0.33) | −1.49 | (0.10) | |
| SMb20946 exoY | Exopolysaccharide production protein, probable sugar transferase | 3.97 | (0.56) | −2.13 | (0.11) | SMb20945, SMb20944 |
| SMb20948 exoU | Glucosyltransferase | 1.74 | (0.29) | −2.13 | (0.15) | |
| SMb20954 exoH | Succinyltransferase | 3.80 | (0.65) | −2.67 | (0.19) | SMb20955, SMb20956, |
| SMb20957, SMb20958, | ||||||
| SMb20959, SMb20960, | ||||||
| SMb20961 | ||||||
| SMb21188 | Acyltransferase, possible surface-saccharide specific acetyltransferase | 2.90 | (0.45) | −4.07 | (0.35) | SMb21189, SMb21190, |
| SMb21191 | ||||||
| SMb21440 | Conserved hypothetical protein, signal peptide | 9.00 | (1.30) | −12.45 | (0.95) | |
| SMc00070 | Conserved hypothetical protein, signal peptide | 2.08 | (0.29) | −2.49 | (0.17) | |
| SMc00084 | Conserved hypothetical protein | 1.80 | (0.26) | −2.20 | (0.13) | |
| SMc00159 | Conserved hypothetical protein, signal peptide | −4.24 | (0.58) | 2.57 | (0.29) | SMc00158, SMc00157 |
| SMc00191 | Hpt domain-containing protein | 1.80 | (0.26) | −1.63 | (0.11) | |
| SMc00404 | Lytic transglycosylase, RlpA-family protein | 2.00 | (0.27) | −3.99 | (0.32) | |
| SMc00658† | Transcription regulator, LuxR family | 2.07 | (0.33) | −1.77 | (0.12) | |
| SMc01580 | Conserved hypothetical protein | 5.20 | (0.73) | −24.81 | (3.27) | SMc01581 |
| SMc01774 | Conserved hypothetical protein, signal peptide | 2.83 | (0.36) | −1.51 | (0.14) | |
| SMc01855 | Lytic transglycosylase, signal peptide | 3.95 | (0.56) | −4.78 | (0.29) | |
| SMc02317 | Conserved hypothetical protein, signal peptide | 1.87 | (0.26) | −1.80 | (0.10) | |
| SMc02832‡ | Taurine, valine, isoleucine and leucine ABC transporter, periplasmic solute-binding component | 1.51 | (0.25) | −1.45 | (0.08) | |
| SMc03046 rem‡ | Transcription regulator of exponential growth motility | −14.92 | (2.52) | 1.79 | (0.22) | SMc03047, SMc03048, |
| SMc03049, SMc03050, | ||||||
| SMc03051, SMc03052, | ||||||
| SMc03053, SMc03054, | ||||||
| SMc03055, SMc03056, | ||||||
| SMc03057 | ||||||
| SMc04236 | Hypothetical protein | 5.93 | (1.13) | −3.39 | (0.26) | |
| SMc04276 | Conserved hypothetical protein, signal peptide | 4.73 | (0.64) | −3.21 | (0.22) | |
| Group 2. Genes identified by chIP with ≥1.4-fold change in expression in one chvI mutant and <1.4-fold reciprocal change in expression in the other chvI mutant by qRT-PCR | ||||||
| SMb20300 cyaF7† | Adenylate cyclase | 2.22 | (0.31) | −1.39 | (0.10) | |
| SMb20829† | Secreted calcium-binding protein | 2.06 | (0.28) | −1.27 | (0.13) | |
| SMb21026 | Hypothetical protein | 2.15 | (0.29) | −1.28 | (0.10) | SMb21025 |
| SMc00354‡ | Outer membrane protein | 1.76 | (0.25) | −1.25 | (0.07) | |
| SMc00604 ropB1 | Outer membrane protein | 1.89 | (0.33) | −1.16 | (0.06) | |
| SMc01469 mcpW‡ | Methyl accepting chemotaxis transmembrane protein W | −7.33 | (0.86) | 1.28 | (0.17) | SMc01468 |
| SMc01556 | Conserved hypothetical protein | 1.76 | (0.24) | −1.26 | (0.07) | |
| SMc02426† | Conserved hypothetical protein | −1.29 | (0.18) | 1.96 | (0.14) | SMc02427 |
| SMc02552 | Hypothetical protein | 2.63 | (0.45) | −1.11 | (0.09) | |
| SMc02560 chvI‡ | Transcriptional regulator | 3.01 | (0.43) | −1.11 | (0.07) | |
| SMc02645 cfa2‡ | Cyclopropane-fatty-acyl synthase | −1.75 | (0.24) | 1.38 | (0.14) | |
| SMc02727† | Conserved hypothetical protein | 2.93 | (0.41) | −1.31 | (0.08) | |
| SMc02941‡ | Conserved hypothetical transmembrane protein, signal peptide | 1.83 | (0.30) | −1.16 | (0.07) | |
| SMc04381 opgC | Succinyltransferase, involved in succinylation of cyclic-beta-1,2-glucans | 2.51 | (0.35) | −1.38 | (0.09) | |
Average fold change in expression in chvI mutants is compared to wild type expression, where wild type is normalized to 1. Increases in expression compared to wild type have positive fold-change values whereas decreases in expression have negative fold-change values. Three biological replicates of WT, chvI(D52E), and chvI(K214T) cDNA were tested in triplicate for each gene. The average fold change in gene expression between each chvI mutant and WT is shown. SEM, Standard error of the mean, is shown in parenthesis. Genes negatively regulated by ChvI are indicated in bold.
Genes not reliably detected in the Chen et al., 2009 transcriptome study.
Genes that did not meet expression change thresholds to be included in the Chen et al., 2009 transcriptome data table.
Operon predictions are from RhizoRegNet. Note: SMc00157 appears to be co-regulated with SMc00159 by qRT-PCR, and SMb21025 appears to be co-regulated with SMb21026 in the Chen et al., 2009 dataset, although these were not predicted to be in operons by RhizoRegNet.
The expression of the remaining 66 of the 115 candidates had not been reliably detected above background in one or more of the GeneChips used in the previous transcriptome study (Supporting Information Table S3). Thus, we used qRT-PCR to examine the expression of these 66 candidates as well.
The ChvI regulon
We define a gene as “directly regulated by ChvI” if the gene’s upstream region is bound by ChvI to control its transcription. For the 115 candidates that were found to overlap or be just downstream of DNA regions enriched by chIP, qRT-PCR was performed to test for ChvI-dependent transcription. From qRT-PCR data with cDNA from a chvI partial LOF mutant, chvI GOF mutant, and wild-type strain, genes were grouped based on the magnitude of their expression change in both chvI mutants compared to wild type (Table 1 and Supporting Information Table S4). The 21 genes in Group 1 had a ≥1.4-fold reciprocal change in expression in both chvI mutants compared to wild type (Table 1). The 14 genes in Group 2 had a ≥1.4-fold change in expression in one chvI mutant and <1.4-fold reciprocal change in expression in the other chvI mutant. Of the 35 genes in Groups 1 and 2, 30 genes were positively regulated and five genes were negatively regulated by ChvI. Twenty-one of the 35 genes had been identified as ChvI-regulated in the previous transcriptome study (Chen et al., 2009). Of the 14 new ChvI direct target genes identified via chIP-chip and qRT-PCR in the present study, eight genes had not shown changes in expression large enough to be defined as a ChvI-regulated in the previous transcriptome study, and six genes had expression that was too low to be reliably detected previously (Chen et al., 2009).
We conclude that all of the genes in Table 1 are direct transcriptional targets of ChvI: all had upstream region bound by ChvI protein as discovered by chIP-chip, and all showed reciprocal changes in expression in the chvI GOF and chvI partial LOF mutants by qRT-PCR. Several of the 35 genes listed in the first column are in operons, thus Table 1 shows a total of 64 ChvI direct target genes when including genes in the second or later positions in operons (“Other genes in operon” column). In agreement with previous knowledge that ExoS/ChvI regulates succinoglycan synthesis, three of the exo operons involved in succinoglycan synthesis, beginning with the exoY, exoU, and exoH genes, respectively, were categorized as Group 1. Defective ExoS/ChvI signaling is also known to affect motility of S. meliloti, and genes with roles in motility or chemotaxis are directly negatively regulated by ChvI: rem, a response regulator of exponential growth motility (Rotter et al., 2006), and other motility genes in its predicted operon (Group 1); and mcpW (SMc01469), a methyl accepting chemotaxis transmembrane protein (Group 2) (Meier et al., 2007).
Interestingly, chvI was identified as a direct transcriptional target of itself (Table 1, Group 2), indicating that ChvI positively regulates its own expression. ChvI had been suggested to autoregulate its transcription based on an in vitro binding assay, but this possibility was not explored further (Bélanger and Charles, 2013). Similar to rem, chvI had not been categorized as ChvI-regulated in the previous transcriptome study because the magnitude of the change in chvI expression in both chvI mutants failed to meet the defined thresholds (Chen et al., 2009). Thus, the identification of chvI as a ChvI direct target gene in the present study was possible due to testing by qRT-PCR and having a category with a lower threshold for gene expression changes (Group 2). Besides chvI, another putative regulator controlled directly by ChvI is SMc00658, encoding a LuxR-family transcription regulator whose expression is cell-cycle regulated (De Nisco et al., 2014).
The qRT-PCR data also revealed 59 genes with ≥1.4-fold change in expression in at least one chvI mutant, and either no change in expression or a non-reciprocal change in expression in the other chvI mutant; these 59 genes were further divided into Groups 3, 4 and 5 based on their expression patterns (Supporting Information Table S4). Since the chIP-chip results indicated that all of these genes had their upstream region bound by ChvI, but a reciprocal change in expression was not observed in both chvI mutants, the Group 3–5 genes may be ChvI direct target genes but are more likely to be controlled by additional transcriptional regulators compared to genes in Groups 1 and 2.
One Group 3 gene of note is exoR (SMc02078), which encodes a periplasmic regulator that inhibits ExoS/ChvI signaling. Our results indicate that exoR expression is increased in the chvI GOF mutant but unchanged in the chvI partial LOF mutant. Of the 59 genes in Groups 3–5, 28 have annotated functions, while the remaining 31 encode proteins with unknown function.
Of the original 115 candidates, 17 genes tested by qRT-PCR failed to show regulation by ChvI (<1.4-fold change in expression for both chvI mutants compared to wild-type), and four genes could not be tested for technical reasons (Experimental Procedures; Supporting Information Table S4).
Identification of a consensus sequence upstream of ChvI direct target genes
To identify a nucleotide sequence important for ChvI binding, we used the MEME algorithm (Bailey et al., 2006) to analyze the upstream sequences of ChvI direct target genes. We used only the Group 1 and 2 genes in the analysis since we had high confidence that these genes were indeed direct targets of ChvI. We were aided by the work of Schlüter et al. (2013), which mapped transcription start sites (TSS) throughout the whole genome of S. meliloti strain Rm1021. We excluded from our MEME analysis the Group 1 and 2 genes with no transcriptional start site identified by Schlüter and colleagues (see Experimental Procedures). In total, we analyzed upstream sequences from 24 positively regulated genes and 5 negatively regulated genes from Groups 1 and 2. For the three genes whose upstream region was previously shown to bind directly to ChvI (SMc01580, SMb21440, and ropB1) via DNase I footprinting or EMSA (Chen et al., 2009), the footprinted or gel-shifted sequence was used. For all the other genes, we initially tried 200 bp sequences upstream of the TSS, but noticed that E-values improved by using 50 bp of sequence upstream of the TSS. MEME did not find a motif within 50 bp upstream of the TSS for three of the negatively regulated genes, so we used 101 bp of sequence centered on the TSS for all the negatively regulated genes, reasoning that ChvI might bind downstream of the TSS to effect negative regulation.
We identified a 15 bp-long motif (Figure 3, Supporting Information Figure S1) that was present in all of the input sequences, with an overall E value of 5.60E-07. Some of the most highly conserved nucleotides in the motif were in direct trinucleotide repeats of GCC near the beginning and end of the motif. Direct repeats are commonly found in the recognition sequences of other members of the OmpR/PhoB subfamily of response regulators, which bind to DNA as a homodimer (Capra and Laub, 2012). For example, the 18 bp-long “Pho box” bound by the PhoB response regulator contains two 7 bp-long direct repeats with a conserved 4 bp spacer (Yuan et al., 2006). Consistent with the spacing of the repeats in the Pho box, the GCC repeats in the ChvI consensus sequence are 11 nt apart (beginning at positions 2 and 13 of the motif). Thus, we identified a consensus sequence upstream of ChvI direct target genes in Groups 1 and 2.
Figure 3. A possible ChvI binding motif.

The motif was found upstream of the 29 Group 1 and 2 genes for which TSS data was available (Schlüter et al., 2013). The input for MEME was the TSS and 50 bp upstream for all positively regulated genes, except for ropB1 and SMb21440, the gel shifted sequence was used, and for SMc01580, the footprinted sequence was used (Chen et al., 2009). For all negatively regulated genes, the input was 101 bp of sequence centered on the TSS. The logo represents a motif that occurs once in each input sequence. Information content at each nucleotide position is shown in bits and the log likelihood ratio of this motif is 194. The E value is 5.6E-07, which is an estimate of the number of 15-bp-long motifs with a log likelihood ratio of 194 or higher that would be expected in a similarly sized set of 29 random input sequences.
The motif is important for ChvI binding to DNA
To test the importance of the consensus sequence for ChvI binding, we mutated the motif upstream of several ChvI direct target genes and performed EMSA. Previously, we had only been able to detect binding via EMSA with three upstream regions (Chen et al., 2009), but recently we found EMSA conditions that allowed us to detect binding upstream of more direct target genes (see Experimental Procedures). In the upstream region of four Group 1 genes (exoY, SMb21440, SMc00084, and SMc01580) and two Group 2 genes (chvI and ropB1), we used site-directed mutagenesis to delete the last 14 bp of the motif (Δ mutation; Figure 4A-F, left panel), encompassing the most highly conserved nucleotides in the motif. For each gene, we also generated a motif substitution mutation in which the three nucleotides in the direct repeats (positions 2–4 and 13–15 in the motif) were replaced by adenines (SUB mutation), altering the most highly conserved bases near the beginning and end of the motif (Figure 4A-F, left panel).
Figure 4. Mutation of the motif decreases ChvI binding.

(Left panel) Sequences of the motif upstream of (A) exoY, (B) SMb21440, (C) SMc00084, (D) SMc01580, (E) chvI, and (F) ropB1 are in black, with 3 nt of flanking sequence shown in grey: WT, no mutations; Δ, deletion of positions 2–15 of the motif; SUB, substitution mutations in the conserved bases at positions 2–4 and 13–15 of the motif (underlined). Distance of each motif from the predicted mTSS is as follows: exoY, 23 bp; SMb21440, 25 bp; SMc00084, 24 bp; SMc01580, 68 bp; chvI, 34 bp; ropB1, 46 bp (Schlüter et al., 2013). (Right panel) EMSA with DNA fragments upstream of (A) exoY, (B) SMb21440, (C) SMc00084, (D) SMc01580, (E) chvI, and (F) ropB1 with WT motif (gel lanes 1–3), Δ mutation (lanes 4–6), or SUB mutation (lanes 7–9). Amounts of ChvI D52E protein added to each binding reaction are indicated under each lane, and 1.25 fmol of DNA was used in each reaction. In WT motif samples, single arrowheads indicate free DNA while double arrowheads indicate a mobility shift. For exoY, SMc00084, and SMc01580, a shifted band of further decreased mobility is observed with 200 ng of protein compared to 100 ng protein, suggesting binding to both half-sites, compared to one half-site, in the motif (A, C, and D, lanes 1–3). The faint upper band in the SMc01580 lanes with the Δ mutation (D, lanes 4–6) appears to be a DNA structure that forms independently of ChvI protein, since that band is observed when no protein is added to the binding reaction (D, lane 4). Results were replicated in at least two independent trials.
We performed EMSA with DNA from the upstream regions of all six genes, with either the WT motif, Δ mutation, or the SUB mutation. Upstream DNA fragments (100–200 bp long; see Experimental Procedures) were PCR amplified, labeled, mixed with purified recombinant ChvI D52E protein, and subjected to electrophoresis on native polyacrylamide gels. The ChvI D52E protein was used since this form of ChvI protein was found to bind to DNA in previous EMSA (Chen et al., 2009). For all six genes, a mobility shift was evident in the binding reactions with the upstream DNA harboring the WT motif and 200 ng of ChvI protein, indicating ChvI binding to the DNA (Figure 4A-F, right panel, lanes 1–3). In contrast, for all six genes, deletion of the motif or substitution mutations in the motif either prevented a mobility shift or strongly decreased the amount of shifted DNA (Figure 4A-F, right panel, lanes 4–9). Thus, deletion or substitution mutations in the motif greatly decrease ChvI binding to the upstream region of ChvI direct target genes.
The motif is important for transcription of ChvI direct target genes
Since mutation of the motif results in decreased in vitro ChvI binding to the upstream region of direct target genes, we would expect that the motif mutations result in decreased ChvI-dependent transcription in vivo. We introduced GUS transcriptional fusions with the same motif deletion and substitution mutations (Figure 4A-F) into the genome of wild-type S. meliloti and measured GUS expression. We found that the motif deletion mutation upstream of all six genes resulted in a large decrease in transcription (Figure 5, black bars) compared to wild-type (Figure 5, white bars), indicating that the motif is important for transcription. In some cases, the expression decrease in the deletion mutant compared to wild-type is extremely large (30–100-fold), as for SMc00084, SMc01580, chvI, and ropB1; in other cases, the decrease is more modest (2-fold), as for exoY and SMb21440. The varying decreases in expression for motif deletion mutants of different genes may be influenced by the presence of additional ChvI-binding motifs upstream of some genes (see Discussion).
Figure 5. Mutation of the motif generally results in decreased expression of ChvI direct target genes.

GUS activity was measured in S. meliloti strains with an integrated transcriptional GUS fusion for exoY, SMb21440, SMc00084, SMc01580, chvI, or ropB1 (WT motif, white bars; Δ mutation, black bars; SUB mutation, grey bars). For each motif, two independent transconjugants were assayed in duplicate, and standard deviations are shown. The experiment was repeated twice with similar results. The GUS activities of the Δ mutation and SUB mutation strains were normalized to the GUS activity of the corresponding strain with the WT motif. The absolute GUS activity in Miller units for each strain with the WT motif was: exoY, 2.67 ± 0.31; SMb21440, 0.49 ± 0.03; SMc00084, 14.38 ± 0.32; SMc01580, 0.88 ± 0.04; chvI, 1.77 ± 0.07; ropB1, 2.38 ± 0.21.
Similar to the deletion mutations, we found that substitution mutations in the motif resulted in a large decrease in expression of exoY, SMb21440, SMc00084, SMc01580, and ropB1 (Figure 5, grey bars). The exception was the substitution mutation in the motif upstream of chvI, which resulted in a 4-fold increase in expression compared to wild-type (Figure 5). This strong increase in expression was seen in five independent transconjugants with the chvI motif substitution mutation, and sequencing of the intergenic region upstream of chvI in these transconjugants did not reveal any additional mutations besides the SUB mutations (data not shown). Since the substitution mutations in the DNA region upstream of chvI resulted in loss of ChvI binding in vitro (Figure 4E, right panel, lanes 7–9), and the motif overlaps the −35 region of chvI (Supporting Information Figure S1), it is possible that the substitution mutations alter the −35 region of chvI to allow strong expression in a ChvI-independent manner.
Taken together, our analysis of the motif deletion and substitution mutations for six ChvI direct target genes indicate that the motif, and especially the highly conserved bases near the beginning and end of the motif, is important for ChvI binding to the DNA upstream of these genes to regulate their transcription.
Occurrence of the motif in the S. meliloti genome
To determine how many other occurrences of the ChvI binding motif were present in the S. meliloti genome, we used the motif found by MEME to search globally upstream of all mTSS identified by Schlüter et al., 2013 (see Experimental Procedures). We reasoned that mTSS are most likely to represent TSS of mRNA (Schlüter et al., 2013). Using the FIMO program (Grant et al., 2011) to search 100 bp upstream of the dominant mTSS represented by the most sequence reads (Schlüter et al., 2013) for each gene (n=2727) returned 3098 motifs upstream of 1774 genes (p-value < 0.01), while extending the search to all predicted mTSS (n=4502) returned 5131 motifs upstream of 1907 genes (Supporting Information Table S5).
We compared the FIMO global motif dataset with the 276 genes identified by chIP (Supporting Information Table S2) to determine how many of these genes were associated with a possible ChvI-binding motif. Since the 276 genes had been identified by chIP, we hypothesized that many of the 276 genes had a ChvI-binding motif upstream of their mTSS. 177 of the 276 genes identified by chIP had predicted mTSS (Schlüter et al., 2013). We found that the majority of these 177 genes (78%; 138/177) had a predicted ChvI-binding motif within 100 bp of their mTSS (Supporting Information Figure S2): about one-third (31%; 54/177) of the genes both had a FIMO motif match and had been categorized as one of the 115 ChvI direct target candidates via transcriptome analysis (Supporting Information Table S3), and about half (47%; 84/177) of the genes had a FIMO motif match but had not been categorized as a ChvI direct target candidate via transcriptome analysis. A small set (8%; 15/177) of the genes had no FIMO match but were categorized as ChvI direct target candidates, and the last 14% (24/177) of the genes neither had a FIMO motif match nor were categorized ChvI direct target candidates (Supporting Information Table S3). This last subset (24/177) included genes that may not be expected to harbor a ChvI-binding motif, such as genes divergently transcribed from a ChvI direct target candidate. Thus, most of the 177 chIP-identified genes with predicted mTSS have potential upstream ChvI-binding motifs.
Some DNA regions enriched by chIP but lacking apparent ChvI-dependent regulation may be attributed to nonspecific binding of ChvI inside S. meliloti cells captured during the chIP procedure. It is also possible that some enriched regions correspond to genes encoding ChvI-regulated small RNAs and antisense RNAs, whose expression was not detectable in our analyses. However, it is also likely that, when narrowing the list to the 115 ChvI direct target candidates after chIP-chip, the single growth condition (exponential phase, LB medium) used for Affymetrix transcriptome and qRT-PCR analyses failed to capture the entire repertoire of ChvI-regulated genes. In summary, our approach illustrates the utility of combining global DNA-binding and gene expression analysis to identify direct targets of a key regulatory pathway in S. meliloti.
Discussion
Using chIP-chip and qRT-PCR, we defined 35 upstream regions under direct transcriptional control of ChvI. Each such region was bound by ChvI protein, as demonstrated by chIP-chip, and the genes immediately downstream of each region showed reciprocal changes in qRT-PCR-assessed transcript abundance in the chvI partial LOF mutant and the chvI GOF mutant compared to wild-type. Since many of these 35 regions are upstream of operons, in total we found 64 genes that are direct transcriptional targets of ChvI (Groups 1 and 2). Our findings confirm and significantly extend our previous work that identified three regions (upstream of SMc01580, ropB1, and SMb21440) directly bound by ChvI using EMSA (Chen et al., 2009), and other previous work that showed that the SMb21188-msbA2 operon is directly targeted by ChvI (Bélanger and Charles, 2013). The use of chIP, in which protein-DNA complexes are crosslinked in vivo prior to immunoprecipitation, enabled us to identify many more ChvI direct target genes compared to our previous study that relied on detecting ChvI binding in vitro (Chen et al., 2009).
Notable ChvI direct targets identified in this study include chvI itself (Group 2), the exoY and exoH operons encoding enzymes involved in succinoglycan synthesis (Group 1), and rem, encoding a key regulator that activates expression of the class II flagellar assembly and motility genes (Group 1) (Rotter et al., 2006). While ChvI had been proposed to play a role in coordinating EPS-I production and motility, evidence that motility/chemotaxis genes were directly regulated by ChvI was lacking until now (Barnett and Long, 2015; Barnett and Long, 2018; Chen et al., 2009; Wells et al., 2007; Yao et al., 2004). The effect of ChvI on rem transcription is negative, rather than positive as on the exo genes, and further work is needed to understand the mechanism by which ChvI down-regulates promoter activity.
Many of the ChvI direct target genes (Groups 1 and 2) encode proteins that are exported from the cytoplasm. PSORT subcellular location analysis (Yu et al., 2011) predicts that 17% of ChvI direct target proteins are located in the periplasm or outer membrane, or are secreted extracellularly, which is nearly four times the proportion predicted for the S. meliloti proteome as a whole (data not shown). For some of these ChvI direct targets, such as ropB1 (Group 2) and SMc00354 (Group 2) encoding outer membrane proteins, functional importance for the cell envelope has been established. An S. meliloti ropB1 transposon mutant is sensitive to the envelope disrupting agent deoxycholate (DOC) (Campbell et al., 2003) and orthologous ropB1 mutants in closely related Rhizobium leguminosarum are sensitive to DOC and sodium dodecyl sulfate (SDS) (Foreman et al., 2010). S. meliloti SMc00354 mutants are sensitive to the cationic peptides NCR247 and polymyxin B (Arnold et al., 2017). The SMc00354 gene is divergently transcribed from exoD (Group 3), which encodes a hydrophobic protein possibly associated with the cytoplasmic membrane. ExoD is required for infection of plant roots, normal EPS-I production, and growth under alkaline conditions (Reed and Walker, 1991a; Reed and Walker, 1991b).
In addition to the exo genes required for EPS-I biosynthesis, ChvI regulates genes involved in synthesis of other cell surface carbohydrates such as kpsF1 (SMb20809; lipopolysaccharide; Group 1), SMb20829 (possibly capsular polysaccharide; Group 2), and SMb21188-SMb21191 (unknown cell surface polysaccharide; Group 1) (Beck et al., 2008; Finan et al., 2001; Griffitts and Long, 2008). ChvI also regulates opgC (SMc04381; Group 2), encoding a succinyltransferase for cyclic β−1,2 glucans (Kawaharada et al., 2010). ChvI regulation of two lytic transglycosylase genes (SMc00404 and SMc01855; both Group 1) and a putative Tol-Pal system gene (SMc02941; Group 2) suggests that ChvI plays a role in peptidoglycan remodeling. Thus, the ChvI regulon comprises numerous genes that encode proteins predicted to affect the cell envelope, consistent with the extensive remodeling of cell envelope proteins seen in bvrR and bvrS mutants of B. abortus (Lamontagne et al., 2007; Viadas et al., 2010). However, most ChvI direct target genes have unknown functions and need further characterization.
Expression changes for many of the ChvI regulon genes have been reported in previous S. meliloti transcriptome studies using various growth conditions and mutants (Barnett and Long, 2015; Calatrava-Morales et al., 2017; Hellweg et al., 2009; Krol and Becker, 2004; Krol et al., 2016; Penterman et al., 2014; Santos et al., 2014). Genes with altered expression due to syrA (exopolysaccharide regulator) overexpression showed especially strong overlap with ChvI regulon genes, which had led us to propose that SyrA may interact with ExoS/ChvI (Barnett and Long, 2015). In comparing the list of ChvI direct target genes (Groups 1 and 2) to all other S. meliloti transcriptome data sets, we also identified significant overlap (using hypergeometric probability tests, data not shown) with the transcriptomes for NCR247-treatment (Penterman et al., 2014) and acid shock (Hellweg et al., 2009), and for podJ (Fields et al., 2012), cbrA (Gibson et al., 2007), ntrY (Calatrava-Morales et al., 2017) and emrR mutants (Santos et al., 2014). Phenotypes common to these four mutants are increased exopolysaccharide production, decreased swimming motility, and increased sensitivity to SDS and/or DOC.
In addition to the ChvI regulon genes in Groups 1 and 2, we have identified 59 other potential ChvI direct target genes in Groups 3, 4, and 5 (Supporting Information Table S4). All the genes in Groups 3–5 showed upstream region binding by ChvI protein in chIP-chip and showed a change in expression of ≥1.4-fold in at least one of the chvI mutants compared to wild-type by qRT-PCR. Group 3 genes showed ≥1.4-fold change in expression for one chvI mutant and no significant change in the other chvI mutant, whereas Group 4 and 5 genes showed a non-reciprocal change in expression in both chvI mutants. Genes in Groups 3–5 could be direct target genes of ChvI, although the failure to detect a reciprocal change in expression for these genes in both chvI mutants implies that the ChvI-dependent expression of these genes is more complex. For example, perhaps some Group 3 genes have no significant change in expression in one of the chvI mutants because S. meliloti must maintain a certain basal level of those genes’ expression for viability. Additional regulators besides ChvI could be involved in controlling the expression of Group 3–5 genes.
Like Groups 1 and 2, Groups 3–5 are enriched for genes encoding proteins with predicted outer membrane, periplasm, and extracellular locations, including genes potentially involved in cell surface polysaccharide production (eglC, Group 3; SMb20810, Group 3; SMb20941, Group 5; SMb21067; Group 3) and peptidoglycan synthesis/remodeling (glmM, Group 5; mrcA1, Group 5; mepA, Group 4; SMc02432, Group 4). Our categorization of exoR (SMc02078), which encodes a periplasmic inhibitor of ExoS/ChvI signaling, in Group 3 is consistent with previous studies showing that exoR expression is negatively regulated by ExoR and thus positively regulated by ExoS/ChvI (Chen et al., 2008; Lu and Cheng, 2010). Located upstream of exoR in the genome, the Group 3 gene fadL (SMc02079) encodes an outer membrane-localized fatty acid transporter that promotes growth on palmitoleic acid and uptake of long-chain acyl-homoserine lactone quorum-sensing signals (Krol and Becker, 2014). Regulation of fadL expression was not previously linked to ExoR or ExoS/ChvI.
Identification of the 35 DNA regions directly regulated by ChvI in Groups 1 and 2 allowed us to identify a recognition sequence for ChvI binding to DNA. This “ChvI box” differs from and represents a major improvement over the previously reported motif (Chen et al., 2009). The previous motif search included indirect as well as direct target genes and was performed prior to the availability of TSS data (Schlüter et al., 2013). Here, we demonstrated by mutational analysis using six ChvI direct target genes (exoY, SMb21440, SMc00084, and SMc01580 from Group 1, and chvI and ropB1 from Group 2) that the newly-discovered ChvI box is important for ChvI binding to DNA and transcriptional regulation of ChvI direct target genes.
Like the motif recognized by other members of the OmpR/PhoB subfamily of response regulators, which recognize DNA as a homodimer (Capra and Laub, 2012), the ChvI box contains two direct repeats. The 18 bp-long “Pho box” has two 7 bp-long direct repeats with a conserved 4 bp spacer (Yuan et al., 2006), and similarly the ChvI box has its conserved GCC repeats spaced 11 nt apart. The 15-bp length, rather than 18-bp length, of the ChvI box implies that the beginning of the first repeat and/or the end of the second repeat is not highly conserved. The Pho box allows two PhoB molecules to bind head to tail to stabilize the binding of σ70 to the −35 region upstream of the TSS, compensating for the lack of the canonical −35 sequence recognized by σ70 (Blanco et al., 2002; Canals et al., 2012; Yuan et al., 2006). Consistent with this model, the ChvI box is found overlapping the −35 regions of many of the ChvI direct target genes in Groups 1 and 2 (Supporting Information Figure S1). However, in some cases the ChvI box is found farther upstream of the TSS, downstream of the TSS, or overlapping the TSS (Supporting Information Figure S1). Furthermore, upstream of the mTSS of many Group 1 and 2 genes, FIMO found more than one possible ChvI box (Supporting Information Table S5). Indeed, although deletion of the motif found by MEME resulted in a decrease in expression for all six ChvI direct target genes tested (Figure 5), we noted that the magnitude of the expression decrease compared to wild-type ranged from 2- to 100-fold, depending on the gene tested. For the two genes with a 2-fold decrease in expression due to the motif deletion (exoY and SMb21440), FIMO found two additional possible ChvI boxes in the 100 bp upstream of their mTSS (Supporting Information Table S5). ChvI binding at these additional ChvI boxes could explain why a larger decrease in expression was not observed in the single motif deletions for these genes (Figure 5). However, the situation is not simple, since FIMO found one additional possible ChvI box upstream of SMc01580, but deletion of just the one motif found by MEME (Supporting Information Table S1) resulted in a 100-fold decrease in expression (Figure 5). Future experiments will be needed to determine the implications of having more than one ChvI box upstream of a particular TSS, the significance of the distance of the ChvI box from the TSS, the importance of specific nucleotide positions in the ChvI box, and the influence of the motif on ChvI action as a positive or negative transcriptional regulator. Further refinement of the ChvI box sequence should also narrow the number of occurrences found in the S. meliloti genome.
Transcriptional regulation by ExoS/ChvI in S. meliloti is critical for symbiosis with plant hosts as well as for free living phenotypes such as exopolysaccharide production, motility, nutrient utilization, biofilm formation, and cell envelope integrity (Bélanger et al., 2009; Doherty et al., 1988; Wang et al., 2010; Wells et al., 2007; Yao et al., 2004). The ChvI regulon identified here, which includes many genes associated with cell envelope functions, will allow us to probe the function of ExoS/ChvI signaling in the free-living and symbiotic forms of the bacteria. Additionally, the identification of the ChvI box consensus sequence bound by ChvI will facilitate more detailed investigation of the mechanisms underlying ExoS/ChvI gene regulation.
Experimental Procedures
Strains, media, growth conditions, and genetic techniques
All strains in this study (Table 2) are derived from Rm1021 (streptomycin [Sm]-resistant derivative of wild-type strain SU47) and were grown at 30°C in LB medium. Antibiotics were used at the following concentrations: Sm, 500 μg ml−1; neomycin (Nm), 50 μg ml−1; ampicillin (Ap), 100 μg ml−1; kanamycin (Km), 30 μg ml−1; chloramphenicol (Cm), 50 μg ml−1; and gentamicin (Gm), 25 μg ml−1 for S. meliloti and 5 μg ml−1 for Escherichia coli. All E. coli plasmids were maintained in DH5α cells. Plasmids (Table 2) were transferred from E. coli to S. meliloti by triparental conjugation using helper plasmid pRK600 (Finan et al., 1986). Oligonucleotides are listed in Supporting Information Table S6.
Table 2.
Strains and Plasmids
| Genotype or relevant characteristics | Reference or source | |
|---|---|---|
| Strains | ||
| Rm1021 | Derivative of RCR2011; Sm | Meade et al., 1982 |
| EC69 | chvI(K214T) (integrated pDW181); Sm Hy | Wells et al., 2007 |
| EC176 | Wild type (integrated pDW181); Sm Hy | Wells et al., 2007 |
| EC220 | chvI(D52E)/chvI+ (integrated pEC177); Sm Hy | Chen et al., 2009 |
| EC409 | chvI(D52E)/chvI+ (integrated pEC405); Sm Sp | Chen et al., 2009 |
| EC412 | chvI(K214T) (integrated pEC406); Sm Sp | Chen et al., 2008 |
| ES147 | chromosomal chvI replaced with chvI-HA in Rm1021; Sm | This study |
| Plasmids | ||
| pAR142 | PSMc00084 (−245 to 90 in ORF) in pVO155; Ap Nm/Km | This study |
| pC91 | pEC570 with motif deletion (−141 to −128 before Start codon); Ap Nm/Km | This study |
| pC92 | pEC570 with motif substitutions (−141 to −139 and −130 to −128 before Start codon); Ap Nm/Km | This study |
| pC164 | PchvI (−341 to −1 before Start codon) in pVO155; Ap Nm/Km | This study |
| pC165 | pC164 with motif substitutions (−119 to −117 and −108 to −106 before Start codon); Ap Nm/Km | This study |
| pC166 | pC164 with motif deletion (−119 to −106 before Start codon); Ap Nm/Km | This study |
| pC173 | pEC340 with motif substitutions (−104 to −102 and −93 to −91 before Start codon); Ap Nm/Km | This study |
| pC174 | pEC340 with motif deletion (−104 to −91 before Start codon); Ap Nm/Km | This study |
| pC196 | pAR142 with motif deletion (−131 to −118 before Start codon); Ap Nm/Km | This study |
| pC197 | pAR142 with motif substitutions (−131 to −129 and −120 to −118 before Start codon); Ap Nm/Km | This study |
| pDW33 | Identical to pVO155; Ap Hy | Cronan and Keating, 2004 |
| pDW181 | PhisB in pDW33; Ap Hy | Wells et al., 2007 |
| pEC78 | PchvI and chvI in pCR2.1 TOPO; Ap | Chen et al., 2009 |
| pEC177 | chvI(D52E) in pDW33; Ap Hy | Chen et al., 2009 |
| pEC340 | PexoY (−773 to −1 before Start codon) in pVO155; Ap Nm/Km | Chen et al., 2009 |
| pEC405 | chvI(D52E) in pMB439; Ap Sp | Chen et al., 2009 |
| pEC406 | PhisB in pMB439; Ap Sp | Chen et al., 2008 |
| pEC570 | PropB1 (−361 to −1 before Start codon) in pVO155; Ap Nm/Km | Chen et al., 2009 |
| pEC616 | PSMb21440 (−309 to 91 in ORF) pVO155; Ap Nm/Km | Chen et al., 2009 |
| pEC618 | PSMc01580 and SMc01580 (−215 before Start codon through Stop codon) in pVO155; Ap Nm/Km | Chen et al., 2009 |
| pES112 | pEC78 with an NheI site inserted just before the Stop codon of chvI; Ap | This study |
| pES119 | pES112 with an HA tag ligated into NheI site; Ap | This study |
| pES129 | chvI (−405 before Start codon through 115 bp past Stop codon) in pJQ200SK; Gm | This study |
| pJQ200SK | Suicide vector with sacB for gene replacements; Gm | Quandt and Hynes, 1993 |
| pMB439 | pBluescript SK(−) derivative with Spr cassette; Ap Sp | Barnett et al., 2000 |
| pNR28 | pEC618 with motif deletion (−112 to −99 before Start codon); Ap Nm/Km | This study |
| pNR29 | pEC618 with motif substitutions (−112 to −110 and −101 to −99 before Start codon); Ap Nm/Km | This study |
| pNR36 | pEC616 with motif deletion (−167 to −154 before Start codon); Ap Nm/Km | This study |
| pNR37 | pEC616 with motif substitutions (−167 to −165 and −156 to −154 before Start codon); Ap Nm/Km | This study |
| pNR39 | pNR28 with additional upstream region (−400 before Start through Stop codon); Ap Nm/Km | This study |
| pNR40 | pNR29 with additional upstream region (−400 before Start through Stop codon); Ap Nm/Km | This study |
| pRF1173 | pET29a plasmid with full-length chvI(D52E) | Chen et al., 2009 |
| pRK600 | Conjugal transfer helper plasmid; Cm | Finan et al., 1986 |
| pVO155 | Terminator and polylinker preceding uidA (encoding GUS) in pUC119 derivative; Ap Nm/Km | Oke and Long, 1999 |
Epitope tagging and gene replacement of chvI
To construct the Haemagglutinin (HA)-tagged chvI allele, the Quikchange (Stratagene) site-directed mutagenesis protocol was used to insert an NheI site just before the translational stop codon (primers OMB35/OMB36) of chvI in pEC78, generating pES112. Complementary oligos (OEC94/OEC95) were hybridized to produce a fragment that encoded the HA tag with flanking NheI sites. The HA tag was ligated into NheI-digested pES112 generating pES119, which was verified by sequencing. The HA-tagged chvI gene (including 405 bp of the intergenic region upstream of the open reading frame through 115 bp past the stop codon) was released from pES119 by digesting with BamHI and XbaI. The fragment was ligated into pJQ200SK, generating pES129, which was transferred into S. meliloti via conjugation. The chvI-HA allele was integrated into the chromosome of Rm1021 by conjugation and gentamicin selection. Transconjugants were grown on LB media with 5% sucrose to counterselect for the plasmid and isolate strains that had undergone a second homologous recombination. The resulting strain (ES147) was tested by PCR using flanking primers (S3A/OEC29) to verify that the wild-type chvI allele had been replaced with the chvI-HA allele. ES147 was also tested by immunoblotting as well as by immunoprecipitation followed by immunoblotting to verify that ChvI-HA had the expected molecular weight (29 kD) and could be immunoprecipitated with anti-HA antibody (F7; Santa Cruz Biotechnology).
Formaldehyde crosslinking, immunoprecipitation (IP), amplification, and DNA microarrays (ChIP-chip)
ChIP-chip protocols were adapted from previously published methods (Barnett et al., 2004; Chen et al., 2008; Laub et al., 2002). 500 OD600 of cells (Rm1021 and ES147a) grown in LB to 1.6–2.0 OD600 ml−1 were diluted with LB to 1 OD600 ml−1, and sodium phosphate and formaldehyde were added to a final concentration of 10 mM and 1%, respectively. After incubation for 3 min at room temperature, cells were harvested by centrifugation and washed one time in TY without CaCl2. Cells were lysed by incubating in 36 ml lysis buffer (50 mM Tris-HCl pH 8, 100 mM NaCl, 20% glycerol, 1 mg ml−1 lysozyme, and protease inhibitors for Histidine-tagged proteins; Sigma-Aldrich) for 20 min on ice followed by the addition of 4 ml of 10% Triton X-100 and an additional incubation on ice for 10 min. The lysates were sonicated on ice in ten 1 min pulses, giving an average DNA fragment size of ~500 bp. Cell debris was pelleted and the supernatant was precleared by incubating with Protein A agarose (Pierce/Thermo Fisher Scientific) for 40 min at 4°C. After removal of the Protein A agarose, 750 μl of the precleared supernatant was set aside to prepare the Total DNA samples, and 38 ml of the precleared supernatant was subjected to ChIP. For the Total DNA samples, DNA was isolated using phenol/chloroform extraction and ethanol precipitation, followed by additional cleanup using the standard protocol in the Qiagen PCR Purification kit. For chIP, the supernatant was incubated with anti-HA antibody (F7; Santa Cruz Biotechnology) and Protein A agarose overnight at 4°C. The beads were washed 5 times with cold IP buffer (50 mM Tris-HCl pH 8, 100 mM NaCl, 20% glycerol) and 2 times with TE. Then, beads were resuspended in TE and incubated for 6 h at 65°C to reverse crosslinks. Beads were removed by centrifugation, and the chIP DNA was amplified using the Round A and B amplification procedures (Rich, 2008). ChIP DNA and Total DNA were fragmented to ~100–200 bp using DNAse (Pierce/Thermo Fisher Scientific) and DNase buffer (USB), 3’ labeled with Biotin-ddUTP, and hybridized onto Affymetrix Symbiosis GeneChip microarrays as previously described (Barnett et al., 2004).
chIP-chip data analysis
Each DNA sample (IP-tag, IP-no tag, and genomic) was hybridized onto Affymetrix Symbiosis GeneChips in triplicate, and data were analyzed using Affymetrix software. The Symbiosis GeneChips have 13 perfect match and 13 1-bp mismatch oligonucleotides for each annotated ORF, as well as even tiling of both strands of intergenic regions >150bp long using 25-nt oligomers, spaced 1 to 7 nt apart. GeneChips were scaled to a target signal intensity of 500 using all probe sets (global scaling) as previously described (Barnett et al., 2004). Thus, comparison of GeneChips with the IP-tag samples vs. the IP-no tag samples yielded nine pairwise comparisons. Similarly, comparison of GeneChips with the IP-tag samples vs. genomic DNA samples also yielded nine pairwise comparisons. A DNA region was categorized as “enriched by chIP” if, in all 18 of the pairwise comparisons (IP-tag vs. IP-no tag and IP-tag vs. genomic DNA), the Affymetrix GeneChip probe set corresponding to a given DNA region had an average signal log ratio (SLR; log2 ratio) increase of ≥0.96 and was evaluated by the software as significantly increased or decreased (P≤0.05).
The genomic context of each of the 489 regions enriched by chIP (Figure 2; Supporting Information Table S1) was inspected manually to identify the genes that overlapped a chIP-enriched region and/or that had at least 1 chIP-enriched region overlapping 300 bp upstream of the gene. The 300 bp upstream distance was chosen because Schlüter et al. (2013) had reported that transcription start sites (TSS) located more than 300 nt away from the start codon were rarely observed, and that 68 nt was the average distance between TSS of mRNAs (mTSS) and the start codon (Schlüter et al., 2013). These genes were compared with operon prediction data from RhizoRegNet (Krol et al., 2011); for genes in an operon, only the first gene was counted. Thus, 276 genes were identified by chIP-chip (Supporting Information Table S2), and these 276 genes corresponded to 462 of the 489 DNA regions enriched by chIP. The remaining 27 of the 489 DNA regions enriched by chIP did not correspond to the 276 genes; those 27 DNA regions were either coding regions of later genes in an operon (when the first gene in the operon or its upstream region was not enriched by chIP), or intergenic regions >300bp upstream of the start codon of a gene.
Next, for these 276 genes, previous transcriptional profiling data (Chen et al., 2009) were used as a guide to identify the 115 candidate target genes whose expression would be checked by qRT-PCR (Supporting Information Table S3). The previous transcriptional profiling experiment had compared gene expression in a chvI gain of function (GOF) mutant [chvI (D52E/chvI+)] to wild-type in triplicate (giving nine pairwise comparisons), and gene expression in a chvI partial loss of function mutant (LOF) [chvI (K214T)] to wild-type in triplicate (also giving nine pairwise comparisons). The chvI (D52E/chvI+) mutant was used as the gain of function strain since the chvI (D52E) allele alone appears to be lethal (Chen et al., 2009); the basis for this apparent lethality is unknown but suggests that too much ChvI activity is deleterious. Forty-nine of the 115 genes were deemed candidates for testing by qRT-PCR since they had an increase or decrease of average SLR ≥ 0.5 (an increase or decrease in expression of ≥1.4-fold) in eight or nine of the pairwise comparisons for at least one of the chvI mutants compared to wild-type in the Chen et al., 2009 dataset. Genes that encoded tRNAs, rRNAs, other noncoding RNAs, and transposases were omitted, since their expression could not be quantified reliably by the procedure that had been used for RNA purification and transcriptional profiling (Chen et al., 2009). In addition, since it was demonstrated that GeneChips could have difficulty evaluating genes with low expression (Barnett et al., 2004), 66 of the 115 candidates tested by qRT-PCR were genes that had low absolute expression or high standard deviation in their signal in the Chen et al., 2009 dataset; specifically, these genes were called “not present” by the software in one or more of the GeneChips, indicating that their expression could not be reliably detected above background on the microarray (Supporting Information Table S3). But if a gene with unreliable expression was in an operon with other genes that were called “present” and had “no change” in expression in the Chen et al., 2009 dataset, these genes were not included in the list of 66 genes to be tested by qRT-PCR.
The Affymetrix GeneChip data have been deposited in the Gene Expression Omnibus (GEO) database under accession numbers GSE112818 (chIP-chip data) and GSE112819 (ChvI transcriptome data (Chen et al., 2009)).
Quantitative reverse transcription PCR (qRT-PCR)
For consistency while following up the transcriptome study used to identify the 115 candidates to test by qRT-PCR (as described above), the RNA samples from the previous transcriptional profiling experiment (Chen et al., 2009) were used to prepare cDNA for qRT-PCR. Three biological replicates of each strain (wild-type, EC176; chvI(K214T) partial LOF mutant, EC69; and chvI(D52E)/chvI+ GOF mutant, EC220) were cultured in separate flasks of LB medium with Sm selection to an optical density at 600 nm of 0.5 to 0.7. RNA was isolated from each sample and single-stranded cDNA was synthesized as previously described (Barnett et al., 2004).
Gene specific primers (Supporting Information Table S6) for qRT-PCR were designed using the Primer3 program (http://frodo.wi.mit.edu/)(Untergasser et al., 2012), with the aim of amplifying fragments ~150 bp long. Primer pairs were evaluated for their reaction efficiency by using four ten-fold dilutions of Rm1021 genomic DNA (1 ng, 0.1 ng, 0.01 ng, and 0.001 ng per reaction) to construct a standard curve. After each reaction was completed, melt curves were constructed (measuring fluorescence while increasing the temperature from 65°C to 95°C in 0.5°C increments) and analyzed to ensure that only one large peak was present, indicating that a single PCR product was produced by each primer pair. To confirm that the proper DNA fragments were amplified, PCR products were run on a gel, excised, and sequenced (Sequetech, Inc., Mountain View, CA; Eton Bioscience, San Diego, CA). Four of the 116 candidate genes (SMb20838, SMc00286, SMc02887, and SMc05001) could not be tested by qRT-PCR due to high sequence identity to other genes in the S. meliloti genome, which precluded design of gene-specific primers.
The nine cDNA samples were each tested in triplicate (“technical triplicates”) using the CFX96 Real Time PCR Detection System (Bio-Rad). To test relative gene expression, each qRT-PCR reaction included 2 ng of cDNA, a gene-specific primer pair, and SYBR Green SensiMix (Bioline). A standard curve was run on the same plate for each primer pair to ensure that each primer pair had a reaction efficiency between 85–110%.
Cycle threshold (Ct) values obtained from qRT-PCR were compared to calculate relative gene expression. Gene expression in the chvI partial LOF mutant and chvI GOF mutant were compared to gene expression in wild type using CFX Manager Software (Bio-Rad) and the Pfaffl method (Pfaffl, 2001), which incorporates reaction efficiency into expression ratios. Gene expression was also normalized using uppS (Smc02097) as a reference gene (Barnett et al., 2004).
Bioinformatics
DNA sequence upstream of the most likely transcriptional start site (TSS) identified in (Schlüter et al., 2013) was downloaded from the S. meliloti genome database (https://iant.toulouse.inra.fr/bacteria/annotation/cgi/rhime.cgi/). For MEME analysis, mTSS from mRNA and lmTSS from leaderless mRNA were used in most cases; pmTSS from putative mRNA were only used in two cases (SMb20300 and SMc02645) where no mTSS or lmTSS had been identified. Group 1 and 2 genes with no TSS identified in Schlüter et al. (SMb20948, SMc00658, SMc04236, SMc04276, SMc01556, and SMc02727) were excluded from the motif search. For the positively-regulated Group 1 and 2 genes, 50 bp of upstream sequence including the TSS was used, with the following exceptions: the published footprinted sequence was used for SMc01580 and the published gel-shifted sequence was used for SMb21440 and ropB1 (Chen et al., 2009); thus, all of the input SMc01580 sequence and part of the ropB1 sequence was more than 50 bp away from the mTSS. For negatively-regulated Group 1 and 2 genes, 101 bp of sequence centered on the TSS was used. Sequences were analyzed for a possible ChvI binding motif using the MEME algorithm (http://meme-suite.org/tools/meme), (Bailey et al., 2006) with the “Zero or one occurrence per sequence” and “motif on given strand” options.
FIMO software (Grant et al., 2011) was used to search for additional ChvI-binding motifs within 100 nt upstream of all mTSS (given strand only) reported in Schlüter et al., 2013 (Supporting Information Table S5). Only mTSS were used for the FIMO search, rather than including lmTSS and pmTSS, because mTSS are more likely to represent TSS of an mRNA (Schlüter et al., 2013). For FIMO, a p-value cutoff of < 0.01 was chosen, so that all of the ChvI-direct target motifs identified by MEME in Supporting Figure S1 were included.
Transcriptional fusion plasmid construction
Transcriptional fusion plasmids for chvI and SMc00084 were constructed by PCR amplifying gene regions with flanking SpeI/XhoI sites and ligating into pVO155. For chvI, pC164 contains the 341-bp intergenic region upstream of the chvI ORF, which was amplified using primer pair OCL125/126. For SMc00084, pAR142 contains −245 before start through 90 in the ORF, which was amplified using primer pair OAR47/48, and digested at the SpeI site introduced by OAR47 and an endogenous XhoI site at bp 90 in the ORF. Construction of transcriptional fusion plasmids for exoY, SMb21440, SMc01580, and ropB1 was described previously (Chen et al., 2009).
Site-directed mutagenesis of ChvI binding motifs
Site directed mutagenesis (SDM) to delete or generate substitution mutations in the motif upstream of exoY, SMb21440, SMc00084, SMc01580, chvI, and ropB1 was performed according to the QuikChange II Site Directed Mutagenesis Kit protocol (Agilent Technologies, Santa Clara, CA). For the substitution mutations, partially overlapping primers were designed (Zheng et al., 2004). For exoY, pEC340 was the template for SDM, and primer pairs OCL132/133 and OCL130/131 were used to construct the motif deletion plasmid pC174 and substitution plasmid pC173, respectively. For SMb21440, pEC616 was the template for SDM, and primer pairs ONR53/54 and ONR55/56 were used to construct the motif deletion plasmid pNR36 and substitution plasmid pNR37, respectively. For SMc00084, pAR142 was the template for SDM, and primer pairs OCL168/169 and OCL170/171 were used to construct the motif deletion plasmid pC196 and substitution plasmid pC197, respectively. For SMc01580, pEC618 was the template for SDM, and primer pairs ONR49/50 and ONR51/52 were used to construct the motif deletion plasmid pNR28 and substitution plasmid pNR29, respectively. Additional upstream region was then cloned into pNR28 and pNR29 (beginning at −400 before the start codon), to facilitate homologous recombination of the plasmid into the S. meliloti genome (see “Transcriptional fusion assays” below); these resulting plasmids were pNR39 and pNR40, respectively. For chvI, pC164 was the template for SDM, and primer pairs OCL134/135 and OCL136/137 were used to construct the motif deletion plasmid pC166 and substitution plasmid pC165, respectively. For ropB1, pEC570 was the template for SDM, and primer pairs OCL52/53 and OCL50/51 were used to construct the motif deletion plasmid pC91 and motif substitution plasmid pC92. All plasmids were verified by sequencing.
Gel mobility shift assays
Using the WT, motif deletion, motif substitution plasmids described in the section above as templates, the following primer pairs were used to amplify DNA fragments for EMSA (lengths shown are for the fragments with the WT and SUB mutations; the Δ mutation fragments are 14 bp shorter): exoY, OCL78/79 (128 bp); SMb21440, OCL 157/158 (115 bp); SMc00084, OCL146/147 (196 bp); SMc01580, OCL43/42 (101 bp); chvI, OCL82/85 (148 bp); and ropB1, OEC199/200 (115 bp). DNA fragments were purified using the Qiagen PCR cleanup kit or Zymo DNA Clean and Concentrator kit. DNA fragments were labeled using the Biotin 3’-End DNA Labeling Kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions, with the following modifications: reactions with half of the recommended volume were prepared and the 37°C incubation was extended to 1–1.5 hours. The labeling efficiency of DNA fragments was assessed by dot blot, following the Chemiluminescent Nucleic Acid Detection Module protocol (Thermo Fisher Scientific) with the addition of an extra wash step. Imaging was performed using the Omega Lum™ G Imaging System (Aplegen, Pleasanton, CA).
As previously described (Chen et al., 2009), recombinant D52E ChvI protein was used for EMSA because we expected that form of the protein to bind more tightly than the wild-type form in vitro, and generally we have had difficulty detecting ChvI binding via EMSA. Recombinant D52E ChvI protein was extracted from BL21(DE3) cells harboring the pRF1173 plasmid (pET29a plasmid with full-length D52E ChvI), using an S-Tag thrombin purification kit (EMD Millipore, Temecula, CA). The protein was stored at −80°C in ChvI storage buffer (20 mM HEPES [pH 7.4], 50 mM KCl, 5 mM Dithiothreitol [DTT], 50% glycerol). Binding reactions (7 μl) consisted of binding buffer (10 mM Tris-HCl [pH 7.5], 10 mM KCl, 0.1 mM DTT, 2.5% glycerol, 25 µg ml−1 bovine serum albumin), 0.5 µg poly dI-dC (Thermo Fisher Scientific), recombinant D52E ChvI protein (0, 100, and 200 ng) and 3’ end biotinylated probes (1.25 fmol). Binding reactions were incubated for 20 minutes at room temperature (20°C), then 3 μl loading buffer (10 mM Tris-HCl [pH 7.5], 10 mM KCl, 0.1 mM DTT, 22.5% glycerol, 25 µg ml−1 bovine serum albumin, 0.5 mg ml−1 bromophenol blue, 0.5 mg ml−1 xylene cyanol FF), was added and samples were loaded onto 7% polyacrylamide (29:1 acrylamide:bis-acrylamide) gels prepared with 1.2X Tris-Borate buffer (107 mM Tris, 107 mM boric acid). The gels had been pre-run with 1.2X Tris-Borate running buffer at 100V for 50 min at 4°C prior to sample loading. Samples were separated by electrophoresis at 200V for 40 min at 4°C. Semi-dry transfer onto Biodyne Nylon membranes (Thermo Fisher Scientific) was performed with 0.5X TBE buffer for 80 minutes at 300mA constant current using a Trans-Blot Semi-Dry Electrophoretic Transfer Cell (Bio-Rad). Membranes were dried then crosslinked twice for 30 seconds each time using ultraviolet crosslinker CL-1000 (UVP Inc., Upland, CA) set at 120,000 microjoules per cm². DNA fragments were detected following the Chemiluminescent Nucleic Acid Detection Module (Thermo Fisher Scientific) protocol with the addition of an extra wash step. Imaging was performed using the Omega Lum™ G Imaging System (Aplegen).
Transcriptional fusion assays
Transcriptional fusion plasmids with WT or mutated motifs (exoY: pEC340, pC174, pC173; SMb21440: pEC616, pNR36, pNR37; SMc00084, pAR142, pC196, C197; SMc01580: pEC618, pNR39, pNR40; chvI, pC164, pC166, pC165; ropB1: pEC570, pC91, pC92) were transferred into Rm1021 by triparental conjugation. Integration of each transcriptional fusion plasmid via single crossover into the genome results in duplication of the gene region that had been cloned into the transcriptional fusion plasmid (see Table 2). Thus, integration of a transcriptional fusion plasmid harboring a mutated motif resulted in one WT upstream region and one mutated upstream region in the genome. From the genome of S. meliloti transconjugants, we PCR amplified (see Supporting Information Table S6) and sequenced the region immediately upstream of the GUS gene to verify that the appropriate sequence (with WT or mutated motif) was upstream of the GUS gene and thus controlled GUS expression. As an additional control, to ensure that the GUS gene was functional in each transcriptional fusion plasmid with a mutated motif, transconjugants that had integrated the plasmid with the WT sequence upstream of the GUS gene were tested to verify that GUS expression was similar to that of the strain with the WT motif transcriptional fusion plasmid integrated (data not shown). Two independent transconjugants were each assayed in duplicate for GUS activity as previously described (Swanson et al., 1993).
Supplementary Material
Acknowledgments
We are grateful to A. Rizzacasa for construction of the pAR142 plasmid. This work was supported by NSF award IOS-0818981 (to E.J.C.), NIH Maximizing Access to Research Careers (MARC) grant 5T34GM008612–20 to CSU Fullerton (C.M.), NIH MARC grant 5T34GM008612–23 to CSU Fullerton (J.A.O.), a California State University Special Fund for Research, Scholarship, and Creative Activity Award (to E.J.C.), and NIH grant R01 GM093628 (to S.R.L.). The authors declare that they have no conflicts of interest.
Footnotes
Supporting information
Additional supporting information may be found in the online version of this article at the publisher’s website.
References
- Altamirano-Silva P, Meza-Torres J, Castillo-Zeledón A, Ruiz-Villalobos N, Zuñiga-Pereira AM, Chacon-Diaz C, Moreno E, Guzmán-Verri C, and Chaves-Olarte E (2018) Brucella abortus senses the intracellular environment through the two-component system BvrR/BvrS allowing the adaptation to its replicative niche. Infect Immun doi: 10.1128/IAI.00713-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold MFF, Penterman J, Shabab M, Chen EJ, and Walker GC (2018) Important late stage symbiotic role of the Sinorhizobium meliloti exopolysaccharide succinoglycan. J Bacteriol doi: 10.1128/JB.00665-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold MFF, Shabab M, Penterman J, Boehme KL, Griffitts JS, and Walker GC (2017) Genome-wide sensitivity analysis of the microsymbiont Sinorhizobium meliloti to symbiotically important, defensin-like host peptides. mBio 8: e01060–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Williams N, Misleh C, and Li WW (2006) MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res 34: W369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett MJ, Bittner AN, Toman CJ, Oke V, and Long SR (2012) Dual RpoH sigma factors and transcriptional plasticity in a symbiotic bacterium. J Bacteriol 194: 4983–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett MJ and Long SR (2015) The Sinorhizobium meliloti SyrM regulon: effects on global gene expression are mediated by syrA and nodD3. J Bacteriol 197: 1792–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett MJ and Long SR (2018) Novel genes and regulators that influence production of cell surface exopolysaccharides in Sinorhizobium meliloti. J Bacteriol 200: e00501–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett MJ, Oke V, and Long SR (2000) New genetic tools for use in the Rhizobiaceae and other bacteria. Biotechniques 29: 240–245. [DOI] [PubMed] [Google Scholar]
- Barnett MJ, Toman CJ, Fisher RF, and Long SR (2004) A dual-genome Symbiosis Chip for coordinate study of signal exchange and development in a prokaryote-host interaction. Proc Natl Acad Sci USA 101: 16636–16641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck S, Marlow VL, Woodall K, Doerrler WT, James EK, and Ferguson GP (2008) The Sinorhizobium meliloti MsbA2 protein is essential for the legume symbiosis. Microbiology 154: 1258–1270. [DOI] [PubMed] [Google Scholar]
- Bélanger L and Charles TC (2013) Members of the Sinorhizobium meliloti ChvI regulon identified by a DNA binding screen. BMC Microbiol 13: 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger L, Dimmick KA, Fleming JS, and Charles TC (2009) Null mutations in Sinorhizobium meliloti exoS and chvI demonstrate the importance of this two-component regulatory system for symbiosis. Mol Microbiol 74: 1223–1237. [DOI] [PubMed] [Google Scholar]
- Blanco AG, Sola M, Gomis-Ruth FX, and Coll M (2002) Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure 10: 701–713. [DOI] [PubMed] [Google Scholar]
- Calatrava-Morales N, Nogales J, Ameztoy K, van Steenbergen B, and Soto MJ (2017) The NtrY/NtrX system of Sinorhizobium meliloti GR4 regulates motility, EPS I production, and nitrogen metabolism but is dispensable for symbiotic nitrogen fixation. Mol Plant Microbe Interact 30: 566–577. [DOI] [PubMed] [Google Scholar]
- Campbell GR, Sharypova LA, Scheidle H, Jones KM, Niehaus K, Becker A, and Walker GC (2003) Striking complexity of lipopolysaccharide defects in a collection of Sinorhizobium meliloti mutants. J Bacteriol 185: 3853–3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals A, Blanco AG, and Coll M (2012) σ70 and PhoB activator: getting a better grip. Transcription 3: 160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra EJ and Laub MT (2012) Evolution of two-component signal transduction systems. Annu Rev Microbiol 66: 325–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles TC and Nester EW (1993) A chromosomally encoded two-component sensory transduction system is required for virulence of Agrobacterium tumefaciens. J Bacteriol 175: 6614–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EJ, Fisher RF, Perovich VM, Sabio EA, and Long SR (2009) Identification of direct transcriptional target genes of ExoS/ChvI two-component signaling in Sinorhizobium meliloti. J Bacteriol 191: 6833–6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EJ, Sabio EA, and Long SR (2008) The periplasmic regulator ExoR inhibits ExoS/ChvI two-component signalling in Sinorhizobium meliloti. Mol Microbiol 69: 1290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HP and Walker GC (1998) Succinoglycan production by Rhizobium meliloti is regulated through the ExoS-ChvI two-component regulatory system. J Bacteriol 180: 20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronan GE and Keating DH (2004) Sinorhizobium meliloti sulfotransferase that modifies lipopolysaccharide. J Bacteriol 186: 4168–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies BW and Walker GC (2007) Identification of novel Sinorhizobium meliloti mutants compromised for oxidative stress protection and symbiosis. J Bacteriol 189: 2110–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nisco NJ, Abo RP, Wu CM, Penterman J, and Walker GC (2014) Global analysis of cell cycle gene expression of the legume symbiont Sinorhizobium meliloti. Proc Natl Acad Sci U S A 111: 3217–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty D, Leigh JA, Glazebrook J, and Walker GC (1988) Rhizobium meliloti mutants that overproduce the R. meliloti acidic calcofluor-binding exopolysaccharide. J Bacteriol 170: 4249–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields AT, Navarrete CS, Zare AZ, Huang Z, Mostafavi M, Lewis JC, Rezaeihaghighi Y, Brezler BJ, Ray S, Rizzacasa AL et al. (2012) The conserved polarity factor podJ1 impacts multiple cell envelope-associated functions in Sinorhizobium meliloti. Mol Microbiol 84: 892–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finan TM, Kunkel B, De Vos GF, and Signer ER (1986) Second symbiotic megaplasmid in Rhizobium meliloti carrying exopolysaccharide and thiamine synthesis genes. J Bacteriol 167: 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finan TM, Weidner S, Wong K, Buhrmester J, Chain P, Vorhölter FJ, Hernandez-Lucas I, Becker A, Cowie A, Gouzy J et al. (2001) The complete sequence of the 1,683-kb pSymB megaplasmid from the N2-fixing endosymbiont Sinorhizobium meliloti. Proc Natl Acad Sci U S A 98: 9889–9894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foreman DL, Vanderlinde EM, Bay DC, and Yost CK (2010) Characterization of a gene family of outer membrane proteins (ropB) in Rhizobium leguminosarum bv. viciae VF39SM and the role of the sensor kinase ChvG in their regulation. J Bacteriol 192: 975–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes BA, González JE, and Oresnik IJ (2014) Exopolysaccharide production in response to medium acidification is correlated with an increase in competition for nodule occupancy. Mol Plant Microbe Interact 27: 1307–1317. [DOI] [PubMed] [Google Scholar]
- Gibson KE, Barnett MJ, Toman CJ, Long SR, and Walker GC (2007) The symbiosis regulator CbrA modulates a complex regulatory network affecting the flagellar apparatus and cell envelope proteins. J Bacteriol 189: 3591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson KE, Kobayashi H, and Walker GC (2008) Molecular determinants of a symbiotic chronic infection. Annu Rev Genet 42: 413–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics 27: 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffitts JS and Long SR (2008) A symbiotic mutant of Sinorhizobium meliloti reveals a novel genetic pathway involving succinoglycan biosynthetic functions. Mol Microbiol 67: 1292–306. [DOI] [PubMed] [Google Scholar]
- Heavner ME, Qiu WG, and Cheng HP (2015) Phylogenetic co-occurrence of ExoR, ExoS, and ChvI, components of the RSI bacterial invasion switch, suggests a key adaptive mechanism regulating the transition between free-living and host-invading phases in Rhizobiales. PloS one 10: e0135655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellweg C, Pühler A, and Weidner S (2009) The time course of the transcriptomic response of Sinorhizobium meliloti 1021 following a shift to acidic pH. BMC Microbiol 9: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KM (2012) Increased production of the exopolysaccharide succinoglycan enhances Sinorhizobium meliloti 1021 symbiosis with the host plant Medicago truncatula. J Bacteriol 194: 4322–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KM, Sharopova N, Lohar DP, Zhang JQ, VandenBosch KA, and Walker GC (2008) Differential response of the plant Medicago truncatula to its symbiont Sinorhizobium meliloti or an exopolysaccharide-deficient mutant. Proc Natl Acad Sci U S A 105: 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaharada Y, Kelly S, Nielsen MW, Hjuler CT, Gysel K, Muszynski A, Carlson RW, Thygesen MB, Sandal N, Asmussen MH et al. (2015) Receptor-mediated exopolysaccharide perception controls bacterial infection. Nature 523: 308–312. [DOI] [PubMed] [Google Scholar]
- Kawaharada Y, Kiyota H, Eda S, Minamisawa K, and Mitsui H (2010) Identification of the Mesorhizobium loti gene responsible for glycerophosphorylation of periplasmic cyclic β−1,2-glucans. FEMS Microbiol Lett 302: 131–7. [DOI] [PubMed] [Google Scholar]
- Kawaharada Y, Nielsen MW, Kelly S, James EK, Andersen KR, Rasmussen SR, Fuchtbauer W, Madsen LH, Heckmann AB, Radutoiu S et al. (2017) Differential regulation of the Epr3 receptor coordinates membrane-restricted rhizobial colonization of root nodule primordia. Nat comm 8: 14534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol E and Becker A (2004) Global transcriptional analysis of the phosphate starvation response in Sinorhizobium meliloti strains 1021 and 2011. Mol Genet Genomics 272: 1–17. [DOI] [PubMed] [Google Scholar]
- Krol E and Becker A (2014) Rhizobial homologs of the fatty acid transporter FadL facilitate perception of long-chain acyl-homoserine lactone signals. Proc Natl Acad Sci U S A 111: 10702–10707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol E, Blom J, Winnebald J, Berhörster A, Barnett MJ, Goesmann A, Baumbach J, and Becker A (2011) RhizoRegNet-A database of rhizobial transcription factors and regulatory networks. J Biotechnology 155: 127–134. [DOI] [PubMed] [Google Scholar]
- Krol E, Klaner C, Gnau P, Kaever V, Essen LO, and Becker A (2016) Cyclic mononucleotide- and Clr-dependent gene regulation in Sinorhizobium meliloti. Microbiology 162: 1840–1856. [DOI] [PubMed] [Google Scholar]
- Lamontagne J, Butler H, Chaves-Olarte E, Hunter J, Schirm M, Paquet C, Tian M, Kearney P, Hamaidi L, Chelsky D et al. (2007) Extensive cell envelope modulation is associated with virulence in Brucella abortus. J Proteome Res 6: 1519–1529. [DOI] [PubMed] [Google Scholar]
- Laub MT, Chen SL, Shapiro L, and McAdams HH (2002) Genes directly controlled by CtrA, a master regulator of the Caulobacter cell cycle. Proc Natl Acad Sci U S A 99: 4632–4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman AP and Long SR (2013) Exopolysaccharides from Sinorhizobium meliloti can protect against H2O2-dependent damage. J Bacteriol 195: 5362–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CW and Murray JD (2016) The role of flavonoids in nodulation host-range specificity: an update. Plants 5: plants5030033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SR (2016) SnapShot: signaling in symbiosis. Cell 167: 582–582 e1. [DOI] [PubMed] [Google Scholar]
- Lu HY and Cheng HP (2010) Autoregulation of Sinorhizobium meliloti exoR gene expression. Microbiology 156: 2092–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HY, Luo L, Yang MH, and Cheng HP (2012) Sinorhizobium meliloti ExoR is the target of periplasmic proteolysis. J Bacteriol 194: 4029–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroti G and Kondorosi E (2014) Nitrogen-fixing Rhizobium-legume symbiosis: are polyploidy and host peptide-governed symbiont differentiation general principles of endosymbiosis? Front microbiol 5: 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade HM and Signer ER (1977) Genetic mapping of Rhizobium meliloti. Proc Natl Acad Sci USA 74: 2076–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier VM, Muschler P, and Scharf BE (2007) Functional analysis of nine putative chemoreceptor proteins in Sinorhizobium meliloti. J Bacteriol 189: 1816–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Williams M, Loewen PC, and Oresnik IJ (2006) Isolation of salt-sensitive mutants of Sinorhizobium meliloti strain Rm1021. Microbiology 152: 2049–2059. [DOI] [PubMed] [Google Scholar]
- Molle V, Fujita M, Jensen ST, Eichenberger P, González-Pastor JE, Liu JS, and Losick R (2003a) The Spo0A regulon of Bacillus subtilis. Mol Microbiol 50: 1683–1701. [DOI] [PubMed] [Google Scholar]
- Molle V, Nakaura Y, Shivers RP, Yamaguchi H, Losick R, Fujita Y, and Sonenshein AL (2003b) Additional targets of the Bacillus subtilis global regulator CodY identified by chromatin immunoprecipitation and genome-wide transcript analysis. J Bacteriol 185: 1911–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J and González JE (2009) The novel genes emmABC are associated with exopolysaccharide production, motility, stress adaptation, and symbiosis in Sinorhizobium meliloti. J Bacteriol 191: 5890–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oke V and Long SR (1999) Bacterial genes induced within the nodule during the Rhizobium-legume symbiosis. Mol Microbiol 32: 837–849. [DOI] [PubMed] [Google Scholar]
- Østerås M, Stanley J, and Finan TM (1995) Identification of Rhizobium-specific intergenic mosaic elements within an essential two-component regulatory system of Rhizobium species. J Bacteriol 177: 5485–5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellock BJ, Teplitski M, Boinay RP, Bauer WD, and Walker GC (2002) A LuxR homolog controls production of symbiotically active extracellular polysaccharide II by Sinorhizobium meliloti. J Bacteriol 184: 5067–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penterman J, Abo RP, De Nisco NJ, Arnold MF, Longhi R, Zanda M, and Walker GC (2014) Host plant peptides elicit a transcriptional response to control the Sinorhizobium meliloti cell cycle during symbiosis. Proc Natl Acad Sci U S A 111: 3561–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quandt J and Hynes MF (1993) Versatile suicide vectors which allow direct selection for gene replacement in gram-negative bacteria. Gene 127: 15–21. [DOI] [PubMed] [Google Scholar]
- Reed JW and Walker GC (1991a) Acidic conditions permit effective nodulation of alfalfa by invasion-deficient Rhizobium meliloti exoD mutants. Genes Dev 5: 2274–2287. [DOI] [PubMed] [Google Scholar]
- Reed JW and Walker GC (1991b) The exoD gene of Rhizobium meliloti encodes a novel function needed for alfalfa nodule invasion. J Bacteriol 173: 664–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich V (2008) Target DNA Amplification and Labeling via the A/B/C Protocol. https://openwetware.org/wiki/DeLong:Lab_Protocols:Microarray_Protocols
- Rotter C, Mühlbacher S, Salamon D, Schmitt R, and Scharf B (2006) Rem, a new transcriptional activator of motility and chemotaxis in Sinorhizobium meliloti. J Bacteriol 188: 6932–6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos MR, Marques AT, Becker JD, and Moreira LM (2014) The Sinorhizobium meliloti EmrR regulator is required for efficient colonization of Medicago sativa root nodules. Mol Plant Microbe Interact 27: 388–399. [DOI] [PubMed] [Google Scholar]
- Schlüter JP, Reinkensmeier J, Barnett MJ, Lang C, Krol E, Giegerich R, Long SR, and Becker A (2013) Global mapping of transcription start sites and promoter motifs in the symbiotic α-proteobacterium Sinorhizobium meliloti 1021. BMC Genomics 14: 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharypova LA, Chataigne G, Fraysse N, Becker A, and Poinsot V (2006) Overproduction and increased molecular weight account for the symbiotic activity of the rkpZ-modified K polysaccharide from Sinorhizobium meliloti Rm1021. Glycobiology 16: 1181–1193. [DOI] [PubMed] [Google Scholar]
- Sola-Landa A, Pizarro-Cerdá J, Grilló MJ, Moreno E, Moriyón I, Blasco JM, Gorvel JP, and López-Goñi I (1998) A two-component regulatory system playing a critical role in plant pathogens and endosymbionts is present in Brucella abortus and controls cell invasion and virulence. Mol Microbiol 29: 125–138. [DOI] [PubMed] [Google Scholar]
- Swanson JA, Mulligan JT, and Long SR (1993) Regulation of syrM and nodD3 in Rhizobium meliloti. Genetics 134: 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, and Rozen SG (2012) Primer3--new capabilities and interfaces. Nucleic Acids Res 40: e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderlinde EM and Yost CK (2012) Mutation of the sensor kinase chvG in Rhizobium leguminosarum negatively impacts cellular metabolism, outer membrane stability, and symbiosis. J Bacteriol 194: 768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viadas C, Rodrîguez MC, Sangari FJ, Gorvel JP, Garcia-Lobo JM, and López-Goñi I (2010) Transcriptome analysis of the Brucella abortus BvrR/BvrS two-component regulatory system. PloS one 5: e10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriezen JA, de Bruijn FJ, and Nüsslein K (2007) Responses of rhizobia to desiccation in relation to osmotic stress, oxygen, and temperature. Appl Environ Microbiol 73: 3451–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Kemp J, Da Fonseca IO, Equi RC, Sheng X, Charles TC, and Sobral BW (2010) Sinorhizobium meliloti 1021 loss-of-function deletion mutation in chvI and its phenotypic characteristics. Mol Plant Microbe Interact 23: 153–160. [DOI] [PubMed] [Google Scholar]
- Wells DH, Chen EJ, Fisher RF, and Long SR (2007) ExoR is genetically coupled to the ExoS-ChvI two-component system and located in the periplasm of Sinorhizobium meliloti. Mol Microbiol 64: 647–64. [DOI] [PubMed] [Google Scholar]
- Yao SY, Luo L, Har KJ, Becker A, Rüberg S, Yu GQ, Zhu JB, and Cheng HP (2004) Sinorhizobium meliloti ExoR and ExoS proteins regulate both succinoglycan and flagellum production. J Bacteriol 186: 6042–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu NY, Laird MR, Spencer C, and Brinkman FS (2011) PSORTdb--an expanded, auto-updated, user-friendly protein subcellular localization database for Bacteria and Archaea. Nucleic Acids Res 39: D241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan ZC, Liu P, Saenkham P, Kerr K, and Nester EW (2008) Transcriptome profiling and functional analysis of Agrobacterium tumefaciens reveals a general conserved response to acidic conditions (pH 5.5) and a complex acid-mediated signaling involved in Agrobacterium-plant interactions. J Bacteriol 190: 494–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan ZC, Zaheer R, Morton R, and Finan TM (2006) Genome prediction of PhoB regulated promoters in Sinorhizobium meliloti and twelve proteobacteria. Nucleic Acids Res 34: 2686–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Baumann U, and Reymond JL (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res 32: e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.