

ABSTRACT
Tissue cells continually monitor anchorage conditions by gauging the physical properties of their underlying matrix and surrounding environment. The Rho and Ras GTPases are essential components of these mechanosensory pathways. These molecular switches control both cytoskeletal as well as cell fate responses to anchorage conditions and are thus critical to our understanding of how cells respond to their physical environment and, by extension, how malignant cells gainsay these regulatory pathways. Recent studies indicate that 2 proteins produced by the SHC1 gene, thought for the most part to functionally oppose each other, collaborate in their ability to respond to mechanical force by initiating respective Rho and Ras signals. In this review, we focus on the coupling of Shc and GTPases in the cellular response to mechanical anchorage signals, with emphasis on its relevance for cancer.
KEYWORDS: Aiolos, Anoikis, cancer, cellular anchorage, focal adhesion kinase (FAK)GEF-H1, matrix, metastasis, p115-RhoGEF, p52Shc, p66Shc, Ras, RhoA, Shc
Rho GTPases and mechanosensory pathways
Normal cells use a variety of mechanisms by which they sense and respond to changes in physical forces exerted upon them. This mechanosensory response to external force also appears to have applications in the mechanism by which cells gauge the stiffness and mechanical integrity of its underlying matrix. Since matrix beds under static conditions may resist but generally do not initiate changes in force vectors, cells are thought to test their physical environment by initiating tension across their anchorage sites and measuring its resistance. Such bidirectional signaling appears to proceed in large part through integrins, which assemble dynamic mechanosensory complexes within the cell. Probing of the extracellular environment in this fashion leads to changes in cell shape and movement. Migratory cells, for instance, travel in the direction of increasing stiffness, a process called durotaxis. Cell fate itself is profoundly influenced by matrix stiffness and cell shape in a cell-specific fashion. Epithelial and endothelial cells are induced to proliferate on firm beds and become cytostatic or apoptotic on softer beds. Further, mesenchymal stem cells with multilineage potential will differentiate along adipogenic, neurogenic, myogenic, or osteogenic lines in response to increasing stiffness and decreasing shape constraints.1,2 In vivo, a comparable mechanosensory process is required for developmental programs such as gastrulation and ventralization; thus, basic mechanotransduction is essential to integrate physical context into complex cellular decisions.
Tissue cells also sense complete loss of mechanical resistance upon detachment from their matrix beds, which can be viewed as one extreme (essentially infinite compliance) on the continuum of matrix stiffness. Faced with the complete loss of matrix anchorage, normal tissue cells undergo a specific form of programmed cell death termed anoikis.3 While the simple loss of outside-in integrin signals has been thought to trigger anoikis, mechanical signals appear to be required to initiate cell death. Endothelium, epithelium, and fibroblasts forced into suspension are not rescued by saturating amounts of soluble matrix proteins or RGD peptides. Such cells round up and undergo anoikis with integrins fully ligated.3,4 Similarly, fibroblasts contracting against collagen undergo cell death when the matrix is mechanically unloaded, despite full contact with the unstressed collagen. In much the same way, matrix detachment can be viewed as a failure of mechanical loading, much like a tug-of-war game in which one side suddenly releases the rope. Indeed, loss of actinomyosin contraction through inhibition of nonmuscle myosin II, ROCK, or MLCK significantly blunts anoikis.5
The sensing of and response to physical forces requires extensive cytoskeletal remodeling and the generation of internal tension, both of which are largely controlled by the small GTPases of the Rho family.2 Rho GTPases are therefore involved in most if not all such mechanosensory pathways. With respect to the mechanical response to matrix, the Rho GTPases initiate both nuclear and cytoskeletal signals. Adhesion of fibroblasts to fibronectin, for instance, causes RhoA activation through the guanyl exchange factors (GEFs) p115 RhoGEF and LARG, leading to stress fiber and focal adhesion formation.6 Tension applied to matrix-coated microbeads similarly initiates cytoskeletal reinforcement and nuclear translocation of transcription factors through RhoA activation.7 The Rho proteins also control cell polarity complexes, which maintain differentiation or activation states through dynamic interactions with the extracellular matrix.
Morphogenic programs are frequently driven by matrix interactions and involve responses to mechanical forces; thus, it is not surprising that Rho GTPases also control developmental events. The earliest example of epithelial-to-mesenchymal transition (EMT) during vertebrate development occurs during gastrulation, during the formation of the mesoderm. Prior to gastrulation, RhoA is activated at the base of ectodermal cells where they attach to basement membrane. Gastrulation is initiated by loss of ectodermal Net1, a RhoA GEF, which reduces RhoA activity at the base of cells and leads to disruption of epithelial-basement membrane interactions and invasion of Net1-deficient cells fated to become the mesoderm. Likewise, during Drosophila development, the purse-string dorsal closure of lateral epithelium requires fly orthologs of Rac1, Cdc42, and RhoA acting in parallel. Tissue morphogenesis in C. elegans also requires tension, in this case promoting hemidesmosome formation through Rac1, to transduce forces which coordinate epithelial morphogenesis.8
The role of Shc in transducing mechanical signals
The SHC1 gene expresses 2 transcripts that encode 3 proteins differing only in the length of their N-termini (p66Shc, p52Shc, and p46Shc). The order of its core binding domains (PTB, CH1, and SH2) defines the mammalian Shc gene family (in humans, SHC1–4). Some Shc proteins such as p66Shc contain an additional N-terminal CH2 while others lack the central CH1 domain. The Shc genes are evolutionarily ancient, and orthologous genes have been identified in both vertebrates and invertebrates based on this modular binding domain organization.
The p52Shc isoform has been most widely studied and is known to link receptor tyrosine kinases, cytokine and G-protein coupled receptors, and phosphoinositide lipids, with downstream Ras and Rac1 signals. p52Shc becomes tyrosine phosphorylated on the conserved tyrosine residues Y317 as well as Y239/Y240, forming binding sites for Grb2 and leading to recruitment of the Ras GEF SOS. Activation of Ras by the p52Shc-Grb2-SOS complex is required for mitogenic receptor signaling as well as the transforming activity of the polyoma middle T antigen, which recruits p52Shc through its PTB domain.9 In addition, the neurotrophin receptors TrkA and TrkB, which also recruit p52Shc through its PTB domain, are highly expressed in a variety of carcinomas and are known to block anoikis and promote metastatic behavior through Ras. Thus, p52Shc in particular is strongly associated with Ras signaling.
Besides coupling Ras to soluble ligands, the Shc proteins appear to play a role in mediating matrix signals, largely through their association with specific integrins. Mice deleted in all 3 Shc forms die at E11.5 from severe defects in cell-cell and cell-matrix contacts in the cardiovascular system, and Shc-deleted fibroblasts show defects in spreading on fibronectin.10 In endothelial cells, the ability to associate with Shc proteins, more specifically p52Shc, defines a class of integrins capable of activating ERK and Rac1, mediating adhesion-dependent survival.11 Integrins which fail to recruit Shc are weak activators of ERK and do not support adhesion-dependent proliferation. Shc proteins have also been found to participate in mechanotransduction. Application of shear stress to endothelial cells induces the association of all 3 Shc isoforms with β1 and αvβ3 integrins.12 In mice, regions of abnormal shear in the aortic arch cause phosphorylation of Shc and its association with the PECAM-1/VE-Cadherin/VEGFR2 mechanosensory complex.13 Further, conditional cardiomyocyte deletion of all 3 Shc isoforms or knock-in of a mutant p52Shc impairs systolic function despite increased cardiomyocyte contractility, revealing Shc's role in mechanical coupling.14 In addition, data derived from other knock-in mouse models indicate that the conserved, phosphorylated tyrosines within the CH1 domain are required for the formation of muscle spindles, the mechanosensory organs within skeletal muscle that regulate motor behavior.15
Most if not all of the Shc functions outlined above have been ascribed to p52Shc; in contrast, the normal physiologic role for p66Shc has until recently been cryptic, and any functional links to p52Shc were completely unknown. p66Shc is the product of a separate, alternatively spliced mRNA transcribed from the SHC1 gene. Like p52Shc, p66Shc binds to receptor tyrosine kinases such as EGFR and HGF and becomes tyrosine phosphorylated; however, p66Shc does not activate ERK and in fact blocks proliferation, possibly through sequestration of Grb216,17. Consequently, unlike p52Shc, p66Shc is not transforming when overexpressed in NIH-3T3 cells.16
A number of observations have been made using a single strain of p66Shc-deleted mice, which were initially reported to live 30% longer and resist the oxidative stressor paraquat.18 These mice have reduced atheroma lesions upon being fed a high-fat diet, and decreased age-dependent endothelial dysfunction.19,20 Other studies implicate p66Shc in vascular dysfunction and amyloid β-peptide induced neuronal loss,21,22 and demonstrate the ability of p66Shc to generate reactive oxidants and induce mitochondrial apoptosis. While such findings implicate p66Shc in causing oxidative stress and aging, this same strain of p66Shc-deleted mice, when larger numbers of animals were studied in 2 separate animal facilities, showed no differences in longevity at all.23 In addition, these mice were found to have a shortened survival compared with controls when exposed to a more natural living environment, suggesting a decrease rather than increase in their stress resistance.24 A second p66Shc null mouse strain on the same background, which, unlike the first has had the Neo selection cassette removed from the ShcA gene, was reported to have an obese phenotype with increased rather than decreased susceptibility to fatty diets and no apparent health benefits.25,26 While the reasons for the discrepant p66Shc phenotypes are not clear, a conserved role for the SHC1 locus overall in stress response and aging may be supported by independent studies of a distant ortholog in C. elegans. Deletion of this gene, which is predicted to encode a single PTB-SH2 domain-containing protein without CH1/CH2 domains, results in accelerated aging and enhanced sensitivity to heat, oxidative stress, and heavy metal stressors.27
While its link to aging seems unclear, p66Shc appears to have a role in promoting the differentiation of matrix- and tension-sensitive cells. Antisense oligonucleotides specific for p66Shc, for instance, block terminal differentiation of myoblasts into myotubes.28 This process entails changes in mechanical properties of the cells; accordingly, this treatment is accompanied by disruption in actin stress fiber structure and resultant cell rounding, suggesting a marked loss of internal tensional forces. p66Shc knockdown in embryonic mouse lung explants prevents epithelial differentiation and increases proliferation but diminishes branching morphogenesis, another process guided by mechanical factors.29 Likewise, knockdown of p66Shc in bovine zygotes causes a marked reduction in blastocyst development with fewer mitotic arrests, demonstrating a role for p66Shc in promoting differentiation and enforcing cell cycle checkpoints at an early developmental stage.30
Overall, especially given the known association between abnormal matrix integrity and cellular senescence, an overarching role for the different Shc isoforms may be to translate mechanical signals from the matrix to alter cell fate decisions ranging from proliferation to differentiation to aging or death. Were this the case, a clear link to the Rho as well as Ras GTPases might be expected as a plausible mechanism. Is there evidence for this link and for some level of coordination between p66Shc and 52Shc?
Sorting of Ras and Rho signals by p52Shc and p66Shc
The divergence of biologic functions performed by p52Shc and p66Shc may explain the SHC1 gene structure, which contains 2 separately regulated transcriptional start sites. The p52Shc promoter is contained within a 1250 base unmethylated CpG island typical of housekeeping genes, and reflects its ubiquitous expression (unpublished). In contrast, the p66Shc transcriptional start site lies downstream within the first intron, and is activated in a lineage-restricted fashion, being highly expressed in anchorage dependent cell types and only minimally in haematopoietic cells. This expression pattern predicts that p66Shc may play an important role specific to adherent tissue cells. Indeed, p66Shc has been found to mediate anoikis, and thus its expression correlates with anchorage dependence. Metastatic cancer cells and other transformed cell lines resist anoikis following detachment and many do not express p66Shc, whereas ectopic expression of p66Shc restores anoikis in these cells and is attenuated by mutations within the N-terminal CH2 domain unique to p66Shc.5,31 Conversely, primary human bronchial epithelial cells or normal endothelial cells are highly anchorage-dependent, express p66Shc, and display robust anoikis which is completely blocked by selective knockdown of p66Shc.31 Thus p66Shc is both necessary and sufficient to impart anchorage dependence upon normal epithelioid cells.
p66Shc reports anchorage status from focal adhesions, large tension-bearing integrin clusters known to be critical for matrix sensing and mechanotransduction. Total internal fluorescence microscopy confirms targeting of both p66Shc and p52Shc to focal adhesions, and mutational analyses indicate that the phosphotyrosine-binding surface of the Shc PTB domain is responsible for focal adhesion targeting.5 Importantly, p66Shc mutants which do not translocate to focal adhesions also fail to restore anoikis when transfected into 293 cells, confirming focal adhesion targeting as a requisite for adhesion sensing.5
Knockdown of RhoA as well as the use of dominant negative and constitutively active mutants also indicate that p66Shc requires RhoA to mediate anoikis.5,31 These studies are consistent with the demonstration that suppression of the RhoA/ROCK pathway is required for anchorage-independent growth.32 As previously mentioned, the inhibition of actinomyosin-induced tension also blocks anoikis, indicating that the p66Shc-RhoA pathway acts upstream of a tension test of cellular adhesion to firm matrix. Thus p66Shc is critical to implementing a mechanosensory loop that reports anchorage to solid matrix (Fig. 1).
Figure 1.
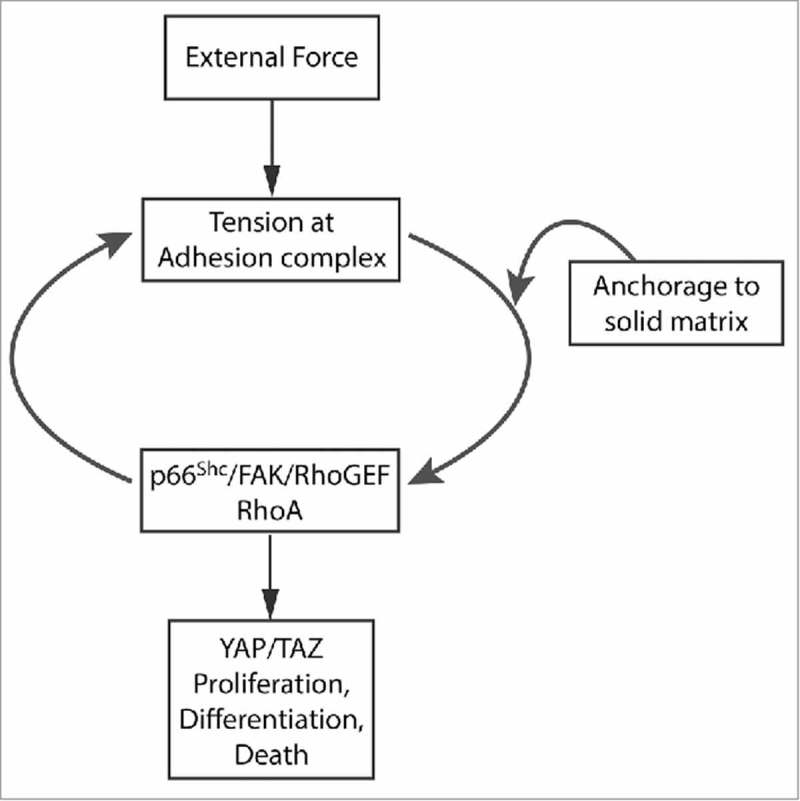
Central role of p66Shc in transducing mechanical signals. Proposed schematic showing a central loop linking the p66Shc/FAK/RhoGEF complex, activation of RhoA, and developed or applied tension at adhesion complexes. The point(s) of entry of matrix testing is unknown, but it appears to feed into the p66Shc/FAK/RhoGEF complex. Disruption of this complex simultaneously impairs mechanosensory pathways involved in matrix stiffness testing, anoikis, and the response to external force.
The molecular complex responsible for processing such mechanical signals has been recently identified by studying the response to tensile forces upon adhesion complexes. Using fibronectin-coated paramagnetic microbeads, both p66Shc and p52Shc were found to translocate to these integrin complexes, especially upon application of magnetic force.33 Proteomic screens have identified focal adhesion kinase (FAK), known to be critical for integrin-dependent mechanotransduction, as the necessary binding partner of both Shc proteins within adhesion complexes.33 These studies further demonstrate that tyrosine phosphorylation of FAK is required for specific binding of the Shc PTB domain, shared by both p66Shc and p52Shc, with the amino-terminal FERM domain of FAK. Of importance, FAK responds to mechanical force by physical separation of this FERM domain from its kinase domain, which is thought to relieve kinase autoinhibition and potentially expose new binding sites on the FERM domain.
The recruitment of the Shc proteins to integrins through FAK is consistent with its ability to orchestrate mitogenic and cytoskeletal responses. In a recent study, the interactome of p52Shc was determined at multiple time points following EGFR activation, revealing 3 sequential waves of p52Shc-associated signaling complexes.34 Within 2 minutes, direct association of p52Shc with EGFR and Grb2 stimulated the recruitment of mitogenic and prosurvival-associated proteins feeding into the Ras/ERK and PI3K pathways. The second cluster, peaking from 2 to 5 minutes, included phosphatases that terminate mitogenic signaling and initiate a switch toward a third cluster of proteins, which reorganize the cytoskeleton. This third cluster of proteins, including SgK269/PEAK1, Asap2, and the RasGAP Dab2ip, control cell shape and movement.
While both p66Shc and not p52Shc translocate to adhesion complexes by targeting FAK, only p66Shc is capable of mediating the RhoA response to mechanical force. p66Shc and FAK are required to recruit the Rho-specific GEFs, p115-RhoGEF and GEF-H1, which become activated upon application of force to the adhesion complex. The p66Shc-FAK complex binds GEF-H1 and p115-RhoGEF, though the exact nature of these interactions is unclear. In contrast, the p52Shc-FAK complex is incapable of assembling such a complex. Thus, p66Shc initiates the RhoA response to tension through FAK-dependent recruitment of Rho-specific GEFs. Of importance, knockdown of GEF-H1 or p115-RhoGEF significantly blunts anoikis, emphasizing the importance of this complex in mediating mechanical anchorage signals.
As might be expected, this p66Shc/FAK/RhoGEF complex is not only important for reporting complete loss of anchorage but is also required for the proper sensing of matrix compliance. Key targets of this mechanosensory pathway are the YAP and TAZ transcription factors, which mediate the proliferative response to stiff matrix beds.35 Notably, YAP and TAZ act downstream of RhoA in this mechanosensory pathway. Accordingly, when cells are plated on polyacrylamide hydrogels of varying stiffness, the p66Shc/FAK/RhoGEF complex which responds to external tension is also required to activate RhoA and transactivate YAP/TAZ as gels stiffen.33 Thus, this same p66Shc/FAK/RhoGEF complex is required to initiate both anoikis following loss of adhesion and proliferation in response to matrix attachment. Since the former is associated with decreases and the latter with increases in YAP/TAZ activity, it seems probable that an unknown intermediate part of the pathway is required to interpret the results of the RhoA-initiated tension test, leading to either inactivation or activation of YAP and TAZ in response to either a compliant or stiff physical environment. In total, these studies suggest that the p66Shc/FAK/RhoGEF complex serves as a general mechanosensory module that mechanistically links pathways which report matrix stiffness, loss of anchorage, and externally applied forces (Fig. 1).
As noted above, p52Shc does not participate in the recruitment or activation of Rho GEFs in response to tension. However, force applied to adhesion complexes simultaneously activates Ras as well as RhoA, and Ras activation requires p52Shc but not p66Shc. p52Shc also forms a complex with FAK, and the p52Shc-FAK complex specifically recruits the Ras GEF SOS1, whereas the p66Shc-FAK complex does not.33 Thus p66Shc and p52Shc each assemble a GTPase activating complex at adhesion sites but bifurcate mechanical signals by differential recruitment of Rho or Ras GEFs in an exclusive fashion (Fig. 2). The importance of having the 2 Shc proteins govern different GTPases is clearly shown in lung epithelial cells which abnormally lose p66Shc expression. Normal bronchial epithelium in which p66Shc is knocked down have excessive Ras and reduced RhoA activity, with consequent loss of anoikis.31 Oppositely, metastatic lung cancer epithelial cells which repress p66Shc have hyperactive wild-type Ras that phenocopies oncogenic mutant Ras. In these malignant cells, reexpression of p66Shc reduces Ras and increases RhoA activity, restoring anchorage dependence.31 This arrangement would suggest that cells may independently titrate the relative Ras and Rho-dependent responses to mechanical force by altering the relative expression of the 2 Shc isoforms (Fig. 3). This careful balance can be expected to control cell-matrix interactions in a lineage-specific fashion in normal cells; thus, disruption of this balance is likely to blur lineage features in cancer.
Figure 2.
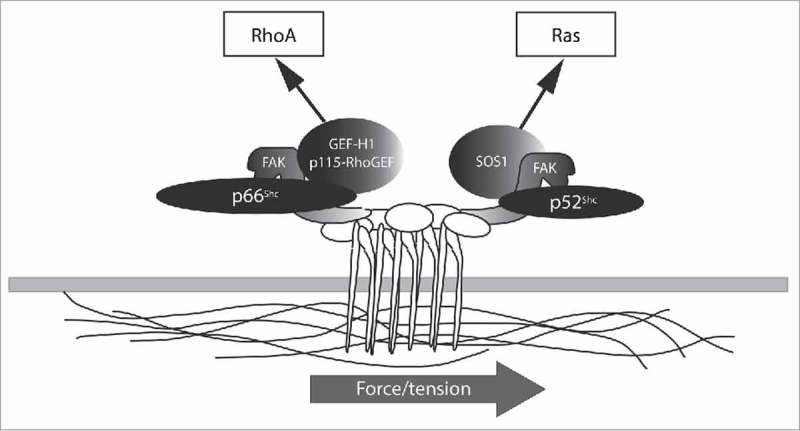
Shc proteins differentially respond to force. Cartoon shows the formation of independent p66Shc/FAK/RhoGEF and p52Shc/FAK/RasGEF complexes. Mechanical force transduction bifurcates into RhoA and Ras pathways through the formation of these distinct Shc complexes.
Figure 3.
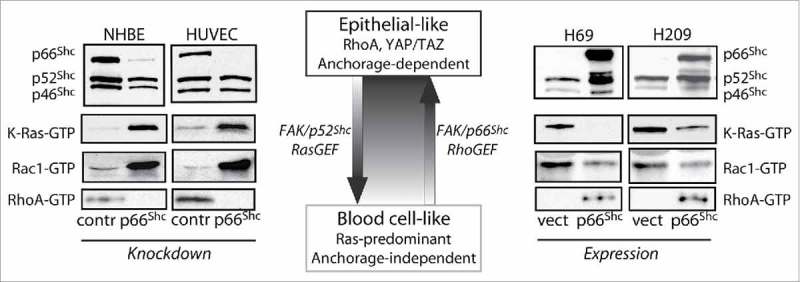
Shc isoforms titrate a GTPase and phenotype switch. The central cartoon summarizes functional studies which demonstrate how p66Shc and p52Shc control an epithelial-like to blood cell-like phenotypic switch that accompanies a Rho-to-Ras GTPase shift. Blots at left demonstrate how knockdown of p66Shc in normal human bronchial epithelium (NHBE) or human umbilical vein endothelium (HUVEC) causes K-Ras and Rac1 hyperactivation and RhoA inactivation, leading to loss of anoikis. In the reverse direction, blots at right show that ectopic expression of p66Shc in metastatic small cell lung cancer cells (H69, H209) reverses K-Ras and Rac1 hyperactivation and restores RhoA activity, reinstating anoikis.
p66Shc and cancer
RhoA and cellular tension have complex effects on malignant progression, with studies demonstrating either tumor suppressing or promoting effects of cellular tension. p66Shc may likewise play divergent roles in cancer development. In primary breast cancers, a high ratio of phosphorylated p52Shc to total p66Shc protein predicts 2-fold higher relapse rates, indicating a repressive role for p66Shc.36 However, overexpression of p66Shc in breast cancer cell lines increases Met-dependent signaling, promoting the expression of mesenchymal genes and a motile phenotype, and p66Shc expression correlates with the expression of EMT markers in primary breast cancers.37 In colon cancers p66Shc expression correlates with worse survival rates.38 The reason for these discrepant results is presently unknown.
In lung cancer, p66Shc has an anti-metastatic effect, consistent with its role enforcing anchorage dependence. In a large panel of lung epithelial cells and cell lines, p66Shc mRNA and protein expression successively decrease from normal bronchial epithelium to non-small cell lung cancer to small cell lung cancer, correlating with increasing metastatic behavior. In human lung primary tumors, low levels of p66Shc staining predict strikingly poor survival outcomes in both early and late stage disease.39 Similarly, in experimental tumors, p66Shc reexpression blocks metastatic behavior, as expected, through loss of RhoA-dependent anoikis.31
In metastatic lung cancer cells, p66Shc repression results from a surprising epigenetic mechanism linked to cell lineage determination, highlighting the connection between SHC1 and cell-type specification. Specifically, p66Shc silencing is triggered by aberrant expression of the lymphocyte-restricted transcription factor Aiolos. Aiolos binds multiple sites on SHC1 including the principal p66Shc enhancer, E2, and causes remodeling of SHC1 chromatin leading to dissociation of E2 from its promoter and p66Shc silencing.39 Interestingly, the normal function of Aiolos and its paralog Ikaros is to initiate chromatin remodeling at a large number of target genes in a process that commits haematopoietic stem cells to the lymphocyte line. Remarkably, deletion of Ikaros from early pre-B cells shifts the morphology of these early lymphoid precursors cells from a typical rounded shape to a flattened epithelioid shape, with an increase in anchorage dependence to bone marrow matrix.40 In a reverse fashion, ectopic expression of Aiolos in A549 epithelial cells causes a broad repression of genes associated with epithelial behavior, particularly in functional gene groups controlling cell-matrix and cell-cell adhesion. Accordingly, Aiolos-expressing lung cancer cells gain certain lymphocyte-like properties such as cell rounding, loss of anchorage dependence and epithelial polarization, and marked increases in metastatic behavior in mice with induction of chemokine and guidance receptors that normally mediate lymphocyte homing to bone marrow, lymph node, lung, and other organs commonly targeted by both immune cells and metastatic tumors.39 Thus Aiolos expression by malignant epithelium appears to initiate epigenetic reprogramming that compromises epithelial lineage integrity and permits lymphocyte mimicry. Repression of p66Shc and the attendant GTPase shift from Rho to Ras-dominated signaling appears to be an important consequence of this reprogramming event (Fig. 3).
Summary
SHC1 is an evolutionarily conserved gene, suggesting that p66Shc and p52Shc are likely to serve a common function. A reasonable shared function may be to transduce environmental signals, both soluble (e.g. growth factors) and physical (e.g., matrix), to the nucleus. Current evidence suggests that the Shc proteins collaborate through the sorting of Ras and Rho GTPase signals, specifically in response to internal or external forces. Dysregulation of Shc, particularly the lineage-restricted p66Shc, can be expected to distort GTPase signals to alter cell fate decisions. In the case of cancer, such dysregulation may result from broader epigenetic reprogramming that leads to the loss of normal interactions with solid matrix, permitting metastasis. Further work in this area will uncover related mechanosensory complexes that can be targeted in cancer therapy.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the American Heart Association, the Cancer Prevention Research Institute of Texas (RP160307), and the NIH (R01-CA208620).
References
- [1].Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell 2006; 126(4):677-89; PMID:16923388; http://dx.doi.org/ 10.1016/j.cell.2006.06.044 [DOI] [PubMed] [Google Scholar]
- [2].McBeath R, Pirone DM, Nelson CM, Bhadriraju K. Chen CS Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell 2004; 6(4):483-95; PMID:15068789; http://dx.doi.org/ 10.1016/S1534-5807(04)00075-9 [DOI] [PubMed] [Google Scholar]
- [3].Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol 1994; 124(4):619-26; PMID:8106557; http://dx.doi.org/ 10.1083/jcb.124.4.619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Re F, Zanetti A, Sironi M, Polentarutti N, Lanfrancone L, Dejana E, Colotta F. Inhibition of anchorage-dependent cell spreading triggers apoptosis in cultured human endothelial cells. J Cell Biol 1994; 127(2):537-46; PMID:7523422; http://dx.doi.org/ 10.1083/jcb.127.2.537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ma Z, Myers DP, Wu RF, Nwariaku FE, Terada LS. p66Shc mediates anoikis through RhoA. J Cell Biol 2007; 179(1):23-31; PMID:17908916; http://dx.doi.org/ 10.1083/jcb.200706097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dubash AD, Wennerberg K, Garcia-Mata R, Menold MM, Arthur WT, Burridge K. A novel role for Lsc/p115 RhoGEF and LARG in regulating RhoA activity downstream of adhesion to fibronectin. J Cell Sci 2007; 120(Pt 22):3989-98; PMID:17971419; http://dx.doi.org/ 10.1242/jcs.003806 [DOI] [PubMed] [Google Scholar]
- [7].Guilluy C, Swaminathan V, Garcia-Mata R, Timothy O'Brien E, Superfine R, Burridge K. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat Cell Biol 2011; 13(6):724-9; http://dx.doi.org/ 10.1038/ncb2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang H, Landmann F, Zahreddine H, Rodriguez D, Koch M, Labouesse M. A tension-induced mechanotransduction pathway promotes epithelial morphogenesis. Nature 2011; 471(7336):99-103; PMID:21368832; http://dx.doi.org/ 10.1038/nature09765 [DOI] [PubMed] [Google Scholar]
- [9].Ong SH, Dilworth S, Hauck-Schmalenberger I, Pawson T, Kiefer F. ShcA and Grb2 mediate polyoma middle T antigen-induced endothelial transformation and Gab1 tyrosine phosphorylation. EMBO J 2001; 20(22):6327-36; PMID:11707404; http://dx.doi.org/ 10.1093/emboj/20.22.6327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lai KM, Pawson T. The ShcA phosphotyrosine docking protein sensitizes cardiovascular signaling in the mouse embryo. Genes Dev 2000; 14(9):1132-45; PMID:10809671 [PMC free article] [PubMed] [Google Scholar]
- [11].Wary KK, Mainiero F, Isakoff SJ, Marcantonio EE, Giancotti FG. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell 1996; 87(4):733-43; PMID:8929541; http://dx.doi.org/ 10.1016/S0092-8674(00)81392-6 [DOI] [PubMed] [Google Scholar]
- [12].Chen KD, Li YS, Kim M, Li S, Yuan S, Chien S, Shyy JY. Mechanotransduction in response to shear stress. Roles of receptor tyrosine kinases, integrins, and Shc. Journal of Biological Chemistry 1999; 274(26):18393-400. [DOI] [PubMed] [Google Scholar]
- [13].Liu Y, Sweet DT, Irani-Tehrani M, Maeda N, Tzima E. Shc coordinates signals from intercellular junctions and integrins to regulate flow-induced inflammation. J Cell Biol 2008; 182(1):185-96; PMID:18606845; http://dx.doi.org/ 10.1083/jcb.200709176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vanderlaan RD, Hardy WR, Kabir MG, Pasculescu A, Jones N, deTombe PP, Backx PH, Pawson T. The ShcA phosphotyrosine docking protein uses distinct mechanisms to regulate myocyte and global heart function. Circ Res 2011; 108(2):184-93; PMID:21148430; http://dx.doi.org/ 10.1161/CIRCRESAHA.110.233924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hardy WR, Li L, Wang Z, Sedy J, Fawcett J, Frank E, Kucera J, Pawson T. Combinatorial ShcA docking interactions support diversity in tissue morphogenesis. Science 2007; 317(5835):251-6; PMID:17626887; http://dx.doi.org/ 10.1126/science.1140114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Migliaccio E, Mele S, Salcini AE, Pelicci G, Lai KM, Superti-Furga G, Pawson T, Di Fiore PP, Lanfrancone L, Pelicci PG. Opposite effects of the p52shc/p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase-fos signalling pathway. EMBO J 1997; 16(4):706-16; PMID:9049300; http://dx.doi.org/ 10.1093/emboj/16.4.706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Okada S, Kao AW, Ceresa BP, Blaikie P, Margolis B, Pessin JE. The 66-kDa Shc isoform is a negative regulator of the epidermal growth factor-stimulated mitogen-activated protein kinase pathway. J Biol Chem 1997; 272(44):28042-9; PMID:9346957; http://dx.doi.org/ 10.1074/jbc.272.44.28042 [DOI] [PubMed] [Google Scholar]
- [18].Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 1999; 402(6759):309-13; PMID:10580504; http://dx.doi.org/ 10.1038/46311 [DOI] [PubMed] [Google Scholar]
- [19].Napoli C, Martin-Padura I, de Nigris F, Giorgio M, Mansueto G, Somma P, Condorelli M, Sica G, De Rosa G, Pelicci P. Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc Natl Acad Sci U S A 2003; 100(4):2112-6; PMID:12571362; http://dx.doi.org/ 10.1073/pnas.0336359100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Francia P, delli Gatti C, Bachschmid M, Martin-Padura I, Savoia C, Migliaccio E, Pelicci PG, Schiavoni M, Luscher TF, Volpe M, Cosentino F. Deletion of p66shc gene protects against age-related endothelial dysfunction. Circulation 2004; 110(18):2889-95; PMID:15505103; http://dx.doi.org/ 10.1161/01.CIR.0000147731.24444.4D [DOI] [PubMed] [Google Scholar]
- [21].Smith WW, Norton DD, Gorospe M, Jiang H, Nemoto S, Holbrook NJ, Finkel T, Kusiak JW. Phosphorylation of p66Shc and forkhead proteins mediates Abeta toxicity. J Cell Biol 2005; 169(2):331-9; PMID:15837797; http://dx.doi.org/ 10.1083/jcb.200410041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kim CS, Jung SB, Naqvi A, Hoffman TA, DeRicco J, Yamamori T, Cole MP, Jeon BH, Irani K. p53 impairs endothelium-dependent vasomotor function through transcriptional upregulation of p66shc. Circ Res 2008; 103(12):1441-50; PMID:18988897; http://dx.doi.org/ 10.1161/CIRCRESAHA.108.181644 [DOI] [PubMed] [Google Scholar]
- [23].Ramsey JJ, Tran D, Giorgio M, Griffey SM, Koehne A, Laing ST, Taylor SL, Kim K, Cortopassi GA, Lloyd KC, et al.. The influence of Shc proteins on life span in mice. J Gerontol A Biol Sci Med Sci 2014; 69(10):1177-85; PMID:24336818; http://dx.doi.org/ 10.1093/gerona/glt198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Giorgio M, Berry A, Berniakovich I, Poletaeva I, Trinei M, Stendardo M, Hagopian K, Ramsey JJ, Cortopassi G, Migliaccio E, et al.. The p66Shc knocked out mice are short lived under natural condition. Aging Cell 2012; 11(1):162-8; PMID:22081964; http://dx.doi.org/ 10.1111/j.1474-9726.2011.00770.x [DOI] [PubMed] [Google Scholar]
- [25].Tomilov AA, Ramsey JJ, Hagopian K, Giorgio M, Kim KM, Lam A, Migliaccio E, Lloyd KC, Berniakovich I, Prolla TA, et al.. The Shc locus regulates insulin signaling and adiposity in mammals. Aging Cell 2011; 10(1):55-65; PMID:21040401; http://dx.doi.org/ 10.1111/j.1474-9726.2010.00641.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tomilov A, Bettaieb A, Kim K, Sahdeo S, Tomilova N, Lam A, Hagopian K, Connell M, Fong J, Rowland D, et al.. Shc depletion stimulates brown fat activity in vivo and in vitro. Aging Cell 2014; 13(6):1049-58; PMID:25257068; http://dx.doi.org/ 10.1111/acel.12267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Neumann-Haefelin E, Qi W, Finkbeiner E, Walz G, Baumeister R, Hertweck M. SHC-1/p52Shc targets the insulin/IGF-1 and JNK signaling pathways to modulate life span and stress response in C. elegans. Genes Dev 2008; 22(19):2721-35; http://dx.doi.org/ 10.1101/gad.478408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Natalicchio A, Laviola L, De Tullio C, Renna LA, Montrone C, Perrini S, Valenti G, Procino G, Svelto M, Giorgino F. Role of the p66Shc isoform in insulin-like growth factor I receptor signaling through MEK/Erk and regulation of actin cytoskeleton in rat myoblasts. J Biol Chem 2004; 279(42):43900-9; PMID:15262993; http://dx.doi.org/ 10.1074/jbc.M403936200 [DOI] [PubMed] [Google Scholar]
- [29].Lee MK, Smith SM, Banerjee MM, Li C, Minoo P, Volpe MV, Nielsen HC. The p66Shc adapter protein regulates the morphogenesis and epithelial maturation of fetal mouse lungs. Am J Physiol Lung Cell Mol Physiol 2014; 306(4):L316-25; PMID:24375794; http://dx.doi.org/ 10.1152/ajplung.00062.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Favetta LA, Madan P, Mastromonaco GF, St John EJ, King WA, Betts DH. The oxidative stress adaptor p66Shc is required for permanent embryo arrest in vitro. BMC Dev Biol 2007; 7:132; PMID:18047664; http://dx.doi.org/ 10.1186/1471-213X-7-132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ma Z, Liu Z, Wu RF, Terada LS. p66Shc restrains Ras hyperactivation and suppresses metastatic behavior. Oncogene 2010; 29:5559-5567; PMID:20676142; http://dx.doi.org/ 10.1038/onc.2010.326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dohn MR, Brown MV, Reynolds AB. An essential role for p120-catenin in Src- and Rac1-mediated anchorage-independent cell growth. J Cell Biol 2009; 184(3):437-50; PMID:19188496; http://dx.doi.org/ 10.1083/jcb.200807096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wu RF, Liao C, Fu G, Hayenga HN, Yang K, Ma Z, Liu Z, Terada LS. p66Shc couples mechanical signals to RhoA through FAK-dependent recruitment of p115-RhoGEF and GEF-H1. Mol Cell Biol 2016; 36:2824-2837; http://dx.doi.org/ 10.1128/MCB.00194-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zheng Y, Zhang C, Croucher DR, Soliman MA, St-Denis N, Pasculescu A, Taylor L, Tate SA, Hardy WR, Colwill K, et al.. Temporal regulation of EGF signalling networks by the scaffold protein Shc1. Nature 2013; 499(7457):166-71; PMID:23846654; http://dx.doi.org/ 10.1038/nature12308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al.. Role of YAP/TAZ in mechanotransduction. Nature 2011; 474(7350):179-83; PMID:21654799; http://dx.doi.org/ 10.1038/nature10137 [DOI] [PubMed] [Google Scholar]
- [36].Davol PA, Bagdasaryan R, Elfenbein GJ, Maizel AL, Frackelton AR Jr.. Shc proteins are strong, independent prognostic markers for both node-negative and node-positive primary breast cancer. Cancer Res 2003; 63(20):6772-83. [PubMed] [Google Scholar]
- [37].Hudson J, Ha JR, Sabourin V, Ahn R, La Selva R, Livingstone J, Podmore L, Knight J, Forrest L, Beauchemin N, et al.. p66ShcA promotes breast cancer plasticity by inducing an epithelial-to-mesenchymal transition. Mol Cell Biol 2014; 34(19):3689-701; PMID:25071152; http://dx.doi.org/ 10.1128/MCB.00341-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Grossman SR, Lyle S, Resnick MB, Sabo E, Lis RT, Rosinha E, Liu Q, Hsieh CC, Bhat G, Frackelton AR Jr., Hafer LJ. p66 Shc tumor levels show a strong prognostic correlation with disease outcome in stage IIA colon cancer. Clin Cancer Res 2007; 13(19):5798-804. [DOI] [PubMed] [Google Scholar]
- [39].Li X, Xu Z, Du W, Zhang Z, Wei Y, Wang H, Zhu Z, Qin L, Wang L, Niu Q, et al.. Aiolos Promotes Anchorage Independence by Silencing p66(Shc) Transcription in Cancer Cells. Cancer Cell 2014; 25(5):575-89; PMID:24823637; http://dx.doi.org/ 10.1016/j.ccr.2014.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Joshi I, Yoshida T, Jena N, Qi X, Zhang J, Van Etten RA, Georgopoulos K. Loss of Ikaros DNA-binding function confers integrin-dependent survival on pre-B cells and progression to acute lymphoblastic leukemia. Nat Immunol 2014; 15(3):294-304; PMID:24509510; http://dx.doi.org/ 10.1038/ni.2821 [DOI] [PMC free article] [PubMed] [Google Scholar]