

ABSTRACT
Peripheral T-cell lymphomas (PTCLs) are rare, heterogeneous tumors with poor response to standard therapy and few targeted treatments available. The identification of mutations in the T-cell receptor (TCR) signaling pathway that either directly or indirectly affect Ras- and Rho-family GTPases is an emerging theme across PTCL subtypes. This review summarizes the role of GTPases in TCR signaling and highlights the constellation of mutations in this pathway among PTCLs. In particular, focus is given to the functional impact of the mutations and opportunities for targeted therapy. These mutations include activating mutations and gene fusions involving the guanine nucleotide exchange factor, VAV1, as well as activating and dominant negative mutations in the GTPases KRAS and RHOA, respectively. In addition to mutations directly affecting the GTPase pathway, TCR signaling mutations indirectly affecting Ras- and Rho-family GTPases involving genes such as CD28, FYN, LCK, and PLCG1 are also reviewed.
KEYWORDS: fusion protein, GTPase, mutation, peripheral T-cell lymphoma, RAC1, RHOA, T cell signaling, VAV1
Introduction
Peripheral T-cell lymphomas (PTCLs) are malignancies of mature T cells encompassing a heterogeneous group of over 20 lymphoma subtypes.1 The frequency of individual subtypes varies widely by geographic region.2,3 In Western countries, the most common subtype is PTCL, not otherwise specified (PTCL, NOS), a highly diverse group of tumors lacking sufficient criteria to be classified into one of the other, more specific entities. Angioimmunoblastic T-cell lymphoma (AITL) and anaplastic large cell lymphoma (ALCL) are other common subtypes, each with unique pathological, molecular, and clinical features. In contrast, adult T-cell leukemia/lymphoma (ATL) and NK/T-cell lymphoma subtypes are more common in Asian countries. Overall, the response to standard chemotherapy is quite poor, and few targeted therapeutic options have been identified for PTCL.
T-cell receptor (TCR) signaling plays a critical physiologic role in T-cell function and its dysregulation has been proposed to be a major factor in PTCL pathogenesis.4,5 Of note, expression of TCR-associated proteins varies among PTCL subtypes.6,7 Recent sequencing of PTCLs has revealed somatic mutations and genomic rearrangements in the TCR signaling pathway, bearing direct or indirect effects on GTPase signaling. Herein, we will summarize the role of GTPases in TCR signaling in normal T cells and detail the functional implications reported to date for genetic alterations occurring in this pathway.
GTPases in T-cell receptor signaling
Signal transduction through GTP-binding proteins is coordinated to regulate multiple aspects of lymphocyte development and action. In particular, the small GTPases, such as those of the Ras and Rho families, play prominent roles in T-cell development, selection, activation, and migration (as reviewed in references 8–10). Small GTPases are 21 kDa “molecular switches” that bind and hydrolyze guanosine triphosphate (GTP). Multiple stimuli promote the loading of GTP onto this family of proteins (Fig. 1). When in the GTP-bound form, GTPases are in the active state and are capable of binding downstream effector proteins. These GTPases have weak intrinsic hydrolase activity, and thus rely on associated GTPase activating proteins (GAPs) to catalyze the hydrolysis of GTP to guanosine diphosphate (GDP). This conversion changes the conformation, thereby inactivating the GTPase and preventing binding to effectors. Conversely, GTPases are activated by guanosine nucleotide exchange factors (GEFs), which facilitate the release of GDP from the GTPase and allow the binding of GTP. This action converts the protein to the GTP-loaded, active conformation, primed to bind effectors and propagate the signal to downstream factors.
Figure 1.
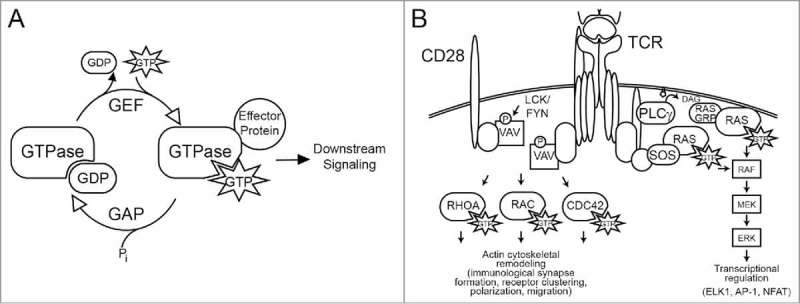
(A) Activation cycle of small GTPases. Ras and Rho family GTPases are activated at the cell membrane by GEFs, which catalyze the release of GDP and promote the loading of GTP, thereby inducing binding to effector proteins. GTPase activating proteins (GAPs) facilitate the hydrolysis of GTP to GDP, thereby inactivating the GTPase. Membrane targeting of GTPases is also regulated by guanine nucleotide dissociation inhibitors (GDIs, not shown). (B) Role of GTPases in T-cell receptor (TCR) signaling. Activation of the TCR stimulates activation of Ras through multiple mechanisms, including PLCγ-mediated generation of DAG, which recruits the Ras guanosine nucleotide exchange factor (GEF) GRP and through recruitment of another Ras GEF, SOS. Rho family GTPases including RAC are activated by multiple GEFs, including VAV1, which is activated by tyrosine phosphorylation (as reviewed in references, 8, 10, 52 and others).
Ras GTPases
The Ras family includes some of the best characterized GTPases, and the corresponding genes are among the most frequently mutated genes in multiple human tumor types. RAS is activated at the cell membrane by diverse stimuli, including activation of both the pre-TCR and mature TCR in T cells. Ligation of the TCR induces the formation of a proximal signaling complex, including phospholipase Cγ (PLCγ; encoded by PLCG1) and the adaptor growth factor receptor-bound protein 2 (GRB2), which are both required for activation of RAS. PLCγ hydrolyzes the lipid species phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] to the second messengers diacylglycerol (DAG) and inositol 3-phosphate (IP3). In T cells, the GEF RASGRP1 binds to DAG, while the GEF Son of Sevenless (SOS) binds GRB2, thereby coupling RAS activation to TCR signaling. RAS activates multiple potent downstream signaling cascades, including the phosphatidylinositol-3-kinase (PI3K) pathway and mitogen-activated protein (MAP) kinase signaling through RAF, MEK, and ERK MAP; these signaling pathways are critical regulators of cell survival and proliferation.11 The DAG produced by PLCγ also activates protein kinase C resulting in CARD11 phosphorylation and subsequent activation of the CARD11-BCL10-MALT1 complex, a key mediator of NF-κB activation. In addition, RAS activates the GTPase RAC12 and cooperates with IP3-regulated calcium signaling to regulate NFAT (nuclear factor of activated T cells) transcription factors.13,14 RAS is also required in T-cell development for positive and negative selection.15
Rho GTPases
The Rho family of GTPases is comprised of 22 members. This review will focus specifically on RAC1, CDC42, and RHOA, which are best characterized as regulators of actin cytoskeletal remodeling. TCR ligation or engagement of the costimulatory protein CD28 is followed by a burst of actin reorganization to induce TCR clustering and formation of the immunological synapse, thereby stabilizing the interaction between T cells and antigen-presenting cells. Active, GTP-bound Rho GTPases bind to specific effector proteins. As many as 70 effector proteins for RHOA, RAC1, or CDC42 have been described, including various proteins involved in actin cytoskeletal organization such as p21-activated kinase (PAK) and WASp/WAVE family proteins.9 These GTPases also regulate cell migration, adhesion, trafficking, and transcription, thereby regulating T-cell differentiation, proliferation, and activation.
A primary activator of RAC in T cells is the haematopoietic -specific GEF VAV1. VAV1 integrates signals from tyrosine kinases to activate RAC, and potently regulates lymphocyte development and activation. Genetic knockout of VAV1 results in dramatic defects in TCR signaling and clustering, reduced calcium flux, and impaired NFAT transcriptional activity.16-19 VAV1 is phosphorylated by Src family kinases, including LCK, which is activated following TCR ligation.20,21 Phosphorylation of critical tyrosine residues near the N-terminus of VAV1 induces a conformational change that relieves autoinhibition and exposes the Dbl homology (DH) domain, which is responsible for GEF function, primarily toward RAC.22-24 In addition, VAV1 also demonstrates GEF-independent activities, likely due to the adaptor function of its Src homology- (SH) 2 and SH3 domains.25,26 Interestingly, VAV1 has been found to be aberrantly expressed in multiple human tumor types, and deletion of key regulatory domains converts VAV1 to an oncogene.27-29 Thus, while critical for lymphocyte function, VAV1 is also a proto-oncogene implicated in human cancers.
Somatic alterations in GTPase-related genes in in peripheral T-cell lymphomas
VAV1
Through an integrated mate-pair DNA sequencing and RNA sequencing approach for fusion detection, we recently discovered recurrent VAV1 fusion genes in PTCL.30 Fluorescence in situ hybridization (FISH) studies demonstrated that chromosomal rearrangements of the VAV1 locus were present with a frequency of 11% in PTCL, NOS and ALCL, but absent in other subtypes (Fig. 2). Both of the VAV1 fusions studied by next generation-sequencing showed identical fusion breakpoints at residue 777, just proximal to the C-terminal SH3 domain and resulting in complete loss of this domain in the fusion protein. The C-terminal SH3 domain is responsible for autoinhibition of VAV1 activation, and deletion of this domain results in constitutive activation.31 Consistent with loss of this autoinhibitory function, we demonstrated in vitro that the VAV1 fusion promoted cell growth and migration. We further showed that RAC1 was the major GTPase activated by the VAV1 fusion protein, and that the resultant cell growth was RAC1-dependent. Importantly, we also showed that the VAV1 fusion was targetable with the clinically available RAC1 inhibitor azathioprine.
Figure 2.
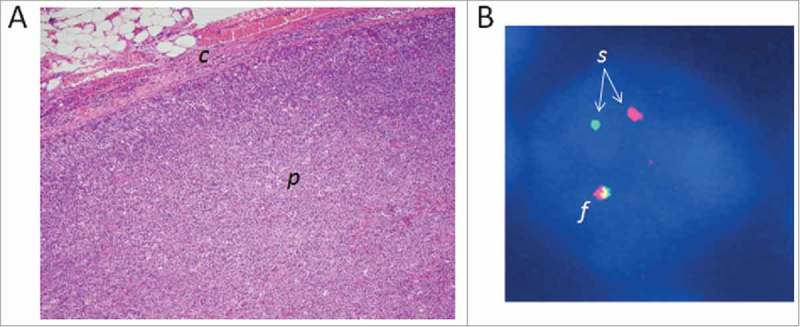
Three-centimeter lymph node from a 55 year-old male with peripheral T-cell lymphoma, not otherwise specified, bearing a chromosomal rearrangement of the GEF gene VAV1. VAV1 rearrangements are seen recurrently in PTCL (see text).30 (A) Photomicrograph of the lymph node biopsy (hematoxylin and eosin stain; original magnification, 10×). The architecture of the lymph node parenchyma (p) has been effaced by the infiltrating lymphoma cells. c, lymph node capsule. (B) Fluorescence in situ hybridization (FISH) image of a single lymphoma cell nucleus (stained blue). The DNA has been hybridized with red and green fluorescent probes flanking the VAV1 locus. One VAV1 allele shows a normal red-green fusion signal (f), whereas the red and green signals of the other allele are split (s), indicating a VAV1 rearrangement.
In further support of an oncogenic role for VAV1 in PTCLs, somatic mutations in the VAV1 gene have also been identified in 18% of ATLs and in occasional cases of AITL and other PTCLs derived from T follicular helper (TFH) cells.5,32 Similar to the role of the breakpoint in VAV1 fusions, most VAV1 mutations in ATL and AITL/TFH-like PTCLs affected the C-terminal SH3 domain, while additional mutation clusters in acidic, pleckstrin homology (PH), and zinc-finger protein domains were also identified in ATL.32 One of the residues in the SH3 domain found to be mutated in both studies, D797, was previously demonstrated to have a role in oncogenic transformation.29 In addition, a mutation occurring in the acidic domain Y174C was predicted to disrupt autoinhibitory interaction with the Dbl homology (DH) domain, and this residue has been shown to negatively regulate VAV1 activity.33 Although functional characterization of these specific mutations in the context of PTCL has not been reported, the clustering of mutations in specific regions of VAV1, in particular the C-terminal SH3 domain, suggest a role for VAV1 activation in T-cell malignancies. To this end, RAC inhibition should be evaluated as a potential targeted therapy approach for PTCLs with activating mutations or structural alterations of VAV1. In addition, the potential effect of these genetic alterations on the non-GEF activity of VAV1, as well as the role of mutations or altered function of other VAV family members,5,34 merits further study.
RHOA
A highly recurrent mutation in the GTP/GDP binding domain of RHOA, G17V, was identified by several groups to be present in both AITL (50% to 71% of cases) and PTCL, NOS (8–18% of cases, particularly those with AITL-like features).35-37 In vitro experiments revealed a dominant negative role for the G17V mutation whereby mutant RHOA prevents binding of GTP by both mutant and wild type RHOA, presumably by sequestering RHOA GEFs.35-37 This dominant negative function was further supported by data demonstrating inhibition of serum response factor-responsive element (SRF-RE) reporter activity and decreased actin stress fiber formation.35,36 In a T-cell model, overexpression of wild type but not mutant RHOA resulted in growth suppression, although mutant RHOA itself did not promote growth over mock-transduced control cells.35 Interestingly, gene set enrichment analysis of PTCLs with the G17V mutation compared to those with wild type RHOA revealed enrichment of the RAC1 pathway as well as T-cell cytokine signaling, NOTCH, and NF-κB pathways.38 On the basis of the reciprocal relationship between RAC1 and RHOA,39 it would be of interest to evaluate RAC1 activation in the RHOA G17V model for potential therapeutic targeting strategies via RAC1 inhibition. In addition, understanding mechanisms for cooperativity between RHOA G17V and other mutations with which it is associated, including those involving IDH2, DNMT3A, and particularly TET2, may elucidate additional potential therapeutic strategies.40
Other RHOA mutations, albeit far less frequent than G17V, have been reported in PTCL, affecting amino acids 16–19 (C16R, G17E, T19I, and K18N) of the GTP/GDP binding domain.5,36 Unlike G17V, the K18N mutation appears to activate rather than inhibit RHOA; hence, its specific role in PTCL tumorigenesis is unclear.
PLCG1/KRAS
In addition to mutations directly or indirectly affecting Rho family GTPases, genetic aberrations have also been reported in genes involved in Ras family GTPase signaling, including PLCG1 and KRAS. Of these, PLCG1 is the most commonly involved and is recurrently mutated in ATL, cutaneous T-cell lymphoma (CTCL), AITL, and PTCL, NOS.5,32,41,42 Mutations in PLCG1 affect regions that encode several protein domains and have been shown to promote MALT1 cleavage and NFAT activity to varying degrees.5,41
The GTPase KRAS is highly mutated across human cancers, most commonly in carcinomas of the pancreas, lung, and colon. Recently, KRAS mutations also were reported in PTCLs.5,43 Most of the KRAS mutations identified in PTCL overlap with previously characterized mutation hotspots, including amino acids 12 and 13 as well as the less frequently mutated amino acids 18 and 146. Although the function of these mutations has not been reported in PTCL, these mutations are well characterized in other tumor types. Primarily, mutations in Ras family genes lead to Ras activation, subsequent activation of targets such as ERK, and promotion of cell growth, invasion, and survival, and represent a promising candidate for targeted therapies.44,45
CD28, LCK, and FYN
Mutations upstream in the TCR pathway may also dramatically impact GTPase activity. The Src-family kinases LCK and FYN play a critical role in the early steps of TCR signaling including phosphorylation of the TCRς chain, leading to recruitment and activation of ZAP-70 and subsequent phosphorylation of adaptor molecules such as LAT, which in turn facilitate downstream kinase signaling and T-cell activation.46 LCK and FYN also are required for CD28 engagement and subsequent VAV1 phosphorylation (Fig. 1).47 Recurrent somatic mutations in CD28 have been reported in AITL (9–11%), ATL, and, less frequently, in PTCL, NOS.5,32,48,49 In vitro study of 2 commonly mutated residues, D124 and T195, revealed activation of TCR signaling.48,49 The D124V mutation, located near the ligand binding site of CD28, conferred greater affinity for its CD86 ligand.48 Likewise, the T195P mutation, situated between the SH2 and SH3 domains, exhibited higher affinity to the adaptor proteins GRB2 and GRAP2.48,49 NF-κB activity also was enhanced by these CD28 mutants, suggesting increased GTPase activity, although GTPases were not measured directly.48,49 CD28 gene fusions, namely CTLA4-CD28 and ICOS-CD28, have been reported across numerous PTCL subtypes and at greater frequency than CD28 mutations.32,48,50,51 Focal amplifications of CD28 also have been reported in PTCLs.32,51 However, further study is needed to clearly define the functional role of these fusions and their possible effect on GTPase signaling.
Several mutations have been identified in FYN, primarily affecting its SH2, SH3, and C-terminal domains in PTCL, NOS, AITL, and ATL, with frequencies of about 3–4%.5,32,36 In a manner similar to that observed for VAV1 and RHOA mutations, mutations in FYN are predicted to interfere with an inhibitory interaction between the FYN SH2 domain and the C-terminal Src kinase (CSK)-phosphorylated Y531 residue.36 In vitro studies of FYN mutations identified by Palomero et al revealed that these mutations indeed activated FYN through loss of the interaction between the SH2 domain and phosphorylated Y531.36 Furthermore, mutant FYN activation was targetable with the kinase inhibitor dasatinib. However, the functional implications of these FYN mutations and specifically their effect on GTPase activity, GTPase-dependent signaling, and downstream functions such as actin structure, migration, and cell growth have not been reported. LCK mutations in the tyrosine kinase domain (N446K, P447R) were recently reported in a single AITL patient, but although predicted to be activating, their recurrence and function have not yet been determined.5
Summary
With the recent progress in mutational profiling of PTCLs, somatic mutations and gene fusions have been reported in several GTPase-related genes, including VAV1, RHOA, KRAS, PLCG1, CD28, FYN, and LCK (Table 1). In addition to the TCR-related genes upstream of GTPases (VAV1, CD28, FYN, LCK, PLCG1) and GTPases themselves (RHOA and KRAS), activating mutations have been reported in downstream TCR signaling genes such as CARD11.5,32 Among TCR-related genes, PLCG1 is the most frequently mutated in PTCL, and PLCG1 mutations often co-occur with one of the other TCR-related mutations.5,32 In contrast, mutations in non-PLCG1 TCR-related genes do not often occur together in the same case.
Table 1.
Summary of mutations and fusion genes involving GTPase and TCR–related genes in peripheral T-cell lymphomas.
| Gene | Mutation | Affected domain/Functional consequence | References |
|---|---|---|---|
| VAV1 | SNV | Activating | 5, 32 |
| Fusion | Truncation of SH3 domain in VAV1-GSS and VAV1-MYO1F fusions; increased RAC activation | 30 | |
| RHOA | SNV | G17V in GTP binding domain; dominant negative function | 35–37 |
| FYN | SNV | SH2 domain; activating through loss of inhibitory SH2 domain and Y531 interaction | 5, 32, 36 |
| CD28 | SNV | D124, T195; activation of NF-κB and TCR signaling | 32, 48, 49 |
| Fusion | CTLA4-CD28 and ICOS-CD28 | 5, 32, 48, 50 | |
| LCK | SNV | Predicted activating | 5 |
| PLCG1 | SNV | MALT1 and NFAT activation | 5, 32, 41 |
| KRAS | SNV | Activating | 5, 43 |
SNV, single nucleotide variant; TCR, T-cell receptor.
With the exception of RHOA G17V, most of these genomic events either have been demonstrated to be activating in vitro or are predicted to be activating. Because RHOA opposes RAC1 activity, the dominant negative RHOA G17V mutation may have some similarities to VAV1 rearrangements; in fact, ectopic expression of both VAV1 fusions and RHOA G17V cause cytoskeletal rearrangement. However, the effect of RHOA G17V on RAC1 has not been reported. Likewise, KRAS has been shown to promote RAC1 activation, although KRAS mutations remain to be studied in T cell models. Hence, GTPases, especially RAC1, may become increasingly recognized for a role in PTCL biology including cell growth, cytoskeletal remodeling, and cell migration. Targeted therapeutic strategies such as RAC inhibition (azathioprine) and tyrosine kinase inhibition (dasatinib) have been shown to target VAV1 fusions and FYN mutations, respectively. It will be important moving forward to evaluate the specific role of other GTPase-related mutations on GTPase signaling and to test these and related drugs for efficacy against these candidate therapeutic targets.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Supported by Award Number R01 CA177734 (ALF), from the National Cancer Institute and by the Fraternal Order of Eagles Cancer Research Fund, Mayo Clinic Cancer Center (RLB, GLR).
References
- [1].Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, et al.. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016; 127:2375-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Armitage JO. The aggressive peripheral T-cell lymphomas: 2015. Am J Hematol 2015; 90:665-73; PMID:26031230; http://dx.doi.org/ 10.1002/ajh.24076 [DOI] [PubMed] [Google Scholar]
- [3].Vose J, Armitage J, Weisenburger D. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol 2008; 26:4124-30; PMID:18626005; http://dx.doi.org/ 10.1200/JCO.2008.16.4558 [DOI] [PubMed] [Google Scholar]
- [4].Wilcox RA. A three-signal model of T-cell lymphoma pathogenesis. Am J Hematol 2016; 91:113-22; PMID:26408334; http://dx.doi.org/ 10.1002/ajh.24203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vallois D, Dobay MP, Morin RD, Lemonnier F, Missiaglia E, Juilland M, Iwaszkiewicz J, Fataccioli V, Bisig B, Roberti A, et al.. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood 2016; 128:1490-502; PMID:27369867; http://dx.doi.org/ 10.1182/blood-2016-02-698977 [DOI] [PubMed] [Google Scholar]
- [6].Agostinelli C, Rizvi H, Paterson J, Shende V, Akarca AU, Agostini E, Fuligni F, Righi S, Spagnolo S, Piccaluga PP, et al.. Intracellular TCR-signaling pathway: novel markers for lymphoma diagnosis and potential therapeutic targets. Am J Surg Pathol 2014; 38:1349-59; PMID:25118816; http://dx.doi.org/ 10.1097/PAS.0000000000000309 [DOI] [PubMed] [Google Scholar]
- [7].Bonzheim I, Geissinger E, Roth S, Zettl A, Marx A, Rosenwald A, Muller-Hermelink HK, Rudiger T. Anaplastic large cell lymphomas lack the expression of T-cell receptor molecules or molecules of proximal T-cell receptor signaling. Blood 2004; 104:3358-60; PMID:15297316; http://dx.doi.org/ 10.1182/blood-2004-03-1037 [DOI] [PubMed] [Google Scholar]
- [8].Bustelo XR. Understanding Rho/Rac biology in T-cells using animal models. Bioessays 2002; 24:602-12; PMID:12111721; http://dx.doi.org/ 10.1002/bies.10107 [DOI] [PubMed] [Google Scholar]
- [9].Bustelo XR, Sauzeau V, Berenjeno IM. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays 2007; 29:356-70; PMID:17373658; http://dx.doi.org/ 10.1002/bies.20558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cantrell DA. GTPases and T cell activation. Immunol Rev 2003; 192:122-30; PMID:12670400; http://dx.doi.org/ 10.1034/j.1600-065X.2003.00028.x [DOI] [PubMed] [Google Scholar]
- [11].Izquierdo M, Leevers SJ, Marshall CJ, Cantrell D. p21ras couples the T cell antigen receptor to extracellular signal-regulated kinase 2 in T lymphocytes. J Exp Med 1993; 178:1199-208; PMID:8376929; http://dx.doi.org/ 10.1084/jem.178.4.1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Genot E, Reif K, Beach S, Kramer I, Cantrell D. p21ras initiates Rac-1 but not phosphatidyl inositol 3 kinase/PKB, mediated signaling pathways in T lymphocytes. Oncogene 1998; 17:1731-8; PMID:9796702; http://dx.doi.org/ 10.1038/sj.onc.1202101 [DOI] [PubMed] [Google Scholar]
- [13].Genot E, Cleverley S, Henning S, Cantrell D. Multiple p21ras effector pathways regulate nuclear factor of activated T cells. Embo J 1996; 15:3923-33; PMID:8670897 [PMC free article] [PubMed] [Google Scholar]
- [14].Woodrow M, Clipstone NA, Cantrell D. p21ras and calcineurin synergize to regulate the nuclear factor of activated T cells. J Exp Med 1993; 178:1517-22; PMID:8228805; http://dx.doi.org/ 10.1084/jem.178.5.1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Swan KA, Alberola-Ila J, Gross JA, Appleby MW, Forbush KA, Thomas JF, Perlmutter RM. Involvement of p21ras distinguishes positive and negative selection in thymocytes. Embo J 1995; 14:276-85; PMID:7835338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fischer KD, Kong YY, Nishina H, Tedford K, Marengere LE, Kozieradzki I, Sasaki T, Starr M, Chan G, Gardener S, et al.. Vav is a regulator of cytoskeletal reorganization mediated by the T-cell receptor. Curr Biol 1998; 8:554-62; PMID:9601639; http://dx.doi.org/ 10.1016/S0960-9822(98)70224-6 [DOI] [PubMed] [Google Scholar]
- [17].Fischer KD, Zmuldzinas A, Gardner S, Barbacid M, Bernstein A, Guidos C. Defective T-cell receptor signalling and positive selection of Vav-deficient CD4+ CD8+ thymocytes. Nature 1995; 374:474-7; PMID:7700360; http://dx.doi.org/ 10.1038/374474a0 [DOI] [PubMed] [Google Scholar]
- [18].Tarakhovsky A, Turner M, Schaal S, Mee PJ, Duddy LP, Rajewsky K, Tybulewicz VL. Defective antigen receptor-mediated proliferation of B and T cells in the absence of Vav. Nature 1995; 374:467-70; PMID:7700358; http://dx.doi.org/ 10.1038/374467a0 [DOI] [PubMed] [Google Scholar]
- [19].Zhang R, Alt FW, Davidson L, Orkin SH, Swat W. Defective signalling through the T- and B-cell antigen receptors in lymphoid cells lacking the vav proto-oncogene. Nature 1995; 374:470-3; PMID:7700359; http://dx.doi.org/ 10.1038/374470a0 [DOI] [PubMed] [Google Scholar]
- [20].Gulbins E, Coggeshall KM, Baier G, Katzav S, Burn P, Altman A. Tyrosine kinase-stimulated guanine nucleotide exchange activity of Vav in T cell activation. Science 1993; 260:822-5; PMID:8484124; http://dx.doi.org/ 10.1126/science.8484124 [DOI] [PubMed] [Google Scholar]
- [21].Han J, Das B, Wei W, Van Aelst L, Mosteller RD, Khosravi-Far R, Westwick JK, Der CJ, Broek D. Lck regulates Vav activation of members of the Rho family of GTPases. Mol Cell Biol 1997; 17:1346-53; PMID:9032261; http://dx.doi.org/ 10.1128/MCB.17.3.1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Crespo P, Schuebel KE, Ostrom AA, Gutkind JS, Bustelo XR. Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature 1997; 385:169-72; PMID:8990121; http://dx.doi.org/ 10.1038/385169a0 [DOI] [PubMed] [Google Scholar]
- [23].Li P, Martins IR, Amarasinghe GK, Rosen MK. Internal dynamics control activation and activity of the autoinhibited Vav DH domain. Nat Struct Mol Biol 2008; 15:613-8; PMID:18488041; http://dx.doi.org/ 10.1038/nsmb.1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yu B, Martins IR, Li P, Amarasinghe GK, Umetani J, Fernandez-Zapico ME, Billadeau DD, Machius M, Tomchick DR, Rosen MK. Structural and energetic mechanisms of cooperative autoinhibition and activation of Vav1. Cell 2010; 140:246-56; PMID:20141838; http://dx.doi.org/ 10.1016/j.cell.2009.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kuhne MR, Ku G, Weiss A. A guanine nucleotide exchange factor-independent function of Vav1 in transcriptional activation. J Biol Chem 2000; 275:2185-90; PMID:10636924; http://dx.doi.org/ 10.1074/jbc.275.3.2185 [DOI] [PubMed] [Google Scholar]
- [26].Miletic AV, Graham DB, Sakata-Sogawa K, Hiroshima M, Hamann MJ, Cemerski S, Kloeppel T, Billadeau DD, Kanagawa O, Tokunaga M, et al.. Vav links the T cell antigen receptor to the actin cytoskeleton and T cell activation independently of intrinsic Guanine nucleotide exchange activity. PLoS One 2009; 4:e6599; PMID:19672294; http://dx.doi.org/ 10.1371/journal.pone.0006599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bustelo XR, Ledbetter JA, Barbacid M. Product of vav proto-oncogene defines a new class of tyrosine protein kinase substrates. Nature 1992; 356:68-71; PMID:1311423; http://dx.doi.org/ 10.1038/356068a0 [DOI] [PubMed] [Google Scholar]
- [28].Katzav S. Flesh and blood: the story of Vav1, a gene that signals in hematopoietic cells but can be transforming in human malignancies. Cancer Lett 2007; 255:241-54; PMID:17590270; http://dx.doi.org/ 10.1016/j.canlet.2007.04.015 [DOI] [PubMed] [Google Scholar]
- [29].Razanadrakoto L, Cormier F, Lauriente V, Dondi E, Gardano L, Katzav S, Guittat L, Varin-Blank N. Mutation of Vav1 adaptor region reveals a new oncogenic activation. Oncotarget 2015; 6:2524-37; PMID:25426554; http://dx.doi.org/ 10.18632/oncotarget.2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Boddicker RL, Razidlo GL, Dasari S, Zeng Y, Hu G, Knudson RA, Greipp PT, Davila JI, Johnson SH, Porcher JC, et al.. Integrated mate-pair and RNA sequencing identifies novel, targetable gene fusions in peripheral T-cell lymphoma. Blood 2016; 128:1234-45; PMID:27297792; http://dx.doi.org/ 10.1182/blood-2016-03-707141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Barreira M, Fabbiano S, Couceiro JR, Torreira E, Martinez-Torrecuadrada JL, Montoya G, Llorca O, Bustelo XR. The C-terminal SH3 domain contributes to the intramolecular inhibition of Vav family proteins. Sci Signal 2014; 7:ra35; PMID:24736456; http://dx.doi.org/ 10.1126/scisignal.2004993 [DOI] [PubMed] [Google Scholar]
- [32].Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, Totoki Y, Chiba K, Sato-Otsubo A, Nagae G, et al.. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet 2015; 47:1304-15; PMID:26437031; http://dx.doi.org/ 10.1038/ng.3415 [DOI] [PubMed] [Google Scholar]
- [33].Bustelo XR. Regulatory and signaling properties of the Vav family. Mol Cell Biol 2000; 20:1461-77; PMID:10669724; http://dx.doi.org/ 10.1128/MCB.20.5.1461-1477.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Colomba A, Courilleau D, Ramel D, Billadeau DD, Espinos E, Delsol G, Payrastre B, Gaits-Iacovoni F. Activation of Rac1 and the exchange factor Vav3 are involved in NPM-ALK signaling in anaplastic large cell lymphomas. Oncogene 2008; 27:2728-36; PMID:17998938; http://dx.doi.org/ 10.1038/sj.onc.1210921 [DOI] [PubMed] [Google Scholar]
- [35].Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, Muto H, Tsuyama N, Sato-Otsubo A, Okuno Y, et al.. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet 2014; 46:171-5; PMID:24413737; http://dx.doi.org/ 10.1038/ng.2872 [DOI] [PubMed] [Google Scholar]
- [36].Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, Carpenter Z, Abate F, Allegretta M, Haydu JE, et al.. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet 2014; 46:166-70; PMID:24413734; http://dx.doi.org/ 10.1038/ng.2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yoo HY, Sung MK, Lee SH, Kim S, Lee H, Park S, Kim SC, Lee B, Rho K, Lee JE, et al.. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet 2014; 46:371-5; PMID:24584070; http://dx.doi.org/ 10.1038/ng.2916 [DOI] [PubMed] [Google Scholar]
- [38].Manso R, Sanchez-Beato M, Monsalvo S, Gomez S, Cereceda L, Llamas P, Rojo F, Mollejo M, Menarguez J, Alves J, et al.. The RHOA G17V gene mutation occurs frequently in peripheral T-cell lymphoma and is associated with a characteristic molecular signature. Blood 2014; 123:2893-4; PMID:24786457; http://dx.doi.org/ 10.1182/blood-2014-02-555946 [DOI] [PubMed] [Google Scholar]
- [39].Sander EE, ten Klooster JP, van Delft S, van der Kammen RA, Collard JG. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J Cell Biol 1999; 147:1009-22; PMID:10579721; http://dx.doi.org/ 10.1083/jcb.147.5.1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chiba S, Enami T, Ogawa S, Sakata-Yanagimoto M. G17V RHOA: Genetic evidence of GTP-unbound RHOA playing a role in tumorigenesis in T cells. Small GTPases 2015; 6:100-3; PMID:26103434; http://dx.doi.org/ 10.4161/21541248.2014.988088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Vaque JP, Gomez-Lopez G, Monsalvez V, Varela I, Martinez N, Perez C, Dominguez O, Grana O, Rodriguez-Peralto JL, Rodriguez-Pinilla SM, et al.. PLCG1 mutations in cutaneous T-cell lymphomas. Blood 2014; 123:2034-43; PMID:24497536; http://dx.doi.org/ 10.1182/blood-2013-05-504308 [DOI] [PubMed] [Google Scholar]
- [42].Manso R, Rodriguez-Pinilla SM, Gonzalez-Rincon J, Gomez S, Monsalvo S, Llamas P, Rojo F, Perez-Callejo D, Cereceda L, Limeres MA, et al.. Recurrent presence of the PLCG1 S345F mutation in nodal peripheral T-cell lymphomas. Haematologica 2015; 100:e25-7; PMID:25304611; http://dx.doi.org/ 10.3324/haematol.2014.113696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zimmermann M, Dasari S, Boddicker RL, Zeng Y, Eckloff B, Cunningham JM, Wu Y, Porcher JC, Link BK, Ansell S, et al.. Mutations Targeting the ErbB Pathway and MSC in Peripheral T-Cell Lymphoma. Blood 2015; 126:2681. [Google Scholar]
- [44].Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, Aziz M, Kassner M, Bryant KL, Pierobon M, et al.. Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell 2016; 29:75-89; PMID:26725216; http://dx.doi.org/ 10.1016/j.ccell.2015.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hobbs GA, Wittinghofer A, Der CJ. Selective targeting of the KRAS G12C mutant: Kicking KRAS when it's down. Cancer Cell 2016; 29:251-3; PMID:26977877; http://dx.doi.org/ 10.1016/j.ccell.2016.02.015 [DOI] [PubMed] [Google Scholar]
- [46].Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 2004; 23:7990-8000; PMID:15489916; http://dx.doi.org/ 10.1038/sj.onc.1208074 [DOI] [PubMed] [Google Scholar]
- [47].Salazar-Fontana LI, Barr V, Samelson LE, Bierer BE. CD28 engagement promotes actin polymerization through the activation of the small Rho GTPase Cdc42 in human T cells. J Immunol 2003; 171:2225-32; PMID:12928366; http://dx.doi.org/ 10.4049/jimmunol.171.5.2225 [DOI] [PubMed] [Google Scholar]
- [48].Rohr J, Guo S, Huo J, Bouska A, Lachel C, Li Y, Simone PD, Zhang W, Gong Q, Wang C, et al.. Recurrent activating mutations of CD28 in peripheral T-cell lymphomas. Leukemia 2016; 30:1062-70; PMID:26719098; http://dx.doi.org/ 10.1038/leu.2015.357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lee SH, Kim JS, Kim J, Kim SJ, Kim WS, Lee S, Ko YH, Yoo HY. A highly recurrent novel missense mutation in CD28 among angioimmunoblastic T-cell lymphoma patients. Haematologica 2015; 100:e505-7; PMID:26405154; http://dx.doi.org/ 10.3324/haematol.2015.133074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sekulic A, Liang WS, Tembe W, Izatt T, Kruglyak S, Kiefer JA, Cuyugan L, Zismann V, Legendre C, Pittelkow MR, et al.. Personalized treatment of Sezary syndrome by targeting a novel CTLA4:CD28 fusion. Mol Genet Genomic Med 2015; 3:130-6; PMID:25802883; http://dx.doi.org/ 10.1002/mgg3.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yoo HY, Kim P, Kim WS, Lee SH, Kim S, Kang SY, Jang HY, Lee JE, Kim J, Kim SJ, et al.. Frequent CTLA4-CD28 gene fusion in diverse types of T-cell lymphoma. Haematologica 2016; 101:757-63; PMID:26819049; http://dx.doi.org/ 10.3324/haematol.2015.139253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tybulewicz VL, Henderson RB. Rho family GTPases and their regulators in lymphocytes. Nat Rev Immunol 2009; 9:630-44; PMID:19696767; http://dx.doi.org/ 10.1038/nri2606 [DOI] [PMC free article] [PubMed] [Google Scholar]