

ABSTRACT
Together with a cyclin-dependent kinase (CDK) partner G1 cyclins control cell cycle entry by phosphorylating a number of nuclear targets and releasing a transcriptional program at the end of G1 phase. Yeast G1 cyclins also operate on cytoplasmic targets involved in the polarization of the cytoskeleton and vesicle trafficking. These processes are mainly controlled by the small GTPase Cdc42, and G1 cyclins regulate the activity of this and other small GTPases through the modulation of their regulators and effectors. This regulation is key for different developmental outcomes in unicellular organisms. In mammalian cells cytoplasmic G1 cyclin D1 has been shown to promote the activity of Rac1 and Ral GTPases and to block RhoA. Regulation of these small GTPases by G1 cyclins may constitute a mechanism to coordinate proliferation with cell migration and morphogenesis, important processes not only during normal development and organogenesis but also for tumor formation and metastasis. Here we briefly review the evidence supporting a role of G1 cyclins and CDKs as regulators of the activity of small GTPases, emphasizing their functional relevance both in budding yeast and in mammalian cells.
Keywords: cell polarity, G1 cyclins, mammals, small GTPases, yeast
Introduction
Cyclin-dependent kinases (CDKs) are a family of serine/threonine protein kinases whose enzymatic activation requires the binding of a regulatory cyclin subunit. Although they constitute the central components of the cell cycle control system, cyclin-CDK complexes are also involved in other cellular processes.1 Broadly, CDKs may be grouped in two classes: those binding multiple cyclins and related to the cell cycle, exemplified by the yeast CDKs Cdc28 and Pho85; and those related to transcription and bound by single cyclins.2 But even those CDKs and cyclins that regulate the cell cycle events show non-canonical functions.3 During the G1 phase of the cell cycle, cyclin expression and localization are precisely regulated by temporal and spatial cues in order to coordinate growth and proliferation. Under favorable circumstances, the nuclear accumulation of G1 cyclins (Cln3-Cdc28 in yeast, cyclins D-CDK4,6 in mammals) triggers the commitment to undergo a new round of cell division: a process called Start in yeast and the Restriction Point (RP) in mammalian cells.4 This involves the induction of a transcriptional program that includes, among other targets, the likewise G1 cyclins Cln1, Cln2, Pcl1 and Pcl2 in yeast and cyclin E in mammals. Cln1 and Cln2 complexed with Cdc28 and cyclin E with CDK2 participate in a positive feedback loop to assure the irreversible execution of Start/RP.4
In the yeast Saccharomyces cerevisiae cell cycle, the G1/S transition encompasses the initiation of DNA replication, the duplication of the spindle pole body (analogous to the mammalian centrosome), and the formation of a bud. This last event entails the polarization of the cytoskeleton and the canalization of secretion toward a point. Mammalian cells do not bud, but they do polarize their cytoskeleton and vesicle trafficking in order to adhere, migrate and acquire specific shapes. These processes are controlled in both yeast and mammals by a number of small GTPases.5,6 In turn, there is mounting evidence that in yeast Pcl cyclins in association with Pho85, and Cln cyclins with Cdc28 are key regulators of these small GTPases, and there is emerging data supporting an analogous view in mammalian cells. Here we briefly review the evidence supporting the role of G1 cyclins and CDKs as regulators of the activity of small GTPases, emphasizing their functional relevance both in budding yeast and mammalian cells.
Yeast
In the yeast Saccharomyces cerevisiae, and other fungi, the Rho family GTPase Cdc42 acts as a master regulator of the process of budding and its associated polarized growth. There are four known GAPs for Cdc42 (Bem2, Bem3, Rga1 and Rga2) but only one GEF (Cdc24). Bud emergence ultimately depends on CDK activity at G1, with no budding when G1 CDKs are lacking, and showing premature induction of budding when CDKs are activated prematurely.6,7 Although it contains a serine residue within a minimal (SP) CDK phosphorylation motif, yeast Cdc42 does not seem to be phosphorylated. Instead the regulation of its activity by G1 CDKs is mediated by its regulators (GAPs and GEF) and effectors. In principle, Cdc24 made a good candidate for CDK regulation because, as Cdc42, it is also required for bud emergence and cytoskeleton polarization,8,9 it is hyperphosphorylated in vivo in a cell cycle-regulated manner that depends on Cdc28-Cln activity,10 and it is an in vitro substrate of G1 CDKs.11,12 However, the expression of a constitutively activated form of Cdc42 is sufficient to induce hyperphosphorylation of Cdc24, even in cln1,2,3 depleted cells, and mutation of six putative CDK consensus sites or up to 35 phospho-sites detected by mass spectrometry has no impact on Cdc24 function.10,13 Hence, whether Cdc24 is phosphorylated by G1 CDKs in vivo and whether this potential phosphorylation may be of any physiological significance remain mysterious. Cdc28 activity in G1 is nonetheless required for Cdc24 release from the nucleus in late G1, a prerequisite for its localization at sites of polarized growth.14,15 During G1 Cdc24 remains sequestered in the nucleus by association with Far1, which acts otherwise as a CDK inhibitor during the mating pheromone response. In the absence of pheromone, Far1 is phosphorylated by Cdc28-Cln activity and targeted for degradation, which allows Cdc24 to be exported to the cytoplasm. This, however, cannot be the sole function of G1 CDKs in triggering cellular polarization as cytoplasmic mutant forms of Cdc24 are unable to induce polarized growth in cells lacking Cdc28-Cln activity.10,14,15
Cdc42-directed GAPs have been detected as putative CDK substrates in a number of systematic studies;16-18 in particular, Rga1 and Bem3 have been associated with Cln2-Cdc28.17 Cln2 interaction with Rga1 likely involves a recently described new docking site in Cln2 because when mutated their interaction is considerably reduced, and it tames the effects of overexpressed Cln2 on polarized growth,19 although other targets may mediate this effect. In addition to Cln2-Cdc28, Rga1 may also be phosphorylated by the Pho85 kinase and its associated cyclins Pcl1 and Pcl2 because when overexpressed in a pcl1Δ pcl2Δ background Rga1 severely inhibits polarized growth.20 Likely, Bem3 - and also possibly Bem2 - is a physiological target of Cln2-Cdc28. The protein gets phosphorylated at bud emergence in a Cdc28-Cln dependent manner and it is phosphorylated in vitro by immunoprecipitated Cln2. Moreover, mutating five CDK consensus sites in the protein renders it immune to phosphorylation in vivo under conditions of high Cdc28-Cln activity, and expression of this nonphosphorylatable mutant is toxic in an otherwise wild type background.21 Another Cdc42 GAP that has been the focus of several studies is Rga2. Overexpression of Rga2 is lethal in a number of conditions where G1 CDK activity is compromised (i.e., cln1,2Δ; pcl1,2Δ; pcl1,9Δ; pho85Δ),22,23 supporting a negative regulation of Rga2 by phosphorylation. Furthermore, Rga2 is hyperphosphorylated in vivo in a Cdc28- and Pho85-dependent manner, and is a specific target of Cln2-Cdc28 and Pho85 complexes in vitro.12,22 It co-immunoprecipitates with Pcl1, Pcl2 and Pcl9, and overexpression of nonphosphorylatable mutants of Rga2 produces clear phenotypes.22 More evidence for Rga2 regulation by G1 CDKs comes from the human pathogen Candida albicans, a filamentous fungus able to grow in a variety of morphological forms: from unicellular budding yeast to long filaments named hyphae.24 During hyphal development polarized growth must be very active, and Rga2 is prevented from localizing to the hyphal tips, where Cdc42 is accumulated.25,26 However, in the absence of Hgc1 (a hypha-specific G1 cyclin associated to Cdc28) full hyphae cannot develop, and Rga2 is enriched at the tip of the incipient hypha. Deletion of Rga2 restores hyphal development in hgc1Δ cells, Rga2 co-immunoprecipitates with and its phosphorylation depends on Hgc1, and mutation of 16 CDK consensus sites abolishes the hyperphosphorylation observed upon induction of hyphal growth.26 Hence, it is likely that Rga2 is regulated by phosphorylation by Hgc1-Cdc28 during hyphal development. Although all these results make a compelling case for a model whereby Cdc42-GTP would be kept low during G1 by high GAP activity that would become inhibited by phosphorylation as CDK activity increases during the G1-S transition, as yet, biochemical evidence that phosphorylation inhibits GAP activity is lacking.
Another level in which G1 CDKs may control the activity of Cdc42 is through the regulation of its effectors and adaptor proteins. These may include Boi1 - and its paralog Boi2 - which interacts in vivo with Cdc24, is phosphorylated in a Cdc28-dependent manner in vivo, and is a Cln2-Cdc28 target in vitro. In addition, the mutation of 12 CDK consensus sites in the protein shows clear defects in polarization.12 The scaffold protein Bem1 is still another likely target of G1 CDKs. Although Bem1 is not necessary to localize Cdc24 to the incipient bud site, it is required to stabilize active Cdc24 at the plasma membrane.10 In a cln1,2Δ background, both deletion or overexpression of Bem1 is lethal.23 In the phytopathogen fungus Ustilago maydis a physical interaction between Bem1 and the Cdk5/Pho85 kinase at the cell tip has been described, which is in turn essential for the interaction between Bem1 and Cdc24 and its accumulation at the cell pole.27 A distinctive feature in U. maydis is that Cdc24 functions as GEF for a Rac1 homolog (not Cdc42), which is nonetheless involved in the regulation of polar growth.28 In S. cerevisiae Cdc42 not only controls polarized growth but it is also involved in vacuolar homotypic fusion. This process is specifically regulated by Cln3-Cdc28 in a Bem1-dependent manner: Bem1 phosphorylation depends on Cdc28, it is phosphorylated in vitro by Cln3-Cdc28, and mutation of a single serine at position 72 in Bem1 abrogates the regulatory effects of Cln3 and abolishes the appearance of a phospho-form of the protein that is present in wild type cells but is also absent in a cln3Δ background.29
Another small GTPase, not essential for polarization, but crucial for bud growth and cell wall biosynthesis is Rho1. The GDP/GTP cycle of Rho1 is cell cycle regulated, and Cln2 overexpression (but not Cln3, Clb2, Clb3 or Clb5) increases the levels of active Rho1.30 The Rho1-directed GEF Tus1 is required for the augmented levels of active Rho1 both during the cell cycle and upon Cln2 overexpression. Tus1 is very likely an in vivo substrate of Cln2-Cdc28 because it is phosphorylated in vivo in a Cdc28-dependent manner and it co-immunoprecipitates with Cln2. Moreover, Tus1 phosphorylation is required for the Cln2-dependent activation of Rho1.30 Recently, it has been described a new docking site in Cln2 that is likely involved in the interaction with Tus1.19 Another Rho1-directed GEF, Rom2, is phosphorylated in vitro by the G1 complex Pcl1-Pho85,31 although the biological significance of this phosphorylation is unknown.
Still another instance of a small GTPase regulated by G1 CDK activity is the case of Sec4 during hyphal growth in Candida. This Rab GTPase mediates the post-golgi transport of secretory vesicles at the growing tip, and their docking with the exocyst before their fusion to the cell membrane; essential processes to maintain polarized growth (see ref. 32 and references therein). Sec4 is activated by its GEF Sec2, and Sudbery and coworkers have shown that phosphorylation of serine 584 in Sec2 is critical for hyphal development. The pattern of phospho-forms of Sec2 during hyphal growth depends on Cdc28 activity. Partial inhibition of cdc28 prevents normal hypha formation but this phenotype is readily rescued by a phosphomimetic S584E form of Sec2. Besides, G1 cyclins Ccn1 and Hgc1 show genetic and physical interactions with Sec2.32 Thus, it is likely that Sec2 is a substrate of G1 cyclins-Cdc28 at least in C. albicans.
Figure 1 summarizes our current knowledge about the regulatory network controlled by G1 cyclins and involving the activities of small GTPases in yeast.
Figure 1.
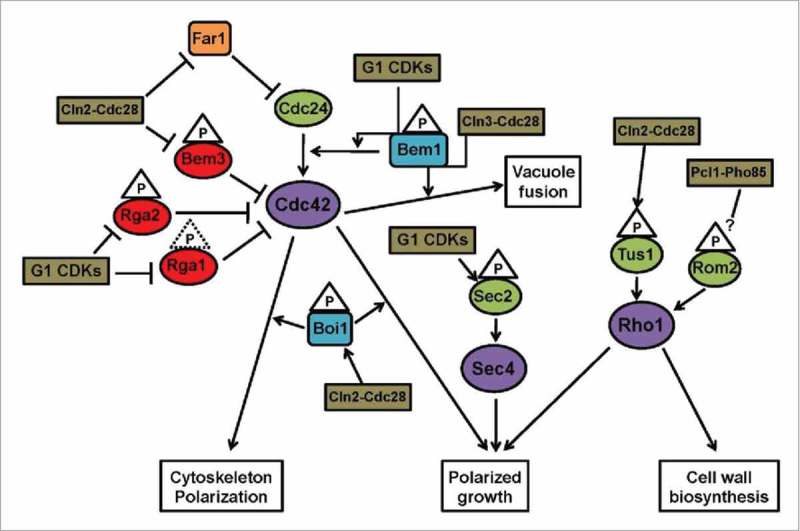
Representative diagram of the regulatory network of small GTPases and G1 cyclins in yeast. When both Cdc28-Cln and Pho85-Pcl complexes participate, we use the notation G1 CDKs. Cln2 is partially redundant with Cln1; we only indicate Cln2 to simplify the diagram. Phosphorylation events are indicated by a triangle; dotted line means poor in vivo evidence. Color code: purple - small GTPases; green - GEFs; red - GAPs; blue - effectors/adaptors; brown - CDK complexes; orange - Far1 inhibitor.
Mammals
In mammalian cells adhesion, migration and morphogenesis require the polarization of vesicle trafficking and the cytoskeleton, which are controlled by several small GTPases.5,6 Here there is recent evidence that also G1 cyclins, specifically cyclin D1, may regulate the activity of these small GTPases through different mechanisms. Admittedly the evidence is sketchier than in yeast and the mechanisms may involve more indirect relationships. An early observation that cyclin D1 may affect both cell adhesion and migration was the correlation of its expression with the metastatic potential of some tumors,33 indicating a role in the regulation of the invasion capacity of tumor cells and not only on their proliferation rate. Accordingly, cyclin D1−/− mouse embryonic fibroblasts (MEFs) present reduced cellular motility, higher levels of spreading, and stronger adhesion to substrate.34 Several studies mechanistically link cyclin D1 with the regulation of adhesion through the small GTPases Rho, Rac and Ral (Fig. 2).
Figure 2.
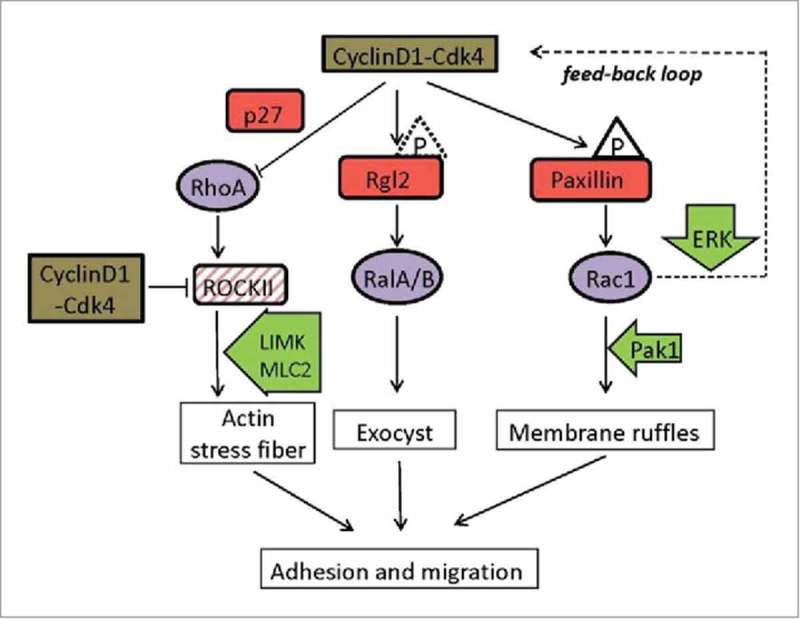
Representative diagram of cyclin D1-Cdk4 cytoplasmic targets. Red squares represent the direct targets and substrates of the cyclin D1-Cdk4 complex. Triangles characterize phosphates but the phosphorylation by Cdk4 has still not been demonstrated in vivo for Rgl2 (dotted line). Purple circles symbolize small GTPases regulated by the complex. Green arrows are other proteins involved in the transduction pathways. Dashed square indicate a target transcriptionally repressed by cyclin D1-Cdk4. A possible positive feed-back loop for Rac1 signaling is depicted (dotted line).
One way cyclin D1 promotes cell motility is by constraining RhoA activity. RhoA is activated in response to tension on integrins, and promotes actin polymerization and stress fiber assembly leading to increased adhesion to the cell matrix. This is accomplished through the activation of the RhoA-activated kinase II (ROCKII), a RhoA downstream effector, that itself activates myosin light chain 2 and the LIM kinase, which in turn phosphorylates and inhibits the actin-depolymerizing protein cofilin.35 Cyclin D1-associated activity represses transcription of ROCKII.34 Accordingly, cyclin D1−/− MEFs show high ROCKII activity, increased stress fiber formation, and low migration efficiency. Another mechanism whereby cyclin D1 regulates RhoA activity involves the CDK inhibitor p27Kip1. This protein interferes with RhoA activation by physically impeding the interaction of the GTPase with its GEFs.36 This cell-cycle independent role of p27 occurs after its sequestration and stabilization in the cytoplasm, which depends upon the phosphorylation of three residues - Ser10, Thr157, and Thr198 – in the protein.37 Cyclin D1 promotes the accumulation of p27 by repressing Skp2, a component of the E3 ubiquitin ligase targeting p27Kip1, and by favoring the phosphorylation of p27 at serine 10.38 Thus, it seems that cyclin D1-associated activity induces cell migration by promoting the accumulation of p27 in the cytoplasm that in turns reduces RhoA activity. Intriguinly, cyclin D1 and p27 have both been found physically associated to activated RhoA.38 Although this result is controversial with an inhibitory role on RhoA activity, it opens the possibility for a more direct implication of cyclin D1 in RhoA regulation.
Another mechanism by which cyclin D1 regulates motility involves the scaffold protein paxillin and the Rac GTPase. Paxillin is a focal adhesion protein that impinges on the regulation of these small GTPases via different mechanisms acting on their GEFs and GAPs.39 For instance, the interaction of cells with substratum induces integrin signaling that promotes FAK (focal adhesion kinase) and SRC activation. Both kinases phosphorylate paxillin at tyrosines 31 and 118 and allow its interaction with the Rac1 GEFs DOCK180 and PIX, which activates Rac1. Also, phosphorylation of paxillin at serine 83 by extracellular signal-regulated kinase (ERK) induces Rac1 activation and cell migration in epithelial cells.40 It is thought that phosphorylation at serine 83 promotes the association of paxillin with FAK, which in turn would induce phosphorylation of paxillin at tyrosine residues leading to Rac1 activation. A similar mechanism is envisioned for the induction of cell migration by the c-Jun N-terminal kinase (JNK)-dependent phosphorylation of paxillin at serine 178.41 However, in these proposed mechanisms the participation of DOCK180 and PIX has not been demonstrated. Cyclin D1-Cdk4 phosphorylates a membrane-associated subpopulation of paxillin at serines 83 and 178.42 Cyclin D1 overexpression triggered Rac1 activation in the presence of wild type paxillin, but not in the presence of a non-phosphorylatable version of the protein (S83A, S178A). Alternatively, the expression of a phosphomimetic version of paxillin (S83E, S178E) caused Rac1 activation even in the absence of cyclin D1. Then, the phosphorylation of paxillin by cyclin D1-Cdk4 gives rise to Rac1 activation and a concomitant promotion of membrane ruffling in MEFs and tumor cells, leading to increased invasion capacity and metastatic potential.42 Rho and Rac activities need to be tightly regulated for proper migration. By using specific FRET biosensors for Rho and Rac activities, it has been demonstrated that Rac1 and RhoA operate antagonistically through spatial separation and precise timing.43 Considering that cyclin D1 can inhibit Rho activity (see above) and induce Rac1 activity, it is tempting to speculate of a possible role for cytoplasmic cyclin D1 in coordinating Rac and Rho activities during migration.
Finally, Ral GTPases bind to several components of the exocyst and regulate vesicle traffic. Exocytosis and vesicle recycling are essential processes required for cell attachment and motility, and accordingly the activation of Ral GTPases is required for cell motility.44 Cytoplasmic cyclin D1-Cdk4 also modulates Ral GTPase activity.45 Cyclin D1 physically interacts with both Ral A and Ral B, it shows extensive cytoplasmic co-localization with these small GTPases, and it was found physically associated to Ral-GTP forms in a pull down assay for activated Ral. In addition, cyclin D1 also interacts with the Ral GEF Rgl2, which is phosphorylated in vitro by the complex. Expression of cyclin D1 increases the levels of activated Ral B in a manner that depends on the assembly of an active kinase complex. Hence it is likely that cyclin D1 regulates the activity of Ral GTPases through the phosphorylation of Rgl2 and possibly through more direct mechanisms. Consistently, the expression of a hyperactive allele of Rgl2 subverts the motility defect of cyclin D1-defective fibroblasts.45
Concluding remarks and perspectives
Developmental outcomes in unicellular organisms as well as tissue and organ formation in multicellular ones require a complex set of molecular mechanisms to coordinate proliferation with morphogenetic processes and differentiation. In this scenario, G1 cyclins may play an important role in cell-fate decision making. On the one hand, through their well-described nuclear function G1 cyclins control cell proliferation, and on the other hand, chiefly through a cytoplasmic activity they regulate cell morphogenesis. Importantly, this effect on morphogenetic processes appears to be achieved mainly, although not exclusively, through the regulation of the activity of several small GTPases. There is abundant evidence for this in yeast, and strong indications that the same is truth in mammalian cells. Because distinct functions of G1 cyclins take place in different cellular compartments the regulation of the localization of G1 cyclin-CDK complexes may constitute an important mechanism in determining cell fate.
In addition to their role during development, alterations in the amount and localization of G1 cyclins in mammalian cells, in particular cyclin D1, are related to oncogenesis and metastasis. While the overexpression of cyclin D1 has unmistakable effects on the proliferation capacity of tumor cells, it also promotes migration, boosting the invasive capacity and metastatic potential of tumor cells. Hence, the characterization of the mechanisms involved in the regulation of small GTPases by G1 cyclins should be of obvious interest in the field of molecular oncology.
Some small GTPases participate in signaling pathways involved in the regulation of the expression and activity of G1 cyclins (not reviewed here). Hence the communication between G1 cyclins and small GTPases in some cases may go in both directions. This allows the existence of positive feed-back loop modules that may enhance different cellular processes, as it is the case for cell migration. Feed-back mechanisms provide plasticity to cell signaling regulation and to GTPase-controlled mechanisms.46 Thus, it will be of interest to elucidate whether such mechanisms are actually into place in the case of cyclin D1 with Rac1, and also with RhoA.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to members of the cell cycle lab for stimulating discussions and for helpful considerations.
Funding
This work was funded by Spanish Ministry of Education and Science (BFU2013–42895) from the Spanish Ministry of Economy and Competitivity and Catalan Government (SGR-559). T. Cemeli and MV Monserrat were supported by predoctoral fellowships from Spanish Ministry of Education and Science (FPU) and from University of Lleida, respectively.
References
- [1].Morgan DO. The cell cycle: principles of control. London: New Science Press Ltd, 2007. [Google Scholar]
- [2].Malumbres M. Cyclin-dependent kinases. Genome Biol [Internet] 2014. [cited 2015September1]; 15:122 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4097832&tool=pmcentrez&rendertype=abstract; http://dx.doi.org/ 10.1186/gb4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hydbring P, Malumbres M, Sicinski P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat Rev Mol Cell Biol [Internet] 2016. [cited 2016April1]; 17:280-92. Available from: http://dx.doi.org/ 10.1038/nrm.2016.27; http://dx.doi.org/ 10.1038/nrm.2016.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Johnson A, Skotheim JM. Start and the restriction point. Curr Opin Cell Biol 2013; 25:717-23; PMID:23916770; http://dx.doi.org/ 10.1016/j.ceb.2013.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature 2002; 420:629-35; PMID:12478284; http://dx.doi.org/ 10.1038/nature01148 [DOI] [PubMed] [Google Scholar]
- [6].Etienne-Manneville S. Cdc42–the centre of polarity. J Cell Sci [Internet] 2004; 117:1291-300. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15020669; http://dx.doi.org/ 10.1242/jcs.01115 [DOI] [PubMed] [Google Scholar]
- [7].Howell AS, Lew DJ. Morphogenesis and the cell cycle. Genetics [Internet] 2012; 190:51-77. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22219508; http://dx.doi.org/ 10.1534/genetics.111.128314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Adams AEM, Pringle JR. Relationship of actin and tubulin distribution to bud growth in wild-type and morphogenetic-mutant Saccharomyces cerevisiae. J Cell Biol 1984; 98:934-45; PMID:6365931; http://dx.doi.org/ 10.1083/jcb.98.3.934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sloat BF, Adams A, Pringle JR. Roles of the CDC24 gene product in cellular morphogenesis during the saccharomyces cerevisiae cell cycle. J Cell Biol 1981; 89:395-405; PMID:7019215; http://dx.doi.org/ 10.1083/jcb.89.3.395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gulli MP, Jaquenoud M, Shimada Y, Niederhäuser G, Wiget P, Peter M. Phosphorylation of the Cdc42 exchange factor Cdc24 by the PAK-like kinase Cla4 may regulate polarized growth in yeast. Mol Cell [Internet] 2000; 6:1155-67. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11106754; http://dx.doi.org/ 10.1016/S1097-2765(00)00113-1 [DOI] [PubMed] [Google Scholar]
- [11].Moffat J, Andrews B. Late-G1 cyclin-CDK activity is essential for control of cell morphogenesis in budding yeast. Nat Cell Biol [Internet] 2004; 6:59-66. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14688790; http://dx.doi.org/ 10.1038/ncb1078 [DOI] [PubMed] [Google Scholar]
- [12].McCusker D, Denison C, Anderson S, Egelhofer TA, Yates JR, Gygi SP, Kellogg DR. Cdk1 coordinates cell-surface growth with the cell cycle. Nat Cell Biol [Internet] 2007; 9:506-15. Available from: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=17417630&retmode=ref&cmd=prlinks\npapers2://publication/doi/10.1038/ncb1568; http://dx.doi.org/ 10.1038/ncb1568 [DOI] [PubMed] [Google Scholar]
- [13].Wai SC, Gerber SA, Li R. Multisite phosphorylation of the guanine nucleotide exchange factor Cdc24 during yeast cell polarization. PLoS One [Internet] 2009; 4:e6563 Available from: http://www.ncbi.nlm.nih.gov/pubmed/19668330; http://dx.doi.org/ 10.1371/journal.pone.0006563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shimada Y, Gulli MP, Peter M. Nuclear sequestration of the exchange factor Cdc24 by Far1 regulates cell polarity during yeast mating. Nat Cell Biol 2000; 2:117-24; PMID:10655592; http://dx.doi.org/ 10.1038/35000073 [DOI] [PubMed] [Google Scholar]
- [15].Nern A, Arkowitz RA. Nucleocytoplasmic shuttling of the Cdc42p exchange factor Cdc24p. J Cell Biol 2000; 148:1115-22; PMID:10725324; http://dx.doi.org/ 10.1083/jcb.148.6.1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Holt LJ, Tuch BB, Villén J, Johnson AD, Gygi SP, Morgan DO. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science [Internet] 2009; 325:1682-6. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2813701&tool=pmcentrez&rendertype=abstract\nhttp://www.ncbi.nlm.nih.gov/pubmed/19779198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Archambault V, Chang EJ, Drapkin BJ, Cross FR, Chait BT, Rout MP. Targeted proteomic study of the cyclin-Cdk module. Mol Cell 2004; 14:699-711; PMID:15200949; http://dx.doi.org/ 10.1016/j.molcel.2004.05.025 [DOI] [PubMed] [Google Scholar]
- [18].Ubersax J a, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO. Targets of the cyclin-dependent kinase Cdk1. Nature 2003; 425:859-64; PMID:14574415; http://dx.doi.org/ 10.1038/nature02062 [DOI] [PubMed] [Google Scholar]
- [19].Bhaduri S, Valk E, Winters MJ, Gruessner B, Loog M, Pryciak PM. A docking interface in the cyclin Cln2 promotes multi-site phosphorylation of substrates and timely cell-cycle entry. Curr Biol [Internet] 2015; 25:316-25. Available from: 10.1016/j.cub.2014.11.069; http://dx.doi.org/ 10.1016/j.cub.2014.11.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].He F, Nie W-C, Tong Z, Yuan S-M, Gong T, Liao Y, Bi E, Gao X-D. The GTPase-activating protein Rga1 interacts with Rho3 GTPase and may regulate its function in polarized growth in budding yeast. PLoS One [Internet] 2015; 10:e0123326 Available from: http://www.ncbi.nlm.nih.gov/pubmed/25860339; http://dx.doi.org/ 10.1371/journal.pone.0123326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Knaus M, Pelli-Gulli M-P, van Drogen F, Springer S, Jaquenoud M, Peter M. Phosphorylation of Bem2p and Bem3p may contribute to local activation of Cdc42p at bud emergence. EMBO J [Internet] 2007; 26:4501-13. Available from: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=17914457&retmode=ref&cmd=prlinks\npapers2://publication/doi/10.1038/sj.emboj.7601873; http://dx.doi.org/ 10.1038/sj.emboj.7601873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sopko R, Huang D, Smith JC, Figeys D, Andrews BJ. Activation of the Cdc42p GTPase by cyclin-dependent protein kinases in budding yeast. EMBO J [Internet] 2007; 26:4487-500. Available from: http://emboj.embopress.org/content/26/21/4487.abstract; http://dx.doi.org/ 10.1038/sj.emboj.7601847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zou J, Friesen H, Larson J, Huang D, Cox M, Tatchell K, Andrews B. Regulation of cell polarity through phosphorylation of Bni4 by Pho85 G1 cyclin-dependent kinases in Saccharomyces cerevisiae. Mol Biol Cell [Internet] 2009; 20:3239-50. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20496754; http://dx.doi.org/ 10.1091/mbc.E08-12-1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sudbery P, Gow N, Berman J. The distinct morphogenic states of Candida albicans. Trends Microbiol 2004; 12:317-24; PMID:15223059; http://dx.doi.org/ 10.1016/j.tim.2004.05.008 [DOI] [PubMed] [Google Scholar]
- [25].Court H, Sudbery P. Regulation of Cdc42 GTPase activity in the formation of hyphae in Candida albicans. Mol Biol Cell [Internet] 2007; 18:265-81. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17538018; http://dx.doi.org/ 10.1091/mbc.E06-05-0411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zheng X-D, Lee RTH, Wang Y-M, Lin Q-S, Wang Y. Phosphorylation of Rga2, a Cdc42 GAP, by CDK/Hgc1 is crucial for Candida albicans hyphal growth. EMBO J [Internet] 2007; 26:3760-9. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1952229&tool=pmcentrez&rendertype=abstract; http://dx.doi.org/ 10.1038/sj.emboj.7601814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Alvarez-Tabarés I, Pérez-Martín J. Cdk5 kinase regulates the association between adaptor protein Bem1 and GEF Cdc24 in the fungus Ustilago maydis. J Cell Sci [Internet] 2008; 121:2824-32. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18682498; http://dx.doi.org/ 10.1242/jcs.026286 [DOI] [PubMed] [Google Scholar]
- [28].Mahlert M, Leveleki L, Hlubek A, Sandrock B, Bölker M. Rac1 and Cdc42 regulate hyphal growth and cytokinesis in the dimorphic fungus Ustilago maydis. Mol Microbiol 2006; 59:567-78; PMID:16390450; http://dx.doi.org/ 10.1111/j.1365-2958.2005.04952.x [DOI] [PubMed] [Google Scholar]
- [29].Han BK, Bogomolnaya LM, Totten JM, Blank HM, Dangott LJ, Polymenis M. Bem1p, a scaffold signaling protein, mediates cyclin-dependent control of vacuolar homeostasis in Saccharomyces cerevisiae. Genes Dev 2005; 19:2606-18; PMID:16230527; http://dx.doi.org/ 10.1101/gad.1361505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kono K, Nogami S, Abe M, Nishizawa M, Morishita S, Pellman D, Ohya Y. G1/S cyclin-dependent kinase regulates small GTPase Rho1p through phosphorylation of RhoGEF Tus1p in Saccharomyces cerevisiae. Mol Biol Cell [Internet] 2008; 19:1763-71. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18256282; http://dx.doi.org/ 10.1091/mbc.E07-09-0950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Pnas 2008; 105:10762-7; PMID:18669648; http://dx.doi.org/ 10.1073/pnas.0805139105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bishop A, Lane R, Beniston R, Chapa-y-Lazo B, Smythe C, Sudbery P. Hyphal growth in Candida albicans requires the phosphorylation of Sec 2 by the Cdc28-Ccn1/Hgc1 kinase. EMBO J [Internet] 2010; 29:2930-42. Available from: 10.1038/emboj.2010.158; http://dx.doi.org/ 10.1038/emboj.2010.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Drobnjak M, Osman I, Scher HI, Fazzari M, Cordon-Cardo C. Overexpression of cyclin D1 is associated with metastatic prostate cancer to bone. Clin Cancer Res 2000; 6:1891-5; PMID:10815912 [PubMed] [Google Scholar]
- [34].Li Z, Wang C, Jiao X, Lu Y, Fu M, Quong A a, Dye C, Yang J, Dai M, Ju X, et al.. Cyclin D1 regulates cellular migration through the inhibition of thrombospondin 1 and ROCK signaling. Mol Cell Biol [Internet] 2006. [cited 2014December12]; 26:4240-56. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1489104&tool=pmcentrez&rendertype=abstract; http://dx.doi.org/ 10.1128/MCB.02124-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Burridge K, Guilluy C. Focal adhesions, stress fibers and mechanical tension. Exp Cell Res [Internet] 2016; 343:14-20. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26519907; http://dx.doi.org/ 10.1016/j.yexcr.2015.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev [Internet] 2004; 18:862-76. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15078817; http://dx.doi.org/ 10.1101/gad.1185504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev [Internet] 2006; 20:47-64. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16391232; http://dx.doi.org/ 10.1101/gad.1384406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li Z, Jiao X, Wang C, Ju X, Lu Y, Yuan L, Lisanti MP, Katiyar S, Pestell RG. Cyclin D1 induction of cellular migration requires p27(KIP1). Cancer Res 2006; 66:9986-94; PMID:17047061; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-1596 [DOI] [PubMed] [Google Scholar]
- [39].Deakin NO, Turner CE. Paxillin comes of age. J Cell Sci [Internet] 2008; 121:2435-44. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18650496; http://dx.doi.org/ 10.1242/jcs.018044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ishibe S, Joly D, Liu Z, Cantley L. Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol Cell [Internet] 2004. [cited 2015January14]; 16:257-67. Available from: http://www.sciencedirect.com/science/article/pii/S109727650400615X; http://dx.doi.org/ 10.1016/j.molcel.2004.10.006 [DOI] [PubMed] [Google Scholar]
- [41].Huang Z, Yan D-P, Ge B-X. JNK regulates cell migration through promotion of tyrosine phosphorylation of paxillin. Cell Signal [Internet] 2008. [cited 2014December12]; 20:2002-12. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18713649; http://dx.doi.org/ 10.1016/j.cellsig.2008.07.014 [DOI] [PubMed] [Google Scholar]
- [42].Fusté NP, Fernández-Hernández R, Cemeli T, Mirantes C, Pedraza N, Rafel M, Torres-Rosell J, Colomina N, Ferrezuelo F, Dolcet X, et al.. Cytoplasmic cyclin D1 regulates cell invasion and metastasis through the phosphorylation of paxillin. Nat Commun [Internet] 2016. [cited 2016June8]; 7:11581 Available from: http://www.nature.com/doifinder/10.1038/ncomms11581; http://dx.doi.org/ 10.1038/ncomms11581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature 2009; 461:99-103; PMID:19693013; http://dx.doi.org/ 10.1038/nature08242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bodemann BO, White MA. Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat Rev Cancer [Internet] 2008; 8:133-40. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18219307; http://dx.doi.org/ 10.1038/nrc2296 [DOI] [PubMed] [Google Scholar]
- [45].Fernández RMH, Ruiz-Miró M, Dolcet X, Aldea M, Garí E. Cyclin D1 interacts and collaborates with Ral GTPases enhancing cell detachment and motility. Oncogene [Internet] 2011; 30:1936-46. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21242975; http://dx.doi.org/ 10.1038/onc.2010.577 [DOI] [PubMed] [Google Scholar]
- [46].Bustelo XR. A transcriptional cross-talk between RhoA and c-Myc inhibits the RhoA/Rock-dependent cytoskeleton. Small GTPases 2010; 1:69-74; PMID:21686122; http://dx.doi.org/ 10.4161/sgtp.1.1.12986 [DOI] [PMC free article] [PubMed] [Google Scholar]