

Abstract
Dozens of genes have been implicated in late onset Alzheimer's disease (AD) risk, but none has a defined mechanism of action in neurons. Here, we show that the risk factor Pyk2 (PTK2B) localizes specifically to neurons in adult brain. Absence of Pyk2 has no major effect on synapse formation or the basal parameters of synaptic transmission in the hippocampal Schaffer collateral pathway. However, the induction of synaptic LTD is suppressed in Pyk2-null slices. In contrast, deletion of Pyk2 expression does not alter LTP under control conditions. Of relevance for AD pathophysiology, Pyk2−/− slices are protected from amyloid-β-oligomer (Aβo)-induced suppression of LTP in hippocampal slices. Acutely, a Pyk2 kinase inhibitor also prevents Aβo-induced suppression of LTP in WT slices. Female and male transgenic AD model mice expressing APPswe/PSEN1ΔE9 require Pyk2 for age-dependent loss of synaptic markers and for impairment of learning and memory. However, absence of Pyk2 does not alter Aβ accumulation or gliosis. Therefore, the Pyk2 risk gene is directly implicated in a neuronal Aβo signaling pathway impairing synaptic anatomy and function.
SIGNIFICANCE STATEMENT Genetic variation at the Pyk2 (PTK2B) locus is a risk for late onset Alzheimer's disease (AD), but the pathophysiological role of Pyk2 is not clear. Here, we studied Pyk2 neuronal function in mice lacking expression with and without transgenes generating amyloid-β (Aβ) plaque pathology. Pyk2 is not required for basal synaptic transmission or LTP, but participates in LTD. Hippocampal slices lacking Pyk2 are protected from AD-related Aβ oligomer suppression of synaptic plasticity. In transgenic AD model mice, deletion of Pyk2 rescues synaptic loss and learning/memory deficits. Therefore, Pyk2 plays a central role in AD-related synaptic dysfunction mediating Aβ-triggered dysfunction.
Keywords: Alzheimer's, amyloid, Pyk2, synapse
Introduction
Alzheimer's disease (AD) afflicts >5 million people in the United States and 45 million worldwide (Alzheimer's Association, 2012). Although the pathology, biomarkers, and early onset dominant cases demonstrate that amyloid-β (Aβ) peptide accumulation triggers downstream neuroinflammation, tau tangle pathology, and eventual cell loss, the biochemistry of AD remains poorly defined (Hardy and Selkoe, 2002; Selkoe, 2011; Heneka et al., 2015; De Strooper and Karran, 2016). Importantly, synapse loss in AD is correlated with progression (Selkoe, 2002; Scheff et al., 2006, 2007).
Genetic studies of late-onset AD (LOAD) document a range of risk factors (Lambert et al., 2013). A number have been linked to neuroinflammation, endocytosis, and protein processing (Holtzman et al., 2011; Guerreiro et al., 2013; Jonsson et al., 2013; Hong et al., 2016; Pimenova et al., 2018). However, implication of risk genes in neuronal pathways proximal to synapse loss has remained nil.
Among LOAD risk genes is Pyk2 (PTK2B, FAK2), a kinase similar to FAK (focal adhesion kinase) (Mitra et al., 2005). FAK is expressed widely, whereas Pyk2 is the most prominent in the CNS (Zhang et al., 1994; Menegon et al., 1999). Previous work has implicated Pyk2 in synaptic plasticity (Huang et al., 2001; Bartos et al., 2010; Hsin et al., 2010; Giralt et al., 2017). Pyk2 protein is activated by intracellular calcium and phosphorylated by Fyn to achieve full activation (Dikic et al., 1996; Qian et al., 1997; Andreev et al., 2001; Park et al., 2004; Collins et al., 2010a,b). Despite proof that Pyk2 variation alters AD risk, its role in Aβ synaptic pathophysiology is unclear. A study of tau pathology suggested that the Drosophila homolog contributes to neurodegeneration (Dourlen et al., 2017) and Pyk2 is reported to interact directly with tau (Li and Götz, 2018).
Studies of AD-related signaling at synapses have revealed a toxic effect of fibril-free soluble Aβ oligomers (Aβo's) (Lambert et al., 1998; Walsh et al., 2002; Shankar et al., 2008) and work from our laboratory demonstrated an Aβo-dependent correlation of Pyk2 activity with AD. Specifically, we described a pathway in which Aβo bind to the cellular prion protein (PrPC), creating a cell surface complex that activates mGluR5 (Laurén et al., 2009; Um et al., 2012, 2013). Transmembrane mGluR5 stimulates intracellular Fyn and increases intracellular calcium. Although PrPC is not required in all experimental Aβ/AD models, pharmacological blockade or genetic deletion of PrPC, mGluR5, or Fyn rescues Aβo and AD transgene phenotypes in multiple experiments (for review, see Salazar and Strittmatter, 2017; Purro et al., 2018). Phenotypes dependent on this pathway include suppressed synaptic plasticity, synapse loss, memory impairment, and tau accumulation, but not Aβ deposition and gliosis. These in vivo findings are consistent with previous in vitro work showing that Aβo inhibition of LTP, enhancement of LTD, and damage to dendritic spines requires Fyn (Lambert et al., 1998). With regard to Pyk2, there is direct interaction and cross activation with Fyn (Qian et al., 1997; Andreev et al., 2001; Collins et al., 2010a; Kaufman et al., 2015; Zhao et al., 2016). Moreover, the transgenic AD mice exhibit elevated Pyk2 function, which is normalized by PrPC deletion, by mGluR5 deletion or inhibition, or by Fyn inhibition in parallel with rescue of synapses and memory (Kaufman et al., 2015; Haas and Strittmatter, 2016; Haas et al., 2016, 2017). It has also been reported that the Pyk2 homolog FAK is activated by soluble Aβo (Zhang et al., 1994).
Based on Pyk2 being a LOAD risk gene with synaptic localization, a molecular link to Aβo signaling and a correlation with transgenic AD models, we sought to characterize synaptic function in mice lacking Pyk2 and to determine whether Pyk2 mediates synaptic phenotypes triggered by Aβo. Transgenic AD model mice lacking Pyk2 are protected from synapse loss and memory impairment. Therefore, the LOAD risk gene Pyk2 is coupled to an Aβo signaling pathway and is a proximal mediator of synapse loss.
Materials and Methods
Animals
All mice were cared for by the Yale Animal Resource Center. Yale's institutional animal care and use committee approved all experiments. The AD model mouse strain used was the APPswe/PSEN1ΔE9 line (APP/PS1) on a C57BL/6J background, originally purchased from The Jackson Laboratory (RRID:MMRRC_034832-JAX) (Jankowsky et al., 2003). The Pyk2−/− mice were generated by Schlessinger and colleagues (Okigaki et al., 2003) (RRID:MGI_3584536) and generously provided on the C57BL/6J background after 10 backcrosses by Dr. David Schlaepfer (University of California–San Diego). All experiments used littermate control mice with no preference for male or female mice. The percentage of female mice in the APP/PS1 and the APP/PS1, Pyk2−/− groups was 42% and 56%, respectively, for Figures 6, 7, and 8.
Figure 6.

Pyk2 mediates learning and memory deficits in APP/PS1 mice. A, Twelve-month-old mice and age-matched litter mate controls were subjected to the MWM test. Latency was calculated as the time to find the hidden platform across six blocks of four trials. APP/PS1 mice (n = 12) took significantly more time to find the hidden platform compared with WT (n = 11, p < 0.0001), Pyk2−/− (n = 9, p = 0.0227), or APP/PS1;Pyk2−/− mice (n = 9, p = 0.0137). Data are graphed as mean ± SEM, two-way repeated-measures ANOVA with Tukey's multiple-comparisons test: *p < 0.05, ****p < 0.0001. B, The probe trial was performed 24 h after the sixth forward swim trial block by removing the hidden platform. Quadrant time (%) is calculated as the percentage time spent in the target quadrant in 1 min. APP/PS1 (n = 12) mice spend significantly less time in the target quadrant compared with WT (n = 11, p = 0.0039), Pyk2−/− (n = 9, p = 0.0201), or APP/PS1;Pyk2−/− mice (n = 9, p = 0.0105). Data are graphed as mean ± SEM, one-way ANOVA with Tukey's multiple-comparisons test: *p < 0.05, **p < 0.01. C, Mice were subjected to the NOR test by familiarizing them to an object and subsequently allowed to explore a novel object and familiar 1 h after exposure to the familiar object. APP/PS1 mice (n = 14, p = 0.9705) did not display a preference for either familiar or novel object compared with WT (n = 11, p < 0.0001), Pyk2−/− (n = 9, p = 0.0336), and APP/PS1;Pyk2−/− mice (n = 10, p = 0.0014), which spent significantly more time with the novel object. Data are graphed as mean ± SEM, two-way ANOVA with Tukey's multiple-comparisons test: *p < 0.05, **p < 0.01, ****p < 0.0001. D, Mice were subjected to PAT and latency to enter the dark chamber was measured initially and 24 h after receiving a mild foot shock in the dark chamber. APP/PS1 mice (n = 16) reentered the dark chamber significantly more rapidly compared with WT (n = 14, p = 0.0010), Pyk2−/− (n = 11, p = 0.0053), or APP/PS1;Pyk2−/− mice (n = 10, p = 0.0089). Data are graphed as mean ± SEM, two-way ANOVA with Tukey's multiple-comparisons test: *p < 0.05, **p < 0.01. For details, see Table 1.
Figure 7.

Deletion of Pyk2 limits astrocytosis but has no effect on microgliosis or Aβ plaque burden. A, Representative immunofluorescent images of anti-GFAP staining in the hippocampus for the indicated genotypes. Scale bar, 50 μm. B, Quantification of percentage area immunoreactive for the astrocyte marker GFAP using an anti-GFAP antibody. APP/PS1 (n = 8 animals) mice have significantly more GFAP-immunoreactive percentage area compared with WT (n = 8, p < 0.0001) and Pyk2−/− (n = 8, p = 0.0002) mice, whereas APP/PS1;Pyk2−/− (n = 6, p = 0.0316) mice have significantly less immunoreactive percentage area than APP/PS1 mice but significantly more than WT mice (p = 0.0198). Data are graphed as mean ± SEM, where each n reflects one mouse (with three 40 μm sections averaged to a single value per mouse), one-way ANOVA with Tukey's multiple-comparisons test: *p < 0.05, ***p < 0.001, ****p < 0.0001. C, Representative immunofluorescent images of Iba1 in the hippocampus for the indicated genotypes. D, Quantification of percentage area immunoreactive for the microglial marker Iba1 using an anti-Iba1 antibody. APP/PS1 (n = 8) mice have significantly more Iba1-immunoreactive percentage area compared with WT (n = 8, p = 0.0090) and Pyk2−/− (n = 8, p = 0.0455) mice. Data are graphed as mean ± SEM, where each n reflects one mouse (with three 40 μm sections averaged to a single value per mouse), one-way ANOVA with Tukey's multiple-comparisons test: *p < 0.05, **p < 0.01. E, Representative immunofluorescent images of immunoreactive Aβ in the hippocampus for the indicated genotypes. Scale bar, 200 μm. F, Quantification of percentage area immunoreactive for Aβ using an anti-Aβ antibody. APP/PS1 (n = 8) and APP/PS1;Pyk2−/− (n = 6) mice do not have significantly different Aβ-immunoreactive percentage area (p = 0.8024). Data are graphed as mean ± SEM, where each n reflects one mouse (with three 40 μm sections averaged to a single value per mouse); Unpaired two-tailed t test. For details, see Table 1.
Figure 8.

Pyk2 mediates synapse loss in APP/PS1 mice A, Representative immunofluorescent images of immunoreactive SV2A in the mouse dentate gyrus for the indicated genotypes at 12 months of age. Scale bar, 10 μm. B, Quantification of percentage area immunoreactive for SV2A using an anti-SV2A antibody. WT mice (n = 8, p = 0.0222), Pyk2−/− mice (n = 8, p = 0.0008), and APP/PS1;Pyk2−/− (n = 8, p < 0.0001) mice have significantly higher SV2A-immunoreactive percentage area compared with APP/PS1 mice (n = 8). Data are graphed as mean ± SEM, where each n reflects one mouse (with three 40 μm sections averaged to a single value per mouse), one-way ANOVA with Tukey's multiple-comparisons test: *p < 0.05, ****p < 0.0001. C, Quantification of percentage area immunoreactive for PSD-95 using an anti-PSD-95 antibody. Pyk2−/− (n = 8, p = 0.0308) and APP/PS1;Pyk2−/− (n = 8, p = 0.0388) mice have significantly higher PSD-95-immunoreactive percentage area compared with APP/PS1 mice (n = 8), with WT (n = 8, p = 0.0831) mice displaying a modest increase in PSD-95 immunoreactivity compared with APP/PS1 mice. Data are graphed as mean ± SEM, where each n reflects one mouse (with three 40 μm sections averaged to a single value per mouse), one-way ANOVA with Holm–Sidak's multiple-comparisons test for the indicated pairs: *p < 0.05. For details, see Table 1.
Aβ1-42 preparation
Synthesized Aβ1-42 peptide was obtained from the Keck Large-Scale Peptide Synthesis Facility (Yale University). Aβo's were prepared in specially formulated glutamate-free F-12 medium to avoid direct stimulation of cultured neurons as described previously (Laurén et al., 2009). Concentrations of Aβo's are expressed in monomer equivalents, with 1 μm total Aβ1-42 peptide corresponding to ∼10 nm oligomeric species (Laurén et al., 2009). Each new preparation of Aβo was confirmed to be >95% high-molecular-weight soluble oligomers by size exclusion chromatography as described previously (Laurén et al., 2009; Um et al., 2012; Kaufman et al., 2015).
Brain tissue collection
Mice were killed and brains were dissected and divided at the midline into two hemispheres; one hemisphere was drop-fixed in 4% paraformaldehyde (PFA) for 24 h and the other hemisphere was flash frozen and stored at −80°C. Following PFA fixation, brains were stored in PBS with 0.05% sodium azide. For immunostaining, brains were cut into 40 μm coronal sections using a Leica WT1000S Vibratome. Sections were stored in PBS with 0.05% sodium azide at 4°C until staining. For biochemical analyses, animals were euthanized via live decapitation. Hippocampi and cortices were dissected on ice and immediately homogenized in 10 volumes (μl/mg tissue) Syn-PER synaptic protein extraction reagent (Thermo Scientific). Crude synaptosome pellets (P2′) were resuspended in 1% Triton X-100 with 150 mm NaCl and 50 mm Tris and 20% was removed, respun, and resolubilized in 1% SDS for Western blotting of the crude synaptosome fraction. The remaining 80% of the resuspended P2′ fraction was used for PSD isolation as described previously (Villasana et al., 2006). Protease and phosphatase inhibitors were used for every step during fractionation.
Immunoblotting
Samples were electrophoresed through 4–20% Tris-glycine gels (Bio-Rad) and transferred onto nitrocellulose membranes (Invitrogen) using an iBlot Gel Transfer Device (Novex-Life Technologies). Loaded sample volumes were normalized to total protein concentration as determined using a Pierce BCA Protein Assay Kit. Membranes were blocked at room temperature for 1 h in blocking buffer for fluorescent Western blotting (Rockland MB-070-010) and incubated in primary antibodies overnight at 4°C. The following primary antibodies were used for immunoblotting: anti-Pyk2 (Cell Signaling Technology, #3480S, 1:1000 RRID:AB_2174093 and Santa Cruz Biotechnology, #SC130077, 1:1000 RRID:AB_2174109), anti-pPyk2 Y402 (Abcam, #ab131543, 1:1000 RRID:AB_11157717), anti-PSD-95 (Millipore, #MAB1596, 1:1000 RRID:AB_2092365 and Synaptic Systems, #124-002, 1:2000 RRID:AB_887760), anti-NMDARB (Cell Signaling Technology, #4207S, 1:1000 RRID:AB_1264223), anti-pNR2B Y1472 (PhosphoSolutions, #p1516-1472, 1:1000 RRID:AB_2492182), anti-Fyn (Cell Signaling Technology, #4023S, 1:1000 RRID:AB_10698604 and Santa Cruz Biotechnology, #sc71133, 1:1000 RRID:AB_1123049), anti-pSFK Y416 (Cell Signaling Technology, #6943S, 1:1000 RRID:AB_10860257 and Cell Signaling Technology, #A2101, 1:1000 RRID:AB_331697), anti-β-actin (Cell Signaling Technology, #3700, 1:10,000 RRID:AB_2242334) and anti-actin (Sigma-Aldrich, #A2066, 1:2000 RRID:AB_476693). After primary antibody incubation, the membranes were washed and applied appropriate secondary antibodies (Odyssey donkey anti-rabbit, anti-mouse, or anti-chicken IRDye 680 or 800 conjugates, LI-COR Biosciences, RRID:AB_2716687, RRID:AB_10953628, RRID:AB_10974977, RRID:AB_621848, RRID:AB_621847, and RRID:AB_1850023) for 1 h at room temperature. The proteins were visualized using a LI-COR Odyssey infrared imaging system and quantified with Image Studio Lite software.
Immunohistology
Free-floating 40 μm sections were washed in a 24-well plate once with 0.1% PBS-Triton X-100 for 5 min (unless otherwise noted for a particular antibody). Subsequently, sections were blocked with 10% normal horse serum (NHS) in PBS for 1 h at room temperature. Sections were then incubated in primary antibody with 4% NHS in PBS overnight at 4°C. We used the following primary antibodies: rabbit anti-β-amyloid (Cell Signaling Technology, #2454, 1:250 RRID:AB_2056585), chicken anti-GFAP (Abcam, #ab4674, 1:500 RRID:AB_304558), rabbit anti-Iba1 (Wako Chemicals, #019-19741, 1:250 RRID:AB_839504), rabbit anti-PSD-95 (Invitrogen, #51-6900, 1:250 RRID:AB_2533914; this staining required antigen retrieval with for 5 min with 1% SDS in PBS at 90°C), and rabbit anti-SV2A (Abcam, #ab32942, 1:250 RRID:AB_778192). Sections were washed three times after primary incubation with PBS for 10 min each and then incubated with fluorescent secondary antibodies (donkey anti-rabbit, donkey anti-rat, or donkey anti-chicken; Invitrogen, Alexa Fluor, 1:500 RRID:AB_141637, RRID:AB_2535792, RRID:AB_2535794, RRID:AB_2535795, and RRID:AB_923386) for 1 h at room temperature. Samples were washed three times in PBS for 10 min each. To quench lipofuscin autofluoresence for Iba1 staining, we incubated samples in 10 mm CuSO4 in ammonium acetate for 15 min (Schnell et al., 1999) after secondary antibody. The samples were washed once with PBS and mounted onto glass slides (Superfrost Plus; Thermo Scientific) and mounted using Vectashield (Vector Laboratories).
Imaging and analysis of immunohistology
A Zeiss 710 confocal microscope with a 63× 1.4 numerical aperture oil-immersion lens was used to analyze PSD-95 and SV2A staining. Three 40 μm hippocampus-containing sections were chosen, each 160 μm apart. Three images of the mossy fibers in the dentate gyrus were obtained for each slice and ImageJ was used to calculate the area occupied by immunoreactive puncta and averaged per mouse. The same microscope was used to image GFAP and Iba1 staining. For GFAP and Iba1 staining, 3 × 2 20× tiled images were taken to capture the entirety of the hippocampus. This was done for each mouse for three sections from each mouse (160 μm apart). ImageJ was used to calculate the glial marker-positive area and averaged per mouse. We used a Zeiss AxioImager ZI fluorescent microscope (5× air-objective lens) to image and analyze Aβ staining and followed the same imaging and analysis procedures. In all cases, n = number of mice where the averaged percentage positive area for each derived from three 40 μm sections. All imaging and analyses were completed by an observer unaware of genotype.
Preparation of acute mouse brain slices
Mouse brains from 2-to 6-month-old WT or Pyk2−/− mice were dissected after rapid decapitation. Acute 400 μm coronal slices were cut in ice-cold artificial CSF (aCSF) containing the following (in mm): 119 NaCl, 2.5 KCl, 1.3 MgSO4, 26.2 NaHCO3, 11 D-glucose, and 1.25 NaH2PO4 using a 1000 Plus Vibratome with steel razor blades. Brain slices were then transferred to brain slice submersion chambers filled with aCSF with 2.4 mm CaCl2 at room temperature and incubated under constant oxygenation with 95% O2 and 5% CO2. Slices were allowed to recover for 45 min before any recording.
Electrophysiology
The 400 μm coronal slices were prepared as described above and submerged in a recording chamber (BSC-PC; Warner Instruments) containing aCSF with 2.4 mm CaCl2 that was continuously oxygenated (95% O2, 5% CO2). After the 45 min recovery period, slices were treated with Aβo (1 μm monomer, 10 nm oligomer estimate) or vehicle for 30 min before induction of LTP, with the identity of treatment and genotype blinded to the electrophysiologist. For PF-719 experiments, slices were preincubated with PF-719 (500 nm) for 30 min before recording. Extracellular field recordings were performed in the stratum radiatum of the hippocampus by stimulating Schaffer collateral fibers using a bipolar tungsten electrode (TM33CCNON; World Precision Instruments). Extracellular field excitatory postsynaptic potentials (fEPSPs) were recorded using a glass microelectrode (2–6 MΩ) (4878; World Precision Instruments) filled with aCSF and placed in the stratum radiatum of CA1 neurons. Test stimuli were induced at 0.033 Hz and the stimulus intensity was set at 50% of maximal fEPSP slope. A stable baseline was recorded for at least 20 min before induction of LTP and slices were excluded if baseline could not be stabilized after 1 h. LTP was induced by theta-burst stimulation (TBS) (10 bursts of 4 shocks at 100 Hz with an interburst interval of 200 ms) given at baseline intensity. LTD experiments were performed by delivering a low-frequency stimulus (LFS) at 300 pulses (5 min) or 900 pulses (15 min) at 1 Hz. Slices were excluded from analysis with preestablished cutoff for poor preparations in which 10 or more data points below 75% of baseline level during the post-TBS recording. For LTD experiments, slices were excluded if potentials drifted by >20% in the plateau period 30–60 min after LFS. The exclusion rates were low and did not differ by genotype: 13% for WT LTP, 10% for Pyk2−/− LTP, 14% for WT LTD, and 17% for Pyk2−/− LTD. fEPSPs were recorded using an Molecular Devices 700B amplifier and a Digidata 1440A digitizer, with data analysis performed by pClamp 10 software (Molecular Devices, RRID:SCR_011323) and Prism software (RRID:SCR_002798). All recording and analyses were completed by an observer unaware of genotype or treatment group.
PF-719 synthesis
The previously described PF-719 (Tse et al., 2012) small molecule was synthesized by Chinglu Pharmaceutical Research. They provided mass spectroscopy data with appropriate parent ions and NMR spectroscopy data confirming the structure of the compound with ∼95% purity. This was validated independently by our laboratory and confirmed the presence of the major peak with an appropriate m/z.
Behavioral testing
The order of behavioral testing was: novel object recognition (NOR), Morris water maze (MWM), and passive avoidance test (PAT) last. The interval between each behavioral test was 1 week. Exclusion criteria were used for each test as described below. Exclusion criteria for one test were independent of any other test. All mouse handling and analyses were completed by an observer unaware of genotype.
NOR.
As described in a previous study (Zhang et al., 2012), mice were handled for 5 min a day for 5 d before the experiment to reduce anxiety. Mice were habituated to clean, rectangular, and empty rat cages in a dimly lit behavioral testing room for 1 h. The cage was centered in front of the experimenter and oriented with the short axis perpendicular to the test administrator. During the acquisition trial, mice were removed from the behavioral cage and two identical objects, either a single 15 ml conical tube with orange cap or wrapped 5 ml plastic syringe (label side down), were placed one inch from the edge of either end of the long axis of the cage perpendicular to the long axis. Object choice was pseudorandom; the object was recorded as the familiar object for each animal. The mouse was then replaced in the cage's center not facing either object and a total timer counted up to 10 min. The mice were allowed to explore the object and the time it took to accumulate 30 s of total orofacial object exploration was recorded. Orofacial was defined as whisking or sniffing. Finally, mice were left for 10 min with the objects in the behavior cage. Then, the objects were removed and discarded and the mice were left in their cages for 1 h. During the test trial, one of both the novel and familiar objects were placed on pseudorandom sides of the cage. Orofacial exploration of each object was timed until a combined total of 30 s was reached. After the trial and acquisition trial of each mouse, cages were cleaned to eliminate scent cues. The experimenter was blinded to object novelty and genotype. Mice that did not explore both objects, that had barbered whiskers, or that failed to reach 30 s of exploration during either trial <6 min were eliminated.
MWM.
Animals were assigned a random code and the experimenter was blinded to genotype. Each mouse was handled for 5 min for the 5 d leading up to any behavioral testing to reduce anxiety. MWM testing was completed in 3 d of training, with the probe trial performed on the fourth day. Mice were repeatedly placed in an open water pool ∼1 m in diameter to find a submerged hidden platform. Clear and colorless water remained at room temperature throughout all aspects of the experiment. The location of the platform remained fixed in the center of one of the quadrants (target quadrant) of the pool throughout the entire testing period. Visual cues remained constant throughout forward and reverse swims. The mice had a total of eight attempts per day to locate the platform and training was divided into two blocks of four. The first block of four attempts was performed in the morning and the second in the afternoon. The order in which the mice were tested remained constant. The mice were gently placed into the pool facing the wall at one of four locations located in the opposite hemisphere from where the platform was and the latency to finding the platform was timed. The order of the four locations used to start the mice varied for each block to ensure that the mice used spatial cues to find the platform. Once a mouse spent 1 s on the platform, the attempt was considered complete and the mouse would be removed from the pool. If a mouse did not find the platform within 60 s, it was guided to the platform and allowed to spend 10 s on the platform, after which it was removed from the pool. This guiding was only performed for trials 1 and 2. Twenty-four hours after the completion of the last block, the mice were tested in a probe trial. The probe trial consisted of returning the mice to the pool to explore for a single trial of 60 s with the hidden platform removed. The start location was the point in the pool farthest from where the platform originally was placed. The latency to platform testing and the probe trials were recorded on a JVC Everio G-series camcorder and tracked by Panlab's Smart software.
PAT.
Following the administration of NOR and MWM, mice were subject to PAT using a box with equally sized, nonelectrified light and electrified dark compartments. The guillotine door between the two collapsed with the mouse's complete movement from the light to the dark side. On day 1 of testing, mice habituated to the light side for 90 s before the door opened. After this, the latency in seconds to enter the dark side was measured for up to 300 s. Mice received a shock lasting 2 s with an intensity of 0.5 mA on the dark side and were left in the dark for 10 s before returning to the home cage, as described previously (Filali and Lalonde, 2009). This was repeated after 5 min for each mouse. On day 2, the shock was lowered to 0 mA and the latency to enter the dark side after habituation was measured once more as a measure of retention of negative association. Perfect retention was considered the maximum latency of 5 min.
Experimental design and statistical analysis
One-way ANOVA with post hoc Tukey's multiple comparisons, one-way ANOVA with Holm–Sidak's multiple-comparisons tests, two-way ANOVA with Sidak multiple-comparisons tests, Kolmogorov–Smirnov test, repeated-measures ANOVA, and Student's t test, as specified in the figure legends, were performed using GraphPad Prism version 5.0d software (RRID:SCR_002798) and SPSS Statistics version 22 software (RRID:SCR_002865). Means ± SEM and specific n values are reported in each figure legend. Data are considered to be statistically significant if p < 0.05. The assumption of Gaussian distribution was checked using D'Agostino–Pearson omnibus test. With the exception of the PAT experiment, all data displayed a Gaussian distribution. For this reason, we performed a Kolmogorov–Smirnov test for the PAT experiment. For the PSD-95 immunohistochemical analyses, we hypothesized that APP/PS1;Pyk2−/− and WT samples were different from APP/PS1 samples based on the SV2A immunohistochemical data and therefore compared these conditions with the Holm–Sidak's multiple-comparisons test. Statistical comparisons for electrophysiological and anatomical experiments were performed with individual data points being the per-animal average. Statistical details are provided in Table 1.
Table 1.
Statistical analysis details of the numerical values, replicates, variance and statistical tests for the data presented in the indicated figures
| Figure | Assay performed | Parameter | Independent variables | n | Descriptive statistics |
Statistical analysis |
|||
|---|---|---|---|---|---|---|---|---|---|
| Average | Error (SD) | Statistical test | ANOVA p-values | Significance | |||||
| 2C | Western blotting | Protein (normalized by WT) | PSD95/actin | WT = 6 mice | 1.0000 | 0.0517 | |||
| Pyk2−/− = 6 mice | 0.9598 | 0.1363 | |||||||
| NR2B/actin | WT = 6 mice | 1.0000 | 0.1080 | ||||||
| Pyk2−/− = 6 mice | 0.9568 | 0.0449 | |||||||
| pNR2B/NR2B | WT = 6 mice | 1.0000 | 0.0653 | ||||||
| Pyk2−/− = 6 mice | 0.9792 | 0.0825 | |||||||
| Fyn/actin | WT = 6 mice | 1.0000 | 0.0904 | ||||||
| Pyk2−/− = 6 mice | 1.0064 | 0.1466 | |||||||
| pSFK/Fyn | WT = 6 mice | 1.0000 | 0.0896 | Unpaired two-tailed t test | p = 0.0378 for WT vs Pyk2−/− | ||||
| Pyk2−/− = 6 mice | 0.8358 | 0.1423 | |||||||
| D | Western blotting | Protein (normalized by WT) | PSD95/actin | WT = 6 mice | 1.0000 | 0.0951 | |||
| Pyk2−/− = 6 mice | 1.1029 | 0.0759 | |||||||
| NR2B/actin | WT = 6 mice | 1.0000 | 0.0867 | ||||||
| Pyk2−/− = 6 mice | 0.9953 | 0.1355 | |||||||
| pNR2B/NR2B | WT = 6 mice | 1.0000 | 0.0828 | ||||||
| Pyk2−/− = 6 mice | 0.9453 | 0.1494 | |||||||
| Fyn/actin | WT = 6 mice | 1.0000 | 0.1370 | ||||||
| Pyk2−/− = 6 mice | 0.9973 | 0.0846 | |||||||
| pSFK/Fyn | WT = 6 mice | 1.0000 | 0.1531 | ||||||
| Pyk2−/− = 6 mice | 0.9495 | 0.1604 | |||||||
| F | Western blotting | Protein (normalized by WT) | PSD95 | WT = 3 mice | 1.0000 | 0.4378 | |||
| Pyk2−/− = 3 mice | 0.8848 | 0.3470 | |||||||
| NR2B/PSD95 | WT = 3 mice | 1.0000 | 0.1528 | ||||||
| Pyk2−/− = 3 mice | 1.0518 | 0.2626 | |||||||
| pNR2B/NR2B | WT = 3 mice | 1.0000 | 0.3200 | ||||||
| Pyk2−/− = 3 mice | 0.7920 | 0.0823 | |||||||
| Fyn/PSD95 | WT = 3 mice | 1.0000 | 0.2094 | ||||||
| Pyk2−/− = 3 mice | 1.0943 | 0.3892 | |||||||
| pSFK/Fyn | WT = 3 mice | 1.0000 | 0.3812 | ||||||
| Pyk2−/− = 3 mice | 0.8294 | 0.3582 | |||||||
| H | Western blotting | Protein (normalized by WT) | PSD95/actin | WT = 6 mice | 1.0000 | 0.6456 | |||
| Pyk2−/− = 6 mice | 1.4363 | 0.9166 | |||||||
| NR2B/actin | WT = 6 mice | 1.0000 | 0.4588 | ||||||
| Pyk2−/− = 6 mice | 1.3994 | 0.6879 | |||||||
| pNR2B/NR2B | WT = 6 mice | 1.0000 | 0.1410 | ||||||
| Pyk2−/− = 6 mice | 0.9372 | 0.1787 | |||||||
| Fyn/actin | WT = 6 mice | 1.0000 | 0.0712 | ||||||
| Pyk2−/− = 6 mice | 1.0291 | 0.0599 | |||||||
| pSFK/Fyn | WT = 6 mice | 1.0000 | 0.0853 | ||||||
| Pyk2−/− = 6 mice | 1.0317 | 0.1116 | |||||||
| I | Western blotting | Protein (normalized by WT) | PSD95/actin | WT = 6 mice | 1.0000 | 0.6240 | |||
| Pyk2−/− = 6 mice | 1.2824 | 0.5764 | |||||||
| NR2B/actin | WT = 6 mice | 1.0000 | 0.6330 | ||||||
| Pyk2−/− = 6 mice | 1.2482 | 0.5186 | |||||||
| pNR2B/NR2B | WT = 6 mice | 1.0000 | 0.1719 | ||||||
| Pyk2−/− = 6 mice | 1.0389 | 0.1452 | |||||||
| Fyn/actin | WT = 6 mice | 1.0000 | 0.1256 | ||||||
| Pyk2−/− = 6 mice | 0.9929 | 0.1440 | |||||||
| pSFK/Fyn | WT = 6 mice | 1.0000 | 0.1089 | ||||||
| Pyk2−/− = 6 mice | 0.9502 | 0.1167 | |||||||
| K | Western blotting | Protein (normalized by WT) | PSD95 | WT = 6 mice | 1.0000 | 0.4375 | |||
| Pyk2−/− = 6 mice | 1.1086 | 0.2102 | |||||||
| NR2B/PSD95 | WT = 6 mice | 1.0000 | 0.2221 | ||||||
| Pyk2−/− = 6 mice | 1.1586 | 0.2256 | |||||||
| pNR2B/NR2B | WT = 6 mice | 1.0000 | 0.0891 | ||||||
| Pyk2−/− = 6 mice | 0.9372 | 0.0900 | |||||||
| Fyn/PSD95 | WT = 6 mice | 1.0000 | 0.3237 | ||||||
| Pyk2−/− = 6 mice | 1.0427 | 0.1791 | |||||||
| pSFK/Fyn | WT = 6 mice | 1.0000 | 0.2279 | ||||||
| Pyk2−/− = 6 mice | 1.0193 | 0.2492 | |||||||
| 3A | Input/output analysis | fEPSP | WT | n = 3 mice | 0.567 | 0.2122 | Two-way ANOVA Sidak's multiple-comparisons test | p > 0.9999 | All rows p > 0.9999 |
| Pyk2−/− | n = 3 mice | 0.4793 | 0.2226 | ||||||
| B | Paired-pulse facilitation | fEPSP | WT: 25 ms | n = 3 mice | 0.9976 | 0.0149 | Two-way ANOVA Sidak's multiple-comparisons test | p > 0.8293 | >0.9999 |
| WT: 50 ms | 1.1108 | 0.0171 | 0.8975 | ||||||
| WT: 100 ms | 1.1656 | 0.0122 | 0.9263 | ||||||
| WT: 200 ms | 1.1071 | 0.0278 | 0.9996 | ||||||
| WT: 300 ms | 1.0603 | 0.0061 | 0.9996 | ||||||
| Pyk2 −/−: 25 ms | n = 3 mice | 1.0013 | 0.0775 | >0.9999 | |||||
| Pyk2 −/−: 50 ms | 1.0728 | 0.0948 | 0.8975 | ||||||
| Pyk2 −/−: 100 ms | 1.1307 | 0.0697 | 0.9263 | ||||||
| Pyk2 −/−: 200 ms | 1.1183 | 0.0409 | 0.9996 | ||||||
| Pyk2 −/−: 300 ms | 1.0713 | 0.0484 | 0.9996 | ||||||
| D | LTD | fEPSP | WT | n = 6 mice | 102 | 16.67 | Student's two tailed t test | WT vs Pyk2−/−: p = 0.5149 | |
| Pyk2−/− | n = 6 mice | 110.6 | 26.5 | ||||||
| F | LTD | fEPSP | WT | n = 7 mice | 81.21 | 24.88 | Student's two tailed t test | WT vs Pyk2−/−: p = 0.0459 | |
| Pyk2−/− | n = 6 mice | 107.9 | 16 | ||||||
| 4B | LTP | fEPSP | WT (vehicle) | n = 19 mice | 143.1 | 21.21 | One-way ANOVA; Tukey's multiple-comparisons test | p = 0.0160 | WT (vehicle) vs WT (Aβo): p = 0.0428 |
| Pyk2 −/− (vehicle) | n = 9 mice | 150.6 | 59.96 | One-way ANOVA; Tukey's multiple-comparisons test | Pyk2 −/− (vehicle) vs WT (Aβo): p = 0.0342 | ||||
| WT (Aβo) | n = 11 mice | 110.3 | 20.58 | One-way ANOVA; Tukey's multiple-comparisons test | |||||
| Pyk2−/− (Aβo) | n = 8 mice | 150.1 | 16.81 | One-way ANOVA; Tukey's multiple-comparisons test | WT (Aβo) vs Pyk2−/− (Aβo): p = 0.0459 | ||||
| 5D | LTP | fEPSP | Vehicle | n = 11 mice | 158.4 | 28.24 | One-way ANOVA; Tukey's multiple-comparisons test | p = 0.0085 | Vehicle vs Aβo: p = 0.0068 |
| PF-719 | n = 9 mice | 142.4 | 27.81 | PF-719 vs Aβo: p = 0.1868 | |||||
| Vehicle + Aβo | n = 8 mice | 117.3 | 18.79 | ||||||
| PF-719 + Aβo | n = 6 mice | 154.9 | 20.32 | PF-719 + Aβo vs Aβo: p = 0.0424 | |||||
| 6A | MWM (forward swim) | Latency (s) | WT | WT = = 11 mice | 26.29 | 11.66 | Two-way ANOVA; Tukey's multiple-comparisons test | 0.0002 | 6th trial (vs APP/PS1): p < 0.0001 |
| Pyk2−/− | Pyk2−/− = = 9 mice | 32.49 | 12.68 | Two-way ANOVA; Tukey's multiple-comparisons test | 6th trial (vs APP/PS1): p = 0.0227 | ||||
| APP/PS1 | APP/PS1 = = 12 mice | 44.98 | 4.531 | Two-way ANOVA; Tukey's multiple-comparisons test | 6th trial (vs WT): p < 0.0001 | ||||
| APP/PS1; Pyk2−/− | APP/PS1;Pyk2−/− = = 9 mice | 35.48 | 11.11 | Two-way ANOVA; Tukey's multiple-comparisons test | 6th trial (vs APP/PS1): p = 0.0137 | ||||
| B | MWM (probe trial) | Quadrant time (%) | WT | WT = = 11 mice | 47.37 | 24.27 | One-way ANOVA; Tukey's multiple-comparisons test | 0.0018 | WT vs APP/PS1: p = 0.0039 |
| Pyk2−/− | Pyk2−/− = = 9 mice | 44.16 | 17.6 | One-way ANOVA; Tukey's multiple-comparisons test | Pyk2−/− vs APP/PS1: p = 0.0201 | ||||
| APP/PS1 | APP/PS1 = = 12 mice | 20.81 | 8.458 | One-way ANOVA; Tukey's multiple-comparisons test | WT vs APP/PS1: p = 0.0039 | ||||
| APP/PS1; Pyk2−/− | APP/PS1;Pyk2−/− = = 9 mice | 46.09 | 15.32 | One-way ANOVA; Tukey's multiple-comparisons test | APP/PS1 vs APP/PS1;Pyk2−/−: p = 0.0105 | ||||
| C | NOR | Exploration time (s) | WT | WT = = 11 mice | familiar = 10.33, novel = 19.67 | familiar: 4.663, novel: 4.663 | Two-way ANOVA; Tukey's multiple-comparisons test | p < 0.0001 | p < 0.0001 for familiar vs novel |
| Pyk2−/− | Pyk2−/− = = 9 mice | familiar = 12.15, novel = 17.85 | familiar: 2.342, novel: 2.342 | p = 0.0336 for familiar vs novel | |||||
| APP/PS1 | APP/PS1 = = 14 mice | familiar = 15.71, novel = 14.28 | familiar: 4.224, novel: 4.237 | p = 0.9705 for familiar vs novel | |||||
| APP/PS1; Pyk2−/− | APP/PS1;Pyk2−/− = = 10 mice | familiar = 11.47, novel = 118.53 | familiar: 2.49, novel: 2.49 | p = 0.0014 for familiar vs novel | |||||
| D | PAT | Latency (s) | WT | n = 14 mice | 253.1 | 90.89 | Kolmogorov–Smirnov test | WT vs APP/PS1: p = 0.0275 | |
| Pyk2−/− | n = 11 mice | 245.3 | 76.03 | Kolmogorov–Smirnov test | Pyk2−/− vs APP/PS1: p = 0.1026 | ||||
| APP/PS1 | n = 16 mice | 138.6 | 114.9 | Kolmogorov–Smirnov test | WT vs APP/PS1: p = 0.0275 | ||||
| APP/PS1; Pyk2−/− | n = 10 mice | 243.0 | 116 | Kolmogorov–Smirnov test | APP/PS1 vs APP/PS1;Pyk2−/−: p = 0.0483 | ||||
| 7B | Immunohistology | GFAP (% area) | WT | n = 8 mice | 1.651 | 0.6078 | One-way ANOVA; Tukey's multiple-comparisons test | p < 0.0001 | WT vs APP/PS1: p < 0.0001 |
| Pyk2−/− | n = 8 mice | 2.38 | 0.8658 | Pyk2−/− vs APP/PS1: p = 0.0002 | |||||
| APP/PS1 | n = 8 mice | 4.588 | 1.16 | APP/PS1 vs APP/PS1;Pyk2−/−: p = 0.0316 | |||||
| APP/PS1; Pyk2−/− | n = 6 mice | 3.168 | 0.8214 | ||||||
| D | Immunohistology | Iba1 (% area) | WT | n = 8 mice | 0.9382 | 0.3263 | One-way ANOVA; Tukey's multiple-comparisons test | p = 0.0026 | WT vs APP/PS1: p < 0.0090 |
| Pyk2 −/− | n = 8 mice | 1.107 | 0.4866 | Pyk2−/− vs APP/PS1: p = 0.0455 | |||||
| APP/PS1 | n = 8 mice | 1.784 | 0.5609 | APP/PS1 vs APP/PS1;Pyk2−/−: p > 0.9999 | |||||
| APP/PS1; Pyk2−/− | n = 6 mice | 1.769 | 0.5515 | ||||||
| F | Immunohistology | Aβ (% area) | APP/PS1 | n = 8 mice | 1.167 | 0.7319 | Student's two-tailed t test | 0.8024 | APP/PS1 vs APP/PS1;Pyk2−/−: p = 0.8024 |
| APP/PS1; Pyk2−/− | n = 6 mice | 1.251 | 0.3801 | ||||||
| 8F | Immunohistology | SV2A (% area) | WT | n = 8 mice | 13.7 | 0.8696 | One-way ANOVA; Tukey's multiple-comparisons test | p < 0.0001 | WT vs APP/PS1: p = 0.0222 |
| Pyk2−/− | n = 8 mice | 15.2 | 3.157 | One-way ANOVA; Tukey's multiple-comparisons test | Pyk2 −/− vs APP/PS1: p = 0.0008 | ||||
| APP/PS1 | n = 8 mice | 10.0 | 1.348 | One-way ANOVA; Tukey's multiple-comparisons test | WT vs APP/PS1: p = 0.0222 | ||||
| APP/PS1; Pyk2−/− | n = 8 mice | 16.3 | 3.078 | One-way ANOVA; Tukey's multiple-comparisons test | APP/PS1 vs APP/PS1;Pyk2−/−: p < 0.0001 | ||||
| G | Immunohistology | PSD-95 (% area) | WT | n = 8 mice | 5.070 | 0.9993 | One-way ANOVA; Holm–Sidak's multiple-comparisons test | p = 0.056 | WT vs APP/PS1: p = 0.0590 |
| Pyk2−/− | n = 8 mice | 5.351 | 1.474 | ||||||
| APP/PS1 | n = 8 mice | 4.012 | 1.094 | One-way ANOVA; Holm–Sidak's multiple-comparisons test | APP/PS1;Pyk2−/− vs APP/PS1: p = 0.0499 | ||||
| APP/PS1; Pyk2−/− | n = 8 mice | 5.288 | 1.083 | One-way ANOVA; Holm–Sidak's multiple-comparisons test | APP/PS1;Pyk2−/− vs APP/PS1: p = 0.0499 | ||||
Results
Pyk2 expression in neurons
We assessed the cellular localization of Pyk2 in brain. Although several studies have supported a neuronal function for Pyk2 (Huang et al., 2001; Bartos et al., 2010; Hsin et al., 2010; Kinoshita et al., 2014; Dourlen et al., 2017; Giralt et al., 2017), others have proposed a non-neuronal role for Pyk2 (Chan et al., 2015; Giralt et al., 2016). We characterized Pyk2 expression in mouse brain by immunohistology, with Pyk2−/− tissue for a control (Okigaki et al., 2003). We observed that the vast majority of Pyk2 is localized to the neuropil (Fig. 1A,B) and Pyk2 colocalizes with MAP2 and PSD95, consistent with a neuronal identity for Pyk2 protein (Fig. 1C,D). Furthermore, little or no Pyk2 is colocalized with the astrocyte marker GFAP or the microglial marker Iba1 (Fig. 1E,F). In addition, there was no overt change in Pyk2 localization in samples from aged transgenic AD mice in the vicinity of Aβ plaque or between them (Fig. 1C–F).
Figure 1.

Pyk2 is expressed by neurons. A, B, Representative immunofluorescent images of Pyk2 immunoreactivity from hippocampal CA1 in WT and Pyk2-null mice at 12 months of age. Scale bar, 10 μm. Confocal images were captured using a 63× objective. C–F, Immunohistology was performed on age-matched littermate controls from WT, Pyk2−/−, and APP/PS1 mice at 12 months of age. Scale bar, 10 μm. Confocal images were captured using a 63× objective. Representative immunofluorescent images of immunoreactive Pyk2-red (same as A) and MAP2-green in CA1 (C), Pyk2-red and PSD95-green in CA1 (D), Pyk2-red and Iba1-green in the stratum radiatum (E), and Pyk2-red and GFAP-green in the stratum radiatum (F).
Characterization of neuronal function in Pyk2−/− mice
We considered whether Pyk2 might be required for synaptic development and anatomy. Multiple synaptic markers are similar in the synaptomsomal and postsynaptic density fractions from Pyk2−/− and WT mouse brain (Fig. 2, Table 1). This is true at 6 months of age (Fig. 2A–F) and 12 months of age (Fig. 2G–K), spanning the time period between the onset of Aβ plaque accumulation and the induction of synaptic loss and impairments in learning and memory in the APPswe/PS1ΔE9 transgenic mice used for the AD-related studies described below. The only significant difference between biochemical markers in the two genotypes is a reduction of the phospho-SFK/Fyn ratio in the cerebral cortex of Pyk2−/− mice at 6–8 months of age. This minor decrease in Fyn activation is consistent with known synergistic interaction of these two tyrosine kinases. By these measures, Pyk2 is nonessential or redundant with regard to basic markers of adult mouse brain synaptic maintenance.
Figure 2.

Normal synaptic markers in Pyk2−/− mice. A, Pyk2 expression in WT and Pyk2−/− mice forebrains. Lysates (1% Triton X-100, 150 mm NaCl, 50 mm Tris, protease and phosphatase inhibitor cocktails) from 6-month-old WT and Pyk2−/− mice forebrains were subjected to immunoblotting with anti-Pyk2 antibody, which recognize N terminus region (∼1–100 aa) of Pyk2. There is no full-length or truncated protein in the null mice. B–D, Synaptic protein expression profile in cortex and hippocampus from 6- to 8-month-old WT and Pyk2−/− mice. Syn-PER and 1% Triton X-100 insoluble cortex and hippocampus crude synaptosome pellets (P2′) were resolubilized in 1% SDS and immunoblotted with anti-Pyk2, anti-PSD-95, anti-pNR2B (pY1472), anti-NR2B, anti-pSFK, anti-Fyn, and anti-actin specific antibodies. Quantification of protein levels by densitometric analysis in cortex (C) and hippocampus (D). Cortical levels of pSFK/Fyn were significantly reduced in Pyk2−/− mice (n = 6, p = 0.038). Protein levels were normalized to WT values. Data are graphed as mean ± SEM (n = 6 mice). *p < 0.05, unpaired two-tailed t test. E, F, Synaptic protein expression profile in hippocampal PSD fraction of 6- to 8-month-old WT and Pyk2−/− mice. PSD fraction (20 μg of protein) was subjected to immunoblotting with indicated antibodies (E) and quantified by densitometric analysis (F). Protein levels were normalized to WT values. Data are graphed as mean ± SEM (n = 3 mice). G–I, Synaptic protein expression profile in cortex and hippocampus from 12-month-old WT and Pyk2−/− mice. Quantification of protein levels by densitometric analysis in cortex (H) and hippocampus (I). Protein levels were normalized to WT values. Data are graphed as mean ± SEM (n = 6 mice). For cortical pSFK/Fyn, n = 5 mice. J, K, Synaptic protein expression profile in hippocampal PSD fraction of 12-month-old WT and Pyk2−/− mice. PSD fraction (2.25 μg of protein) subjected to immunoblotting with indicated antibodies (J) and quantified by densitometric analysis (K). Protein levels were normalized to WT values. Data are graphed as mean ± SEM (n = 6 mice). For C, D, F, H, I, and K, all comparisons were nonsignificant by unpaired two-tailed t test unless otherwise specified (p > 0.05). For details, see Table 1.
We sought to define the electrophysiological function of synaptic networks lacking Pyk2, focusing on fEPSPs at Schaffer collaterals of the CA3–CA1 hippocampal circuit. As a first step, we assessed baseline input/output curves and paired pulse facilitation in WT and Pyk2−/− slices and observed no differences between genotypes (Fig. 3A,B). Pyk2 has previously been implicated in LTD (Hsin et al., 2010), but this has not been assessed in the Pyk2-null condition. To more fully characterize the role(s) of Pyk2 in synaptic plasticity, we monitored hippocampal fEPSPs while inducing LTD with LFS protocols (Li et al., 2009) (Fig. 3C–F). A weak LTD protocol (300 pulses at 1 Hz) does not induce LTD in either WT or Pyk2-null slices, whereas a more robust LTD protocol (900 pulses at 1 Hz) induces LTD within the last 20 min of recording for WT slices, but not Pyk2−/− slices (Fig. 3D,E). These data suggest that Pyk2 is required for the induction of LTD when using a 15 min LFS.
Figure 3.

Pyk2 mediates LTD in the CA1 of hippocampal slices. fEPSPs were recorded from the CA3–CA1 circuit in slices of mouse hippocampus from 2- to 6-month-old mice. A, Input/output (I/O) responses were graphed as fEPSPs (mV/ms) with respect to stimulus intensity (V) at 40 μs duration. We observed no change in baseline synaptic transmission from WT (9 slices from n = 3 mice) and Pyk2−/− (9 slice from n = 3 mice) slices by two-way ANOVA with Sidak's multiple-comparisons test (p > 0.9999 for all comparisons). Data are analyzed and graphed as mean ± SEM, where each n reflects one mouse with data from multiple slices averaged to a single value per mouse. B, Paired-pulse ratios (fEPSP2slope/fEPSP1slope) in WT (9 slices from n = 3 mice) and Pyk2−/− (9 slices from n = 3 mice) slices were not significantly different by two-way ANOVA with Sidak's multiple-comparisons test (25 ms p > 0.9999, 50 ms p = 0.8975, 100 ms p = 0.9263, 200 ms p = 0.9996, and 300 ms p = 0.9996). Statistics were calculated using per-animal data. C, Acute brain slices from WT or Pyk2−/− mice were used to record fEPSPs from the CA3–CA1 hippocampal circuit. The slope of the fEPSPs is plotted as a function of time. Representative traces before a 5 min LFS in black (black arrowhead at time = 0) and at 60 min after LFS in red are superimposed (average, 6 sweeps). D, WT (8 slices from n = 6 mice) slices did not show a significant difference in fEPSP during the last 20 min of recording compared with Pyk2−/− slices (7 slices from n = 6 mice). Data are graphed as mean ± SEM, unpaired two-tailed t test. Statistics were calculated using per-animal data. E, Similar to C, hippocampal slices were used to record fEPSPs. Representative traces before a 15 min LFS in black (black arrowhead at time = 0) and at 60 min after LFS in red are superimposed (average, 6 sweeps). F, WT (13 slices from n = 7 mice) slices displayed a significant decrease in fEPSP during the last 20 min of recording compared with Pyk2−/− slices (12 slices from n = 6 mice). Data are graphed as mean ± SEM, unpaired two-tailed t test. All statistics were calculated using per-animal average data. *p < 0.05. For details, see Table 1.
To test our central hypothesis that Pyk2 mediates AD related synaptic dysfunction and loss, we measured LTP in acute hippocampal slices exposed to Aβo. Acute treatment with Aβo is documented to suppress LTP in acute hippocampal slices (Lambert et al., 1998; Walsh et al., 2002; Laurén et al., 2009; Haas et al., 2016). Here, we measured LTP by recording fEPSPs in the CA3–CA1 hippocampal circuit from WT and Pyk2−/− hippocampal slices treated with vehicle and Aβo. Specifically, we measured fEPSPs before and after TBS and quantified the average of the last 10 min of recording to assess LTP. Importantly, Pyk2−/− slices treated with vehicle are not different from WT vehicle-treated slices, showing that Pyk2 is not required for the induction of LTP (Fig. 4A,B,D). Furthermore, WT slices treated with Aβo display a significant decrease in fEPSP slope during the last 15 min of recording after TBS compared with WT slices treated with vehicle (Fig. 4A–C). Strikingly, Pyk2−/− slices treated with Aβo did not display a decrease in fEPSPs compared with Pyk2−/− vehicle-treated slices and fEPSPs were similar to that of WT vehicle-treated slices (Fig. 4A–D).
Figure 4.

Pyk2 mediates Aβo-dependent deficit in LTP. A, C, D, Acute brain slices from 2- to 6-month-old WT or Pyk2−/− mice were treated with vehicle or Aβo and used to record fEPSPs from the CA3–CA1 hippocampal circuit. The slope of the fEPSPs is plotted as a function of time. Representative traces before TBS in black (black arrowhead at time = 0) and at 60 min after TBS in red are superimposed (average, 6 sweeps) in the respective graphs. Data are graphed as mean ± SEM, where each n reflects one mouse with multiple slices averaged to a single value per mouse. Slices were treated with vehicle (F12 without: glutamate, glycine, and phenol red) or Aβo (1 μm monomer, 10 nm oligomer estimate) for 30 min before TBS. B, WT slices treated with Aβo (13 slices from n = 11 mice) showed a significant decrease in fEPSP compared with WT slices treated with vehicle (22 slices from n = 19 mice), Pyk2−/− slice treated with vehicle (11 slices from n = 9 mice), or Pyk2−/− slices treated with Aβo (10 slices from n = 8 mice) in the last 15 min of recording. Data are graphed as mean ± SEM, one-way ANOVA with Tukey's multiple-comparisons test. All statistics were calculated using per-animal average data. *p < 0.05. For details, see Table 1.
The Pyk2−/− slices lack the kinase constitutively and might therefore have chronic compensatory changes. To acutely inhibit Pyk2, we used the selective inhibitor PF-719 (Tse et al., 2012). In particular, we sought to determine whether acute Pyk2 kinase inhibition might rescue Aβo-dependent deficits in LTP. We observed a significant increase in LTP with WT Aβo slices pretreated with PF-719 compared with Aβo alone (Fig. 5). Overall, these findings suggest that Pyk2 has a physiological role in LTD-based synaptic plasticity even though synaptic markers of baseline hippocampal electrophysiology and LTP do not require Pyk2. Most critically, even though Pyk2 is not required for the induction of LTP, Pyk2 is essential for the Aβo-dependent deficits observed in LTP.
Figure 5.
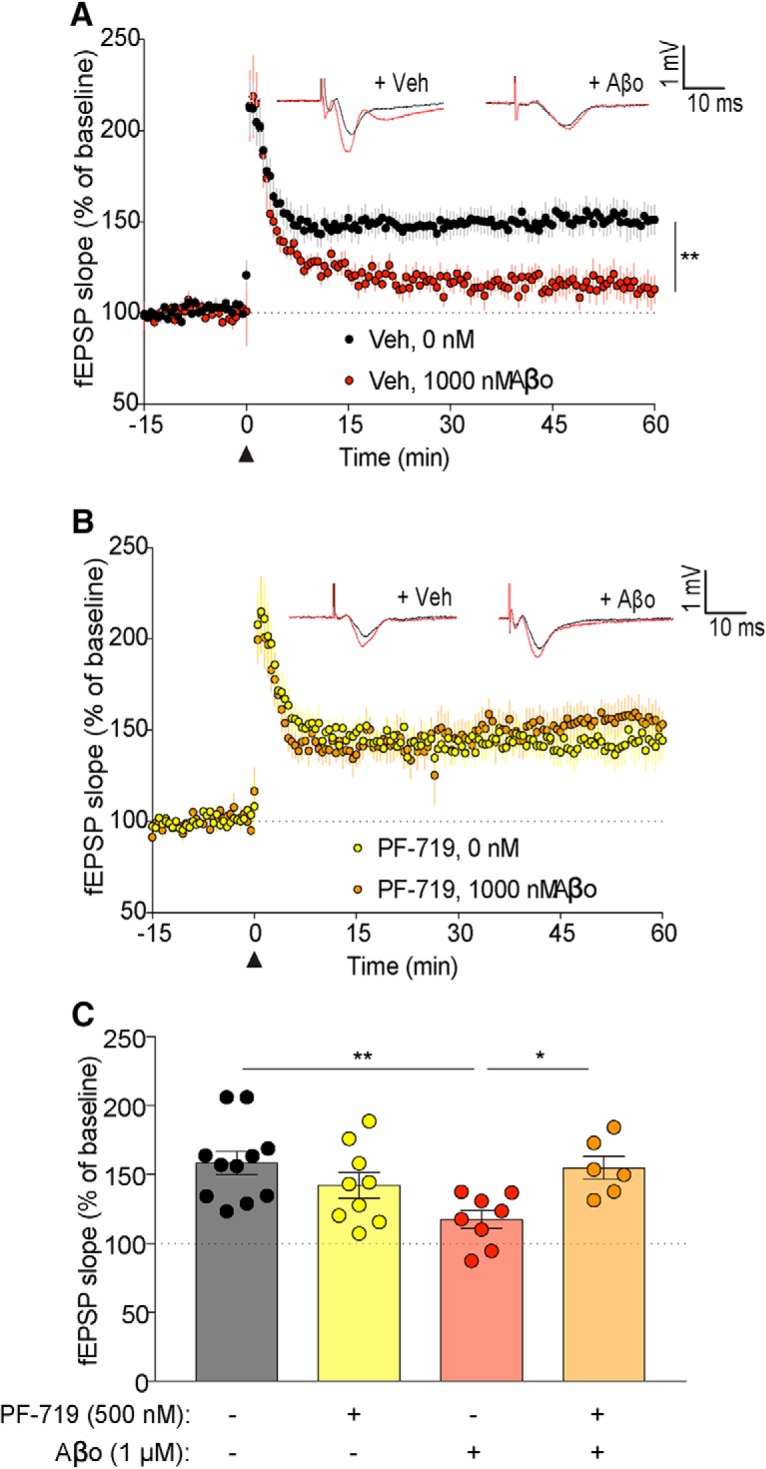
Aβo-dependent deficits in LTP are fully rescued by PF-719. A–C, Acute brain slices from WT 2- to 6-month-old mice were pretreated with vehicle or PF-719 (500 nm) for 30 min before treatment with vehicle or Aβo and used to record fEPSPs from the CA3–CA1 hippocampal circuit. The slope of the fEPSPs is plotted as a function of time. Representative traces before TBS in black (black arrowhead at time = 0) and at 60 min after TBS in red are superimposed (average, 6 sweeps) in the respective graphs. Data are graphed as mean ± SEM, where each n reflects one mouse with multiple slice averaged to a single value per mouse. A, C, WT slices treated with Aβo (8 slices from n = 8 mice) showed a significant decrease in fEPSP compared with WT slices treated with vehicle (11 slices from n = 11 mice) in the last 15 min of recording. Data are graphed as mean ± SEM, n = 1 slice, one-way ANOVA with Tukey's multiple-comparisons test: **p < 0.01. B, C, fEPSPs from WT slices pretreated with PF-719 and subsequently treated with Aβo (6 slices from n = 6 mice) were not significantly different from WT slices pretreated with PF-719 and subsequently treated with vehicle (9 slices from n = 9 mice) during the last 15 min of recording. Data are graphed as mean ± SEM, one-way ANOVA with Tukey's multiple-comparisons test. C, Quantification of the last 15 min of recording was compared between groups. Data are graphed as mean ± SEM, one-way ANOVA with Tukey's multiple-comparisons test: *p < 0.05, **p < 0.01. For details, see Table 1.
Pyk2 mediates learning and memory deficits in APP/PS1 mice
To examine in vivo evidence for Pyk2 in mediating synaptic dysfunction in AD, we used APP/PS1 mice, a previously described familial AD mouse model that displays a robust aging-dependent learning and memory deficit (Jankowsky et al., 2004; Park et al., 2006; Gimbel et al., 2010). We crossed APP/PS1 and Pyk2−/− mice (Okigaki et al., 2003) and produced the following genotypes: WT, Pyk2−/−, APP/PS1, and APP/PS1;Pyk2−/−. APP/PS1 mice display plaque pathology by 6 months (Jankowsky et al., 2004) and display learning and memory deficits at 10 months of age (Park et al., 2006; Gimbel et al., 2010; Salazar et al., 2017). For this reason, we examined cohorts of mice by MWM test at 12 months of age, when deficits are well established (Fig. 6A). Although previous work raised the possibility that Pyk2 is necessary for learning and memory (Huang et al., 2001; Bartos et al., 2010; Hsin et al., 2010; Giralt et al., 2017), the aged Pyk2−/− mice were able to find the hidden platform as quickly as WT mice during both the training and probe phases (Fig. 6B). As expected, APP/PS1 mice took significantly more time to find the hidden platform than WT mice. Strikingly, APP/PS1;Pyk2−/− mice took less time to find the hidden platform than APP/PS1 mice with Pyk2 and the APP/PS1;Pyk2−/− mice perform similar to WT mice. A probe trial was conducted 24 h after the last swim to assess memory retention. APP/PS1 mice spent significantly less time than WT mice in the target quadrant, approximately the amount of time attributed to chance. In contrast, Pyk2−/− and APP/PS1;Pyk2−/− mice spent as much time in the target quadrant as WT mice (Fig. 6B). Therefore, constitutive Pyk2 deletion does not affect healthy learning and memory and deletion of Pyk2 rescues learning and memory deficits in this transgenic mouse model of AD.
To further characterize the role of Pyk2 in APP/PS1-mediated phenotypes, we performed NOR tests on the same cohort of mice (Fig. 6C). Pyk2−/− mice preferred to explore the novel object to a similar degree as WT mice, whereas APP/PS1 mice showed no preference for the novel item. Deleting Pyk2 in APP/PS1 mice fully rescued the APP/PS1 deficit (Fig. 6C). As a distinct aversive learning task, the PAT was assessed for this group of mice. All groups of mice rapidly entered the dark chamber initially, wherein they received a mild foot shock, with the same protocol repeated 5 min later. At 24 h after the initial foot shock, the APP/PS1 mice showed significantly less learned delay to enter the dark chamber compared with the WT, Pyk2−/−, and APP/PS1;Pyk2−/− mice, which all delayed longer to enter the dark chamber, such that most mice never entered the dark chamber within the 5 min observation period (Fig. 6D). Together, these data demonstrate that Pyk2 is essential for APP/PS1 mice to exhibit age-dependent learning and memory deficits.
Pyk2 does not mediate Aβ accumulation or gliosis in APP/PS1 mice
Mechanistically, Pyk2 deletion is predicted to rescue behavioral outcomes by removing a postsynaptic signaling pathway downstream of Aβ accumulation. For this reason, upstream Aβ accumulation and glial reaction are not expected to be altered even though behavior is recovered to WT levels. Astrocytosis was examined as anti-GFAP immunoreactive area in brain sections. APP/PS1 mice displayed significantly greater GFAP immunoreactivity than WT mice, whereas Pyk2 deletion did not alter the WT level (Fig. 7A,B). The APP/PS1;Pyk2−/− mice have an intermediate phenotype with significantly less GFAP immunoreactivity than APP/PS1 mice, but significantly more than WT mice. Microgliosis detected by anti-Iba1 immunoreactive area is increased in APP/PS1 brain sections relative to WT and Pyk2−/−. The APP/PS1;Pyk2−/− sections displayed as much microgliosis as do APP/PS1 mice with Pyk2, not the intermediate phenotype observed for GFAP (Fig. 7C,D). Similarly, plaque load remained unchanged by deletion of Pyk2 (Fig. 7E,F). We conclude that microgliosis and Aβ plaque load are independent of Pyk2, whereas astrocytic reaction partially depends on Pyk2 in this mouse AD model.
Pyk2 is required for synapse loss in APP/PS1 mice
Because Pyk2 is necessary for the Aβo-dependent suppression of LTP in hippocampal slices and AD transgene-mediated memory deficits, we sought to determine whether AD-related synaptic loss in vivo requires Pyk2. We examined brain tissue from the four genotypes of aged mice described above by immunohistology. Sections of dentate gyrus were stained for SV2A (a presynaptic marker) and PSD-95 (a postsynaptic marker) and the immunoreactive area was measured in confocal images for each respective marker. The APP/PS1 mice showed a significant decrease in percentage of SV2A immunoreactive area compared with WT mice, whereas Pyk2−/− mice did not differ from WT mice (Fig. 8A,B). Critically, the APP/PS1;Pyk2−/− hippocampus has the same presynaptic marker positive area as WT and significantly more than the APP/PS1 samples. Similarly, the APP/PS1 deficit in postsynaptic PSD-95 area is fully rescued by Pyk2 deletion while Pyk2 deletion has no effect in the absence of AD-related pathology (Fig. 8C). Therefore, the synaptic marker loss in aged APP/PS1 mice requires the presence of the LOAD risk gene Pyk2.
Discussion
The present study demonstrates that the AD risk gene product Pyk2 is expressed in neurons and contributes to synaptic LTD, but is not required for basal synaptic function or LTP. With regard to AD mechanisms, Pyk2 is essential for LTP suppression in slices by Aβo and for memory impairment plus synapse loss in AD model transgenic mice. Therefore, inhibition of Pyk2 activation may be a target for disease-modifying AD intervention.
Genetic analysis of rare and common variants that modify LOAD risk provides insight into the basis of bona fide human disease, as opposed to discovery techniques using imperfect animal models. From multiple GWAS studies of common variants and sequence studies of rare alleles, approximately two dozen genes have been implicated as modifiers of AD (Holtzman et al., 2011; Guerreiro et al., 2013; Jonsson et al., 2013; Lambert et al., 2013; Pimenova et al., 2018). The challenge has been linking genetic factors mechanistically to the disease process and, most importantly, to AD-related synapse loss, which is central to clinical symptoms. Secreted ApoE is the common gene variant with the largest effect size (Holtzman et al., 2011). Despite being the first risk gene identified, ApoE's action in AD remains unsure and many hypotheses involve altered Aβ metabolism (Corder et al., 1993; Selkoe, 2011; Zhao et al., 2018). Other risk factors have implicated lipid metabolism inflammation/microglia and endosomal pathways in the LOAD pathophysiology (Nixon, 2013; Karch and Goate, 2015; De Strooper and Karran, 2016). However, few risk genes are directly linked to synaptic function and none has a clear validated basis for explaining synaptic loss in AD.
As described above, Pyk2 is unusual among the LOAD risk factors in possessing evidence for synaptic function in healthy brain. Indeed, our study of Pyk2-null slices shows that it is essential for normal LTD, although synaptic density and hippocampal LTP plasticity are identical to WT. Using a separate line disrupting Pyk2, others have reported a widespread failure of synaptic development and loss of postsynaptic markers even in the heterozygous state (Giralt et al., 2017), although this might relate to disruption of additional genes and/or to dominant effects of expressed fragments for that line. With regard to AD, our previous biochemical studies defined a Aβo–PrPC–mGluR5–Fyn pathway that activates Pyk2 (Laurén et al., 2009; Gimbel et al., 2010; Um et al., 2012, 2013; Kaufman et al., 2015; Haas et al., 2016; Heiss et al., 2017; Salazar and Strittmatter, 2017; Salazar et al., 2017). For these reasons, the current study sought to define Pyk2's role in synapses and AD. Pyk2 expression by neurons, subcellular localization to postsynaptic regions, and regulation by Aβo are all consistent with local synaptic action of Pyk2 as a risk gene in AD.
We have observed that Pyk2 is activated in mouse models of AD and, functionally, Pyk2 is required for multiple AD-related phenotypes here. Without Pyk2, hippocampal slices are resistant to Aβo suppression of LTP and mice with familial AD transgenes maintain synaptic markers and learning/memory behaviors despite Aβ pathology. These data all implicate Pyk2 gain-of-function in AD risk. Consistent with these data, the SNP associated with AD has been shown to increase Pyk2 expression in peripheral blood samples (Chan et al., 2015). While this manuscript was in preparation, one study reported decreased pY402-Pyk2 in 5XFAD-transgenic mice with a behavioral benefit from Pyk2 overexpression (Giralt et al., 2018). These conclusions are opposite to previous findings (Kaufman et al., 2015; Haas et al., 2016, 2017) and from those documented here from acute signaling events and from age-dependent transgenic phenotypes. Differences in the Pyk2-null strains, the AD transgenic lines, and mouse age may be reasons for the observed differences. The APPswe/PS1ΔE9 model shows little or no detectable phenotype until 6 months into adulthood, whereas the 5XFAD strain is more aggressive, with the onset of symptoms overlapping with neurodevelopment. Targeting Pyk2 as a therapeutic approach for AD may have unintended behavioral side effects, as suggested by the role of Pyk2 in hippocampal LTD. Although spatial memory, object recognition memory, and passive avoidance learning are not impaired by Pyk2 deletion, other behaviors not tested here might possibly be altered by loss of Pyk2 function. Future development of a Pyk2-based treatment approach will need to consider the therapeutic window for Pyk2 inhibition across a range of behavioral outcomes.
Our previous studies of Aβo signaling started with an unbiased genome-wide search for high-affinity binding sites to identify PrPC (Laurén et al., 2009). The essential role of PrPC in a number of Aβo and AD transgene signaling events implicated a known partner, Fyn kinase, and allowed a postsynapse-specific search for coupling proteins that identified mGluR5 (Um et al., 2012, 2013). Each of these steps is linked intimately to Pyk2. Fyn is a known physical partner for Pyk2 and extensive phosphorylation and activation by Fyn occurs (Dikic et al., 1996; Qian et al., 1997; Park et al., 2004). Pyk2 associates with the PrPC complex in a manner that requires mGluR5 and is regulated by Aβo (Haas and Strittmatter, 2016; Haas et al., 2016). Furthermore, the model systems in which the PrPC/mGluR5/Fyn pathway mediates AD-related phenotypes exhibit Pyk2 activation (Kaufman et al., 2015; Haas and Strittmatter, 2016; Haas et al., 2016, 2017). Genetically, mGluR5 and PrPC interact to mediate Pyk2 activation by Aβo and AD transgenes (Haas et al., 2016). Therefore, upstream signaling events impinge upon the risk gene Pyk2. Deletion of Pyk2 rescues the same AD-related phenotypes of synapse loss and memory impairment as does interruption of PrPC or mGluR5 or Fyn function (Kaufman et al., 2015; Haas and Strittmatter, 2016; Haas et al., 2016, 2017).
Neither perturbation in Pyk2 nor these other signaling complex members alters the accumulation or metabolism of Aβ itself. In general, the glial reaction to Aβ pathology, including both astrocytic reaction and microgliosis, is not altered by interruption of this postsynaptic signaling Pyk2 cascade. Here, we did observe a partial reduction in astrocytic reaction in APP/PS1 mice lacking Pyk2. This might be explained directly by reports of astrocytic Pyk2 function (Giralt et al., 2016) or it may be indirect with protection of synapses reducing astrocytic reaction.
Over time, brain dysfunction triggered by Aβ accumulation in AD leads to tau pathology with prion-like spreading of tauopathy as the disease progresses (Sanders et al., 2014; Kaufman et al., 2016). We have not studied tau here, because tau pathology is minimal in APP/PS1-transgenic models and with acute Aβo exposure. However, a Drosophila homolog of Pyk2 contributes to phenotypes generated by overexpression of mutant tau in flies and the same study found Pyk2 colocalized with tau in human AD (Dourlen et al., 2017). A connection between Pyk2 and tau might be explained by interaction with Fyn, which also interacts physically with both proteins, and/or by the reported ability of Pyk2 to activate Gsk3β, a tau kinase contributing to the protein's hyperphosphorylated status in aggregates (Dikic et al., 1996; Hartigan et al., 2001; Sayas et al., 2006; Ittner et al., 2010; Usardi et al., 2011). Recently, Pyk2 was reported to phosphorylate tau directly (Li and Götz, 2018). Future work will explore the downstream effects of Pyk2 signaling in AD.
The studies here suggest that Pyk2 inhibition has potential as a target for disease-modifying AD treatment. First, human genetic risk studies validate its relevance. Second, the biochemical analysis here shows that Pyk2 activity couples to a postsynaptic signaling pathway for synapse loss that is central to brain dysfunction in AD. Slowing the progression of AD by Pyk2 inhibition does not require alteration of Aβ or tau levels, but would be expected to be synergistic with successful approaches to these targets.
Footnotes
This work was supported by the Falk Medical Research Trust (S.M.S.) and the National Institutes of Health (S.V.S. and S.M.S.). We thank Stefano Sodi for expert technical assistance.
The authors declare no competing financial interests.
References
- Alzheimer's Association (2012) 2012 Alzheimer's disease facts and figures. Alzheimers Dement 8:131–168. 10.1016/j.jalz.2012.02.001 [DOI] [PubMed] [Google Scholar]
- Andreev J, Galisteo ML, Kranenburg O, Logan SK, Chiu ES, Okigaki M, Cary LA, Moolenaar WH, Schlessinger J (2001) Src and Pyk2 mediate G-protein-coupled receptor activation of epidermal growth factor receptor (EGFR) but are not required for coupling to the mitogen-activated protein (MAP) kinase signaling cascade. J Biol Chem 276:20130–20135. 10.1074/jbc.M102307200 [DOI] [PubMed] [Google Scholar]
- Bartos JA, Ulrich JD, Li H, Beazely MA, Chen Y, Macdonald JF, Hell JW (2010) Postsynaptic clustering and activation of Pyk2 by PSD-95. J Neurosci 30:449–463. 10.1523/JNEUROSCI.4992-08.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan G, White CC, Winn PA, Cimpean M, Replogle JM, Glick LR, Cuerdon NE, Ryan KJ, Johnson KA, Schneider JA, Bennett DA, Chibnik LB, Sperling RA, Bradshaw EM, De Jager PL (2015) CD33 modulates TREM2: convergence of Alzheimer loci. Nat Neurosci 18:1556–1558. 10.1038/nn.4126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins M, Bartelt RR, Houtman JC (2010a) T cell receptor activation leads to two distinct phases of Pyk2 activation and actin cytoskeletal rearrangement in human T cells. Mol Immunol 47:1665–1674. 10.1016/j.molimm.2010.03.009 [DOI] [PubMed] [Google Scholar]
- Collins M, Tremblay M, Chapman N, Curtiss M, Rothman PB, Houtman JC (2010b) The T cell receptor-mediated phosphorylation of Pyk2 tyrosines 402 and 580 occurs via a distinct mechanism than other receptor systems. J Leukoc Biol 87:691–701. 10.1189/jlb.0409227 [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261:921–923. 10.1126/science.8346443 [DOI] [PubMed] [Google Scholar]
- De Strooper B, Karran E (2016) The cellular phase of Alzheimer's disease. Cell 164:603–615. 10.1016/j.cell.2015.12.056 [DOI] [PubMed] [Google Scholar]
- Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J (1996) A role for Pyk2 and src in linking G-protein-coupled receptors with MAP kinase activation. Nature 383:547–550. 10.1038/383547a0 [DOI] [PubMed] [Google Scholar]
- Dourlen P, et al. (2017) Functional screening of Alzheimer risk loci identifies PTK2B as an in vivo modulator and early marker of tau pathology. Mol Psychiatry 22:874–883. 10.1038/mp.2016.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filali M, Lalonde R (2009) Age-related cognitive decline and nesting behavior in an APPswe/PS1 bigenic model of Alzheimer's disease. Brain Res 1292:93–99. 10.1016/j.brainres.2009.07.066 [DOI] [PubMed] [Google Scholar]
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA, Strittmatter SM (2010) Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci 30:6367–6374. 10.1523/JNEUROSCI.0395-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giralt A, Coura R, Girault JA (2016) Pyk2 is essential for astrocytes mobility following brain lesion. Glia 64:620–634. 10.1002/glia.22952 [DOI] [PubMed] [Google Scholar]
- Giralt A, Brito V, Chevy Q, Simonnet C, Otsu Y, Cifuentes-Díaz C, de Pins B, Coura R, Alberch J, Ginés S, Poncer JC, Girault JA (2017) Pyk2 modulates hippocampal excitatory synapses and contributes to cognitive deficits in a Huntington's disease model. Nat Commun 8:15592. 10.1038/ncomms15592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giralt A, de Pins B, Cifuentes-Díaz C, López-Molina L, Farah AT, Tible M, Deramecourt V, Arold ST, Ginés S, Hugon J, Girault JA (2018) PTK2B/Pyk2 overexpression improves a mouse model of Alzheimer's disease. Exp Neurol 307:62–73. 10.1016/j.expneurol.2018.05.020 [DOI] [PubMed] [Google Scholar]
- Guerreiro R, et al. (2013) TREM2 variants in Alzheimer's disease. N Engl J Med 368:117–127. 10.1056/NEJMoa1211851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas LT, Strittmatter SM (2016) Oligomers of amyloid beta prevent physiological activation of the cellular prion protein-metabotropic glutamate receptor 5 complex by glutamate in Alzheimer disease. J Biol Chem 291:17112–17121. 10.1074/jbc.M116.720664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas LT, Salazar SV, Kostylev MA, Um JW, Kaufman AC, Strittmatter SM (2016) Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer's disease. Brain 139:526–546. 10.1093/brain/awv356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas LT, Salazar SV, Smith LM, Zhao HR, Cox TO, Herber CS, Degnan AP, Balakrishnan A, Macor JE, Albright CF, Strittmatter SM (2017) Silent allosteric modulation of mGluR5 maintains glutamate signaling while rescuing Alzheimer's mouse phenotypes. Cell Rep 20:76–88. 10.1016/j.celrep.2017.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356. 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- Hartigan JA, Xiong WC, Johnson GV (2001) Glycogen synthase kinase 3beta is tyrosine phosphorylated by PYK2. Biochem Biophys Res Commun 284:485–489. 10.1006/bbrc.2001.4986 [DOI] [PubMed] [Google Scholar]
- Heiss JK, Barrett J, Yu Z, Haas LT, Kostylev MA, Strittmatter SM (2017) Early activation of experience-independent dendritic spine turnover in a mouse model of Alzheimer's disease. Cereb Cortex 27:3660–3674. 10.1093/cercor/bhw188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, et al. (2015) Neuroinflammation in Alzheimer's disease. Lancet Neurol 14:388–405. 10.1016/S1474-4422(15)70016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM (2011) Alzheimer's disease: the challenge of the second century. Sci Transl Med 3:77sr71. 10.1126/scitranslmed.3002369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–716. 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsin H, Kim MJ, Wang CF, Sheng M (2010) Proline-rich tyrosine kinase 2 regulates hippocampal long-term depression. J Neurosci 30:11983–11993. 10.1523/JNEUROSCI.1029-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Lu W, Ali DW, Pelkey KA, Pitcher GM, Lu YM, Aoto H, Roder JC, Sasaki T, Salter MW, MacDonald JF (2001) CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron 29:485–496. 10.1016/S0896-6273(01)00220-3 [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J (2010) Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell 142:387–397. 10.1016/j.cell.2010.06.036 [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Xu G, Fromholt D, Gonzales V, Borchelt DR (2003) Environmental enrichment exacerbates amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Neuropathol Exp Neurol 62:1220–1227. 10.1093/jnen/62.12.1220 [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR (2004) Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet 13:159–170. 10.1093/hmg/ddh019 [DOI] [PubMed] [Google Scholar]
- Jonsson T, et al. (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 368:107–116. 10.1056/NEJMoa1211103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, Goate AM (2015) Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry 77:43–51. 10.1016/j.biopsych.2014.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman AC, Salazar SV, Haas LT, Yang J, Kostylev MA, Jeng AT, Robinson SA, Gunther EC, van Dyck CH, Nygaard HB, Strittmatter SM (2015) Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol 77:953–971. 10.1002/ana.24394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman SK, Sanders DW, Thomas TL, Ruchinskas AJ, Vaquer-Alicea J, Sharma AM, Miller TM, Diamond MI (2016) tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron 92:796–812. 10.1016/j.neuron.2016.09.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita Y, Hunter RG, Gray JD, Mesias R, McEwen BS, Benson DL, Kohtz DS (2014) Role for NUP62 depletion and PYK2 redistribution in dendritic retraction resulting from chronic stress. Proc Natl Acad Sci U S A 111:16130–16135. 10.1073/pnas.1418896111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, et al. (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 45:1452–1458. 10.1038/ng.2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL (1998) Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 95:6448–6453. 10.1073/pnas.95.11.6448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457:1128–1132. 10.1038/nature07761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Götz J (2018) Pyk2 is a novel tau tyrosine kinase that is regulated by the tyrosine kinase fyn. J Alzheimers Dis 64:205–221. 10.3233/JAD-180054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D (2009) Soluble oligomers of amyloid beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62:788–801. 10.1016/j.neuron.2009.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegon A, Burgaya F, Baudot P, Dunlap DD, Girault JA, Valtorta F (1999) FAK+ and PYK2/CAKbeta, two related tyrosine kinases highly expressed in the central nervous system: similarities and differences in the expression pattern. Eur J Neurosci 11:3777–3788. 10.1046/j.1460-9568.1999.00798.x [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD (2005) Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 6:56–68. 10.1038/nrm1549 [DOI] [PubMed] [Google Scholar]
- Nixon RA. (2013) The role of autophagy in neurodegenerative disease. Nat Med 19:983–997. 10.1038/nm.3232 [DOI] [PubMed] [Google Scholar]
- Okigaki M, Davis C, Falasca M, Harroch S, Felsenfeld DP, Sheetz MP, Schlessinger J (2003) Pyk2 regulates multiple signaling events crucial for macrophage morphology and migration. Proc Natl Acad Sci U S A 100:10740–10745. 10.1073/pnas.1834348100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HJ, Westin CF, Kubicki M, Maier SE, Niznikiewicz M, Baer A, Frumin M, Kikinis R, Jolesz FA, McCarley RW, Shenton ME (2004) White matter hemisphere asymmetries in healthy subjects and in schizophrenia: a diffusion tensor MRI study. Neuroimage 23:213–223. 10.1016/j.neuroimage.2004.04.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Widi GA, Gimbel DA, Harel NY, Lee DH, Strittmatter SM (2006) Subcutaneous nogo receptor removes brain amyloid-beta and improves spatial memory in Alzheimer's transgenic mice. J Neurosci 26:13279–13286. 10.1523/JNEUROSCI.4504-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimenova AA, Raj T, Goate AM (2018) Untangling genetic risk for Alzheimer's disease. Biol Psychiatry 83:300–310. 10.1016/j.biopsych.2017.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purro SA, Nicoll AJ, Collinge J (2018) Prion protein as a toxic acceptor of amyloid-beta oligomers. Biol Psychiatry 83:358–368. 10.1016/j.biopsych.2017.11.020 [DOI] [PubMed] [Google Scholar]
- Qian D, Lev S, van Oers NS, Dikic I, Schlessinger J, Weiss A (1997) Tyrosine phosphorylation of Pyk2 is selectively regulated by fyn during TCR signaling. J Exp Med 185:1253–1259. 10.1084/jem.185.7.1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar SV, Strittmatter SM (2017) Cellular prion protein as a receptor for amyloid-beta oligomers in Alzheimer's disease. Biochem Biophys Res Commun 483:1143–1147. 10.1016/j.bbrc.2016.09.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar SV, Gallardo C, Kaufman AC, Herber CS, Haas LT, Robinson S, Manson JC, Lee MK, Strittmatter SM (2017) Conditional deletion of Prnp rescues behavioral and synaptic deficits after disease onset in transgenic Alzheimer's disease. J Neurosci 37:9207–9221. 10.1523/JNEUROSCI.0722-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, Miller TM, Grinberg LT, Seeley WW, Diamond MI (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82:1271–1288. 10.1016/j.neuron.2014.04.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayas CL, Ariaens A, Ponsioen B, Moolenaar WH (2006) GSK-3 is activated by the tyrosine kinase Pyk2 during LPA1-mediated neurite retraction. Mol Biol Cell 17:1834–1844. 10.1091/mbc.e05-07-0688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Mufson EJ (2006) Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging 27:1372–1384. 10.1016/j.neurobiolaging.2005.09.012 [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ (2007) Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 68:1501–1508. 10.1212/01.wnl.0000260698.46517.8f [DOI] [PubMed] [Google Scholar]
- Schnell SA, Staines WA, Wessendorf MW (1999) Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J Histochem Cytochem 47:719–730. 10.1177/002215549904700601 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. (2002) Alzheimer's disease is a synaptic failure. Science 298:789–791. 10.1126/science.1074069 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. (2011) Alzheimer's disease. Cold Spring Harb Perspect Biol 3:a004457. 10.1101/cshperspect.a004457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ (2008) Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 14:837–842. 10.1038/nm1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse KW, Lin KB, Dang-Lawson M, Guzman-Perez A, Aspnes GE, Buckbinder L, Gold MR (2012) Small molecule inhibitors of the Pyk2 and FAK kinases modulate chemoattractant-induced migration, adhesion and akt activation in follicular and marginal zone B cells. Cell Immunol 275:47–54. 10.1016/j.cellimm.2012.03.002 [DOI] [PubMed] [Google Scholar]
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM (2012) Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates fyn to impair neurons. Nat Neurosci 15:1227–1235. 10.1038/nn.3178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, Gunther EC, Nygaard HB, Strittmatter SM (2013) Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79:887–902. 10.1016/j.neuron.2013.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usardi A, Pooler AM, Seereeram A, Reynolds CH, Derkinderen P, Anderton B, Hanger DP, Noble W, Williamson R (2011) Tyrosine phosphorylation of tau regulates its interactions with fyn SH2 domains, but not SH3 domains, altering the cellular localization of tau. FEBS J 278:2927–2937. 10.1111/j.1742-4658.2011.08218.x [DOI] [PubMed] [Google Scholar]
- Villasana LE, Klann E, Tejeda-Simon MV (2006) Rapid isolation of synaptoneurosomes and postsynaptic densities from adult mouse hippocampus. J Neurosci Meth 158:30–36. 10.1016/j.jneumeth.2006.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416:535–539. 10.1038/416535a [DOI] [PubMed] [Google Scholar]
- Zhang C, Lambert MP, Bunch C, Barber K, Wade WS, Krafft GA, Klein WL (1994) Focal adhesion kinase expressed by nerve cell lines shows increased tyrosine phosphorylation in response to Alzheimer's A beta peptide. J Biol Chem 269:25247–25250. [PubMed] [Google Scholar]
- Zhang R, Xue G, Wang S, Zhang L, Shi C, Xie X (2012) Novel object recognition as a facile behavior test for evaluating drug effects in AbetaPP/PS1 Alzheimer's disease mouse model. J Alzheimers Dis 31:801–812. 10.3233/JAD-2012-120151 [DOI] [PubMed] [Google Scholar]
- Zhao M, Finlay D, Zharkikh I, Vuori K (2016) Novel role of Src in priming Pyk2 phosphorylation. PLoS One 11:e0149231. 10.1371/journal.pone.0149231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, Liu CC, Qiao W, Bu G (2018) Apolipoprotein E, receptors, and modulation of Alzheimer's disease. Biol Psychiatry 83:347–357. 10.1016/j.biopsych.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]