

Abstract
Anti-neutrophil cytoplasmic antibodies (ANCA) vasculitides are immune-mediated disorders that primarily affect small blood vessels of the airway and kidneys. Lung involvement, one of the hallmarks of microscopic polyangiitis and granulomatosis with polyangiitis, is associated with increased mortality and morbidity. In recent years, several retrospective series and case reports have described the association of interstitial lung disease (ILD) and ANCA vasculitis, particularly those positive for ANCA specific for myeloperoxidase. In the majority of these patients pulmonary fibrosis occurs concurrently or predates the diagnosis of ANCA vasculitis. More importantly, these studies have shown that ILD has an adverse impact on the long-term prognosis of ANCA vasculitis. This review focuses on the main clinical and radiologic features of pulmonary fibrosis associated with anti-neutrophil cytoplasmic antibodies. Major histopathology features, prognosis and therapeutic options are summarized.
Keywords: ANCA, Interstitial lung disease, Microscopic polyangiitis, Pulmonary fibrosis
1. Introduction
Anti-neutrophil cytoplasmic antibodies (ANCA) are autoantibodies specific for antigens located in the cytoplasmic granules of neutrophils and lysosomes of monocytes [1]. The two major autoantigen targets are myeloperoxidase (MPO-ANCA) and proteinase 3 (PR3-ANCA). ANCA vasculitides (ANCA-V) are multisystem diseases characterized by necrotizing vasculitis that predominantly affects small vessels [2]. The major clinicopathologic variants of ANCA-V are granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), eosinophilic granulomatosis with polyangiitis (EGPA), and single-organ ANCA-V, including renal-limited ANCA vasculitis (RLV). Pulmonary manifestations are a characteristic feature of ANCA-V [3], with the lung being involved in 85–90% of GPA patients during the course of their disease [4–6]. The frequency in MPA is slightly lower, with reported prevalence of 25–55% [7–9]. In both disorders, lung involvement has been associated with unfavorable outcomes [10–12].
During the last few years, an increasing number of publications have reported the association between interstitial lung disease (ILD) and ANCA vasculitis. The objectives of this review are 1) to describe the main clinical and radiologic manifestations of pulmonary fibrosis (PF) associated with ANCA, 2) to detail major histopathological findings and hypothesize physiopathogenic mechanisms involved in the development of this condition, and 3) to summarize the outcome and therapeutic options for affected patients.
We performed a MEDLINE search for English language articles published between January 1970 and September 2016. The search strategy combined the following terms: “vasculitis, ANCA, granulomatosis with polyangiitis, Wegener’s granulomatosis, microscopic polyangiitis, eosinophilic granulomatosis with polyangiitis, Churg-Strauss syndrome, myeloperoxidase or proteinase 3” and “interstitial lung disease, lung fibrosis, pulmonary fibrosis, lung manifestations, thoracic manifestations, interstitial pneumonia or computed tomography (CT)”. Full text of relevant articles were retrieved and reviewed. Additional references quoted in these publications were checked. Data of >200 patients included in case series [13–27] and case reports [28–42] constitute the basis of this review.
2. Epidemiology
Interstitial lung disease associated with ANCA is usually observed in patients older than 65 years old. The average age at presentation seems to be higher in MPA patients with PF than in those with this systemic vasculitis described in general cohorts (66 vs 55 years, respectively) [7,8,13–27], but similar to that reported in cases of idiopathic PF (IPF) [43,44]. Regarding gender, some series have reported a slight predomi-nance of men (60–65%), although this has not been confirmed in other studies [13–26].
Pulmonary fibrosis occurs concurrently or antedates MPA in the majority of affected individuals (Table 1). In previous reports, ILD precedes full vasculitis syndrome in 14–85% of patients, appeared simultaneously with other organ involvement in 36–67%, and was reported after ANCA-associated disease in 8–21% [13–20,40,42,45–50]. In those cases where PF was diagnosed before MPA, the time between both events ranges from a few months to 12 years. It must be noted that in most series PF was not biopsy-proven, and diagnosis was based on the combination of clinical findings, pulmonary function tests (PFT) and CT images.
Table 1.
Characteristics of main series describing the Association of ANCA and Interstitial Lung Disease.
| Country, year Reference center | Patients | Age (mean, range) | Male (%) | ANCA pattern | Follow-up (mean, range) | Chronology of PF-ANCA-V | Full vasculitis development | Outcome |
|---|---|---|---|---|---|---|---|---|
| [26] Japan, 2004 Respiratory | 31 PF ANCA+ | 69 yr (45–87) | 55 | MPO 100% | 120 Mo | Preceding 100% | MPA 8/31 (25%) | Dead 13/31 (41%) 5-yr survival 50% |
| [13] France, 2008 Respiratory | 29 PF: 17 ANCA+ | 66 yr (55–77) | 88 | P-ANCA 82% C-ANCA 18% | 57 Mo | Preceding 76% Concomitant 24% | MPA 7/17 (41%) | Dead 10/17 (58%) 5-yr survival 60% |
| [16] Japan, 2009 Respiratory | 53 PF: 19 ANCA+ | 69 yr (52–80)* | 37 | MPO 89% PR3 11% | From 1 to 90 Mo | Preceding 100% | MPA 4/19 (21%) | Dead 6/19 (31%) 5-yr survival 60% |
| [27] Japan, 2012 Respiratory/Pathology | 224 PF: 13 ANCA+ | 62.1 yr | 67 | MPO 69% PR3 31% | 39.1 Mo (19 to 72) | None | None | Dead 4/13 (30%) |
| [58] Japan, 2013 Internal Medicine | 61 PF: 9 ANCA+ | 69 yr (57–75) | 100 | MPO 100% | 40 Mo (1−121)* | Preceding 100% | MPA 2/9 (22%) | Dead 6/9 (66%) |
| [49] China, 2014 Respiratory | 8 PF ANCA+ | 72.6 yr (60–80) | 50 | MPO 75% PR3 12.5% P- and C-ANCA12.5% | NS | NS | NS | NS |
| [50] Japan, 2015 Respiratory | 504 PF: 36 ANCA+ | 72 yr | 61 | MPO 55% PR3 45% | 2.42 yr (1.38–4.92)* | Preceding 100% | MPA 3/36 (8%) | 5-yr survival 40% |
| [59] Japan, 2016 Respiratory | 120 PF: 12 ANCA+ | 65.2 yr (48–74)* | 66 | MPO 100% | 72 Mo (14–195)* | Preceding 100% | MPA 3/12 (25%) | Dead 5/12 (41%) |
| [48] China, 2014 Respiratory |
19 MPA + PF | 63.6 yr (44–75) | 42 | MPO 100% | 29.9 Mo (8–93)* | Preceding 68% Concomitant 32% | MPA 100% | Dead 6/19 (31%) |
| [14] United States, 1990 Nephrology/Internal Medicine | 3 MPA + PF | 73 yr | 33 | P-ANCA 67% | 120 Mo | Preceding 33% Concomitant 67% | MPA 100% | Dead 3/3100% |
| [22] Japan, 2000 Internal Medicine | 4 MPA + PF | 67 yr | 50 | MPO 100% | NS | Preceding 100% | MPA 100% | Dead 3/4 (75%) |
| [21] Canada, 2003 Respiratory | 6 MPA + PF | 70 yr (63–78) | 50 | P-ANCA 100% | 36 Mo | Preceding 100% | MPA 100% | Dead 5/8 (83%) |
| [15] France, 2009 Internal Medicine | 12 ANCA-V + PF | 70.7 yr (64–78) | 75 | MPO 100% | 49.2 Mo (7–116) | Preceding 25% Concomitant 66% Posterior 8% | MPA 10/12 83% GPA 2/12 17% | Dead 5/12 (41%) |
| [17] Greece, 2010 Pathophysiology |
33 MPA: 13 MPA + PF | 57 yr | 69 | P-ANCA 85% P- and C-ANCA 8% | 38 Mo | Preceding 53% | MPA 100% | Dead 6/13 (46%) 5-yr survival 60% |
| [18] UK, 2011 Respiratory/ Nephrology | 194 MPA: 14 MPA + ILD | 67.3 yr (51–85) | 72 | MPO 100% (tested 13/13) | 7.5 yr | Preceding 14% Concomitant 64% Posterior 21% | MPA 100% | Dead 10/14 (71%) |
| [51] France, 2014 Internal medicine | 49 ANCA-V + PF | 68 yr (58–74)* | 61 | MPO 88% PR3 4% Other 8% | 48 Mo (14–88)* | Preceding 45% Concomitant 43% Posterior 12% | MPA 40/49 (82%) GPA 9/49 (18%) | Dead 18/49 (36%) |
| [19] Mexico, 2015 Respiratory | 40 MPA: 17 MPA + PF | 54.2 yr | 53 | MPO 90% entire cohort | 43 Mo (11−213)* | Preceding 82% | MPA 100% | Dead 9/17 (41%) |
| [20] Argentina, 2015 Respiratory | 28 MPA: 9 MPA + PF | 60 yr | 56 | MPO 100% | 61.2 Mo | Preceding 55% Concomitant 45% | MPA 100% | Dead 4/9 (44%) |
| All series | 991 PF: 145 ANCA + 149 ANCA-V + PF |
68.1 yr 64.7 yr |
65.5 56 |
MPO 83%, PR3 15% MPO 92% |
– – |
Preceding or concomitant 100% Preceding or concomitant 89% |
MPA 20% MPA 96.5% |
Dead 41% Dead 55% |
ANCA-V-ANCA vasculitis; GPA-granulomatosis with polyangiitis; MPA-microscopic polyangiits; MPO-myeloperoxidase; PF-pulmonary fibrosis; PR3-proteinase 3.
Median (IQR)
Prevalence of ILD is higher in MPA than in GPA [15,18,51]. PF has been reported in 23% of GPA cases [52] and in 2.7–45% of MPA patients. In these cases, interstitial disease was usually identified at disease onset [7,11,17–20,48,53,54]. Importantly, there is a significant predominance of MPO-ANCA in published series, i.e., 46–71% of all cases compared to 0–29% for PR3-ANCA [13,27,51,52,54–56].
On the other hand, prevalence of ANCA in cohorts of individuals who initially presented with isolated pulmonary fibrosis ranges between 4%–36% for MPO-ANCA and 2–4% for PR3-ANCA [13,16,27,46,48,50,56–59]. During follow-up, 5–10% of ANCA negative patients developed autoantibodies against MPO or PR3 (incidence of 12 cases per 1000 person years) [13,16,46,50,58]. In the latter group, presence of other autoantibodies, such as rheumatoid factor, erythrocyte sedimentation rate (ESR) >40 mm/h, bronchoalveolar lavage (BAL) eosinophilia and increased areas of low attenuation on CT have been suggested as factors predicting the conversion to positive ANCA [58]. In large cohorts of patients with idiopathic PF, 1.7–25.7% developed full MPA (Table 1) [16,50,58]. Prospective studies are needed to clarify the precise prevalence and incidence of ILD in ANCA vasculitis.
Of relevance, frequency of ILD in ANCA vasculitis seems to be higher in Japan than in Western populations [17,18,20,54,55,60]. Reasons for this particular association may include a higher prevalence of MPO-ANCA positivity and increased frequency of lung involvement and diffuse alveolar hemorrhage [11,61,62].
3. Clinical manifestations
Major symptoms of patients with ANCA-positive isolated pulmonary fibrosis are progressive dyspnea (50–73%) and nonproductive cough (21–60%) [16,26,49,59]. Other manifestations such as pulmonary hemorrhage and hemoptysis (5%) or constitutional symptoms, i.e., fever (31%) and weight loss (5%) were observed less frequently [16,49,58]. In previous studies, ANCA titers did not correlate with the severity of PF [16]. In a small case series, clinical presentation of patients with PF and positive ANCA did not differ from their counterpart with negative autoantibodies [16,49,59].
In contrast, patients with ILD who fulfilled MPA criteria typically manifested with constitutional symptoms (approximately 80%) and extra-pulmonary disease (70–100%) [15]. In these cases, malaise (31–63%), fever (52–90%) and weight loss (52–58%) are frequently detected at diagnosis [17,18,48,51]. In addition, vasculitic involvement is common in the skin (8–31%), peripheral nervous system (8–53%), joints and muscle (23–31%) or kidneys (57–100%) [13,17,18,48,51]. Pulmonary manifestations include progressive breathlessness (30–100%), alveolar hemorrhage (21–49%) and chronic cough (23–84%) [15,17,18, 20,48,51]. Of note, when comparing MPA patients with and without PF, some authors have reported that the first usually exhibited less severe systemic inflammatory response, manifested as lower ESR, higher levels of hemoglobin and importantly, reduced frequency of clinical evident diffuse alveolar hemorrhage, peripheral nerve and kidney involvement [19,20].
4. Laboratory findings and pulmonary function test
In patients with MPA and ILD, ESR and C-reactive protein (CRP) were increased at disease onset in 95% and 73–79%, respectively [48,51]. As expected, >60% of these cases also have abnormal urinalysis [48,50]. In contrast, marked elevations of ESR and CRP were not described in ANCA-positive PF. In this sense, most studies have found similar CRP levels in patients with initially isolated lung fibrosis irrespective of their ANCA status [16,56,58,59].
Measurement of lung function is an important part of the evaluation of individuals with ILD. These ancillary tests are used for assessing the pattern and severity of pulmonary involvement. In both lung-limited ANCA-associated PF and MPA patients with PF, a restrictive pattern with reduction in total lung capacity is the most frequent pattern reported in spirometry (62–80%) [13,15,16,18,20,48,50,51]. Co-existing airflow obstruction can be observed in one third of pulmonary function tests [18]. Other frequent findings include reduction of the diffusing capacity (DLCO) and mild hypoxemia at rest (50% of cases) [13,15,18,20, 21,48,49]. During follow-up, PFT tend to deteriorate as ILD progresses. This was evidenced in a previous study of patients with ILD and ANCA vasculitis, where after a mean of 5 years, basal values of forced expiratory volume in 1 s (FEV1), vital capacity and DLCO were reduced in 29%, 23% and 46%, respectively [18].
BAL features include an increased cell count in 60% of patients, with neutrophilia being the most frequent finding (40–87% of cases) [13,16, 51]. Lymphocytosis and eosinophilia were reported in 20% and 26%, respectively [13,16]. Evidence of acute or chronic alveolar hemorrhage was observed in half of the samples [13]. When compared to idiopathic PF, some authors have reported that those cases associated with ANCA exhibited an increased percentage of BAL neutrophils and eosinophils [58,59]; however, other studies have disputed these results [13,63].
5. Imaging
Interstitial lung lesions are a frequent finding in radiologic studies of ANCA-V patients [64]. In a cohort of 150 MPA individuals, CT images obtained before treatment showed that 97% presented at least one lung abnormality, including interstitial lung changes in 66% [64]. Similarly, in another study that included 62 MPO-ANCA cases (51 with MPA), 82% had an abnormal CT scan, 94% of which displayed findings suggestive of interstitial involvement [65]. Most of these interstitial lesions are non-specific and only a small number of patients will actually show a CT pattern corresponding to some specific idiopathic interstitial pneumonia, such as usual interstitial pneumonia (UIP) or non-specific interstitial pneumonia (NSIP) [64,66].
The major CT features of ILD associated with MPA include ground glass opacities (23–94%) (histologically corresponding to interstitial chronic inflammation, alveolar hemorrhage and areas of fibrosis), reticular shadowing (41–77%), interlobular septal thickening (41–71%), consolidations (23–78%) and honeycombing (23–52%) [17,18,48,64,65]. Airway abnormalities are reported in the form of bronchiolitis (55%), bronchial wall thickening (44%) or bronchiectasis (32–38%) [17,64]. With respect to distribution, interstitial involvement was reported to be symmetrical in 50–100% of patients, affecting predominantly the lung periphery and lower areas [15,18,48,65]. In one study, interstitial disease affected >40% of pulmonary parenchyma in >60% of patients [17].
Using the ATS/ERS International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias criteria [66,67], the most frequent radiological pattern is that of UIP (50–57%), followed by NSIP (7–31%), and desquamative interstitial pneumonia (14%) (Fig. 1) [15,17,18,20,51]. Combined pulmonary fibrosis with emphysema has been recently reported in MPA patients [35,51,64,68]. Importantly, 4–40% of studied cases will not fit any specific CT-pattern [15,17,18,20,51].
Fig. 1.
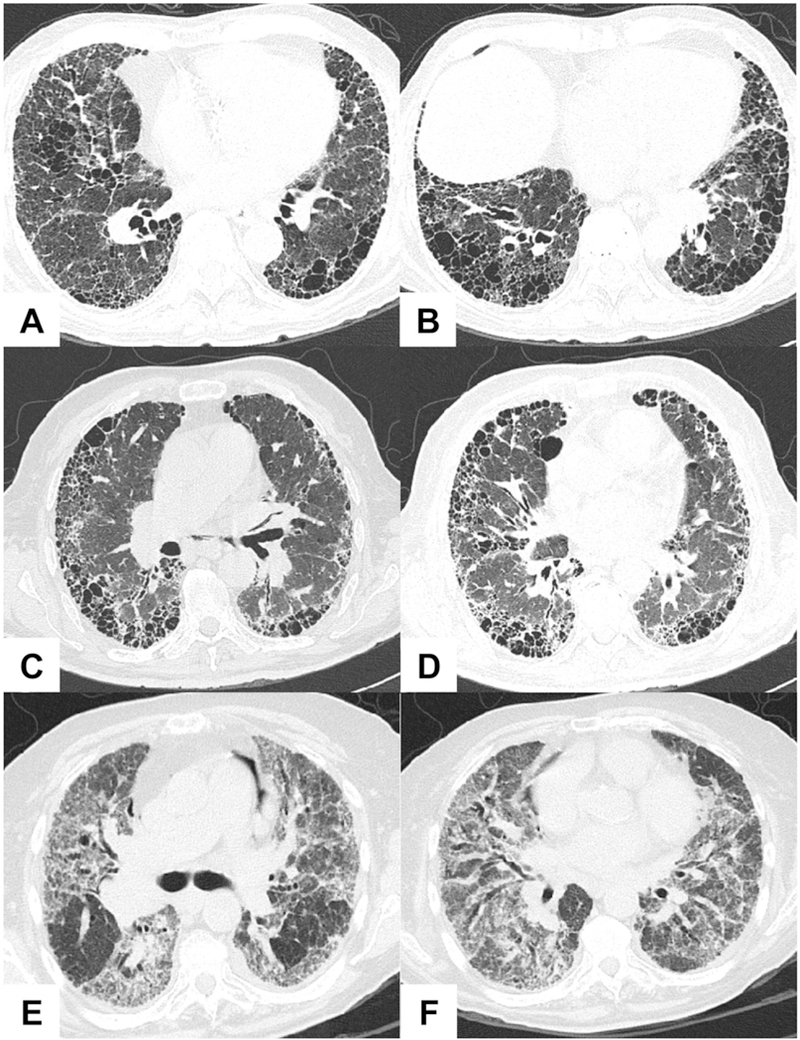
High-resolution computed tomography images of the two major patterns of interstitial pneumonia associated with ANCA. Representative features of usual interstitial pneumonia (UIP) are shown in A-D: honeycombing, cysts, and reticular opacities with basal and subpleural predominance. Nonspecific interstitial pneumonia (NSIP), characterized by patchy ground-glass opacities and traction bronchiectasis, is depicted in E-F. Courtesy of Dr. Lya Pensado.
During follow-up of treated patients, some of the interstitial changes may resolve partially or completely, i.e., ground glass opacities (GGO), reticular pattern, interlobular septal thickening and consolidations [64]. On the other hand, honeycombing, usually consistent with late-stage interstitial fibrosis, tend to remain unchanged or progressed [17, 21,63,65]. These data suggested that both fibrosing and inflammatory processes usually coexist in diverse ratios [64].
In cases with MPO-ANCA positive ILD but without generalized involvement, UIP is also the most common abnormal pattern [13,16,27,46,58,63]. With regard to PR3-ANCA PF, a small series reported UIP or possible UIP in 38% and NSIP in 31% [56]. In these patients, CT may show honeycombing (37–100%), GGO (25–100%), consolidations (0–78%), nodules (0–45%), cyst (27%), traction bronchiectasis (80–100%), thickening of bronchovascular bundles (51%) or interlobular septal thickening (45%) [13,16,26,27,49,58]. In the reviewed studies, fibrotic changes were usually extensive, subpleural and commonly affected the lower lung fields [16,26]. Similarly to that described above for clinical manifestations, no major significant differences in CT images were found between ANCA-positive and ANCA-negative PF [16,58,59]. Overall, there are wide ranges of presentations of pulmonary involvement in the available small studies of ANCA-V lung disease.
6. Histopathology
The histologic features of pulmonary involvement associated with ANCA vasculitis have been described in detail in previous reports [45, 69]. In these studies, interstitial lesions were a common feature of C-ANCA (usually PR3-ANCA) and P-ANCA (usually MPO-ANCA) patients, observed in 100% and 57% of specimens, respectively [45]. In particular, interstitial fibrosis was documented in 16–48% of the reported patients, being focal in most of the analyzed samples [45,69]. In 6% of cases, interstitial fibrosis was the dominant pathologic finding [69].
In studies that specifically described the characteristics of patients with ILD associated with MPA and those with ANCA-positive PF, UIP was the most frequent histological pattern (46–100%) (Fig. 2) [13,15–18,26,27,48,56,58,63]. In these specimens, marked dense fibrosis and honeycombing areas were common [17,26,27]. Of note, although NSIP was the main pattern in <15% of samples, most cases with UIP also showed NSIP-like areas as a minor component [26,27]. When comparing patients with UIP and MPO-ANCA and those with idiopathic UIP, more prominent interstitial inflammation, presence of lymphoid follicles and small airway involvement were more frequent in the former [27,59]. Of note, the presence of active capillaritis or vasculitis has been rarely reported in specimens with ANCA-positive PF [13,26,27, 58,59].
Fig. 2.
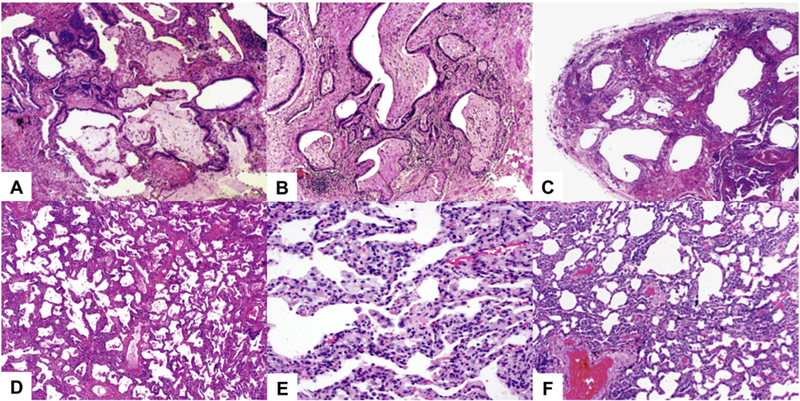
Histopathology findings of interstitial lung disease associated ANCA. Usual interstitial pneumonia hallmarks are shown in A-C: marked variation in histologic appearance, fibroblast foci (A), patchy interstitial fibrosis (B), and honeycomb changes (C). Nonspecific interstitial pneumonia is characterized by diffuse alveolar wall thickening produced by fibrosis (D) and interstitial inflammation without fibrosis (E). Nonspecific interstitial pneumonia does not have significant fibroblastic foci or honeycombing (F). H&E stain. Courtesy of Dr. Rosa María Rivera-Rosales.
7. Pathophysiology
In complex autoimmune diseases such as ANCA vasculitis, diverse mechanisms operate in leading to different disease features, such as glo-merulonephritis or pulmonary involvement. Regarding ILD, there are several potential mediators that may contribute to the development of this complication in ANCA vasculitis. Pulmonary fibrosis might be the end result of iterative episodes of intra-alveolar hemorrhage [30,36, 70]. In this sense, previous series have reported histologic evidence of acute or chronic hemorrhage in more than half of lung biopsies of ANCA patients [69]. Of relevance, most of these individuals did not present overt manifestations of diffuse pulmonary bleeding, indicating that subclinical episodes are common and frequently overlooked [20,69, 71]. In the same line, hemosiderin-laden macrophages, markers of chronic alveolar bleeding, were found to be increased in BAL fluid of pa-tients with pulmonary involvement secondary to ANCA-V but not in ILD-associated with other autoimmune diseases, i.e., systemic lupus er-ythematosus, inflammatory myopathies or systemic sclerosis [70]. In addition, some of the initial CT images of consolidation consistent with alveolar bleeding evolved into areas of honeycombing during long-term follow-up of ANCA patients [65].
Although intra-alveolar hemorrhage may be an important event in the development of ILD, additional factors should be of relevance, as pulmonary fibrosis does not appear to be common in anti-glomerular basement membrane disease, an immune-complex vasculitis clinically characterized by pulmonary hemorrhage [18]. In this regard, it has been suggested that MPO-ANCA per se may play a direct role in the pathogenesis of lung fibrosis [71]. A previous study reported that the activation of MPO by anti-MPO ANCA resulted in the production of major oxidant products. One of these reactive oxygen species, i.e., hypochlorous acid, was able to trigger fibroblast proliferation in vitro. In addition, MPO-ANCA may contribute to pulmonary tissue injury after local release of proteloytic enzymes of activated neutrophils [72]. In fact, one of these enzymes, i.e., elastase, is able to induce pulmonary fibrosis in experimental mice [73,74].
Damage induced by eosinophils and neutrophil extracellular traps (NETs) could also play a role. Extensive tissue eosinophilia has been reported in specimens of ANCA pulmonary tissue with marked interstitial fibrosis [69]. Previously, it was shown that lung fibroblasts might influence function and survival of eosinophils [75]. Additionally, BAL eosinophilia has been suggested as a marker of progressive pulmonary fibrosis [76]. On the other hand, it is well known that NETs are released by ANCA-activated neutrophils [77]. These extracellular traps have the ability to activate lung fibroblasts and promote their differentiation into myofibroblasts [78], prominent cells recruited intensively toward fibrotic areas in ANCA specimens [39]. Other factors that may be relevant include tobacco toxicity and chronic lung parenchymal ischemia [13,20,39,79].
Although the histologic pattern (UIP) observed in MPA seems to be the same as in IPF, some relevant pathogenic mechanisms described in the latter have not been demonstrated in ANCA-V. For example, the key role of epithelial apoptosis, which arises from continued alterations of the epithelium, and of epithelial mesenchymal transformation in IPF has not been shown in ANCA-V-related lung fibrosis [39,80]. In contrast, the role of neutrophils and the complement system in IPF seems negligible as opposed to their important role in the pathogenesis of the ANCA disease.
In summary, it seems probable that repeated episodes of inflammatory alveolar capillary injury causes generation of a reactive fibrotic state, characteristic of ANCA-associated PF [39,81,82]. The lack of appropriate animal models exploring ANCA-induced lung pathology, especially long-term injury when PF develops, has hampered advances in this field.
8. Treatment
Treatment of ANCA-V involves induction of remission using highly potent immunosuppressive drugs, followed by a maintenance phase to prevent relapses and chronic damage [83]. Although regimen options for initial therapy of ANCA patients are well established [84], there is limited data on how to treat the particular subset of cases with ILD. In fact, this specific lung problem is not mentioned in recent publications providing recommendations for the management of ANCA-V [84–86].
Most of the available information for treatment of ILD in ANCA-V de-rives from retrospective studies as no controlled clinical trials have ad-dressed this issue. In the reviewed case series, administered therapy was different in patients with or without extra-pulmonary manifesta-tions. For those with a diagnosis of MPA, treatment for remission induction usually followed standard recommendations and included systemic glucocorticoids (88–100%) in combination with cyclophosphamide (16–92%) or rituximab (10%) [15,18,37,48,51]. Azathioprine or mycophenolate mofetil were used as maintenance therapy [15].
In contrast, patients with ILD and positive ANCA who did not fulfill MPA criteria received non-uniform therapy that included small dosages of corticosteroids alone or in combination with azathioprine, cyclosporine, colchicine, infliximab, interferon-gamma, mizorbine or N-acetylcysteine (44–80%) [13,26,58]. Progressive respiratory failure was the main indication for immunosuppressive drugs in this last group of patients [58].
The effect of treatment in ILD progression is controversial. On the one hand, some studies have showed clinical or radiological improvement in 80% of patients while on immunosuppressive therapy [16,28, 59]. In a series of 49 ANCA-V patients with PF, 1-and 5-year survival rates were significantly better for those that received glucocorticoids in combination with cyclophosphamide or rituximab as induction ther-apy than in those who were treated with corticosteroids alone [51]. In addition, immunosuppressive therapy might be associated with a reduction in the development of full-blown MPA in patients initially pre-sented as isolated PF with positive MPO ANCA [50,58,59]. On the other hand and in sharp contrast, most of the retrospective publications re-ported that the disease remained unchanged or deteriorated in the majority of patients, even in presence of clinically quiescent systemic vasculitis [15,18,26]. The lack of reliable tissue biopsies confirming the type of ILD and the absence of a definitive staging system for assessing the severity of ILD are undoubtedly some of the main reasons for these contradictory results. In addition, radiological findings caused by an inflammatory abnormality, like GGO, tend to resolve completely after treatment whereas chronic forms of lung fibrosis (honeycombing), usually remain unchanged [30]. Although no clear conclusions can be drawn about the initial treatment of ILD-associated with ANCA, it is recommended that general measures are instituted as required: supplemental oxygen, pulmonary rehabilitation and influenza and pneumo-coccal vaccination.
Evaluation of efficacy and safety of immunosuppressive drugs in well-controlled clinical trials is an unmet need in this field. Based on the experience obtained for the treatment of ILD-associated with other autoimmune disorders, such as systemic sclerosis (SSc) or inflam-matory myopathies, mycophenolate mofetil and rituximab could be emerging options for patients with PF associated with ANCA. In system-ic sclerosis, results of a recent clinical trial and observational studies showed that mycophenolate mofetil was associated with stabilization or modest improvement of PFT and dyspnea in patients with progressive ILD [87–89]. Similar results have been reported in polymyositis/dermatomyositis where additionally, this drug exhibited a corticosteroid sparing effect [89]. Treatment with monoclonal antibody rituximab has been associated with stabilization of lung function in small case series of patients with refractory systemic sclerosis ILD, autoimmune myopathies and mixed connective tissue disorder [90–92].
In the future, novel anti-fibrotic therapies (pirfenidone and nintedanib) designed for idiopathic pulmonary fibrosis might be tested in ANCA related ILD. Pirfenidone, an oral pyridine with anti-inflammatory, and anti-fibrotic effects seems to slow progression and to improve survival of patients with IPF [93,94]. In addition, treatment with nintedanib, an oral tyrosine kinase inhibitor has been associated stabili-zation of lung fibrosis [95]. Both drugs reveal good safety profile.
Finally, it is important to include the following items into a research agenda: a) The establishment of an international registry of ILD in ANCA vasculitis. This is of relevance as the ANCA-V are infrequent diseases, with different prevalence according to geographical regions. Such inter-national efforts have been fruitful in defining disease spectra around the world; b) If possible, the creation of adequate biobanks with tissue and individual cells samples at early and late disease phases, in order to mechanistically study these diseases at different time points; c) Pro-spective randomized clinical trials which incorporate not only immuno-suppressants, but also other pharmacologic and non-pharmacologic measures which could provide better outcomes, d) More objective mea-surements of severity of lung disease with spirometry and potentially DLCO, and e) Adequate, standardized follow-up.
9. Outcome
ILD has an adverse impact on the long-term prognosis of MPA patients. Most of the series that compare MPA patients with and without PF reported that ILD was associated with poor prognosis and reduced survival [13,15,17,26,96]. In two recent publications, mortality was 2 to 4 times higher in MPA patients with PF [7,19]; although this has not been confirmed by others [18].
In the analyzed series of MPA patients with PF, one-third developed chronic respiratory insufficiency requiring long-term oxygen therapy [51] and 31–85% died during follow-up (Table 1) [13,18,20,26,48,58]. Mean time from presentation to death was 3.5–6 years [13,17,18,58]. In addition, 5-year survival was only 29–60% [13,18,19,55]. Primary causes of death include acute exacerbations, sepsis and progressive lung involvement with end stage respiratory failure [15,17,18,51,58].
In patients with PF and no systemic vasculitis, ANCA-positive and ANCA-negative patients have similar survival rates [13,16,56,58,59,54]. In contrast, in a large cohort of 504 patients with isolated pulmonary fi-brosis, 5-and 10-year mortality rates were significant lower in ANCA-positive patients (61% and 86% vs 38% and 70% for ANCA-negative cases) [50]. In this study, PR3-ANCA (vs MPO), age >65 years and base-line DLCO <70% were associated with increased mortality [50]. In addi-tion, a correlation between ANCA titer and mortality has been reported by some authors [16,58]. As in the case of generalized MPA, causes of death in this group included progressive deterioration of pulmonary function and infections [26].
10. Conclusions
ILD is an uncommon complication of MPO-ANCA and PR3-ANCA vasculitis that is associated with poor prognosis. Most of the time, pulmonary fibrosis develops before or occurs simultaneously with onset of systemic vasculitis symptoms. ANCA vasculitis should be included in the differential diagnosis of “idiopathic” pulmonary fibrosis. ANCA testing should be included in the diagnostic workup of interstitial pneumonia and pulmonary fibrosis both at onset and periodically during the disease course.
Large cohorts of patients with ANCA-associated interstitial pneumonia and fibrosis should be established (preferably by international col-laboration, and with an accompanying tissue biobank) to a) document geographic differences in incidence and prevalence, b) identify factors that may be involved in the development of PF in ANCA-associated vasculitis (e.g. synergistic environmental factors such as air pollution), c) determine the clinical course including transition to systemic vasculitis, perform controlled clinical trials to create optimum treatment protocols for this special subpopulation of ANCA patients.
Take-home messages.
Diffuse interstitial lung disease and pulmonary fibrosis may be clinical manifestations of anti-neutrophil cytoplasmic antibodies (ANCA) vasculitis.
The most common ANCA associated with ILD are those directed against myeloperoxidase (MPO-ANCA).
Pulmonary fibrosis occurs concurrently or precedes to ANCA vasculitis in the majority of affected individuals.
Usual interstitial pneumonia (UIP) is the most frequent radiological pattern in ANCA positive disease.
Interstitial lung disease has an adverse impact on the long-term prognosis of ANCA vasculitis.
Footnotes
Declaration of interest
The authors declare no conflicts of interest.
References
- [1].Falk RJ, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med 1988;318:1651–7. [DOI] [PubMed] [Google Scholar]
- [2].Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65:1–11. [DOI] [PubMed] [Google Scholar]
- [3].Nachman PH, Henderson AG. Pathogenesis of lung vasculitis. Semin Respir Crit Care Med 2011;32:245–53. [DOI] [PubMed] [Google Scholar]
- [4].Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992;116:488–98. [DOI] [PubMed] [Google Scholar]
- [5].Anderson G, Coles ET, Crane M, Douglas AC, Gibbs AR, Geddes DM, et al. Wegener’s granuloma. A series of 265 British cases seen between 1975 and 1985. A report by a sub-committee of the British Thoracic Society Research Committee. Q J Med 1992; 83:427–38. [PubMed] [Google Scholar]
- [6].Comarmond C, Cacoub P. Granulomatosis with polyangiitis (Wegener): clinical aspects and treatment. Autoimmun Rev 2014;13:1121–5. [DOI] [PubMed] [Google Scholar]
- [7].Schirmer JH, Wright MN, Vonthein R, Herrmann K, Nolle B, Both M, et al. Clinical presentation and long-term outcome of 144 patients with microscopic polyangiitis in a monocentric German cohort. Rheumatology (Oxford) 2016;55:71–9. [DOI] [PubMed] [Google Scholar]
- [8].Guillevin L, Durand-Gasselin B, Cevallos R, Gayraud M, Lhote F, Callard P, et al. Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999;42:421–30. [DOI] [PubMed] [Google Scholar]
- [9].Villiger PM, Guillevin L. Microscopic polyangiitis: clinical presentation. Autoimmun Rev 2010;9:812–9. [DOI] [PubMed] [Google Scholar]
- [10].Hassan TM, Hassan AS, Igoe A, Logan M, Gunaratnam C, McElvaney NG, et al. Lung involvement at presentation predicts disease activity and permanent organ damage at 6, 12 and 24 months follow-up in ANCA-associated vasculitis. BMC Immunol 2014;15:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Furuta S, Chaudhry AN, Hamano Y, Fujimoto S, Nagafuchi H, Makino H, et al. Comparison of phenotype and outcome in microscopic polyangiitis between Europe and Japan. J Rheumatol 2014;41:325–33. [DOI] [PubMed] [Google Scholar]
- [12].Reinhold-Keller E, Beuge N, Latza U, de Groot K, Rudert H, Nolle B, et al. An interdisciplinary approach to the care of patients with Wegener’s granulomatosis: longterm outcome in 155 patients. Arthritis Rheum 2000;43:1021–32. [DOI] [PubMed] [Google Scholar]
- [13].Foulon G, Delaval P, Valeyre D, Wallaert B, Debray MP, Brauner M, et al. ANCA-associated lung fibrosis: analysis of 17 patients. Respir Med 2008;102:1392–8. [DOI] [PubMed] [Google Scholar]
- [14].Nada AK, Torres VE, Ryu JH, Lie JT, Holley KE. Pulmonary fibrosis as an unusual clinical manifestation of a pulmonary-renal vasculitis in elderly patients. Mayo Clin Proc 1990;65:847–56. [DOI] [PubMed] [Google Scholar]
- [15].Hervier B, Pagnoux C, Agard C, Haroche J, Amoura Z, Guillevin L, et al. Vasculitis study, pulmonary fibrosis associated with ANCA-positive vasculitides. Retrospective study of 12 cases and review of the literature. Ann Rheum Dis 2009;68:404–7. [DOI] [PubMed] [Google Scholar]
- [16].Nozu T, Kondo M, Suzuki K, Tamaoki J, Nagai A. A comparison of the clinical features of ANCA-positive and ANCA-negative idiopathic pulmonary fibrosis patients. Respiration 2009;77:407–15. [DOI] [PubMed] [Google Scholar]
- [17].Tzelepis GE, Kokosi M, Tzioufas A, Toya SP, Boki KA, Zormpala A, et al. Prevalence and outcome of pulmonary fibrosis in microscopic polyangiitis. Eur Respir J 2010; 36:116–21. [DOI] [PubMed] [Google Scholar]
- [18].Arulkumaran N, Periselneris N, Gaskin G, Strickland N, Ind PW, Pusey CD, et al. Interstitial lung disease and ANCA-associated vasculitis: a retrospective observational cohort study. Rheumatology (Oxford) 2011;50:2035–43. [DOI] [PubMed] [Google Scholar]
- [19].Flores-Suarez LF, Ruiz N, Saldarriaga Rivera LM, Pensado L. Reduced survival in microscopic polyangiitis patients with pulmonary fibrosis in a respiratory referral centre. Clin Rheumatol 2015;34:1653–4. [DOI] [PubMed] [Google Scholar]
- [20].Fernandez Casares M, Gonzalez A, Fielli M, Caputo F, Bottinelli Y, Zamboni M. Microscopic polyangiitis associated with pulmonary fibrosis. Clin Rheumatol 2015;34: 1273–7. [DOI] [PubMed] [Google Scholar]
- [21].Eschun GM, Mink SN, Sharma S. Pulmonary interstitial fibrosis as a presenting manifestation in perinuclear antineutrophilic cytoplasmic antibody microscopic polyangiitis. Chest 2003;123:297–301. [DOI] [PubMed] [Google Scholar]
- [22].Hiromura K, Nojima Y, Kitahara T, Ueki K, Maezawa A, Kawai H, et al. Four cases of anti-myeloperoxidase antibody-related rapidly progressive glomerulonephritis during the course of idiopathic pulmonary fibrosis. Clin Nephrol 2000;53: 384–9. [PubMed] [Google Scholar]
- [23].Nakabayashi K, Arimura Y, Yoshihara K, Fukuoka T, Karube M, Yamato T, et al. Classification of clinical subtypes, patient survival, kidney prognosis, and relapse in patients with MPO-ANCA-associated vasculitis: a single-center experience. Mod Rheumatol 2009;19:420–6. [DOI] [PubMed] [Google Scholar]
- [24].Nakabayashi K, Fujioka Y, Nagasawa T, Kimura T, Kojima K, Arimura Y, et al. Dual myeloperoxidase-antineutrophil cytoplasmic antibody-and antiglomerular basement membrane antibody-positive cases associated with prior pulmonary fibrosis: a report of four cases. Clin Exp Nephrol 2011;15:226–34. [DOI] [PubMed] [Google Scholar]
- [25].Shiraki A, Ando M, Shindoh J, Abe T, Wakahara K, Makino Y, et al. Prevalence of myeloperoxidase-anti-neutrophil cytoplasmic antibody (MPO-ANCA) in patients with interstitial pneumonia. Nihon Kokyuki Gakkai Zasshi 2007;45:921–6. [PubMed] [Google Scholar]
- [26].Homma S, Matsushita H, Nakata K. Pulmonary fibrosis in myeloperoxidase antineutrophil cytoplasmic antibody-associated vasculitides. Respirology 2004;9: 190–6. [DOI] [PubMed] [Google Scholar]
- [27].Tanaka T, Otani K, Egashira R, Kashima Y, Taniguchi H, Kondoh Y, et al. Interstitial pneumonia associated with MPO-ANCA: clinicopathological features of nine patients. Respir Med 2012;106:1765–70. [DOI] [PubMed] [Google Scholar]
- [28].Becker-Merok A, Nossent JC, Ritland N. Fibrosing alveolitis predating microscopic polyangiitis. Scand J Rheumatol 1999;28:254–6. [DOI] [PubMed] [Google Scholar]
- [29].Bhanji A, Karim M. Pulmonary fibrosis-an uncommon manifestation of antimyeloperoxidase-positive systemic vasculitis? NDT Plus 2010;3:351–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Birnbaum J, Danoff S, Askin FB, Stone JH. Microscopic polyangiitis presenting as a “pulmonary-muscle” syndrome: is subclinical alveolar hemorrhage the mechanism of pulmonary fibrosis? Arthritis Rheum 2007;56:2065–71. [DOI] [PubMed] [Google Scholar]
- [31].Mansi IA, Opran A, Sondhi D, Ayinla R, Rosner F. Microscopic polyangiitis presenting as idiopathic pulmonary fibrosis: is anti-neutrophilic cytoplasmic antibody testing indicated? Am J Med Sci 2001;321:201–2. [DOI] [PubMed] [Google Scholar]
- [32].Fernandez Casares M, Gonzalez A, Caputo F, Bottinelli Y, Nastavi P, Zamboni M. Pulmonary fibrosis associated with anti-neutrophil cytoplasmic antibody positive vasculitis. Medicina (B Aires) 2012;72:329–31. [PubMed] [Google Scholar]
- [33].Streho M, Sable-Fourtassou R, Brion MC, Bourotte I, Valeyre D, Brauner M, et al. Churg-Strauss syndrome and pulmonary fibrosis: an unusual association. Presse Med 2006;35:1259–62. [DOI] [PubMed] [Google Scholar]
- [34].Takato H, Yasui M, Waseda Y, Sakai N, Wada T, Fujimura M. A case of microscopic polyangiitis following mycoplasma infection in a patient with MPO-ANCA positive pulmonary fibrosis. Allergol Int 2011;60:93–6. [DOI] [PubMed] [Google Scholar]
- [35].Tzouvelekis A, Zacharis G, Oikonomou A, Koulelidis A, Steiropoulos P, Froudarakis M, et al. Combined pulmonary fibrosis and emphysema associated with microscopic polyangiitis. Eur Respir J 2012;40:505–7. [DOI] [PubMed] [Google Scholar]
- [36].Pineton de Chambrun M, Nunes H, Brocheriou I, Hertig A. Idiopathic lung fibrosis and anti myeloperoxidase glomerulonephritis: the tree that hides the forest. BMC Pulm Med 2015;15:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tomas Chaume L, Martinez Escude A, Ortiz-Santamaria V. Pulmonary fibrosis in patients with positive neutrophil cytoplasmic antibodies vasculitis. Clin Exp Rheumatol 2016;34:145. [PubMed] [Google Scholar]
- [38].Sugimoto T, Kanasaki K, Koyama T, Yokomaku Y, Yasuda H, Kashiwagi A, et al. A case of myeloperoxidase-antineutrophil cytoplasmic antibody positive-polyarteritis nodosa complicated by interstitial pneumonia and rapidly progressive renal failure. Clin Rheumatol 2007;26:429–32. [DOI] [PubMed] [Google Scholar]
- [39].Gindre D, Peyrol S, Raccurt M, Sommer P, Loire R, Grimaud JA, et al. Fibrosing vasculitis in Wegener’s granulomatosis: ultrastructural and immunohistochemical analysis of the vascular lesions. Virchows Arch 1995;427:385–93. [DOI] [PubMed] [Google Scholar]
- [40].Souid M, Terki NH, Nochy D, Hillion D. Myeloperoxidase anti-neutrophil cytoplasmic autoantibodies (MPO-ANCA)-related rapidly progressive glomerulonephritis (RPGN) and pulmonary fibrosis (PF) with dissociated evolution. Clin Nephrol 2001;55:337–8. [PubMed] [Google Scholar]
- [41].Shields O, Shah A, Mann B. Pyrexia of unknown origin and pulmonary fibrosis as a presentation of MPO-ANCA associated vasculitis. BMJ Case Rep 2011;2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Eleftheriou D, Katsenos S, Zorbas S, Griveas I, Psathakis K. Pulmonary fibrosis presenting as an early manifestation of microscopic polyangiitis. Monaldi Arch Chest Dis 2012;77:141–4. [DOI] [PubMed] [Google Scholar]
- [43].Esposito DB, Lanes S, Donneyong M, Holick CN, Lasky JA, Lederer D, et al. Idiopathic pulmonary fibrosis in United States automated claims. Incidence, prevalence, and algorithm validation. Am J Respir Crit Care Med 2015;192:1200–7. [DOI] [PubMed] [Google Scholar]
- [44].Kekevian A, Gershwin ME, Chang C. Diagnosis and classification of idiopathic pulmonary fibrosis. Autoimmun Rev 2014;13:508–12. [DOI] [PubMed] [Google Scholar]
- [45].Gaudin PB, Askin FB, Falk RJ, Jennette JC. The pathologic spectrum of pulmonary lesions in patients with anti-neutrophil cytoplasmic autoantibodies specific for anti-proteinase 3 and anti-myeloperoxidase. Am J Clin Pathol 1995; 104:7–16. [DOI] [PubMed] [Google Scholar]
- [46].Kono M, Nakamura Y, Enomoto N, Hashimoto D, Fujisawa T, Inui N, et al. Usual interstitial pneumonia preceding collagen vascular disease: a retrospective case control study of patients initially diagnosed with idiopathic pulmonary fibrosis. PLoS One 2014;9:e94775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Flores-Suarez LF. Limited pulmonary MPA, a new MPA entity? A rheumatologist’s perspective. Clin Exp Nephrol 2013;17:672–5. [DOI] [PubMed] [Google Scholar]
- [48].Huang H, Wang YX, Jiang CG, Liu J, Li J, Xu K, et al. A retrospective study of microscopic polyangiitis patients presenting with pulmonary fibrosis in China. BMC Pulm Med 2014;14:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yu AP, Chang JX, Liu YJ, Qu QR. Computed tomography image analysis before and after treatment of anti-neutrophil cytoplasmic antibody-associated pulmonary interstitial fibrosis in 8 patients. Clin Ther 2014;36:2064–71. [DOI] [PubMed] [Google Scholar]
- [50].Kagiyama N, Takayanagi N, Kanauchi T, Ishiguro T, Yanagisawa T, Sugita Y. Antineutrophil cytoplasmic antibody-positive conversion and microscopic polyangiitis development in patients with idiopathic pulmonary fibrosis. BMJ Open Respir Res 2015;2:e000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Comarmond C, Crestani B, Tazi A, Hervier B, Adam-Marchand S, Nunes H, et al. Pulmonary fibrosis in antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitis: a series of 49 patients and review of the literature. Medicine (Baltimore) 2014; 93:340–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chen M, Yu F, Zhang Y, Zhao MH. Antineutrophil cytoplasmic autoantibody-associated vasculitis in older patients. Medicine (Baltimore) 2008;87:203–9. [DOI] [PubMed] [Google Scholar]
- [53].Takahashi K, Hayashi S, Ushiyama O, Sueoka N, Fukuoka M, Nagasawa K. Development of microscopic polyangiitis in patients with chronic airway disease. Lung 2005;183:273–81. [DOI] [PubMed] [Google Scholar]
- [54].Katsumata Y, Kawaguchi Y, Yamanaka H. Interstitial lung disease with ANCA-associated Vasculitis. Clin Med Insights Circ Respir Pulm Med 2015;9:51–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hirayama K, Kobayashi M, Usui J, Arimura Y, Sugiyama H, Nitta K, et al. Pulmonary involvements of anti-neutrophil cytoplasmic autoantibody-associated renal vasculitis in Japan. Nephrol Dial Transplant 2015;30(Suppl. 1):i83–93. [DOI] [PubMed] [Google Scholar]
- [56].Hozumi H, Enomoto N, Oyama Y, Kono M, Fujisawa T, Inui N, et al. Clinical implication of proteinase-3-antineutrophil cytoplasmic antibody in patients with idiopathic interstitial pneumonias. Lung 2016;194:235–42. [DOI] [PubMed] [Google Scholar]
- [57].Kang BH, Park JK, Roh JH, Song JW, Lee CK, Kim M, et al. Clinical significance of serum autoantibodies in idiopathic interstitial pneumonia. J Korean Med Sci 2013;28: 731–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ando M, Miyazaki E, Ishii T, Mukai Y, Yamasue M, Fujisaki H, et al. Incidence of myeloperoxidase anti-neutrophil cytoplasmic antibody positivity and microscopic polyangitis in the course of idiopathic pulmonary fibrosis. Respir Med 2013;107: 608–15. [DOI] [PubMed] [Google Scholar]
- [59].Hosoda C, Baba T, Hagiwara E, Ito H, Matsuo N, Kitamura H, et al. Clinical features of usual interstitial pneumonia with anti-neutrophil cytoplasmic antibody in comparison with idiopathic pulmonary fibrosis. Respirology 2016;21:920–6. [DOI] [PubMed] [Google Scholar]
- [60].Katsuyama T, Sada KE, Makino H. Current concept and epidemiology of systemic vasculitides. Allergol Int 2014;63:505–13. [DOI] [PubMed] [Google Scholar]
- [61].Azuma A, Hagiwara K, Kudoh S. Basis of acute exacerbation of idiopathic pulmonary fibrosis in Japanese patients. Am J Respir Crit Care Med 2008;177:1397–8 [author reply 1398]. [DOI] [PubMed] [Google Scholar]
- [62].Watts RA, Scott DG, Jayne DR, Ito-Ihara T, Muso E, Fujimoto S, et al. Renal vasculitis in Japan and the UK—are there differences in epidemiology and clinical phenotype? Nephrol Dial Transplant 2008;23:3928–31. [DOI] [PubMed] [Google Scholar]
- [63].Yamada H ANCA: associated lung fibrosis. Semin Respir Crit Care Med 2011;32: 322–7. [DOI] [PubMed] [Google Scholar]
- [64].Yamagata M, Ikeda K, Tsushima K, Iesato K, Abe M, Ito T, et al. Prevalence and responsiveness to treatment of lung abnormalities on chest computed tomography in patients with microscopic polyangiitis: a multicenter, longitudinal, retrospective study of one hundred fifty consecutive hospital-based Japanese patients. Arthritis Rheum 2016;68:713–23. [DOI] [PubMed] [Google Scholar]
- [65].Ando Y, Okada F, Matsumoto S, Mori H. Thoracic manifestation of myeloperoxidaseantineutrophil cytoplasmic antibody (MPO-ANCA)-related disease. CT findings in 51 patients. J Comput Assist Tomogr 2004;28:710–6. [DOI] [PubMed] [Google Scholar]
- [66].Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013;188:733–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].S. American Thoracic, S. European Respiratory. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus classification of the idiopathic interstitial pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS executive committee, June 2001. Am J Respir Crit Care Med 2002;165:277–304. [DOI] [PubMed] [Google Scholar]
- [68].Gocho K, Sugino K, Sato K, Hasegawa C, Uekusa T, Homma S. Microscopic polyangiitis preceded by combined pulmonary fibrosis and emphysema. Respir Med Case Rep 2015;15:128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Travis WD, Hoffman GS, Leavitt RY, Pass HI, Fauci AS. Surgical pathology of the lung in Wegener’s granulomatosis. Review of 87 open lung biopsies from 67 patients. Am J Surg Pathol 1991;15:315–33. [DOI] [PubMed] [Google Scholar]
- [70].Schnabel A, Reuter M, Csernok E, Richter C, Gross WL. Subclinical alveolar bleeding in pulmonary vasculitides: correlation with indices of disease activity. Eur Respir J 1999;14:118–24. [DOI] [PubMed] [Google Scholar]
- [71].Guilpain P, Chereau C, Goulvestre C, Servettaz A, Montani D, Tamas N, et al. The oxidation induced by antimyeloperoxidase antibodies triggers fibrosis in microscopic polyangiitis. Eur Respir J 2011;37:1503–13. [DOI] [PubMed] [Google Scholar]
- [72].Foucher P, Heeringa P, Petersen AH, Huitema MG, Brouwer E, Tervaert JW, et al. Antimyeloperoxidase-associated lung disease. An experimental model. Am J Respir Crit Care Med 1999;160:987–94. [DOI] [PubMed] [Google Scholar]
- [73].Lucattelli M, Bartalesi B, Cavarra E, Fineschi S, Lunghi B, Martorana PA, et al. Is neutrophil elastase the missing link between emphysema and fibrosis? Evidence from two mouse models. Respir Res 2005;6:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Gehrig S, Duerr J, Weitnauer M, Wagner CJ, Graeber SY, Schatterny J, et al. Lack of neutrophil elastase reduces inflammation, mucus hypersecretion, and emphysema, but not mucus obstruction, in mice with cystic fibrosis-like lung disease. Am J Respir Crit Care Med 2014;189:1082–92. [DOI] [PubMed] [Google Scholar]
- [75].Vancheri C, Gauldie J, Bienenstock J, Cox G, Scicchitano R, Stanisz A, et al. Human lung fibroblast-derived granulocyte-macrophage colony stimulating factor (GM-CSF) mediates eosinophil survival in vitro. Am J Respir Cell Mol Biol 1989;1:289–95. [DOI] [PubMed] [Google Scholar]
- [76].Peterson MW, Monick M, Hunninghake GW. Prognostic role of eosinophils in pulmonary fibrosis. Chest 1987;92:51–6. [DOI] [PubMed] [Google Scholar]
- [77].Chrysanthopoulou A, Mitroulis I, Apostolidou E, Arelaki S, Mikroulis D, Konstantinidis T, et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J Pathol 2014;233:294–307. [DOI] [PubMed] [Google Scholar]
- [78].Yoshida M, Yamada M, Sudo Y, Kojima T, Tomiyasu T, Yoshikawa N, et al. Myeloperoxidase anti-neutrophil cytoplasmic antibody affinity is associated with the formation of neutrophil extracellular traps in the kidney and vasculitis activity in myeloperoxidase anti-neutrophil cytoplasmic antibody-associated microscopic polyangiitis. Nephrology (Carlton) 2016;21:624–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Churg A, Zay K, Shay S, Xie C, Shapiro SD, Hendricks R, et al. Acute cigarette smoke-induced connective tissue breakdown requires both neutrophils and macrophage metalloelastase in mice. Am J Respir Cell Mol Biol 2002;27:368–74. [DOI] [PubMed] [Google Scholar]
- [80].Yan Z, Kui Z, Ping Z. Reviews and prospectives of signaling pathway analysis in idiopathic pulmonary fibrosis. Autoimmun Rev 2014;13:1020–5. [DOI] [PubMed] [Google Scholar]
- [81].Mark EJ, Matsubara O, Tan-Liu NS, Fienberg R. The pulmonary biopsy in the early diagnosis of Wegener’s (pathergic) granulomatosis: a study based on 35 open lung biopsies. Hum Pathol 1988;19:1065–71. [DOI] [PubMed] [Google Scholar]
- [82].Yoshikawa Y, Watanabe T. Pulmonary lesions in Wegener’s granulomatosis: a clinicopathologic study of 22 autopsy cases. Hum Pathol 1986;17:401–10. [DOI] [PubMed] [Google Scholar]
- [83].Holle JU, Gross WL. Treatment of ANCA-associated vasculitides (AAV). Autoimmun Rev 2013;12:483–6. [DOI] [PubMed] [Google Scholar]
- [84].Yates M, Watts RA, Bajema IM, Cid MC, Crestani B, Hauser T, et al. EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis. Ann Rheum Dis 2016;75:1583–94. [DOI] [PubMed] [Google Scholar]
- [85].McGeoch L, Twilt M, Famorca L, Bakowsky V, Barra L, Benseler SM, et al. CanVasc recommendations for the management of antineutrophil cytoplasm antibody-associated vasculitides. J Rheumatol 2016;43:97–120. [DOI] [PubMed] [Google Scholar]
- [86].Ntatsaki E, Carruthers D, Chakravarty K, D’Cruz D, Harper L, Jayne D, et al. BSR and BHPR guideline for the management of adults with ANCA-associated vasculitis. Rheumatology (Oxford) 2014;53:2306–9. [DOI] [PubMed] [Google Scholar]
- [87].Tashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D, Kleerup EC, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med 2016;4:708–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Liossis SN, Bounas A, Andonopoulos AP. Mycophenolate mofetil as first-line treatment improves clinically evident early scleroderma lung disease. Rheumatology (Oxford) 2006;45:1005–8. [DOI] [PubMed] [Google Scholar]
- [89].Fischer A, Brown KK, Bois RM Du, Frankel SK, Cosgrove GP, Fernandez-Perez ER, et al. Mycophenolate mofetil improves lung function in connective tissue disease-associated interstitial lung disease. J Rheumatol 2013;40:640–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Bosello SL, De Luca G, Rucco M, Berardi G, Falcione M, Danza FM, et al. Long-term efficacy of B cell depletion therapy on lung and skin involvement in diffuse systemic sclerosis. Semin Arthritis Rheum 2015;44:428–36. [DOI] [PubMed] [Google Scholar]
- [91].Keir GJ, Maher TM, Ming D, Abdullah R, de Lauretis A, Wickremasinghe M, et al. Rituximab in severe, treatment-refractory interstitial lung disease. Respirology 2014; 19:353–9. [DOI] [PubMed] [Google Scholar]
- [92].Lepri G, Avouac J, Airo P, Anguita Santos F, Bellando-Randone S, Blagojevic J, et al. Effects of rituximab in connective tissue disorders related interstitial lung disease. Clin Exp Rheumatol 2016;34(Suppl. 100):181–5. [PubMed] [Google Scholar]
- [93].Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011;377:1760–9. [DOI] [PubMed] [Google Scholar]
- [94].Nathan SD, Albera C, Bradford WZ, Costabel U, Glaspole I, Glassberg MK, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017;5:33–41. [DOI] [PubMed] [Google Scholar]
- [95].Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med 2011;365: 1079–87. [DOI] [PubMed] [Google Scholar]
- [96].Booth AD, Almond MK, Burns A, Ellis P, Gaskin G, Neild GH, et al. Outcome of ANCA-associated renal vasculitis: a 5-year retrospective study. Am J Kidney Dis 2003;41: 776–84. [DOI] [PubMed] [Google Scholar]