

Abstract
Proper localization of membrane proteins is essential for the function of biological membranes and for the establishment of organelle identity within a cell. Molecular machineries that mediate membrane protein biogenesis need to not only achieve a high degree of efficiency and accuracy, but also prevent off-pathway aggregation events that can be detrimental to cells. The posttranslational targeting of tail-anchored proteins (TAs) provides tractable model systems to probe these fundamental issues. Recent advances in understanding TA-targeting pathways reveal sophisticated molecular machineries that drive and regulate these processes. These findings also suggest how an interconnected network of targeting factors, cochaperones, and quality control machineries together ensures robust membrane protein biogenesis.
Keywords: tail-anchored protein, protein targeting, ATPase, membrane protein biogenesis, chaperones, protein quality control
OVERVIEW
Membrane proteins account for ~35% of the proteins encoded by the genome. The proper functioning of biological membranes requires all newly synthesized membrane proteins to be localized to and inserted into the appropriate membrane destinations. The process of membrane protein biogenesis presents several fundamental challenges for the cell. Given the multiple membranes in a eukaryotic cell, how are newly synthesized membrane proteins recognized and sorted to the correct target membranes? What are the energy requirements in these pathways? How are the hydrophobic transmembrane domains (TMDs) on nascent proteins effectively protected from aggregation as they traverse aqueous cellular environments? How are the TMDs efficiently inserted into the phospholipid bilayer? Finally, have cells evolved correction mechanisms for cases in which targeting and insertion fail?
The best-studied pathway for membrane protein targeting and insertion utilizes the signal recognition particle (SRP). SRP recognizes TMDs near the N terminus of nascent proteins and delivers them to translocation machineries on the eukaryotic endoplasmic reticulum (ER) or the bacterial plasma membrane during translation; this cotranslational mode of targeting effectively minimizes the aggregation of membrane proteins in the cytosol (Akopian et al. 2013). Nevertheless, numerous membrane proteins cannot use SRP and must be targeted via posttranslational pathways, the mechanisms of which are far less well understood. A salient example is the class of tail-anchored proteins (TAs), which contain a single TMD near the C terminus. TAs compose 3–5% of the eukaryotic membrane proteome and mediate diverse cellular processes, including protein translocation across organelle membranes, vesicular transport, apoptosis, and protein quality control (Chartron etal. 2012a, Hegde & Keenan 2011). Because the C-terminal TMDs of TAs are obscured by the ribosome during translation, it was predicted early on that these proteins would be targeted by posttranslational mechanisms (Kutay et al. 1993). In support of this hypothesis, synaptobrevin 2 (Syb2), a tail-anchored SNARE protein, can be targeted to and inserted into the ER after release from the ribosome (Kutay et al. 1995). Recent work has uncovered multiple pathways that posttranslationally target TAs to different cellular membranes and has begun to elucidate the molecular mechanisms underlying these processes.
Studies of TA biogenesis have provided extensive information to help address how cellular machineries overcome multiple challenges during posttranslational membrane protein targeting. These studies begin to elucidate the nature of the targeting signals that direct TAs to diverse cellular membranes; the pathways, driving forces, and molecular mechanisms of their targeting; and the interconnection of the protein-targeting pathway with cellular chaperones and quality control machineries. Here, we summarize recent advances on these fronts from cell biological, genetic, structural, and biochemical studies.
TARGETING SIGNALS
Early microscopy studies of green fluorescent protein (GFP) fused to targeting sequences showed that C-terminal sequences encompassing the TMD are necessary for and sufficient to direct the proper localization of TAs to diverse organelles such as the ER, mitochondria, peroxisomes, and secretory membranes (Egan et al. 1999, Masaki et al. 2003, Mullen & Trelease 2000, Yagita et al. 2013). The notion of C-terminal TMDs as targeting signals, however, raises challenging questions as to how specific organelle information is encoded. The observation that Fission 1 (Fisl)-like proteins can localize to multiple organelles [mitochondria, peroxisomes, and chloroplasts (Ruberti et al. 2014)] highlights the promiscuity that could result from such degenerate topogenic signals.
Sequence comparisons and mutational analyses showed that the targeting information is encoded, in part, by a combination of physicochemical properties in the TMD (Figure 1). On average, TAs on the outer mitochondrial membrane (OMM) have shorter and less hydrophobic TMDs than do TAs that traffic to or through the ER (Figure 1) (Beilharz et al. 2003, Lee et al. 2014, Rao et al. 2016). The TMDs of OMM TAs also exhibit lower helical contents than do the TMDs of ER-destined TAs (Rao et al. 2016). Proteins that harbor isoforms or splice variants that localize to either the ER or the OMM, such as cytochrome b5 (Cytb5) and sarcolemmal membrane-associated protein (SLMAP), provided models for better-controlled analyses. In the cases of both Cytb5 and SLMAP, the TMD of the OMM TA variant has lower hydrophobicity, and substitution of moderately hydrophobic residues with the more hydrophobic and helix-promoting leucine relocalized the variant to the ER (Byers et al. 2009, Hwang et al. 2004). Likewise, replacement of three glycines with leucines in the Fis1 TMD relocalized Fis1 from the OMM to the ER (Beilharz et al. 2003). Recently, hydrophobic residues in the TMD of Bos1, a vesicular TA, were systematically replaced with increasing numbers of alanine and glycine. Correlation analysis of this set of variants further showed that the helical propensity of the TMD plays an important role in engaging ER-targeting factors (Rao et al. 2016; see the section titled Substrate Selection by the GET Pathway, below). Thus, the hydrophobicity and helical propensity of the TMD together dictate TA localization to the ER versus other organelles.
Figure 1.
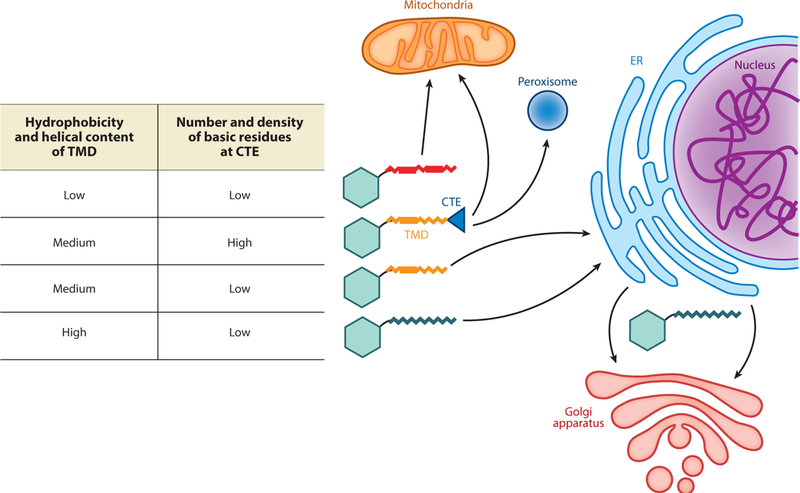
A combination of physicochemical properties in the transmembrane domain (TMD) and C-terminal element (CTE) directs tail-anchored proteins (TAs) to distinct cellular organelles. TAs with weakly hydrophobic TMDs or those with a moderately hydrophobic TMD followed by a basic CTE are targeted to mitochondria or peroxisomes. TAs with moderately to strongly hydrophobic TMDs are targeted to the ER. TAs with long and strongly hydrophobic TMDs further enter vesicular trafficking pathways to reach secretory vesicles, the Golgi apparatus, and the plasma membrane.
Some mitochondrial TAs are enriched in basic residues immediately C terminal to the TMD; this positively charged C-terminal element (CTE) (Figure 1) is important for specifying the OMM localization of these TAs (Horie et al. 2002, Kuroda et al. 1998, Marty et al. 2014, Rao et al. 2016). Mutation of basic residues in the CTE of multiple OMM TAs reduced or abolished their mitochondria targeting and, in the cases of tung Cytb5 and Fis1, led to dual ER and mitochondria localization (Horie et al. 2002, Kuroda et al. 1998, Rao et al. 2016). A systematic variation of the CTE showed that a minimum of four basic residues is necessary and sufficient for specific mitochondrial localization of Fis1 (Rao etal. 2016). In addition, increasing the spacing between the TMD and basic CTE of TAs reduces their mitochondrial targeting (Horie et al. 2002, Marty et al. 2014), and bioinformatic analysis showed that a dibasic motif immediately following the TMD is a strong predictor of OMM TAs in plant cells (Marty et al. 2014). Intriguingly, the combination of a moderately hydrophobic TMD followed by a basic CTE also directs TAs to peroxisomes (Chen et al. 2014a, Halbach et al. 2006, Yagita et al. 2013), although how mitochondrial and peroxisomal TAs are distinguished is unclear (Figure 1). Finally, the observation that insertion of leucines into the TMD of Fis1 overrides the CTE and allows for efficient ER targeting (Beilharz et al. 2003) indicates that TA localization is specified by a combination of physicochemical properties from both the TMD and the CTE.
The ER serves as the gateway from which proteins enter vesicular trafficking pathways to arrive at the Golgi apparatus, secretory granules, or the plasma membrane. Sequence analyses showed that, compared to ER-resident TAs, the TAs localized on secretory vesicles and the plasma membrane have longer and more hydrophobic TMDs (Pedrazzini et al. 1996, Rao et al. 2016). Consistent with this observation, lengthening the TMD of Cytb5 from 17 to 22 amino acids led to recruitment of the mutant protein to ER exit sites and its export to the plasma membrane via the secretory pathway (Honsho et al. 1998, Pedrazzini et al. 1996, Ronchi et al. 2008). Although these observations need to be generalized to additional TAs, it is tempting to propose that the length and hydrophobicity of the TMD dictate whether TAs are retained in the ER or further enter vesicular trafficking (Figure 1).
PATHWAYS AND MECHANISMS FOR TARGETING OF TAIL-ANCHORED PROTEINS
The GET Pathway for Targeting of Tail-Anchored Proteins to the ER
Much recent progress on TA biogenesis was driven by the discovery of the GET (guided entry of TA) pathway. The structure, dynamics, and interactions of GET components have been extensively characterized, providing the highest-resolution understanding of a TA-targeting pathway thus far.
Summary of the pathway.
Early studies showed that TA insertion into the ER is ATP dependent and requires machineries distinct from those that mediate cotranslational protein targeting (Kutay et al. 1995). Subsequently, cross-linking studies in reticulocyte lysate identified a 40-kDa ATPase, TRC40, as a key targeting factor for TAs (Favaloro et al. 2008, Stefanovic & Hegde 2007). The yeast homolog of TRC40, Get3, was genetically linked to two integral membrane proteins on the ER, Get1 and Get2 (Schuldiner et al. 2005), which were shown to form the receptor complex for Get3 (Schuldiner et al. 2008). An additional protein complex, comprising Get4 and Get5, was found to participate in the pathway on the basis of the genetic interactions of Get4 and Get5 with Get1, Get2, and Get3; reduced TA targeting in Δget5 lysates; and the physical association of the Get4/5 complex with Get3 (Jonikas et al. 2009). Biochemical reconstitutions showed that Get4/5 facilitates TA loading onto Get3 from the upstream cochaperone Sgt2 (Wang et al. 2010). Homologs or functional orthologs of all components of the yeast GET pathway have been identified in mammalian cells (Table 1) (Colombo et al. 2016; Mock et al. 2015; Vilardi et al. 2011, 2014; Xu et al. 2012; Yamamoto & Sakisaka 2012). Collectively, these works define a conserved pathway in eukaryotic cells that mediates the targeted delivery and insertion of TAs into the ER.
Table 1.
Components of the GET pathway in yeast and mammalian cells
| Cell type | Upstream cochaperone |
Scaffolding complex | Cytosolic ATPase | Membrane receptors | |
|---|---|---|---|---|---|
| Yeast | Sgt2 | Get4/5 | Get3 | Getl | Get2 |
| Mammal | SGTA | TRC35/UBL4A/BAG6 | TRC40 | WRB | CAML |
This early work, together with subsequent mechanistic studies, defined the major molecular events in the GET pathway (Figure 2). After a nascent TA is synthesized and released from the ribosome, it is captured by Sgt2 through a yet unknown mechanism (step ➊). The Get4/5 complex, via its abilities to bridge between Sgt2 and Get3 and to regulate the conformation of Get3, stimulates the transfer of TA substrate from Sgt2 to ATP-bound Get3 (step ➋). The Get3 •TA complex dissociates from Get4/5, and the TA substrate stimulates ATP hydrolysis on Get3 (step ➌). The Get3 •TA complex then engages the Get1/2 receptor complex at the ER membrane (step ➍), where Get1/2 releases TA from Get3 and facilitates TA insertion into the membrane (step ➎). Finally, Get3 is released from Get1 and is returned to the cytosol through binding of ATP and Get4/5 (step ➏). Our current understanding of the molecular mechanisms of these events is further discussed below.
Figure 2.
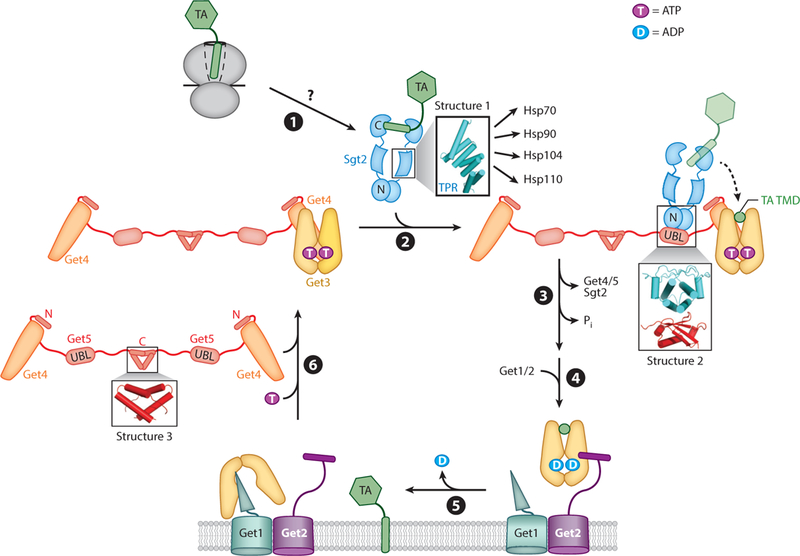
Major steps in the yeast GET pathway. (➊) A nascent tail-anchored protein (TA) is captured by Sgt2 after translation by the ribosome. Structure 1 (PDB 3SZ7) shows the Sgt2 tetratricopeptide repeat (TPR) domain, which binds various chaperones. (➋) Sgt2 transfers the TA to Get3, a process stimulated by the Get4/5 complex. Structure 2 (PDB 2LXC) shows the N-terminal domain of Sgt2 bound to the ubiquitin-like (UBL) domain of Get5. (➌) The Get3•TA complex dissociates from Get4/5, and ATP hydrolysis is activated. (➍) The Get2 subunit in the Get1/2 receptor captures the Get3•TA complex. (➎) Following ADP release, Getl interacts with and disassembles the Get3•TA complex, and the TAis inserted into the membrane through an unknown mechanism. (➏) ATP and Get4/5 together drive the release of Get3 from Getl, recycling Get3 for additional rounds of targeting. Structure 3 (PDB 2LNZ) shows the Get5 homodimerization domain. TMD denotes transmembrane domain. Individual proteins are denoted by the following colors: Getl, cyan; Get2, violet; Get3, yellow; Get4, orange; Get5, red; Sgt2, blue; TA, green.
Structure and function of Sgt2.
The most upstream factor identified thus far in the GET pathway is Sgt2, which captures TAs after their translation. Sgt2 contains multiple protein interaction domains: an N-terminal homodimerization domain (NTD) that binds Get5, a tetratricopep-tide repeat (TPR) domain that interacts with chaperones, and a glutamine-and methionine-rich C-terminal domain (CTD) (Figure 2). Immunoprecipitation experiments demonstrated that the Sgt2 CTD forms the substrate-binding site that selectively captures the TMDs of ER-destined TAs (Rao et al. 2016, Wang et al. 2010). The molecular basis of this recognition is unclear but was speculated to be analogous to how the methionine-rich domain of SRP recognizes hydrophobic signal sequences in substrate proteins (Wang et al. 2010). As the individual domains of Sgt2 are connected by flexible linkers, the relative positions of these domains have not been defined. Small-angle X-ray scattering (SAXS) data suggested that the global conformation of Sgt2 is extended but likely flexible (Chartron et al. 2011), which raises possibilities of regulation by its diverse binding partners.
The Sgt2 NTD is both a homodimerization domain and interaction platform for the ubiquitin-like (UBL) domain of Get5, linking this cochaperone with the rest of the GET pathway (Chang et al. 2010, Chartron et al. 2010, Liou et al. 2007, Wanget al. 2010). Nuclear magnetic resonance (NMR) and crystallographic analyses of the Saccharomyces cerevisiae Sgt2 NTD bound to the Get5 UBL domain elucidated the molecular details of their interaction (Figure 2,Structure2)(Chartron et al. 2012b, Simon et al. 2013, Tung et al. 2013). The Sgt2 homodimer interface is formed by a four-helix bundle, which is contributed by two N-terminal helices from each Sgt2 NTD and is stabilized by hydrophobic interactions. In contrast, the interaction of the Sgt2 NTD with the Get5 UBL domain is electrostatically driven. Conserved residues on the second helices of the Sgt2 NTD form an acidic surface that interacts with conserved basic and hydrophobic residues on a single Get5 UBL domain via charge and shape complementarity (Figure 2, Structure 2). The Sgt2-Get5 interaction is stable but occurs with fast association and dissociation kinetics (Chartron et al. 2012b, Simon et al. 2013), which may allow Get4/5 to rapidly sample Sgt2 molecules.
Get4/5: a scaffold that bridges Sgt2 and Get3.
After capture by Sgt2, the TA substrate is transferred to Get3 in a Get4/5-dependent process (Wang et al. 2010). Get5 is a modular protein composed of an NTD that interacts with Get4, a UBL domain that binds the Sgt2 NTD as described above, and a CTD that mediates homodimerization (Figure 2, Structure 3) (Chartron etal. 2010). A minimal Get4/5N complex, formed between Get4 and the Get5NTD, was sufficient to recognize Get3 in a specific conformation and nucleotide state (Gristick et al. 2014, 2015) and was hence subjected to extensive biochemical and structural studies. Get4 forms an a2-solenoid fold composed of 14 right-handed helical coils (Bozkurt et al. 2010, Chang et al. 2010, Chartron et al. 2010). The N-terminal helix of Get5 docks into a hydrophobic groove formed by helices α12 and α13 and the (β-tongue of Get4, forming an extremely stable Get4-Get5 interface (Chang et al. 2010, Chartron et al. 2010). On the other side of the Get4/5N complex, the N-terminal helices of Get4 provide a combination of acidic and hydrophobic residues to mediate interaction with Get3 (further discussed in the section titled The Get3 ATPase Cycle, below).
Multiple groups have reconstituted Get4/5-mediated stimulation of TA transfer from Sgt2 to Get3 (Mateja et al. 2015, Rao et al. 2016, Wang et al. 2010). Nevertheless, the precise mechanism(s) underlying the stimulatory effect of Get4/5 is not completely understood. By bringing Sgt2 and Get3 into close proximity, Get4/5 could enable a facile route for relay of a TA substrate while minimizing cytosolic exposure of the substrate TMD (Figure 2). In support of this model, the TA is protected from external traps during the transfer, and mutations disrupting the Sgt2-Get5 interaction resulted in significantly less TA transfer and less insertion-competent Get3•TA complexes (Mateja et al. 2015, Shao et al. 2017). As discussed below in the section titled The Get3 ATPase Cycle, the Get4/5 complex also regulates the conformation of the Get3 ATPase, which could further promote substrate capture by Get3. It is also plausible that Get5 induces rearrangements in Sgt2 that facilitate TA release. The relative contributions of these mechanisms to the substrate handover event remain to be defined.
The Get5 CTD forms a stable dimer interface mediated by hydrophobic interactions (Chartron et al. 2012c). SAXS data also showed that Get4 and Get5 form an elongated heterotetramer (2:2) spanning 240 Å (see architecture of the heterotetrameric complete Get4/5 complex in Figure 2) (Chartron et al. 2010). Nevertheless, the functional relevance of Get5 homodimerization is unclear. The residues that mediate Get5 homodimerization are not conserved in its mammalian homolog (see UBL4A in Figure 5a, below). Furthermore, there appears to be an intriguing asymmetry in the Get4/5 complex such that only one copy of Get4 in this heterotetramer binds Get3 at physiological protein concentrations (Gristick et al. 2015, Mateja et al. 2015). The mechanism of this asymmetric interaction and the evolutionary relevance of the Get5 homodimerization domain remain to be determined.
Figure 5.
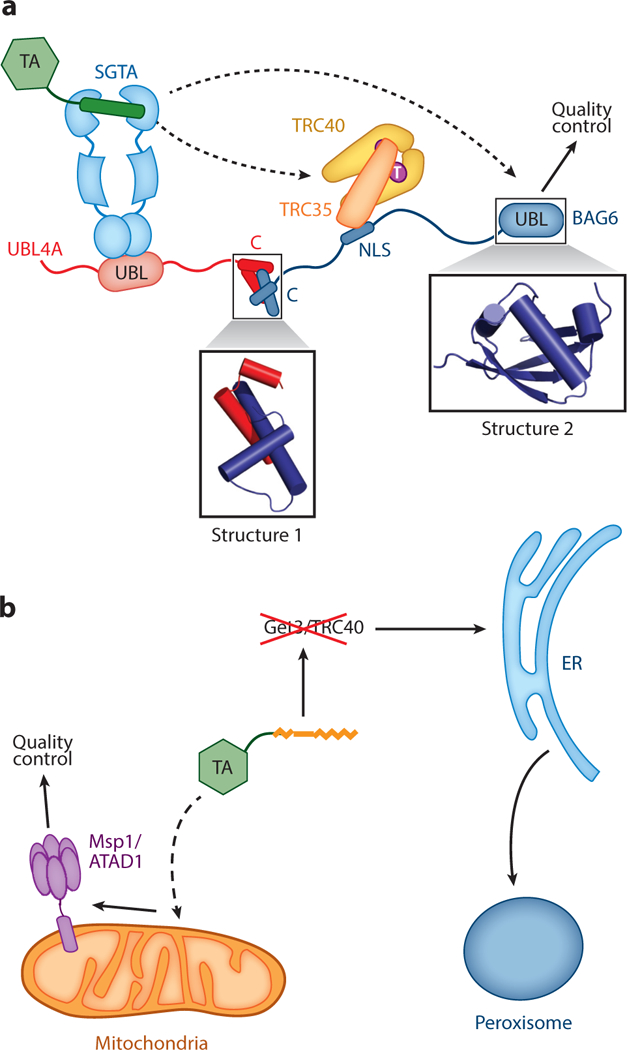
Connection of tail-anchored protein (TA) targeting to quality control pathways. (a) Architecture of the mammalian TA transfer complex, in which SGTA and TRC40 are linked by the heterotrimeric BAG6 complex comprising UBL4A, TRC35, and BAG6. Dashed lines depict the two potential fates of the TA in this complex: transfer to TRC40 for targeting to the ER and transfer to BAG6 for ubiquitylation and degradation. Structure 1 (PDB 4WWR) shows the crystal structure of the complex between the C-terminal helices of UBL4A and BAG6. Structure 2 (PDB 4DWF) shows the crystal structure of the N-terminal UBL domain of BAG6. (b) A local quality control mechanism at mitochondria based on the AAA-ATPase Mspl on the outer mitochondrial membrane, which degrades Pex15 mislocalized to mitochondria when the GET pathway is disabled. Other abbreviations: NLS, nuclear localization sequence; UBL, ubiquitin like.
The Get3 ATPase cycle: Nucleotide-, effector-, and substrate-induced conformational changes drive the targeting pathway.
Central to the GET pathway is the ATPase Get3, which uses its ATPase cycle to capture and deliver TAs to the ER membrane (Favaloro et al. 2010, Stefanovic & Hegde 2007). Early crystallographic work showed that Get3 undergoes ATP-dependent rearrangements that can be coupled to substrate binding (Figure 3, step ➊). Get3 is an obligate homodimer bridged by a tightly coordinated Zn2+ ion. Each Get3 subunit contains a nucleotide hydrolase domain structurally and functionally coupled to a helical domain. Apo-Get3 crystallizes in an open conformation, in which the two helical domains are apart (Figure 3, Structure 1). In contrast, nonhydrolyzable ATP analogs induce readjustments at the dimer interface that bring the helical domains closer to one another (Figure 3, Structure 2) (Bozkurt et al. 2009, Hu et al. 2009, Mateja et al. 2009, Suloway et al. 2009). Importantly, closing of Get3 brings together conserved hydrophobic residues in the helical domains to form a hydrophobic groove that provides the binding site for the TA TMD (Mateja et al. 2015). Nevertheless, Get3 has been observed in a variety of conformations that differ in the degree of opening or closing, suggesting the presence of more than two defined states. Furthermore, molecular dynamics simulations suggest that Get3 dynamically samples multiple conformations (wide open, open, semiopen, semiclosed, and closed) in different nucleotide states (Wereszczynski & McCammon 2012). The number of conformational states in Get3 and how they are regulated by nucleotides remain to be defined.
Figure 3.
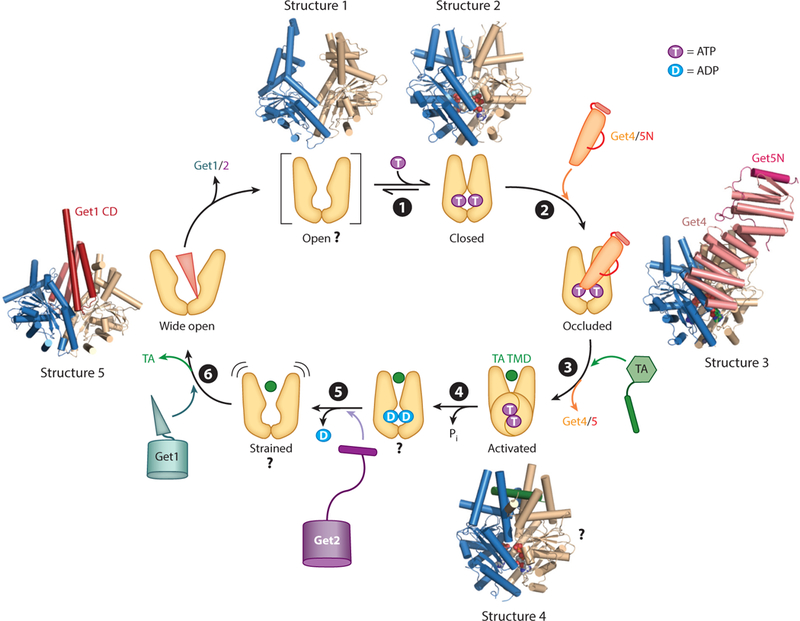
The Get3 ATPase cycle is driven by nucleotides, effectors, and tail-anchored protein (TA) substrates. Structure 1 shows apo-Get3 in an open conformation (PDB 3H84), although whether this species exists in vivo is unclear. (➊) ATP binding induces Get3 into a closed conformation (Structure 2; PDB 2WOJ). (➋) Get4/5 preferentially binds closed Get3 and inhibits its ATPase activity, generating an occluded state (Structure 3; PDB 4PWX). (➌) TA binding induces Get3 into an activated state, and Get3 dissociates from Get4/5. The crystal structure of Get3 bound with a transmembrane domain (TMD) peptide is shown in Structure 4 (PDB 4XTR), although the structural basis for the TA-induced activation of Get3 is unclear. (➍) Activated Get3 hydrolyzes ATP, and the ADP-bound Get3TA complex can bind Get2. (➎) ADP release induces additional rearrangements in the Get3 •TA complex, which enable it to interact with Get1. (➏) The strong preference of Get1 for a wide-open Get3 (Structure 5; PDB 3SJB) drives the release of TA from Get3. CD denotes cytosolic domain. The two subunits in the Get3 homodimer are in blue and tan in all structures.
Indeed, subsequent biochemical, enzymatic, and structural analyses uncovered additional con-formational states in Get3 that are regulated by the TA substrate and other GET components. A major regulator is the Get4/5 complex. Pulldown (Chartron et al. 2010, Gristick et al. 2014) and fluorescence (Rome et al. 2014) studies showed that Get4 preferentially binds ATP-bound Get3, and reciprocally, Get4/5 stabilizes ATP binding to Get3 (Rome et al. 2013). As ATP stabilizes closed Get3, the synergy between ATP and Get4/5 in binding Get3 strongly suggests that Get4/5 also stabilizes Get3 in a closed conformation. In contrast, Get4/5 inhibits the ability of Get3 to hydrolyze ATP (Rome et al. 2013), suggesting that the global closing of Get3 can be uncoupled from catalytic activation at the ATPase site. This Get4/5-induced new state of Get3, termed occluded, was visualized crystallographically using Get4/5N bound to a hydrolysis-deficient mutant of Get3, Get3 (D57V), loaded with ATP (Gristick etal. 2014). A single Get4 molecule bridges the Get3 dimer interface and interacts with both subunits of Get3 (Figure 3, Structure 3). The interaction with one Get3 subunit consists of both electrostatic and hydrophobic contacts, generating an anchoring interface for high-affinity binding of Get4/5 to ATP-bound Get3 (Gristick et al. 2014). Additional residues in Get4 establish a regulatory interface with the other Get3 subunit, at which a putative salt bridge between Get3 K69 and Get4 D74 is critical for ATPase inhibition (Gristick et al. 2014). Together, these results show that Get4/5 primes Get3 into the optimal conformation and nucleotide state for capturing the TA substrate.
In contrast to Get4/5, a TA substrate induces a rapid round of ATP hydrolysis on Get3 (Rome et al. 2013). This observation led to the proposal that the TA substrate induces Get3 into an activated conformation and leads to the ATP hydrolysis event after TA loading on Get3 (Figure 3, steps ➌ and ➍). Upon TA loading, the interaction of Get3 with Get4/5 is weakened at least tenfold in the presence of ATP, and the interaction becomes undetectable with a nucleotide-free Get3•TA complex (Rome et al. 2014). These findings suggest that the TA substrate also helps drive the dissociation of Get3 from Get4/5. Finally, the TA substrate significantly slows nucleotide exchange on Get3. Compared to free Get3, ADP release from the Get3•TA complex is 200-fold slower, and ATP rebinding to the Get3•TA complex is 10,000-fold slower and rate limited by a conformational change at physiological ATP concentrations (Rome et al. 2013). Slower nucleotide exchange provides extended time windows of ~14 and ~12 s for Get3•TA complexes in the ADP-bound and nucleotide-free states, respectively, during which these complexes can interact with the Get1/2 receptor complex (see the section titled The Get1/2 Membrane Receptor Complex Remodels the Targeting Complex, below).
The Get1/2 membrane receptor complex remodels the targeting complex.
Get1/2 provides the receptor complex for the Get3•TA complex at the ER membrane (Schuldiner et al. 2008). Biochemical reconstitution with proteoliposomes validated that these two proteins (and their mammalian homologs) are necessary and sufficient for TA targeting and insertion (Mari-appan et al. 2011, Vilardi et al. 2014, F. Wang et al. 2011, Yamamoto & Sakisaka 2012). Both Getl and Get2 contain three predicted TMDs via which they assemble into a complex (Mari-appan et al. 2011, Wang et al. 2014). Both proteins also contain large cytosolic domains (CDs) (the N-terminal CD in Get2 and the TM1-TM2 loop in Get1) that interact with Get3. Get2 CD contains two amphiphilic helices connected by a glycine linker, and helix a1 electrostatically contacts one of the subunits in the Get3 dimer via conserved basic residues in the 14RERR motif (Mariappan et al. 2011, Stefer et al. 2011, F. Wang et al. 2011). Whereas the Get2 CD cocrystallized with closed, nucleotide-bound Get3 (Mariappan et al. 2011, Stefer et al. 2011), the Get1 CD cocrystallized with apo-Get3 in the most open conformation observed thus far (Figure 3, Structure 5). The Get1 CD consists of two helices that form a coiled coil, which inserts like a wedge into the Get3 dimer interface and contacts both Get3 subunits in the dimer (Kubota et al. 2012, Mariappan et al. 2011, Stefer et al. 2011). Contacts with one Get3 subunit occur at its nucleotide-binding domain, where an extensive interface is formed by both aromatic and charged residues. Contacts with the other Get3 subunit are smaller and involve helix α4 in its helical domain. Finally, Get1 and Get2 share overlapping interaction surfaces, notably the 303DELYED motif on helix α11, on Get3 (Mariappan et al. 2011, Stefer et al. 2011). Both receptor subunits also share overlapping binding sites on Get3 with Get4/5 (Gristick et al. 2014). Thus, these upstream and downstream GET proteins compete for interaction with Get3 during the targeting cycle.
These structural data, together with the following observations, strongly suggest that the Get3•TA complex is first captured by Get2 and then transferred to Get1. Get2 contains a >100-amino-acid linker that connects its Get3-binding helices to its TMDs, which may allow Get2 to search for Get3•TA complexes further away from the ER. Surface plasmon resonance (SPR) and fluorescence analyses show that the Get2 CD can bind Get3 and Get3•TA complexes in nucleotide-bound states, whereas the Get1 CD binds only the nucleotide-free Get3•TA complex and strongly prefers apo-Get3 in which the TA-binding groove is disrupted (Mariappan et al. 2011, Rome et al. 2014, Stefer et al. 2011). Finally, high concentrations of the Get1 CD can displace TA from Get3, whereas the Get2 CD cannot (Mariappan et al. 2011, F. Wang et al. 2011). To-gether, these results support a model in which the Get3•TA complex bound with ADP is initially recruited to the membrane by Get2; upon ADP release, the Get1 CD initiates interaction with and remodels the Get3•TA complex, leading to the release of TA from Get3 and to its insertion into the ER membrane (Figure 2, steps ➍ and ➎, and Figure 3, steps ➎ and ➏).
These data also predict that a stable complex between the Get1 CD and apo-Get3 accumulates at the ER membrane at the end of the targeting cycle (Figure 2, end of step ➎). The tip of Get1 remodels both switch I and switch II loops at the Get3 ATPase site, inducing these loops into a conformation incompatible with nucleotide binding (Kubota et al. 2012, Mariappan et al. 2011, Stefer et al. 2011). Consistent with the structures, SPR and fluorescence measurements showed that ATP and the Get1 CD strongly antagonize one another for binding to Get3 (Kubota et al. 2012, Mariappan et al. 2011, Rome et al. 2014, Stefer et al. 2011). Furthermore, addition of ATP accelerated the release of Get3 from the Get1 CD and vice versa, suggesting a release mechanism involving active displacement (Kubota et al. 2012, Rome et al. 2014). Finally, with full-length Get1/2 proteoliposomes or ER microsomes, Get4/5 was also needed to promote the facile release of Get3 from the membrane (Rome et al. 2014). Thus, the recycling of Get3 in the GET pathway is an elaborate event driven by both ATP and Get4/5 (Figure 2, step ➏).
Despite extensive progress, multiple questions remain for the GET pathway. First, the structural basis for the TA-induced changes in Get3 activity is unclear. Although a cocrystal structure of Get3 (D57N) bound to a TMD peptide (Mateja et al. 2015) is available, the conformation of Get3 in this structure is similar to the conformations in the Get3•Get4/5 complex and in ADP•AlF4-bound Get3. The structure, dynamics, and mechanism of regulation of the Get3•TA complex remain to be elucidated at the molecular level (Figure 3, question marks). In addition, the structures of important intermediates in the pathway, such as Get2 and/or Get1 bound to the Get3•TA complex, are still unavailable. Whether Get2 acts passively to bring the Get3•TA complex to Get1 or plays a more active role is unclear. Although Get1 and Get2 bind at overlapping sites on Get3, their cobinding has been observed by NMR (Stefer et al. 2011); the stoichiometry and relationship of the interaction of Get3•TA with full-length Get1/2 remain to be resolved. Finally, the molecular mechanisms by which Get1/2 inserts the TA into the membrane are still unclear (see further discussion in the section titled Outstanding Questions, below).
Substrate selection by the GET pathway.
The GET pathway provides an opportunity to decipher the mechanism by which targeting machineries specifically select TAs destined to the appropriate organelle. Systematic variation of the targeting sequence in model TAs, coupled with biochemical dissections, revealed at least two selection filters that distinguish GET-dependent from GET-independent substrates (Rao et al. 2016). First, Sgt2 preferentially binds TMDs that have higher hydrophobicity and helical content, a preference that is paralleled by Get3. After TAs are loaded onto Get3, substrates containing a highly basic CTE (which characterizes mitochondrial TAs; see the section titled Targeting Signals, above) are inserted more slowly into the ER membrane and are thus more likely to partition into off-pathway processes, such as dissociation from Get3 and aggregation in the cytosol (Rao et al. 2016). As this second selection occurs after ATP hydrolysis by the Get3^TA complex, it provides a mechanism akin to kinetic proofreading during tRNA selection by the ribosome (Rodnina & Wintermeyer 2001). Additional mechanisms, such as competition by other cellular chaperones and quality control machineries, could further enhance the selectivity of the GET pathway; these possibilities remain to be explored.
Alternative Pathways of Targeting Tail-Anchored Proteins to the ER
In addition to the GET pathway, there is strong evidence for other pathways that deliver TAs to the ER. In yeast lysate, TA variants containing a Fis1 TMD insert into ER microsomes independently of Get3 (Rao et al. 2016). The ER localization of Cytb5 is independent of GET components in mammalian cells (Favaloro et al. 2008, 2010). In vitro, purified Cytb5 and protein tyrosine phosphatase 1B are targeted and inserted into liposomes independently of protein or nucleotides (Brambillasca et al. 2005, 2006; Colombo et al. 2009). Despite these apparently spontaneous reactions, Cytb5 associates with protein factors, including Hsc70, in lysates (Colombo et al. 2009). Given the propensity of TMDs to aggregate in the crowded cytosolic environment, newly synthesized TAs probably associate with cellular chaperones even if their TMDs are moderately hydrophobic. Whether chaperone functions are important for the targeting of these TAs, whether TA targeting in vivo requires additional protein factors, and the number of TAs utilizing these alternative pathways remain open questions.
The recently discovered SRP-independent (SND) proteins likely provide redundant routes for TA targeting to the ER (Aviram et al. 2016). A high-content screen in S. cerevisiae identified three proteins, Snd1, Snd2, and Snd3, whose deletions are synthetically lethal with deletion of Get3 or Get1, suggesting that the SND and GET components provide redundant functions in yeast cells. Furthermore, overexpression of SND proteins partially rescues the loss of SRP components, suggesting that SND proteins also provide a backup pathway(s) for the targeting of some SRP-dependent substrates. Notably, proximity ribosome profiling and variation of the TMD position in reporter proteins showed that the SND proteins have the largest impact on substrates whose TMDs reside at neither the N nor the C terminus. These results suggest that the SRP, SND, and GET components provide partially redundant routes for the targeting of a broad range of membrane proteins, and together these targeting factors provide a robust, interlinked network of pathways for delivering proteins to the ER in yeast cells.
Localization of Tail-Anchored Proteins to Peroxisomes
Analogous to other peroxisomal membrane proteins, TAs are targeted to peroxisomes via one of two pathways: direct targeting from the cytosol or trafficking through the ER (Figure 4). Both pathways are dependent on two targeting factors, the cytosolic Pex19 and its membrane receptor Pex3, which are crucial for peroxisomal biogenesis. Pex19 contains an NTD rich in intrinsically disordered segments and a helical CTD (Hattula et al. 2014, Schueller et al. 2010). Pex3 contains a short N-terminal sequence, a single TMD, and a cytosolic CTD that forms a twisted six-helix bundle (Figure 4, Structure 1). A peptide corresponding to the N-terminal segment of Pex19 docks into a hydrophobic groove at the membrane-distal end of the Pex3 CD (Figure 4, Structure 1) (Sato et al. 2010, Schmidt et al. 2010). Conserved leucines and aromatic residues in this segment are important for Pex3 binding and peroxisomal biogenesis (Sato et al. 2010). In addition, hydrogen/deuterium exchange experiments showed that the C terminus of Pex19 became more protected upon Pex3 binding, suggesting a conformational change at this site (Hattula et al. 2014). As the Pex19 CTD contains its substrate recognition site, the Pex3-induced rearrangement could regulate substrate binding; this hypothesis remains to be tested.
Figure 4.
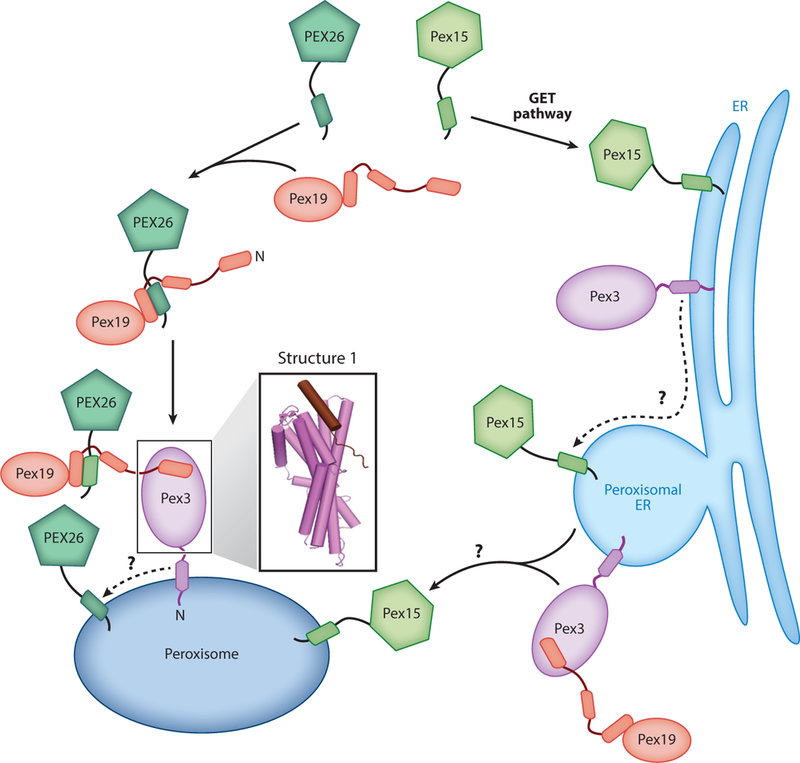
Two distinct pathways target tail-anchored proteins to peroxisomes. In the direct pathway (left path), Pex19 captures PEX26 in the cytosol and delivers it to peroxisomes via interaction with Pex3. PEX26 is then inserted into the peroxisomal membrane via an unknown mechanism. In the ER-dependent pathway (right path), Pex15 is first targeted to the ER membrane and is then sorted to peroxisomal ER. Exit of Pex15 from the ER occurs via budding of preperoxisomal vesicles that eventually fuse with peroxisomes, and this process requires Pex3 and Pex19. Structure 1 (PDB 3AJB) shows the crystal structure of the Pex3 cytosolic domain bound to the Pex19 N-terminal peptide.
The import of peroxisomal membrane proteins via the ER-dependent pathway (Figure 4, right path) was more extensively studied in yeast cells. Microscopy analyses showed that a subset of peroxisomal TAs, including yeast Pex15 and plant ascorbate peroxidase, are first targeted to the ER by the GET machinery (Lisenbee et al. 2003, Mullen & Trelease 2000, Schuldiner et al. 2008, van der Zand et al. 2010). Exit of Pex15 from the ER involves the formation of a specialized subdomain in the ER membrane, termed peroxisomal ER, followed by budding of preperoxisomal vesicles from this region and fusion of the vesicle with existing peroxisomes (Smith & Aitchison 2013). Microscopy studies and in vitro budding assays showed that generation of preperoxisomal vesicles from the ER requires Pex3 and Pex19, ATP, and unidentified cytosolic factors (Lam et al. 2010, van der Zand et al. 2010). Studies on other peroxisomal membrane proteins showed that Pex3 plays two important roles in the post-ER processes. First, its luminal sequence provides the signal that sorts Pex3 to peroxisomal ER. Second, its TMD is required for subsequent transport of the vesicle to existing peroxisomes (Fakieh et al. 2013). Whether these lessons pertain to peroxisomal TAs remains to be determined.
A mammalian TA, PEX26, is directly targeted from the cytosol to peroxisomes and is thus a model substrate for investigations of this pathway (Figure 4, left path). PEX26 is first captured by Pex19, which recognizes both the TMD and basic CTE on the TA and maintains its solubility in the cytosol (Chen et al. 2014a, Halbach et al. 2006, Yagita et al. 2013). The helical CTD of Neurospora crassa Pex19is sufficient to prevent PEX26 aggregation (Chen et al. 2014a), suggesting that the Pex19 CTD contains the substrate-binding site. PEX26is delivered to peroxisomes via the interaction of Pex19 with Pex3 (Chen et al. 2014a, Yagita et al. 2013). Biochemical reconstitutions further identified two elements in the targeting complex, an amphiphilic segment adjacent to the Pex19 helical domain and a membrane-proximal hydrophobic surface on Pex3, that are important for the subsequent membrane insertion of PEX26 (Chen et al. 2014a). It was speculated that a sequential handoff of the TMD, from the Pex19 helical domain to the Pex3 membrane-proximal and finally to the lipid bilayer, provides a pathway for the insertion of PEX26 into the peroxisomal membrane (Chen et al. 2014a). This hypothesis and its mechanistic details remain to be studied.
CONNECTION OF TAIL-ANCHORED PROTEIN TARGETING TO QUALITY CONTROL MACHINERIES
The BAG6 Complex Directs Membrane Proteins to the Ubiquitin-Proteasome Pathway
Protein targeting and translocation are not perfectly efficient, which necessitates degradation mechanisms to clear membrane proteins inappropriately exposed in the cytosol. In mammalian cells, the homologs of Sgt2 (SGTA) and Get3 (TRC40) are physically linked to a multifunctional BAG6 (BCL2-associated athanogene) complex, which could both enhance TA loading onto TRC40 (Mariappan et al. 2010) and direct mislocalized membrane proteins to quality control machineries (Hessa et al. 2011). The heterotrimeric BAG6 complex comprises BAG6 bound to TRC35 and UBL4A, the mammalian homologs of Get4 and Get5, respectively (Figure 5a). Analogous to their yeast homologs, the SGTA NTD recognizes the UBL domain of UBL4A (Figure 5a) (Darby et al. 2014, Xu et al. 2012). Nevertheless, UBL4A lacks residues in the Get5 CTD that mediates homodimerization and instead forms a heterodimer with the C-terminal helices of BAG6 (Figure 5a, Structure 1) (Mock et al. 2015). TRC35 also lacks the β-loop involved in the Get4-Get5 interface and instead binds to the BAG6 nuclear localization sequence (NLS) (Figure 5a) (Mock et al. 2015). Biochemical reconstitutions showed that a minimal BAG6 fragment containing its C-terminal helices and NLS is sufficient to stimulate TA transfer from SGTA to TRC40 (Mock et al. 2015, Shao et al. 2017), mimicking the function of yeast Get4/5 in enhancing TA transfer from Sgt2 to Get3. In addition to participating in the GET pathway, recombinant BAG6 can cross-link to and chaperone substrate proteins that expose hydrophobic sequences (Rodrigo-Brenni et al. 2014, Q. Wang et al. 2011). Moreover, an N-terminal UBL domain of BAG6 (Figure 5a, Structure 2) recruits the ubiquitin ligase RNF126, which mediates polyubiq-uitylation of client proteins for degradation by the proteasome (Rodrigo-Brenni et al. 2014). The BAG6 complex appears to provide a general quality control complex for mislocalized proteins, including membrane proteins inappropriately exposed in the cytosol and retrotranslocated proteins en route to ER-associated degradation (Hessa et al. 2011, Q. Wang et al. 2011).
Intriguingly, the UBL domains of BAG6 and UBL4A recognize the same interaction surface on SGTA (Darby et al. 2014, Leznicki et al. 2013). Thus, TAs bound to SGTA have two potential fates: transfer to TRC40 for targeting to the ER and transfer to BAG6 for ubiquitylation and degradation (Figure 5a). The two paths compete with one another in vitro and in vivo, but transfer toTRC40 appears to dominate under physiological conditions (Leznicki & High 2012, Shao etal. 2017, Wunderley et al. 2014). In addition to BAG6, SGTA has emerged as a key regulator in protein quality control, as its TPR domain also interacts with heat-shock proteins (Scheufler et al. 2000), the proteasome-associated ubiquitin receptor Rpn13 (Leznicki et al. 2015, Thapaliya et al. 2016), and a variety of hormone receptors. Many molecular details of the SGTA-BAG6 cycle remain to be understood, including how SGTA and BAG6 distinguish newly synthesized proteins from mislocalized proteins, what dictates the partition of substrate proteins between the targeting and degradation pathways, and how SGTA participates in additional quality control pathways.
AAA-ATPases Promote Degradation of Tail-Anchored Proteins Mislocalized to Mitochondria
In addition to the cytosolic BAG6 complex, cells have evolved degradation machineries to clear mistargeted TAs. Using ER-and peroxisome-destined Pex15 and Gos1 as model TAs, two recent studies found thatMsp1 (or human ATAD1)—a conserved AAA-ATPase (an ATPase associated with a variety of cellular activities) anchored on the OMM—is essential for sensing and degrading TAs mistargeted to mitochondria in yeast and mammalian cells. When the GET pathway is impaired, loss ofMsp1 results in accumulation of Pex15 and Gos1 on mitochondria, severe mitochondrial damage, and severe cell growth defects (Chen et al. 2014b, Okreglak & Walter 2014). Msp1 also associates with and enhances the degradation of a Pex15 variant misdirected to the OMM (Okreglak & Walter 2014). These findings suggest that, in addition to protein-targeting machineries, Msp1 and potentially other machineries provide local surveillance and quality control mechanisms to further enhance the fidelity of TA localization to specific organelles (Figure 5b).
OUTSTANDING QUESTIONS
Mechanism of Insertion of Tail-Anchored Proteins at the Membrane
How TAs insert into biological membranes is not well understood. Even basic questions, such as whether insertion occurs spontaneously or is protein assisted, remain unclear. This situation differs from that of polytopic membrane proteins and proteins with a TMD near the N terminus, whose insertion is strongly dependent on translocation complexes such as Sec61p (or SecYEG in bacteria) and/or the YidC/Oxa1/Alb3 family of translocases (Dalbey & Kuhn 2014, Rapoport 2007). In contrast, TAs with moderately hydrophobic TMDs can insert into protein-free liposomes, as described above (see the section titled Alternative Pathways of Targeting Tail-Anchored Proteins to the ER), and surprisingly long domains ofvarious sequences (up to 85 residues) and charges that are C terminal to the TMD can be translocated in these apparently unassisted insertion reactions (Brambillasca et al. 2006, Sakamoto et al. 2012). That more hydrophobic TMDs could not be inserted in this mechanism was attributed to their aggregation in solution (Brambillasca et al. 2006). These in vitro experiments suggest that, if a TA is maintained in a soluble, translocation-competent state, its spontaneous insertion into membrane can be efficient and stable. Nevertheless, the structure and dynamics of biological membranes are strongly affected by the proteins embedded in or associated with them, and whether spontaneous TA insertion occurs in vivo is unclear.
The GET pathway provides an example wherein the insertion of a single TMD into the lipid bilayer requires facilitation by membrane-embedded protein factors. Mutation of each TMD in a covalently linked Get1-Get2 construct disrupted TA insertion both in vitro and in vivo without affecting the function of the Get1/2 CDs (Wang et al. 2014). In addition, cross-links were observed between the TMDs of Get1 and the TMD of the model TA Sec22, suggesting a potential TA docking site formed by the TMDs of Get1/2 (Wang et al. 2014). Nevertheless, how the Get1/2 complex facilitates TA insertion into membrane is the least understood aspect of the GET pathway. Many questions remain to be addressed, including the site(s) for TA docking in the Get1/2 receptor, the conformation and orientation of TA when bound at the translocase site, and the pathway by which the substrate TMD accesses and integrates into the lipid bilayer.
Connection of Tail-Anchored Protein-Targeting Pathways with Cellular Chaperones
Both Get3 and Sgt2 (and their mammalian homologs) contain well-defined sites for binding hydrophobic TMDs, and their binding is critical for shielding TAs from the cytosol during targeting. Nevertheless, whether these factors are sufficient for the efficient capture and chaperoning of TAs, and the mechanism by which TAs are loaded onto Sgt2 (or SGTA), remains unclear. Although earlier work observed associations of Get4/5 and the BAG6 complex with ribosomes (Fleischer et al. 2006, Mariappan et al. 2010), the causal relationship between these putative ribosome interactions and TA loading onto Sgt2 has not been established. Further confounding this question is the association of Sgt2 (or SGTA) with molecular chaperones. In yeast lysate, Sgt2 coimmunoprecipitates with members of multiple chaperone families, including Hsp70, Hsp90, Hsp100, and Ybr137w(Chartron et al. 2011, Liou et al. 2007, Wanget al. 2010). Analogous chaperone interactions were observed with mammalian SGTA (Roberts et al. 2015). The chaperone interaction site was mapped to the Sgt2 TPR domain, in which a dicarboxylate clamp binds the charged C-terminal tails of different chaperones (Figure 2, Structure 1) (Chartron et al. 2011, Wang et al. 2010). Despite this structural information, the involvement of chaperones in TA targeting is unclear. Early work found that TAs associate with Hsp40/Hsc70 in cell lysates (Abell et al. 2007), and supplementing Hsc70 enhanced the efficiency of TA targeting to the ER in reconstituted reactions (Rabu et al. 2008). Hsp70 also associates with mitochondrial TAs (Wang et al. 2010) and with Cytb5 (Colombo et al. 2009), which are targeted to the ER independently of GET components. The precise roles of Hsp/Hsc70 chaperones in TA targeting remain to be understood.
Targeting of Tail-Anchored Proteins to Mitochondria and Chloroplasts
The mechanism by which TAs are selectively targeted to mitochondria remains unknown. The targeting and insertion of several OMM TAs have been examined and exhibited no dependencies on nucleotides, electrochemical potential, or components of the translocase on the OMM (Kemper et al. 2008, Setoguchi et al. 2006). Intriguingly, the targeting of Bak and Bcl-XL, two OMM TAs with roles in apoptosis, is dependent on cytosolic factors in the cell lysate, but this requirement could be bypassed when a well-folded GFP was fused to the targeting sequences of these proteins (Setoguchi et al. 2006). This finding led to the hypothesis that OMM TAs require chaperones, depending on the folding competence of their CDs, but a chaperone requirement for the C-terminal TMD has not been established. The only exceptions are Tom5, whose targeting was compromised by knockdown of Tom40, and Bak, whose targeting depends on its binding partner, VDAC2, on the OMM (Setoguchi et al. 2006). Dedicated factors that provide a common pathway for the targeted delivery of OMM TAs remain to be identified. In the absence of this information, whether the targeting of OMM TAs is spontaneous or protein assisted cannot be resolved.
Chloroplasts contain at least eight identified TAs, but the targeting of only a few of these has been characterized (Dhanoa et al. 2010, Lee et al. 2014). Among these TAs are two GTPases that compose the translocase on the outer chloroplast membrane, Toc33 and Toc34, and an outer envelope protein, OEP9. All three proteins require the Arabidopsis ankyrin repeat-containing protein ARK2A for specific targeting to chloroplasts (Dhanoa et al. 2010). ARK2Ais an essential cytosolic factor for OEP biogenesis; pulldown studies and localization assays showed that it acts as a chaperone to prevent the aggregation of a variety of nascent OEPs in the cytosol and facilitates their targeting to chloroplasts (Bae et al. 2008). Whereas the TMD and CTE of OEP9 are sufficient to direct its localization (Dhanoa et al. 2010), the targeting of Toc33 andToc34 also requires their cytosolic GTPase domains. In addition, OEP9 requires different protein factors on the chloroplast membrane for integration relative to Toc33 and Toc34 (Dhanoa et al. 2010). These observations suggest that at least two protein-assisted pathways mediate the targeting and insertion of TAs into chloroplasts. The components and molecular events involved in these pathways remain to be defined.
SUMMARY POINTS.
-
1.
A combination of physicochemical properties in the transmembrane domain and C-terminal elements dictates TA sorting to different organelles.
-
2.
A cascade of protein interactions in the GET pathway mediates TA targeting and insertion into the ER.
-
3.
Nucleotide-, substrate-, and effector-driven conformational changes during the Get3 ATPase cycle drive TA targeting in the GET pathway.
-
4.
Two distinct pathways mediate TA localization to peroxisomes.
-
5.
Quality control machineries in the cytosol and on mitochondria provide clearance mechanisms for mislocalized membrane proteins.
FUTURE ISSUES.
-
1.
How are TAs inserted into biological membranes?
-
2.
How are posttranslational membrane protein-targeting pathways linked to the cellular chaperone network?
-
3.
How are TAs targeted to mitochondria and chloroplasts?
ACKNOWLEDGMENT
This work was supported by NIH grant GM107368 to S.-o.S.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Abell BM, Rabu C, Leznicki P, Young JC, High S. 2007. Post-translational integration of tail-anchored proteins is facilitated by defined molecular chaperones. J. Cell Sci. 120:1743–51 [DOI] [PubMed] [Google Scholar]
- Akopian D, Shen K, Zhang X, Shan SO. 2013. Signal recognition particle: an essential protein-targeting machine. Annu. Rev. Biochem. 82:693–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviram N, Ast T, Costa EA, Arakel EC, Chuartzman SG, et al. 2016. The SND proteins constitute an alternative targeting route to the endoplasmic reticulum. Nature 540:134–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae W, Lee YJ, Kim DH, Lee J, Kim S, et al. 2008. AKR2A-mediated import of chloroplast outer membrane proteins is essential for chloroplast biogenesis. Nat. Cell Biol. 10:220–27 [DOI] [PubMed] [Google Scholar]
- Beilharz T, Egan B, Silver PA, Hofmann K, Lithgow T. 2003. Bipartite signals mediate subcellular targeting of tail-anchored membrane proteins in Saccharomyces cerevisiae. J. Biol. Chem. 278:8219–23 [DOI] [PubMed] [Google Scholar]
- Bozkurt G, Stjepanovic G, Vilardi F, Amlacher S, Wild K, et al. 2009. Structural insights into tail-anchored protein binding and membrane insertion by Get3. PNAS 106:21131–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozkurt G, Wild K, Amlacher S, Hurt E, Dobberstein B, Sinning I. 2010. The structure of Get4 reveals an alpha-solenoid fold adapted for multiple interactions in tail-anchored protein biogenesis. FEBS Lett. 584:1509–14 [DOI] [PubMed] [Google Scholar]
- Brambillasca S, Yabal M, Makarow M, Borgese N 2006. Unassisted translocation of large polypeptide domains across phospholipid bilayers. J. Cell Biol. 175:767–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambillasca S, Yabal M, Soffientini P, Stefanovic S, Makarow M, et al. 2005. Transmembrane topogenesis of a tail-anchored protein is modulated by membrane lipid composition. EMBOJ. 24:2533–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers JT, Guzzo RM, Salih M, Tuana BS. 2009. Hydrophobic profiles of the tail anchors in SLMAP dictate subcellular targeting. BMC Cell Biol. 10:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YW, Chuang YC, Ho YC, Cheng MY, Sun YJ, et al. 2010. Crystal structure of Get4-Get5 complex and its interactions with Sgt2, Get3, and Ydj1. J. Biol. Chem. 285:9962–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartron JW, Clemons WM Jr., Suloway CJ 2012a. The complex process of GETting tail-anchored membrane proteins to the ER. Curr. Opin. Struct. Biol. 22:217–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartron JW, Gonzalez GM, Clemons WM Jr. 2011. A structural model of the Sgt2 protein and its interactions with chaperones and the Get4/Get5 complex. J. Biol. Chem. 286:34325–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartron JW, Suloway CJ, Zaslaver M, Clemons WM Jr. 2010. Structural characterization of the Get4/Get5 complex and its interaction with Get3. PNAS 107:12127–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartron JW, VanderVelde DG, Clemons WM Jr. 2012b.. Structures of the Sgt2/SGTAdimerization domain with the Get5/UBL4AUBL domain reveal an interaction that forms a conserved dynamic interface. Cell Rep. 2:1620–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartron JW, VanderVelde DG, Rao M, Clemons WM Jr. 2012c.. Get5 carboxyl-terminal domain is a novel dimerization motif that tethers an extended Get4/Get5 complex. J. Biol. Chem. 287:8310–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Pieuchot L, Loh RA, Yang J, Kari TM, et al. 2014a.. Hydrophobic handoff for direct delivery of peroxisome tail-anchored proteins. Nat. Commun. 5:5790. [DOI] [PubMed] [Google Scholar]
- Chen YC, Umanah GK, Dephoure N, Andrabi SA, Gygi SP, et al. 2014b.. Msp1/ATAD1 maintains mitochondrial function by facilitating the degradation of mislocalized tail-anchored proteins. EMBOJ.33: 1548–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo SF, Cardani S, Maroli A, Vitiello A, Soffientini P, et al. 2016. Tail-anchored protein insertion in mammals: function and reciprocal interactions of the two subunits of the TRC40 receptor. J. Biol. Chem. 291:15292–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo SF, Longhi R, Borgese N. 2009. The role of cytosolic proteins in the insertion of tail-anchored proteins into phospholipid bilayers. J. Cell Sci. 122:2383–92 [DOI] [PubMed] [Google Scholar]
- Dalbey RE, Kuhn A. 2014. How YidC inserts and folds proteins across a membrane. Nat. Struct. Mol. Biol. 21:435–36 [DOI] [PubMed] [Google Scholar]
- Darby JF, Krysztofinska EM, Simpson PJ, Simon AC, Leznicki P, et al. 2014. Solution structure of the SGTA dimerisation domain and investigation of its interactions with the ubiquitin-like domains of BAG6 and UBL4A. PLOS ONE 9:e113281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanoa PK, Richardson LG, Smith MD, Gidda SK, Henderson MP, et al. 2010. Distinct pathways mediate the sorting of tail-anchored proteins to the plastid outer envelope. PLOS ONE 5:e10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan B, Beilharz T, George R, Isenmann S, Gratzer S, et al. 1999. Targeting of tail-anchored proteins to yeast mitochondria in vivo. FEBS Lett. 451:243–48 [DOI] [PubMed] [Google Scholar]
- Fakieh MH, Drake PJ, Lacey J, Munck JM, Motley AM, Hettema EH 2013. Intra-ER sorting of the peroxisomal membrane protein Pex3 relies on its luminal domain. Biol. Open 2:829–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaloro V, Spasic M, Schwappach B, Dobberstein B. 2008. Distinct targeting pathways for the membrane insertion of tail-anchored (TA) proteins. J. Cell Sci. 121:1832–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaloro V, Vilardi F, Schlecht R, Mayer MP, Dobberstein B. 2010. Asna1/TRC40-mediated membrane insertion of tail-anchored proteins. J. Cell Sci. 123:1522–30 [DOI] [PubMed] [Google Scholar]
- Fleischer TC, Weaver CM, McAfee KJ, Jennings JL, Link AJ. 2006. Systematic identification and functional screens of uncharacterized proteins associated with eukaryotic ribosomal complexes. GenesDev.20:1294–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gristick HB, Rao M, Chartron JW, Rome ME, Shan SO, Clemons WM Jr. 2014. Crystal structure of ATP-bound Get3-Get4-Get5 complex reveals regulation of Get3 by Get4. Nat. Struct. Mol. Biol. 21:437–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gristick HB, Rome ME, Chartron JW, Rao M, Hess S, et al. 2015. Mechanism of assembly of a substrate transfer complex during tail-anchored protein targeting. J. Biol. Chem. 290:30006–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbach A, Landgraf C, Lorenzen S, Rosenkranz K, Volkmer-Engert R, et al. 2006. Targeting of the tail-anchored peroxisomal membrane proteins PEX26 and PEX15 occurs through C-terminal PEX19-binding sites. J. Cell Sci. 119:2508–17 [DOI] [PubMed] [Google Scholar]
- Hattula K, Hirschberg D, Kalkkinen N, Butcher SJ, Ora A. 2014. Association between the intrinsically disordered protein PEX19 and PEX3. PLOS ONE 9:e103101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde RS, Keenan RJ. 2011. Tail-anchored membrane protein insertion into the endoplasmic reticulum. Nat. Rev.Mol. CellBiol. 12:787–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessa T, Sharma A, Mariappan M, Eshleman HD, Gutierrez E, Hegde RS. 2011. Protein targeting and degradation are coupled for elimination of mislocalized proteins. Nature 475:394–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honsho M, Mitoma JY, Ito A. 1998. Retention of cytochrome b5 in the endoplasmic reticulum is transmembrane and luminal domain-dependent. J. Biol. Chem. 273:20860–66 [DOI] [PubMed] [Google Scholar]
- Horie C, Suzuki H, Sakaguchi M, Mihara K. 2002. Characterization of signal that directs C-tail-anchored proteins to mammalian mitochondrial outer membrane. Mol. Biol. Cell 13:1615–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Li J, Qian X, Denic V, Sha B. 2009. The crystal structures of yeast Get3 suggest a mechanism for tail-anchored protein membrane insertion. PLOS ONE 4:e8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang YT, Pelitire SM, Henderson MP, Andrews DW, Dyer JM, Mullen RT. 2004. Novel targeting signals mediate the sorting of different isoforms of the tail-anchored membrane protein cytochrome b5 to either endoplasmic reticulum or mitochondria. Plant Cell 16:3002–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, et al. 2009. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science 323:1693–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper C, Habib SJ, Engl G, Heckmeyer P, Dimmer KS, Rapaport D. 2008. Integration of tail-anchored proteins into the mitochondrial outer membrane does not require any known import components. J. Cell Sci. 121:1990–98 [DOI] [PubMed] [Google Scholar]
- Kubota K, Yamagata A, Sato Y, Goto-Ito S, Fukai S. 2012. Get1 stabilizes an open dimer conformation of Get3 ATPase by binding two distinct interfaces. J. Mol. Biol. 422:366–75 [DOI] [PubMed] [Google Scholar]
- Kuroda R, Ikenoue T, Honsho M, Tsujimoto S, Mitoma JY, Ito A 1998. Charged amino acids at the carboxyl-terminal portions determine the intracellular locations of two isoforms of cytochrome £5. J. Biol. Chem. 273:31097–102 [DOI] [PubMed] [Google Scholar]
- Kutay U, Ahnert-Hilger G, Hartmann E, Wiedenmann B, Rapoport TA 1995. Transport route for synapto-brevin via a novel pathway of insertion into the endoplasmic reticulum membrane. EMBOJ. 14:217–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutay U, Hartmann E, Rapoport TA 1993. A class of membrane proteins with a C-terminal anchor. Trends CellBiol. 3:72–75 [DOI] [PubMed] [Google Scholar]
- Lam SK, Yoda N, Schekman R. 2010. A vesicle carrier that mediates peroxisome protein traffic from the endoplasmic reticulum. PNAS 107:21523–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim DH, Hwang I. 2014. Specific targeting ofproteins to outer envelope membranes ofendosymbiotic organelles, chloroplasts, and mitochondria. Front. Plant Sci. 5:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leznicki P, High S. 2012. SGTA antagonizes BAG6-mediated protein triage. PNAS 109:19214–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leznicki P, Korac-Prlic J, Kliza K, Husnjak K, Nyathi Y, et al. 2015. Binding of SGTA to Rpn13 selectively modulates protein quality control. J. Cell Sci. 128:3187–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leznicki P, Roebuck QP, Wunderley L, Clancy A, Krysztofinska EM, et al. 2013. The association of BAG6 with SGTA and tail-anchored proteins. PLOS ONE 8:e59590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou ST, Cheng MY, Wang C. 2007. SGT2 and MDY2 interact with molecular chaperone YDJ1 in Saccharomyces cerevisiae. Cell Stress Chaperones 12:59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisenbee CS, Heinze M, Trelease RN. 2003. Peroxisomal ascorbate peroxidase resides within a subdomain of rough endoplasmic reticulum in wild-type Arabidopsis cells. Plant Physiol. 132:870–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariappan M, Li X, Stefanovic S, Sharma A, Mateja A, et al. 2010. A ribosome-associating factor chaperones tail-anchored membrane proteins. Nature 466:1120–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariappan M, Mateja A, Dobosz M, Bove E, Hegde RS, Keenan RJ. 2011. The mechanism of membrane-associated steps in tail-anchored protein insertion. Nature 477:61–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty NT, Teresinski HJ, Hwang YT, Clendening EA, Gidda SK, et al. 2014. New insights into the targeting of a subset of tail-anchored proteins to the outer mitochondrial membrane. Front. Plant Sci. 5:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki R, Kameyama K, Yamamoto A. 2003. Post-translational targeting of a tail-anchored green fluorescent protein to the endolpasmic reticulum. J. Biochem. 134:415–26 [DOI] [PubMed] [Google Scholar]
- Mateja A, Paduch M, Chang HY, Szydlowska A, Kossiakoff AA, et al. 2015. Protein targeting. Structure of the Get3 targeting factor in complex with its membrane protein cargo. Science 347:1152–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateja A, Szlachcic A, Downing ME, Dobosz M, Mariappan M, et al. 2009. The structural basis of tail-anchored membrane protein recognition by Get3. Nature 461:361–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mock J-Y, Chartron JW, Zaslaver MA, Xu Y, Ye Y, Clemons WM. 2015. Bag6 complex contains a minimal tail-anchor-targeting module and a mock BAG domain. PNAS 112:106–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen RT, Trelease RN. 2000. The sorting signals for peroxisomal membrane-bound ascorbate peroxidase are within its C-terminal tail. J. Biol. Chem. 275:16337–44 [DOI] [PubMed] [Google Scholar]
- Okreglak V, Walter P. 2014. The conserved AAA-ATPase Msp1 confers organelle specificity to tail-anchored proteins. PNAS 111:8019–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrazzini E, Villa A, Borgese N. 1996. A mutant cytochrome b5 with a lengthened membrane anchor escapes from the endoplasmic reticulum and reaches the plasma membrane. PNAS 93:4207–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabu C, Wipf P, Brodsky JL, High S. 2008. A precursor-specific role for Hsp40/Hsc70 during tail-anchored protein integration at the endoplasmic reticulum. J. Biol. Chem. 283:27504–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao M, Okreglak V, Chio US, Cho H, Walter P, Shan SO. 2016. Multiple selection filters ensure accurate tail-anchored membrane protein targeting. eLife 5:e21301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport TA 2007. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450:663–69 [DOI] [PubMed] [Google Scholar]
- Roberts JD, Thapaliya A, Martinez-Lumbreras S, Krysztofinska EM, Isaacson RL. 2015. Structural and functional insights into small, glutamine-rich, tetratricopeptide repeat protein alpha. Front. Mol. Biosci. 2:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnina MV, Wintermeyer W. 2001. Ribosome fidelity: tRNA discrimination, proofreading and induced fit. Trends Biochem. Sci. 26:124–30 [DOI] [PubMed] [Google Scholar]
- Rodrigo-Brenni MC, Gutierrez E, Hegde RS. 2014. Cytosolic quality control of mislocalized proteins requires RNF126 recruitment to Bag6. Mol. Cell 55:227–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rome ME, Chio US, Rao M, Gristick H, Shan SO 2014. Differential gradients of interaction affinities drive efficient targeting and recycling in the GET pathway. PNAS 111:E4929–4935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rome ME, Rao M, Clemons WM, Shan SO. 2013. Precise timing of ATPase activation drives targeting of tail-anchored proteins. PNAS 110:7666–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchi P, Colombo S, Francolini M, Borgese N. 2008. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J. Cell Biol. 181:105–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruberti C, Costa A, Pedrazzini E, Lo Schiavo F, Zottini M. 2014. FISSION1A, an Arabidopsis tail-anchored protein, is localized to three subcellular compartments. Mol. Plant 7:1393–96 [DOI] [PubMed] [Google Scholar]
- Sakamoto Y, Miura M, Takeuchi F, Park SY, Tsubaki M. 2012. Interaction of modified tail-anchored proteins with liposomes: effect of extensions of hydrophilic segment at the COOH-terminus of holo-cytochromes b5. J. Biosci. Bioeng. 113:322–31 [DOI] [PubMed] [Google Scholar]
- Sato Y, Shibata H, Nakatsu T, Nakano H, Kashiwayama Y, et al. 2010. Structural basis for docking of peroxisomal membrane protein carrier Pex19p onto its receptor Pex3p. EMBOJ. 29:4083–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, et al. 2000. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 101:199–210 [DOI] [PubMed] [Google Scholar]
- Schmidt F, Treiber N, Zocher G, Bjelic S, Steinmetz MO, et al. 2010. Insights into peroxisome function from the structure of PEX3 in complex with a soluble fragment of PEX19. J. Biol. Chem. 285:25410–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueller N, Holton SJ, Fodor K, Milewski M, Konarev P, et al. 2010. The peroxisomal receptor Pex19p forms a helical mPTS recognition domain. EMBOJ. 29:2491–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner M, Collins R, Thompson NJ, Denic V, Bhamidipati A, et al. 2005. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 123:507–19 [DOI] [PubMed] [Google Scholar]
- Schuldiner M, Metz J, Schmid V, Denic V, Rakwalska M, et al. 2008. The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell 134:634–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setoguchi K, Otera H, Mihara K. 2006. Cytosolic factor-and TOM-independent import of C-tail-anchored mitochondrial outer membrane proteins. EMBOJ. 25:5635–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Rodrigo-Brenni MC, Kivlen MH, Hegde RS. 2017. Mechanistic basis for a molecular triage reaction. Science 355:298–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon AC, Simpson PJ, Goldstone RM, Krysztofinska EM, Murray JW, et al. 2013. Structure of the Sgt2/Get5 complex provides insights into GET-mediated targeting of tail-anchored membrane proteins. PNAS 110:1327–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Aitchison JD. 2013. Peroxisomes take shape. Nat. Rev. Mol. Cell Biol. 14:803–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovic S, Hegde RS. 2007. Identification of a targeting factor for posttranslational membrane protein insertion into the ER. Cell 128:1147–59 [DOI] [PubMed] [Google Scholar]
- Stefer S, Reitz S, Wang F, Wild K, Pang YY, et al. 2011. Structural basis for tail-anchored membrane protein biogenesis by the Get3-receptor complex. Science 333:758–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway CJ, Chartron JW, Zaslaver M, Clemons WM Jr. 2009. Model for eukaryotic tail-anchored protein binding based on the structure of Get3. PNAS 106:14849–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapaliya A, Nyathi Y, Martinez-Lumbreras S, Krysztofinska EM, Evans NT, et al. 2016. SGTA interacts with the proteasomal ubiquitin receptor Rpn13 via a carboxylate clamp mechanism. Sci. Rep 6:36622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung JY, Li YC, Lin TW, Hsiao CD. 2013. Structure of the Sgt2 dimerization domain complexed with the Get5 UBL domain involved in the targeting of tail-anchored membrane proteins to the endoplasmic reticulum. Acta Crystallogr. D Biol. Crystallogr. 69:2081–90 [DOI] [PubMed] [Google Scholar]
- van der Zand A, Braakman I, Tabak HF. 2010. Peroxisomal membrane proteins insert into the endoplasmic reticulum. Mol. Biol. Cell 21:2057–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardi F, Lorenz H, Dobberstein B. 2011. WRB is the receptor for TRC40/Asna1-mediated insertion of tail-anchored proteins into the ER membrane. J. Cell Sci. 124:1301–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardi F, Stephan M, Clancy A, Janshoff A, Schwappach B 2014WRB and CAML are necessary and sufficient to mediate tail-anchored protein targeting to the ER membrane. PLOS ONE 9:e85033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Brown EC, Mak G, Zhuang J, Denic V 2010. A chaperone cascade sorts proteins for posttranslational membrane insertion into the endoplasmic reticulum. Mol. Cell 40:159–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Chan C, Weir NR, Denic V 2014. The Get1/2 transmembrane complex is an endoplasmic-reticulum membrane protein insertase. Nature 512:441–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Whynot A, Tung M, Denic V. 2011. The mechanism of tail-anchored protein insertion into the ER membrane. Mol. Cell 43:738–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Liu Y, Soetandyo N, Baek K, Hegde R, Ye Y 2011. Aubiquitin ligase-associated chaperone holdase maintains polypeptides in soluble states for proteasome degradation. Mol. Cell 42:758–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wereszczynski J, McCammon JA. 2012. Nucleotide-dependent mechanism of Get3 as elucidated from free energy calculations. PNAS 109:7759–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderley L, Leznicki P, Payapilly A, High S. 2014. SGTA regulates the cytosolic quality control of hydrophobic substrates. J. Cell Sci. 127:4728–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Cai M, Yang Y, Huang L, Ye Y. 2012. SGTA recognizes a noncanonical ubiquitin-like domain in the Bag6-Ubl4A-Trc35 complex to promote endoplasmic reticulum-associated degradation. Cell Rep. 2:1633–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagita Y, Hiromasa T, Fujiki Y 2013. Tail-anchored PEX26 targets peroxisomes via a PEX19-dependent and TRC40-independent class I pathway. J. Cell Biol. 200:651–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Sakisaka T. 2012. Molecular machinery for insertion of tail-anchored membrane proteins into the endoplasmic reticulum membrane in mammalian cells. Mol. Cell 48:387–97 [DOI] [PubMed] [Google Scholar]