

Abstract
The advent of antibody drug conjugates as pharmaceuticals has fueled a need for reliable methods of site-selective protein modification that furnish homogeneous adducts. While bioorthogonal methods that utilize engineered amino acids often provide an elegant solution to the question of selective functionalization, achieving homogeneity using native amino acids remains a challenge. Herein, we explore visible-light mediated single-electron transfer as a novel mechanism towards enabling site- and chemoselective bioconjugation. Specifically, we demonstrate the use of photoredox catalysis as a platform to selectivity wherein the discrepancy in oxidation potentials between internal vs. C-terminal carboxylates can be exploited towards obtaining C-terminal functionalization exclusively. This oxidation potential-gated technology is amenable to endogenous peptides and has been successfully demonstrated on the protein insulin.
The last two decades have witnessed tremendous growth in research to address the challenge of site- and chemoselective protein modification.1 Propelled by a high demand for technologies that furnish homogeneously modified protein adducts, chemical biologists have successfully delivered a number of robust methods that achieve site-selective protein functionalization via protein engineering and the incorporation of non-natural, bioorthogonal amino acids.2 Chief amongst these methods are ‘click’ and Staudinger ligation strategies, wherein highly uniform products can be obtained by genetically encoding an azide reporter in a site- and number-specific fashion.3,4
While pre-engineering of the protein scaffold has proven to be an indispensable technology for selective protein modification, bioconjugation strategies that harness native amino acids remains highly attractive yet equally elusive. Traditionally, these native-modification methods mainly make use of two of the twenty canonical amino acid residues – cysteine and lysine – that incorporate heteroatom lone-pair nucleophiles. However, obtaining homogeneous products wherein only a single residue at a single site has undergone reaction has proven challenging.5 For example, selective modification of a specific lysine residue is difficult given their high abundance on protein surfaces.6 One solution has been to target proteins wherein one lysine residue is more solvent-accessible to biotinylation than other Lys groups, as demonstrated by Chen, et al.7 In a similar vein, this approach has been applied by Bader, et al. for the selective lipidation of C-terminal cysteines.8 More recently, elegant work from several groups has addressed this problem by extending the scope of natural amino acids employed in bioconjugation to tyrosine, tryptophan, and methionine residues.9,10,11,12,13,14,15,16 These methods take advantage of the inherently low abundance of Tyr, Met, and Trp residues on protein surfaces, thereby achieving a higher degree of site-selectivity.
Over the last several years, our laboratory has employed photoredox catalysis as a platform for activating native functional groups towards C–C and C–X bond formation. One such activation mode has focused on the use of naturally abundant carboxylic acids as latent carbon-centered radicals.17 These transient intermediates, generated through single-electron transfer (SET) and subsequent CO2 extrusion, have been shown to undergo successful coupling with a wide range of electrophilic partners including Michael acceptors18, vinyl sulfones19, and nickel complexes.20 Recently, we questioned whether this technology could be applied to more complex architectures that incorporate multiple carboxylic acids, such as endogenous proteins (Figure 1). Indeed, carboxylic acids are naturally present in proteins due to their incorporation in aspartate, glutamate, and C-terminal residues. Despite the abundance of these carboxylic acid bearing residues, we reasoned that the innate difference in oxidation potentials between side chain alkyl carboxylates (i.e. aspartate and glutamate)21 and C-terminal α-amino carboxylates17 should permit a high degree of site-selectivity -- with decarboxylation-functionalization occurring at the more readily oxidized C-terminus. It is also important to note that traditional methods for carboxylic acid bioconjugation typically fall within the realm of amide bond couplings and esterification with diazo compounds, two technologies that often suffer from indiscriminate regioselectivity.22,23,24,25,26
Figure 1: Photoredox-catalyzed decarboxylative functionalization as a novel electron transfer mechanism towards site- and chemoselective bioconjugation.
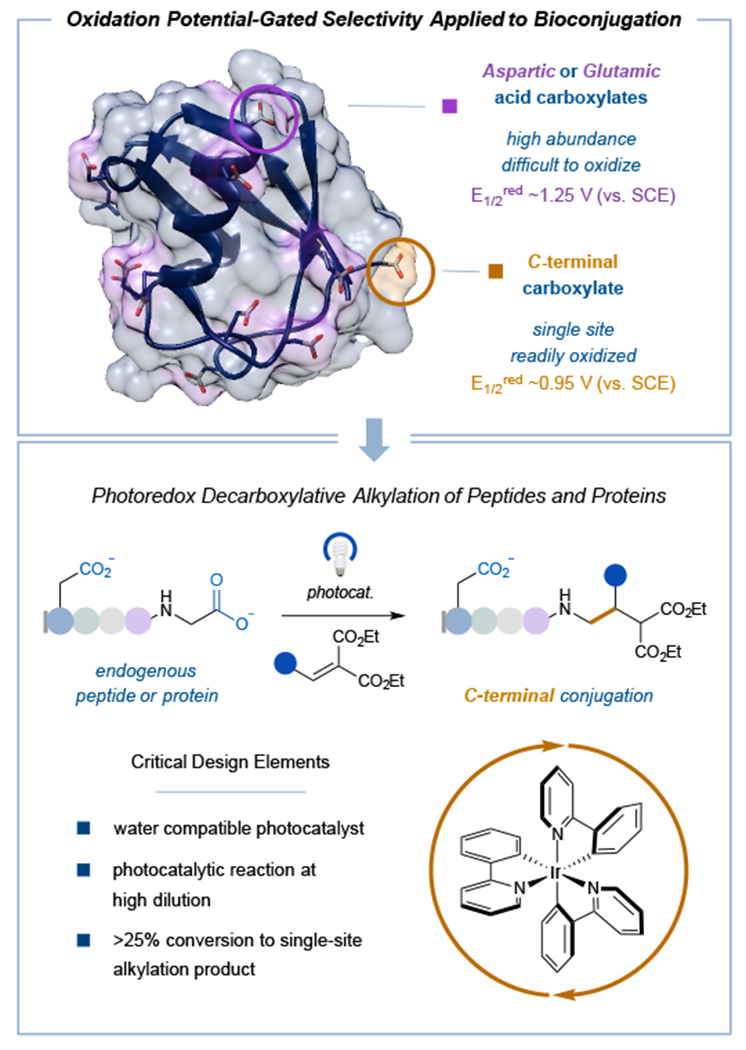
Current methods targeting natural amino acid motifs rely on the intrinsic availability of targeted residues on the protein surface to achieve homogeneously modified products. We postulated that photoredox-catalyzed decarboxylation could capitalize on the inherent differences in oxidation potentials between the more abundant Asp and Glu residues vs. the lone terminal carboxylate to obtain exclusive C-terminal-functionalized products.
In contrast, we reasoned that photoredox C-terminal decarboxylative functionalization might present a general strategy to target proteins in a site-selective manner regardless of its intrinsic topological features. Moreover, we hypothesized that the presence of only one C-terminus position on most protein structures should effectively enable single site modification using only canonical amino acid residues.
Design Plan.
Based on previous studies conducted in our laboratory, we recognized the stability of the resulting radical intermediate following decarboxylation is inherently linked to its ground state oxidation potential.27 Thus, we would expect internal Asp and Glu residues to have higher barriers to oxidation relative to the C-terminal carboxylic acid due to formation of an unstabilized carbon-centered primary radical vs. a heteroatom-stabilized α-amino radical at the protein terminus. In addition to these selectivity considerations, we recognized the need to develop reaction conditions compatible with biological substrates – namely, using aqueous buffer, high dilution, and mild temperatures. By virtue of operating through one-electron redox processes, photoredox catalysis presents a potentially valuable platform for selective reaction design under aqueous conditions. More specifically, one-electron transfer mechanisms are less susceptible to many of the challenges faced when translating cationic or anionic mechanisms to an aqueous environment (due to competitive trapping or attenuated nucleophilicities). As such, we recognized that a key element for the successful execution of these ideals would be the design of a water-compatible photocatalyst. Furthermore, we elected to use α,β-unsaturated carbonyls as the electrophile component given their known ability to readily engage with carbon-centered radicals under photoredox conditions in the presence of water.28 While the cysteine thiol group is known to react with highly electrophilic Michael acceptors, we rationalized that the judicious selection of a less reactive α,β-unsaturated carbonyl would allow for selective entrapment of the relatively high-energy C-terminal radical. Last, to be competitive with existing methodologies that achieve single-site modification with naturally occurring residues (e.g., Tyr, Trp, and N-termini),10,11,13,14,29,30 we recognized that synthetic efficiencies in the range of 25% conversion or greater would render this system a useful bioconjugation technology.
As a model system, we selected diethyl ethylidenemalonate as the Michael acceptor (5) and the N-terminal acetylated tetramer Ac-AGFP-OH as a representative short peptide sequence.18 Initial evaluations using Ir[dF(CF3)ppy]2(dtbbpy)+ (dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-(trifluorome1hyl)pyridme, dtbbpy = 4,4’-di-tert-butyl-2,2’-bipyridine) as the photocatalyst with 10 mM pH 7 buffer and 5% v/v glycerol (overall concentration of 1 mM relative to peptide), proved to be less than fruitful. Moreover, all attempts to employ common photocatalysts that are water soluble (ruthenium-based catalysts, organic dyes) likewise proved ineffective (<20% yield, see Supplementary Information). At this stage we next began to consider strategies that employ biocatalytic cofactors that are known to readily operate in water. More specifically, given that flavins have been shown to mediate acetate decarboxylation (albeit by a two-electron pathway), we hypothesized that these cofactors might function as suitable photocatalysts for single-electron transfer in aqueous bioconjugation processes.31 Gratifyingly, a survey of catalytic flavins (see Supplementary Information) revealed that 30 mol% of riboflavin tetrabutyrate (photocatalyst 1a, Figure 2) was capable of producing the decarboxylative conjugate addition adduct in 79% yield (Figure 3, entry 3).
Figure 2: Proposed mechanism for the C-terminal-selective photoredox decarboxylative conjugate addition.
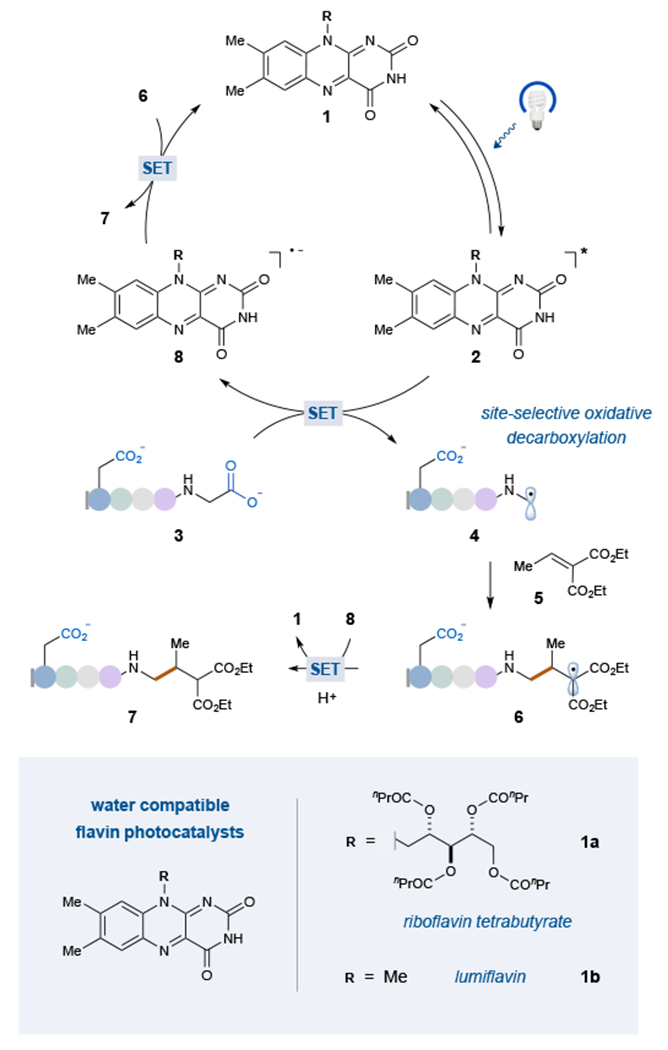
After optimization studies, the most effective photocatalysts are riboflavin tetrabutyrate, (1a), and lumiflavin, (1b). The mechanism is proposed to proceed via one-electron oxidation of 3 by the excited photocatalyst, 2, to furnish α-amino radical 4 after decarboxylation. Addition into Michael acceptor 5 provides adduct 6, which is reduced by reduced photocatalyst 8 and protonated to give C-terminal modified product 7.
Figure 3: Survey of functional group tolerance for the C-terminal-selective photoredox decarboxylative conjugate addition.
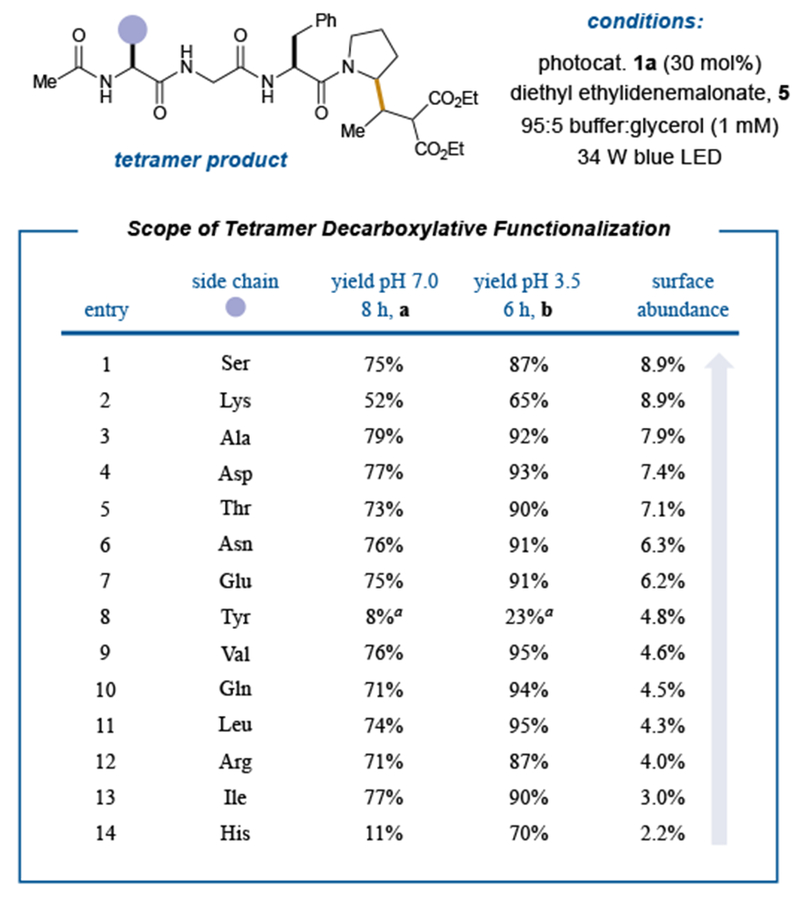
The scope of decarboxylative functionalization proves quite general with respect to the various functional groups present in the canonical amino acids at physiological pH. Importantly, no reaction was detected at the Asp or Glu site in tetramers containing these competing acid functionalities. Enhanced reactivity is observed using a pH 3.5 buffer. a30 mol% photocatalyst 1b. Yields reported as a % conversion as determined from reverse phase HPLC and are an average of three independent trials.
Mechanistically, we propose that the flavin photocatalyst 1 is initially promoted to its singlet excited state by excitation with a 34 W blue light and undergoes subsequent intersystem crossing (quantum yield, ΦISC = 0.38 for riboflavin in water at pH 7).32 The resulting triplet excited photocatalyst 2 is a strong single-electron oxidant (E1/2red = 1.5 V vs. saturated calomel electrode (SCE), in water)33 and should undergo facile SET with C-terminal carboxylate 3 (E1/2red = 1.3 V vs. SCE for Ac-AGFP-OH in water).34 Subsequent loss of CO2 from 3 furnishes α-amino radical 4, a species that is stabilized by the adjacent nitrogen. This transient intermediate then undergoes open-shell addition into Michael acceptor 5 to provide an α-acyl radical 6, which upon reduction by the photocatalyst would generate the corresponding enolate.35 Subsequent protonation would provide the bioconjugation product 7 while the α-acyl radical reduction step would regenerate the ground state of photocatalyst 1 to complete the catalytic cycle.
Results.
With these SET bioconjugative decarboxylative conditions in hand, we next evaluated the functional group tolerance of this technology with various peptides that incorporate the amino acid residues most commonly found on the surface of proteins (i.e. we selected residues that are found with ≥ 2% surface abundance).6 Using solid phase peptide synthesis, we systematically altered the N-terminal residue of the parent Ac-XGFP-OH template to afford 14 peptides that incorporate these most abundant amino acids. To our delight, a significant number of these peptide sequences were able to participate in the desired decarboxylative conjugation at pH 7 to furnish the desired adducts in yields between 71–79% (Figure 3b, entries 1a, 3a–7a, and 9a–13a). However, certain residues performed less efficiently, namely Lys (entry 2a), Tyr (entry 8a), and His (entry 14a). In these cases, we rationalized the decreased yields could be attributed to deleterious oxidation of these residues over the C-terminal carboxylate. With respect to Lys and His, we hypothesized that decreasing the pH of the buffer should ameliorate oxidative side-reactivity by increasing the equilibrium concentrations of ammonium and imidazolium respectively, as these species should be resistant to oxidation.36 Indeed, lowering the pH to 3.5 using a cesium formate buffer resulted in 65% yield for the Lys derivative (entry 2b) and 70% yield for the His derivative (entry 14b). Gratifyingly, use of pH 3.5 buffer universally improved the yields for all tetrapeptide sequences that we evaluated, delivering conjugation adducts in yields ranging from 87–95% (entries 1b, 3b–7b, and 9b–13b).
With respect to the Tyr-containing system, although it is known that Tyr oxidation is less facile at lower pH,37,38 initial trials employing riboflavin tetrabutyrate (1a) gave poor yields. To address this issue, we next examined lumiflavin (Figure 2a, photocatalyst 1b), a less oxidizing flavin photocatalyst.39 From a structural perspective, the absence of the electron-withdrawing ribityl side chain renders the isoalloxazine carbonyls as stronger H-bond acceptors with water, resulting in greater stabilization of the charge separated excited state relative to the ground state.40 This phenomenon serves to render lumiflavin less oxidizing as an excited-state photocatalyst. Indeed, application of lumiflavin with our Tyr-bearing peptide did in fact raise the efficiency of this process to 23% (entry 8b). Perhaps most important, tetramers incorporating Asp and Glu (both of which exhibit primary carboxylic acids), undergo decarboxylation exclusively at the C-terminal position, as confirmed by MS/MS analysis (see Supplementary Information). Indeed, these substrates exhibit perfect selectivity for C-terminal conjugation and high yields at pH 7 (77% and 75%, entries 4a and 7a respectively) and pH 3.5 (93% and 91%, entries 4b and 7b respectively). Moreover, regardless of pH, we did not observe any coupling products arising from hetero-conjugate additions of nucleophilic residues such as Lys, Ser, Thr, and His. Given these type of heteroatom functionalization protocols represent a common strategy in bioconjugation chemistry, we feel this outcome reveals both the kinetic and thermodynamic benefits of employing electron-transfer and open-shell mechanisms in lieu of closed shell, nucleophile trapping protocols.41
Having assessed the functional group tolerance of our reaction on a large, representative group of tetrapeptides, we next decided to examine the applicability of this photocatalytic methodology to endogenous peptides, including several with biological activity. At the outset, we selected commercial peptides ranging in size from 8–10 amino acids. Among those examined were a cardiovascular regulator, angiotensin II, and an inflammation inhibitor, bradykinin. These peptides underwent C-terminal decarboxylative alkylation with the desired mono-functionalization selectivity in yields of 40% and 53% respectively (Figure 4b, entries 15 and 16). Moving forward, we next examined mid-range, endogenous peptides consisting of 11-15 amino acid residues. As highlighted in Figure 4b, myelin basic protein (87–99), type II collagen, and fibronectin binding inhibitor peptide (modified only by appending an N-terminal Phe for ease of spectroscopic analysis whilst developing a quantitative HPLC assay) were all found to give excellent yields under our optimized reaction conditions (41–66%, entries 17–19). As a testament to the levels of site selectivity of our new bioconjugation protocol we also demonstrated a successful alkylation of bivalirudin (Angiomax), an antithrombotic icosapeptide. Despite an initially lower yield of 28% (entry 20), we observed exclusive C-terminal functionalization in the presence of five additional carboxylic acids. HPLC analysis of the crude reaction mixture revealed the remaining mass balance to be unreacted starting material. As such, a second round of photocatalytic activation following re-isolation of the unmodified peptide increased the overall yield to 33%. We hypothesize that the reaction halts at 28% conversion (with no further loss of starting material) due to photocatalyst deactivation as evidenced by UV-Vis time course studies (see Supplementary Information).
Figure 4. Scope of the photoredox decarboxylative conjugate addition applied to endogenous peptides.
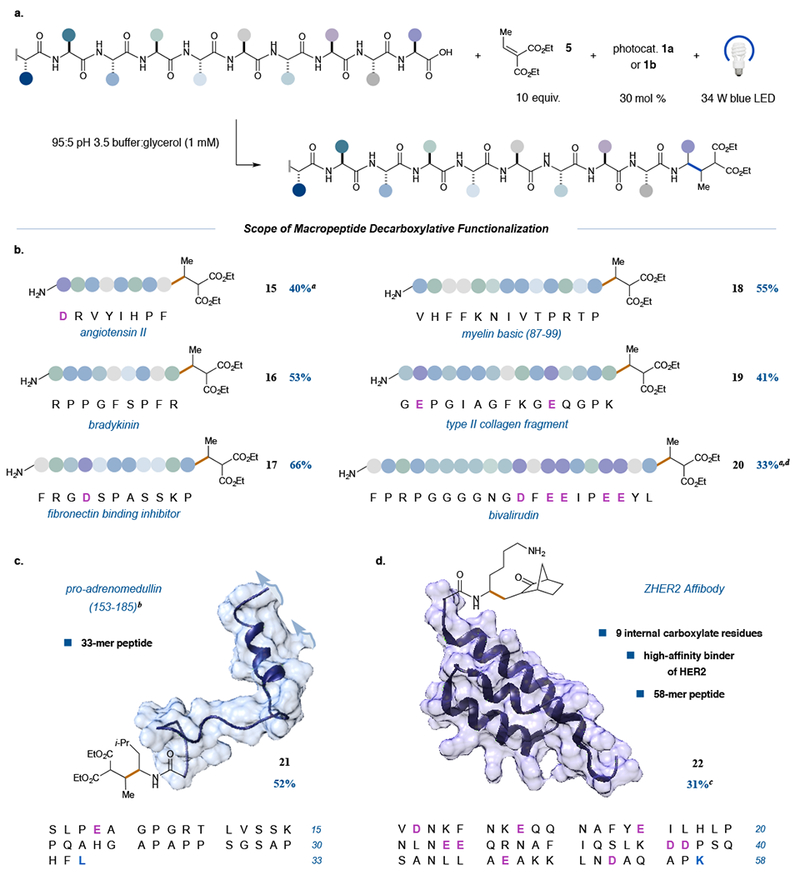
A variety of fully unprotected peptides from 8-mers to 58-mers can be site-selectively modified in this transformation, a, Generalized reaction scheme; b—d, substrate scope. All peptides are commercially available. In the case of the fibronectin binding inhibitor peptide (entry 17), the N-terminal Phe residue was added only for ease of analysis when initially developing the quantitative HPLC assay. Notably, longer peptides with nascent secondary structure (c) and peptides bearing high ratios of internal Asp and Glu residues (d) furnish C-terminal modified products exclusively. For entries 15–19 and 21–22, yields are reported as a % conversion as determined from reverse phase HPLC and are an average of three independent trials. aUsing photocatalyst lb. bRepresentative structure derived from the crystal structure of the full-length protein preproadrenomedullin. c3 equiv. photocatalyst lb and 3-methylene-2-norbomanone as the Michael acceptor. dYield reported as a combined % conversion after re-subjection of recovered starting material (see Supplementary Information).
Having demonstrated the selectivity of our reaction for the terminal alkylation of linear peptides (4–20 amino acids), we next decided to examine sequences with known secondary structure. For this purpose, we selected commercially available pro-adrenomedullin (fragment 153-185) and the ZHER2 affibody derived from immunoglobulin binding protein A. To our delight, proadrenomedullin selectively delivered the C-terminal modified product in 52% yield (entry 21) using our standard conditions. To our surprise, with the ZHER2 affibody, use of the diethyl ethylidenemalonate Michael acceptor resulted in the formation of the desired adduct along with side chain conjugate addition adducts via a competing Lys-addition pathway. At this stage, we rationalized that use of α,β-unsaturated carbonyls that i) are less susceptible to 2e− nucleophile pathways than diethyl ethylidenemalonate, yet ii) are still able to participate in radical additions, might overcome this issue. Indeed, the implementation of the strained, monocarbonyl-containing compound, 3-methylene-2-norbornanone, an established radicalphile under photoredox conditions,42 eliminated any undesired side reactivity, furnishing the modified product in 31% yield (entry 22). In this case recovered starting material accounted for the remaining mass balance.
A true test of any bioconjugation technology lies in its capacity to perform site-selective modification of peptides that exhibit tertiary structure. However, the structural linkages that confer these higher order architectures (e.g. disulfide bridges) can themselves be susceptible to chemical modification,43 thereby often changing the structure-function relationship of any given higher-order proteins. To test the viability of this new photoredox protocol in this context, we selected insulin as a suitable molecular platform to examine the chemical selectivity of electron transfer and open-shell mechanisms with a molecule that contains a variety of functional groups. Structurally, insulin is composed of two parent chains linked by two disulfide bridges, with a third disulfide bond in the A-chain backbone. Insulin also contains four tyrosines, residues that proved particularly challenging during our assessment of tetrameric peptides. Furthermore, and unlike the peptide substrates described thus far, insulin bears two C-terminal carboxylic acids in addition to having two Glu residues on each of the A- and B-chains. In the event, subjecting native insulin to our optimized reaction conditions (with 3-methylene-2-norbomanone as the electrophile) resulted in formation of a monoalkylated adduct in 44% isolated yield wherein modification occurred exclusively at the A-chain C-terminus. A minor adduct (less than 5%) was detected wherein both the A and B-chain C-termini were functionalized (see Supplementary Information for more details). Importantly, product analysis revealed that all three disulfide linkages remained intact and no side-chain decarboxylative or heteroatom conjugate addition was detected. The selectivity for A chain monoalkylation is particularly noteworthy as current technologies generally offer selective functionalization of insulin’s B chain through covalent modification at His10,44 Tyr26,45 and Lys29.46 This photoredox methodology therefore offers a new technology that not only selectively targets a specific C-terminal carboxylic acid, but also modifies the light chain, a structural component that heretofore has not been susceptible to bioconjugation. Moreover, expanding the scope of Michael acceptors to norbornanones that bear bioorthogonal tags (i.e. an alkyne) have also demonstrated promising results (see Supporting Information). We fully expect this Michael acceptor will be able to incorporate other biorthogonal handles (biotin and azides) and work is ongoing in this area.
In conclusion, we present a photoredox bioconjugation strategy that selectively targets C-terminal carboxylic acids in lieu of other functional groups found in protein structures. To our knowledge, this transformation represents an unprecedented approach to site-selective bioconjugation that can provide facile access to homogeneous alkylation adducts by virtue of the inherent presence of only one C-terminal carboxylate in most peptide and protein structures. Work is ongoing to apply this photoredox bioconjugation strategy to the selective functionalization of a wide range of proteins, enzymes, and antibodies.
Supplementary Material
Figure 5: Photoredox-mediated decarboxylative functionalization of human insulin.
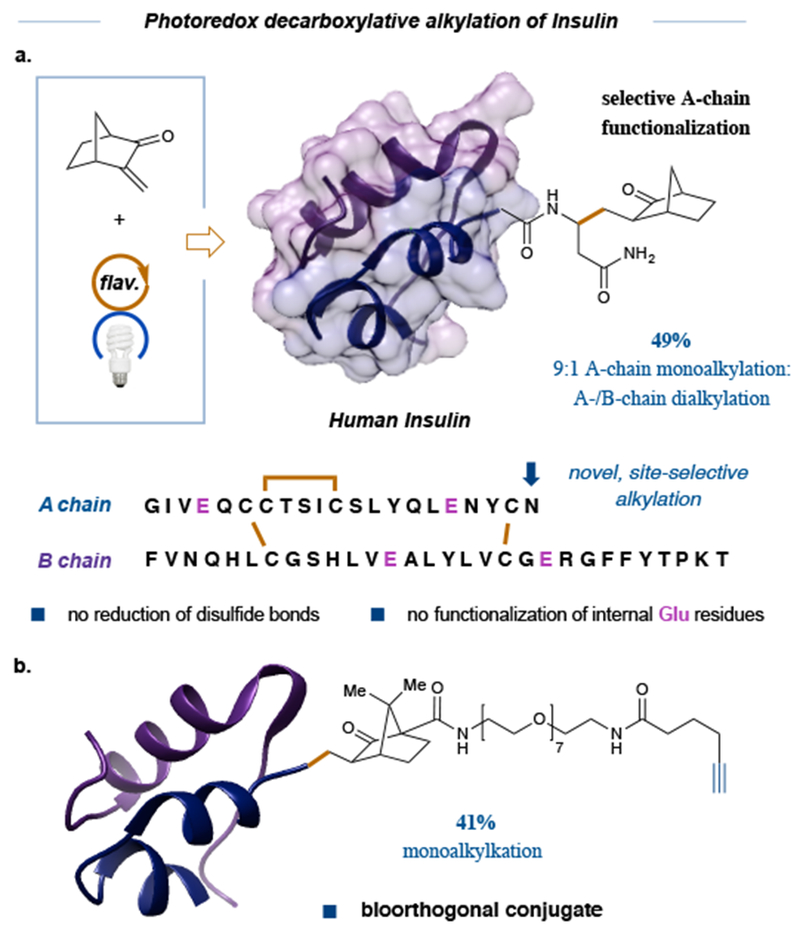
a, Human insulin was functionalized using our decarboxylative photoredox methodology to furnish highly selective mono-alkylation at the C-terminus of the A chain exclusively. b, Functionalization of human insulin with a Michael acceptor incorporating desthiobiotin as an affinity tag. Reaction conditions: 1 equiv. insulin (500 nmol), 10 equiv. of the respective Michael acceptor, 3 equiv. photocatalyst 1b, 95:5 pH 3.5 cesium formate buffer:glycerol (1 mM), 34 W blue LED, 8 h. Yield is reported as a % conversion as determined from reverse-phase HPLC. Current work exploring alkyne and azide bearing Michael acceptors is on-going.
Acknowledgements
The authors are grateful for financial support provided by the NIHGMS (R01 01GM093213-04) and kind gifts from Merck and BMS. The authors are also grateful to Professor Tom Muir, Dr. Zachary Brown, Dr. Robert Thompson and members of the Muir Lab for their advice and analytical support. The authors would also like to thank Dr. István Pelczer and Ken Conover for assistance with NMR spectroscopy.
Footnotes
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature
References
- 1.Boutureira O; Bernardes GJL Advances in chemical protein modification. Chem. Rev. 115, 2174–2195 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Krall N; da Cruz FP; Boutureira O; Bernardes GJL Site-selective protein-modification chemistry for basic biology and drug development. Nat. Chem. 8, 103–113 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Sletten EM; Bertozzi CR Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 48, 6974–6998 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saxon E; Bertozzi CR Cell surface engineering by a modified Staudinger reaction. Science 287, 2007–2010 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Baslé E; Joubert N; Pucheault M Protein chemical modification on endogenous amino acids. Chem. Biol. 17, 213–227 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Miller S; Janin J; Lesk AM; Chothia C Interior and surface of monomeric proteins. J. Mol. Biol. 196, 641–656 (1987). [DOI] [PubMed] [Google Scholar]
- 7.Chen X; Muthoosamy K; Pfisterer A; Neumann B; Weil T Site-selective lysine modification of native proteins and peptides via kinetically controlled labelling. Bioconjugate Chem. 23, 500–508 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Bader B; Kuhn K; Owen DJ; Waldmann H; Wittinghofer A; Kuhlmann J Bioorganic synthesis of lipid-modified proteins for the study of signal transduction. Nature, 403, 223–226 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Romanini DW; Francis MB Attachment of peptide building blocks to proteins through tyrosine bioconjugation. Bioconjugate Chem. 19, 153–157 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Tilley SD; Francis MB Tyrosine-selective protein alkylation using π-allylpalladium complexes. J. Am. Chem. Soc. 128, 1080–1081 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Joshi NS; Whitaker LR; Francis MB A three-component Mannichtype reaction for selective tyrosine bioconjugation. J. Am. Chem. Soc. 126, 15942–15943 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Ban H; Nagano M; Gavrilyuk J; Hakamata W; Inokuma T; Barbas CF III. Facile and stable linkages through tyrosine: bioconjugation strategies with the tyrosine-click reaction. Bioconjugate Chem. 24, 520–532 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antos JM; McFarland JM; Iavarone AT; Francis MB Chemoselective tryptophan labeling with rhodium carbenoids at mild pH. J. Am. Chem. Soc. 131, 6301–6308 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antos JM; Francis MB Selective tryptophan modification with rhodium carbenoids in aqueous solution. J. Am. Chem. Soc. 126, 10256–10257 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Seki Y; Ishiyama T; Sasaki D; Abe J; Sohma Y; Oisaki K; Kanai M Transition metal-free tryptophan-selective bioconjugation of proteins. J. Am. Chem. Soc. 138, 10798–10801 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Lin S; Yang X; Jia S; Weeks AM; Hornsby M; Lee PS; Nichiporuk RV; Iavarone AT; Wells JA; Toste FD; Chang CJ Redox-based reagents for chemoselective methionine bioconjugation. Science, 355, 597–602 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zuo Z; MacMillan DWC Decarboxylative arylation of α-amino acids via photoredox catalysis: a one-step conversion of biomass to drug pharmacophore. J. Am. Chem. Soc. 136, 5257–5260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chu L; Ohta C; Zuo Z; MacMillan DWC Carboxylic acids as a traceless activation group for conjugate additions: a three-step synthesis of (±)-pregabalin. J. Am. Chem. Soc. 136, 10886–10889 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noble A; MacMillan DWC Photoredox-mediated α-vinylation of α-amino acids and N-aryl amines. J. Am. Chem. Soc. 136, 11602–11605 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Merging photoredox with nickel catalysis: coupling of α-carboxyl sp3-carbons with aryl halides. Science 345, 437–440 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galicia M; González FJ Electrochemical oxidation of tetrabutylammonium salts of aliphatic carboxylic acids in acetonitrile. J. Electrochem. Soc. 149, D46–D50 (2002). [Google Scholar]
- 22.Hu Q-Y; Berti F; Adamo R Towards the next generation of biomedicines by site-selective conjugation. Chem. Soc. Rev. 45, 1691–1719 (2016). [DOI] [PubMed] [Google Scholar]
- 23.McGrath NA; Andersen KA; Davis AKF; Lomax JE; Raines RT Diazo compounds for the bioreversible esterification of proteins. Chem. Sci. 6, 752–755 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rajagopalan TG; Stein WH; Moore S The inactivation of pepsin by diazoacetylnorleucine methyl ester. J. Biol. Chem. 241, 4295–4297 (1966). [PubMed] [Google Scholar]
- 25.Delpierre GR; Fruton JS Specific inactivation of pepsin by a diazo ketone. Proc. Natl. Acad. Sci. U.S.A. 56, 1817–1822 (1966). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Totaro KA; Liao X; Bhattacharya K; Finneman JI; Sperry JB; Massa MA; Thorn J; Ho SV; Pentelute BL Systematic investigation of EDC/sNHS-mediated bioconjugation reactions for carboxylated peptide substrates. Bioconjugate Chem. 27, 994–1004 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Noble A; McCarver SJ; MacMillan DWC Merging photoredox and nickel catalysis: decarboxylative cross-coupling of carboxylic acids with vinyl halides. J. Am. Chem. Soc. 137, 624–627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slutskyy Y; Overman LE Generation of the methoxycarbonyl radical by visible-light photoredox catalysis and its conjugate addition with electron-deficient olefins. Org. Lett. 18, 2564–2567 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seki Y; Ishiyama T; Sasaki D; Abe J; Sohma Y; Oisaki K; Kanai M Transition metal-free tryptophan-selective bioconjugation of proteins. J. Am. Chem. Soc. 138, 10798–10801 (2016). [DOI] [PubMed] [Google Scholar]
- 30.MacDonald JI; Munch HK; Moore T; Francis MB One-step site-specific modification of native proteins with 2-pyridinecarboxyaldehydes. Nat. Chem. Bio 11, 326–331 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Novak M; Miller A; Bruice TC; Tollin G The mechanism of flavin 4a substitution which accompanies photolytic decarboxylation of α-substituted acetic acids. Carbanion vs. radical intermediates. J. Am. Chem. Soc. 102, 1465–1467 (1980). [Google Scholar]
- 32.Islam SDM; Penzkofer A; Hegemann P Quantum yield of triplet formation of riboflavin in aqueous solution and of flavin mononucleotide bound to the LOV1 domain of Phot1 from Chlamydomonas reinhardtii. Chem. Phys. 291, 97–114 (2003). [Google Scholar]
- 33.Lu C; Lin W; Wang W; Han Z; Yao S; Lin N Riboflavin (VB2) photosensitized oxidation of 2’-deoxyguanosine-5’-monophosphate (dGMP) in aqueous solution: a transient intermediates study. Phys. Chem. Chem. Phys. 2, 329–334 (2000). [Google Scholar]
- 34.See Supplementary Information for experimental details.
- 35.Bortolamei N; Isse AA; Gennaro A Estimation of standard reduction potentials of alkyl radicals involved in atom transfer radical polymerization. Electrochim. Acta 55, 8312–8318 (2010). [Google Scholar]
- 36.Huvaere K; Skibsted LH Light-induced oxidation of tryptophan and histidine. Reactivity of aromatic N-heterocycles toward triplet-excited flavins. J. Am. Chem. Soc. 131, 8049–8060 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Harriman A Further comments on the redox potentials of tryptophan and tyrosine. J. Phys. Chem. 91, 6102–6104 (1987). [Google Scholar]
- 38.Stubbe J; van der Donk WA Protein radicals in enzyme catalysis. Chem. Rev. 98, 705–762 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Sikorska E; Khmelinskii IV; Prukala W; Williams SL; Patel M; Worrall DR; Bourdelande JL; Koput J Sikorski M, Spectroscopy and photophysics of lumiflavins and lumichromes. J. Phys. Chem. A 108, 1501–1508 (2004). [Google Scholar]
- 40.Koziol J Studies on flavins in organic solvents – I. Spectral characteristics of riboflavin, riboflavin tetrabutyrate, and lumichrome. Photochem. Photobiol. 5, 41–54 (1966). [Google Scholar]
- 41.Garbaccio RM Chemistry of antibody-small molecule drug conjugates. From Comprehensive Organic Synthesis II, 9, 438–462 (2014). [Google Scholar]
- 42.Ravelli D; Zema M; Mella M; Fagnoni M; Albini A Benzoyl radicals from (hetero) aromatic aldehydes. Decatungstate photocatalyzed synthesis of substituted aromatic ketones. Org. Biomol. Chem. 8, 4158–4164 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Wagner A; Koniev O Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem. Soc. Rev. 44, 5495–5551, (2015). [DOI] [PubMed] [Google Scholar]
- 44.Uchida K; Stadtman ER Modification of histidine residues in proteins by reaction with 4-hydroxynonenal. Proc. Natl. Acad. Sci. USA 89, 4544–4548 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y; Luo Y; Zhang R Investigation on insulin tyrosine modification mediated by peroxynitrite. IEEE/ICME International Conference on Complex Chemical Engineering, Beijing, 1813. –1816. (2007). [Google Scholar]
- 46.Lindsay DG; Shall S The acetylation of insulin. Biochem. J. 121, 737–745 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.