

Abstract
Purpose
Mutations in the megalin-encoding gene, LRP2, cause high myopia as seen in patients suffering from Donnai-Barrow/facio-oculo-acoustico-renal syndrome. Megalin is present in both the nonpigmented epithelium of the ciliary body and in the RPE. In this study, we set out to establish an animal model to study the mechanisms underlying the ocular phenotype and to establish if high myopia/megaophthalmos is induced by postnatal megalin-deficiency in the RPE.
Methods
Postnatal RPE-specific deletion of megalin was generated by crossing mice bearing a homozygous loxP-flanked Lrp2 allele with transgenic mice expressing the Cre recombinase driven by the BEST1 promotor. The model was investigated by immunohistologic techniques, and transmission electron microscopy.
Results
Mice with postnatal RPE-specific loss of megalin developed a megaophthalmos phenotype with dramatic increase in ocular size and severe retinal thinning associated with compromised vision. This phenotype was present at postnatal day 14, indicating rapid development in the period from onset of BEST1 promotor activity at postnatal day 10. Additionally, RPE melanosomes exhibited abnormal size and morphology, suggested by electron tomography to be caused by fusion events between multiple melanosomes.
Conclusions
Postnatal loss of megalin in the RPE induces dramatic and rapid ocular growth and retinal degeneration compatible with the high myopia observed in Donnai-Barrow patients. The morphologic changes of RPE melanosomes, believed to be largely inert and fully differentiated at birth, suggested a continued plasticity of mature melanosomes and a requirement for megalin to maintain their number and morphology.
Keywords: megalin, retinal pigment epithelium, macromelanosomes, high myopia, megaophthalmos, retinal degeneration
Mutation of the LRP2 gene causes the very rare Donnai-Barrow syndrome (DBS) also known as facio-oculo-acustico-renal syndrome (OMIM # 222448).1,2 DBS patients suffer from a diverse range of developmental and functional abnormalities including hypertelorism, agenesis of the corpus callosum, diaphragmatic hernia as well as sensorineural hearing loss, low-molecular-weight proteinuria, and high myopia.1 Despite great phenotypic variation in these patients, high grade myopia with refractive errors ranging from −12.5 to 22.0 diopters (D) have been consistently observed.1,3–6 Additional ocular manifestations including large protruding eyes, retinal dystrophy, retinal detachment, pigment changes, and cataract have also been frequently reported in these patients.1,6
The LRP2 gene encodes the 600 kDa type 1, transmembrane protein megalin. Numerous studies have established megalin as an endocytic receptor with a long list of structurally and functionally diverse ligands including vitamin carriers, plasma proteins, enzymes, and enzyme inhibitors as well as hormones and signaling molecules.7 Megalin has been extensively studied in renal proximal tubules. Here, megalin mediates reabsorption of filtered plasma proteins, rescuing vital nutrients from urinary excretion.7 Besides the kidney, megalin is also expressed in a number of other specialized epithelia throughout the body including type II pneumocytes of the lung, the choroid plexus epithelium, and placental cytotrophoblasts.8–11 Notably, megalin is also expressed in the non-pigmented ciliary body epithelium as well as the RPE in the adult mammalian eye.10,12,13
The ocular phenotype of four animal models (zebrafish and mice) with genetic ablation of the megalin-encoding gene have recently been described.13–16 These models are all based on prenatal genetic ablation of megalin in all ocular structures normally expressing megalin. In parallel to observations from DBS patients, high myopia/megaophthalmos, retinal dystrophy, and pigment changes were reported in these animal models suggesting that the mechanisms underlying the development of the ocular abnormalities observed in DBS patients may be investigated using these animal models. However, although very valuable for histologic and functional investigations of ocular megalin-deficiency in general, none of these models allow determination of whether the megaophthalmos is caused by early ocular developmental defects and/or by megalin dysfunction in the RPE specifically. We therefore used the previously reported Cre recombinase under control of the Best1 promoter17,18 to obtain a selective genetic ablation of megalin in the RPE during the early postnatal period. Here, we report that mice with RPE-specific genetic ablation of megalin in the early postnatal period develop rapid megaophthalmos and abnormal RPE pigmentation, resulting in severe retinal dystrophy associated with compromised vision.
Materials and Methods
Animals
Animal experiments and breeding were approved by the Danish Animal Experiments Inspectorate and performed in the animal facility of Department of Biomedicine, Aarhus University, Denmark. All animal procedures were carried out in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Mice with homozygous conditional inactivation of the Lrp2 gene in RPE cells were generated by breeding Tg(BEST1-Cre)1Jdun/J17 transgenic mice with mice bearing a loxP-flanked Lrp2 allele (Lrp2tm1Tew)19 to create first generation BEST1-Cre, Lrp2 heterozygous mice (Lrp2flox/+) on a C57BL/6 background. Heterozygous BEST1-Cre, Lrp2 (C57BL/6) were subsequently crossed with mice bearing loxP-flanked Lrp2 alleles on a 129S6/SvEvTac (Taconic Biosciences, model # 129SVE-F) background to obtain homozygous BEST1-Cre, Lrp2 on a mixed C57BL/6-129/Svj background. Cre-negative littermates served as controls in all experiments.
Tissue Preparation and Microscopy
Mice were euthanized by isoflurane sedation before cervical dislocation and decapitation. For immnohistochemical analyses, eyes were enucleated and fixed by immersion in 4% formaldehyde in 0.1 M cacodylate buffer, pH 7.4 for 1 hour prior to dehydration and paraffin embedding. We cut 5-μm paraffin sections on a microtome (Leica RM 2165; Leica, Ballerup, Denmark), and processed sections for immunohistochemistry using fluorophore-coupled secondary antibodies as previously described.13 Examination and image acquisition were performed using an inverted confocal microscope (Zeiss LSM 510 META; Carl Zeiss AG, Jena, Germany) and processed using commercial software (Axiovision 4.8; Carl Zeiss Microscopy, Thornwood, NY, USA, and Adobe Photoshop 8.0, Adobe, Inc., San Jose, CA, USA).
For immunohistochemical analyses of sections using alkaline phosphatase-coupled secondary antibodies, sections where rehydrated and subsequently heated in Tris-EGTA buffer for antigen retrieval in a microwave for approximately 10 minutes, cooled, and incubated in 50 mM NH4Cl in 0.05 M TBS (pH 7.4) for 30 minutes. Sections were then subsequently incubated with primary antibodies in 0.05 M TBS (pH 7.4) with 0.1% bovine serum albumin and 0.3 % Triton X-100 followed by incubation with secondary antibody. Phosphatase labeling was visualized by incubation with FAST RED substrate (Kem-En-Tec Diagnostics, Taastrup, Denmark) and counterstained with Meier's hematoxylin stain for examination using a microscope (Leica DMR; Leica Microsystems GmbH, Wetzlar, Germany) equipped with a camera (DFC320; Leica Microsystems GmbH) and processed using graphic editing software (Adobe, Inc.).
For general histologic examination, sections were counterstained with Meier's haematoxylin stain and images acquired using a Leica DMR microscope equipped with a Leica DFC320 camera or the microscope (Eclipse 80i; Nikon Corp., Tokyo, Japan) equipped with a motorized stage (H138E50; Prior, Cambridge, UK), a digital camera (DP72; Olympus, Tokyo, Japan), and processed using graphic editing software (Adobe, Inc.).
For ultrastructural analyses, enucleated mouse eyes were fixed in 2% formaldehyde and 2% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4. After fixation eyes were dissected into small pieces of retinal tissue. Tissue pieces were subsequently postfixed for one hour in 1% OsO4 in 0.1 M cacodylate buffer, stained for 1 hour with 0.5% uranyl acetate in 0.05 M Maleate buffer pH 6.0, dehydrated in graded alcohols, and embedded in Epon (TAAB resin 812; VWR - Bie & Berntsen A/S, Soeborg, Denmark). Ultrathin sections of approximately 60 nm were obtained with a cryoultramicrotome (Leica EM FC6; Leica Microsystems GmbH), collected on 100 mesh nickel grids and stained with uranyl acetate and led citrate. Sections were examined using a transmission electron microscope (JEOL JEM-1400+; JEOL, Freising, Germany).
Electron Tomography
We cut and stained 200-nm thick sections with lead citrate in addition to a 10-nm gold particle solution (BBI Solutions, Cardiff, UK). Images were acquired on an electron microscope (FEI T12 Tecnai Spirit; FEI, OR, USA), using tomography (iTEM; ResAlta Research Technologies, Golden, CO, USA) and a camera (Morada; Olympus SIS, Münster, Germany). Tilt series were collected over a tilt range of −60° to +60° in 2° increments around 2 orthogonal axes using a dual-axis tomography holder (Fishione Instruments, Pittsburgh, PA, USA). The images were processed to generate tomograms using the IMOD package.20
Antibodies
(Primary) Sheep anti rat megalin,21 rabbit monoclonal Cre recombinase (D7L7L; Cell Signaling Technology Inc., BioNordika A/S, Herlev, Denmark)
(Secondary) Donkey anti sheep Alexa Fluor-488 antibody (Invitrogen Taastrup Denmark), alkaline phosphatase-coupled swine anti rabbit (DAKO)
(Nuclear stain) Fluoroshield mounting medium with propidium iodide (Sigma-Aldrich Corp., Copenhagen, Denmark).
Results
RPE-Specific Ablation of Megalin
To investigate the physiologic role of megalin in the retina we produced mice with RPE-selective loss of megalin in the early postnatal period by combining mice bearing floxed Lrp2 alleles (Lrp2 flox/flox)19 with mice transgenic for BEST1-Cre.17
Approximately a quarter of the mice with selective RPE megalin deletion showed remarkably large and protruding eyes compared to their Cre-negative littermates (n > 50; Figs. 1A, 1B). Immunohistochemical evaluation of residual RPE megalin expression on mutant whole eye cross-sections revealed that a megalin knock out degree of >90% correlated to the severe megaophthalmos phenotype observed in a quarter of the mice. In order to mimic the full megalin-deficient RPE cells in DBS patients we selected the mice with a distinct megaophthalmos phenotype and a knockout degree >90% for further detailed histologic examinations.
Figure 1.

Overall eye size in control and mutant mice. Control (A) and mutant (B) eyes shown in vivo before enucleation and preparation. Mutant eyes are clearly protruding and enlarged compared to control eyes. Representative histologic cross-sections of enucleated eyes from 14-day-old (C, D) and adult (14–17.5 months old; E, F) normal (C, E) and mutant eyes (D, F) shows a large difference in the overall eye size between the two groups. Scale bars: C, D, E, and F = 1 mm.
Histologic examination of whole-eye cross-sections from these mice (n = 3–6 eyes in each group) confirmed a megaophthalmos phenotype observable already at postnatal day (PND) 14 (Figs. 1C, 1D), only 4 days after Cre-recombinase expression is present,17,18 suggesting a very fast progressing phenotype and furthermore that megalin harbors a very important postnatal role in the RPE. Adult mutant mice (aged 14–17.5 months) also showed a pronounced megaophthalmos phenotype, with the difference in eye size between affected Cre-positive mutant and Cre-negative controls appearing to be even greater (Figs. 1E, 1F). Detailed histologic examination of the retinas from PND14 and adult (aged 14–17.5 months) mutant mice furthermore revealed severe thinning of all retinal layers (Figs. 2B, 2F) as well as hypopigmentation and abnormally large RPE melanosomes (macromelanosomes, Figs. 2D, 2H). Such observations were never seen in control animals (Figs. 2A, 2C, 2E, 2G; n = 3–6 eyes in each group). Subsequent immunohistochemical detection of the Cre recombinase protein in histologic sections from mutant eyes furthermore showed that the presence of RPE macromelanosomes correlated with nuclear expression of Cre recombinase in the RPE cells (Fig. 2I).
Figure 2.

Retinal morphology in 14-day-old and adult normal and mutant eyes. Hematoxylin-eosin–stained histologic cross-section of control (CTRL, left panel) and mutant (KO, right panel) mouse eyes shows severe thinning of all retinal layers in both 14-day-old (B) and adult (14–17.5 months old) (F) mutant mice. Enlargements of the RPE in these mice are given directly below (C, D, G, H) and shows that RPE melanosomes of mutant retinas are abnormally large and reduced in number (melanosomes highlighted by red squares). (I) Immunohistochemical detection of the Cre recombinase in mutant retinas showed a mosaic expression pattern (Cre recombinase positive nuclei are red stained, highlighted by green squares) and also that expression of the Cre recombinase correlates with the presence of macromelanosomes. Scale bars: A, B, E, F = 25 μm and C, D, G, H, I = 5 μm.
Our findings of severe retinal thinning, loss of photoreceptor outer segments (Figs. 2F, 2H) and a gross increase in visual axis identified in the mutant mice with a megaophthalmos phenotype suggests that postnatal megalin-deficiency in the RPE is incompatible with normal visual function.
Interestingly, immunohistochemical analyses of ocular megalin showed a cytoplasmic staining of all RPE cells in control mice (Lrp2 flox/flox) but only a scarce RPE expression in BEST1-Cre positive mutant mice (Lrp2 flox/flox, BEST1-Cre; Figs. 3A, 3B; n = 3 eyes in each group). In contrast, strong megalin expression was observed in both genotypes in the adjacent ciliary marginal zone (Figs. 3C, 3D) supporting the RPE-specific nature of our model.
Figure 3.
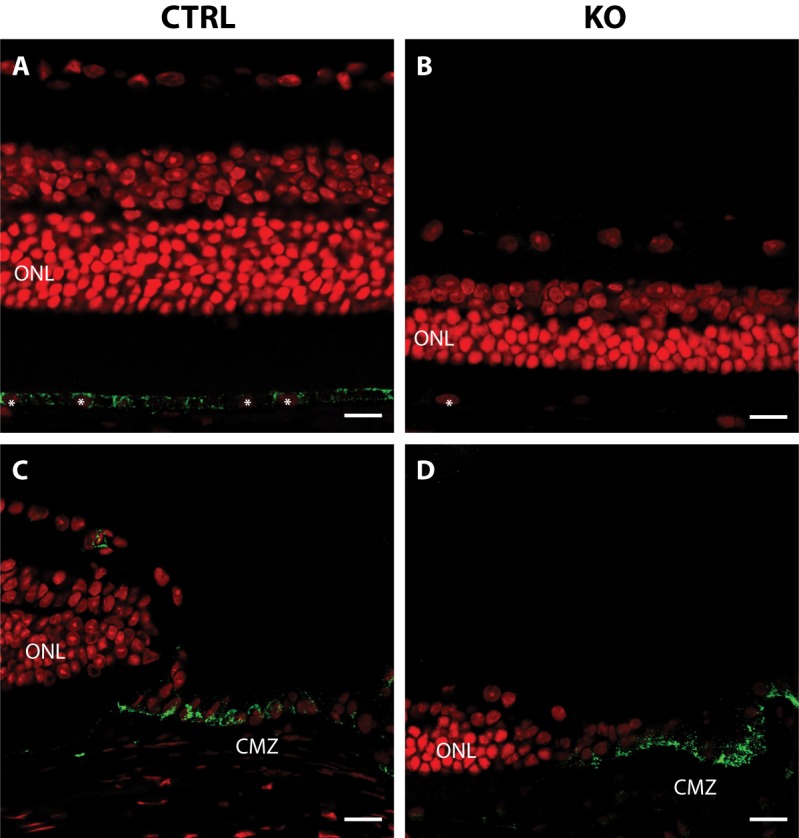
Immunohistochemical localization of megalin in control and mutant eyes. (A–D) Whole eye sections labeled with anti-megalin (green) and propidium iodide (red). Megalin is detected throughout the retina pigment epithelium in control mouse eyes (A) but only in a mosaic pattern in mutant eyes (B). Strong megalin expression is seen in the CMZ of both control (C) and mutant (D) eyes. RPE nuclei are indicated with white asterisks. ONL, outer nuclear layer; CMZ, ciliary marginal zone. Scale bars: A, B, C, and D = 10 μm.
Development of Macromelanosomes
To analyze the development of RPE macromelanosomes in megalin-deficient RPE cells we performed ultrastructural analyses of retinas from normal and mutant mice at PND5 and PND14. As the Cre-recombinase is present from day P1017,18, analyses of melanosomes at PND5 did not show any apparent differences, as expected (n = 3–5; Figs. 4A, 4B). In contrast, histologic examination of mutant retinas from PND14 mice, confirmed a reduction in the number of total melanosomes and an overt increase in the overall size of the melanosomes (n = 3–5; Figs. 4C, 4D), as previously seen on our paraffin sections and as reported in mice with global megalin deficiency.13 The macromelanosomes did not display any apparent difference in melanosome maturation stage but did show shapes that deviated substantially from the normal ellipsoid shape of RPE melanosomes (Fig. 4D). Interestingly, we observed numerous macromelanosomes with multi-spherical shapes indicating fusion events of smaller melanosomes (Figs. 5A, 5B). As electron micrographs are a 2-dimensional (2D) representation of a 3-dimensional (3D) object, it can be challenging to distinguish between overlapping and composite structures. To circumvent this, we performed electron tomography analyses on thick epon sections of abnormal melanosomes from mutant P14 retina (Fig. 5C). Within the 3D tomographic reconstruction (see Supplementary Video 1), the atypically shaped macromelanosome can be seen to be comprised of three separate melanosomes, each identifiable by the perimeters of the packed melanin cores (Figs. 5C, 5D). The melanosome membranes thus appear to have fused together resulting in a single limiting membrane encompassing the entire structure.
Figure 4.

RPE ultrastructure in control and mutant mice. Transmission electron micrographs of the outermost retinal layers in control (CTRL, left panel) and mutant mice (KO, right panel) shows no apparent ultrastructural differences in 5-day-old mice (A, B). In contrast, we observed a large number of ultrastructural abnormalities in 14-day-old mutant mice (D) including thinning of the photoreceptor outer segment layer, macromelanosomes, melanolipofuscin-like structures, as well as a loss of basal infoldings. Scale bars: A, B, C, and D = 2 μm.
Figure 5.

RPE melanosome ultrastructure in 14-day-old mutant mice. High magnification transmission electron micrographs showing the abnormal size and multispherical shapes of selected macromelanosomes from 14-day-old mutant RPE cells (A, B; n = 4). Still frame from a 3D tomographic analysis (see Supplementary Video 1) of a thick epon section from a 14-day-old mutant RPE cell (C). The multispherical shape of the macromelanosome is surrounded by a single limiting membrane encompassing the entire structure (highlighted in green) and is comprised of three separate melanosomes, each identifiable by the perimeters of individual packed melanin cores (highlighted in red [D]). Scale bars: A, B = 1 μm and C, D = 200 nm.
Our ultrastructural investigations of mutant P14 RPE cells (n = 3–4 eyes in each group) furthermore revealed a number of additional cellular abnormalities. Specifically, we observed loss of basal infoldings (Fig. 6B), large melanolipofuscin-like structures (Figs. 6C, 6D), distended and fragmented endoplasmic reticulum throughout the cytoplasm (Figs. 6B, 6E), scattered ribosomes (Fig. 6E), an apparent increase in the number of Golgi apparatus (Fig. 6F) as well as an absence of clathrin-coated vesicles, compared to the examination of control samples (Figs. 6A, 6C, 6D).
Figure 6.

RPE cell ultrastructure in control and mutant 14-day-old mice. Transmission electron micrographs of RPE cells from control (A, C, [zoom of region in A] and D) and mutant (B, E, F, G) 14-day-old mice illustrating the ultrastructural abnormalities identified in mutant mice including loss of basal infoldings (B), large melanolipofuscin-like structures (B, E), distended and fragmented endoplasmic reticulum throughout the cytoplasm (B, F, G), scattered and detached ribosomes (example indicated by small black box [F, G]), apparent increased number of Golgi apparatus (G) as well as a general absence of clathrin-coated vesicles (B, F, G). BI, basal infoldings; BM, basal membrane; ccv, clathrin-coated vesicles; dER, dilated endoplasmic reticulum; G, Golgi apparatus; M, melanosome; Mit, mitochondrion; ML, melanolipofuscin-like; POS, photoreceptor outer segment; Pr, polyribosome. Scale bars: A, B, G = 1 μm, C, D = 500 nm; E, F = 200 nm.
Discussion
Megalin is a very large, multiligand endocytic receptor expressed in multiple ocular structures, which greatly complicates studies of its physiologic and morphologic function in individual cell types. To investigate a potential role of megalin in supporting retinal health, we established a transgenic mouse model with selective, postnatal loss of megalin in the RPE using Cre recombinase controlled by the BEST1 promoter.17 Cre-expression controlled by the BEST1 promotor was recently reported to be greatly influenced by genetic background.17 On a pure C57BL/6 background the number of Cre-expressing RPE cells varied between 50% and 90% compared to a more consistent observation of approximately 90% of Cre-expressing RPE cells on a mixed B6/129 background. Although our model was also bred on a mixed B6/129 background we did not observe the same success in RPE Cre-expression, potentially due to variations of the genetic backgrounds of the mice used in the two studies. The influence of genetic background, combined with an apparent megalin knockout threshold for development of the megaophthalmos phenotype, are most likely the major determining factors leading to a relatively low percentage of mice developing a distinct megaophthalmos phenotype. Using the BEST1 promoter to drive ocular expression of the Cre recombinase, we identified a correlation between low residual megalin RPE expression and an early onset, rapidly developing megaophthalmos phenotype with severe retinal thinning and compromised vision. Striking early-onset myopic/megaophthalmos phenotypes have been reported in a number of megalin-deficient mouse models.13,15,16 Despite observations of increased intraocular pressure in megalin-deficient zebrafish14 mice with combined megalin inactivation in the neural retina, RPE, and ciliary body did not show an increase in intraocular pressure.15 Neither are any reports of increased intraocular pressure in DBS patients known to the authors. It is thus very unlikely that development of an apparently similar megaophthalmos phenotype in our RPE-specific model should be caused by elevated intraocular pressure.
In the previously reported megalin-deficient mouse models, genetic ablation of megalin was introduced during embryonic development and not in a cell-specific manner as reported here. Based on investigations of compound heterozygous global megalin-deficient mice,16 it was recently speculated that aberrant sonic hedgehog signaling leading to abnormal proliferation of the retinal marginal zone are the underlying causes for the megaophthalmos phenotype observed in mice and patients with megalin defects. However, immunohistochemical analyses of megalin in our BEST1-driven mutant mice showed a RPE-selective ablation, with sustained normal expression of megalin in the ciliary marginal zone confirming previous reports of RPE-specific Cre-expression.17,18 Despite the postnatal RPE-specific deletion of Lrp2, we observed a highly similar, early-onset (PND14) megaophthalmos phenotype with severe retina thinning in our mutant mice. This suggests that absence of megalin in the RPE is an essential factor causing or contributing to the high myopia/megaophthalmos phenotype observed in patients, mice, and fish with megalin-deficiency. Furthermore, the Cre recombinase expression is reported to initiate at PND10 when controlled by the BEST1 promoter (Iacovelli et al.,17 Zhao et al.18) and megaophthalmos is present already at PND14 in our mutant mice, indicating that megaophthalmos can develop very rapidly in the early postnatal period and that megalin appears to harbor a key role in preventing this from happening.
The RPE monolayer is situated between the neuroretina and the choroid, serving multiple roles including metabolic support of the retina, recycling of retinal chromophores, absorption of scattered light, and phagocytosis of shed photoreceptor outer segments.22 The RPE is furthermore a significant source of cytokines and growth factors23 and known to express receptors for many of the factors known to be involved in regulation of eye growth and myopia.24 Thus, the RPE occupies an ideal anatomic location for conveying growth regulatory signals from the retina to the choroid and sclera. Although our understanding of retinoscleral signaling in myopia and eye growth is still in its infancy, accumulating evidence suggests the RPE holds a central role.24
Recent studies associated polymorphisms of the gene encoding insulin-like growth factor-1 (IGF-1) with high myopia in Egyptian25 and Chinese26 patients. Interestingly, megalin has been shown to bind to the IGF-1 receptor and suggested to mediate transcytosis of IGF-1 in the choroid plexus epithelium.27,28 The IGF-1 receptor is a transmembrane receptor belonging to the large class of tyrosine kinase receptors. From detailed studies of the epidermal growth factor receptor (EGFR), another member of the tyrosine kinase receptors, it is evident that endocytosis and intracellular sorting of this receptor complex may be key for signal duration.29 Thus, it could be speculated that, if megalin facilitates internalization of the IGF-1-receptor complex, megalin could possibly also downregulate IGF-1 cell signaling by targeting the IGF-1-receptor complex for lysosomal degradation as seen for many of the established megalin ligands.7 An absence of megalin in the RPE could thus potentially lead to increased IGF-1 signaling in the eye and aberrant eye growth with the associated development of high myopia.
The secreted glycoprotein sonic hedgehog (Shh)30 is important for eye morphogenesis,31 but is also a key morphogen and signaling molecule during embryonic development in general. Similarities of neuronal abnormalities of mice deficient in either Shh or megalin, led to investigations of a functional relationship of the two.32 In vitro studies showed that megalin is able to bind and internalize Shh suggesting that megalin may be involved in modulating Shh signaling in megalin-expressing tissues. Interestingly, investigations of form-deprivation myopia in mice and guinea pigs showed that increased Shh signaling influenced myopia development,33,34 as well as postnatal eye growth.33 Taken together, this might cause one to speculate that absence of megalin in the RPE could lead to increased retinoscleral Shh signaling, thus contributing to dysregulated eye growth and development of myopia. Although very interesting, a potential role for megalin in IGF-1 and/or Shh signaling remains purely speculative at this point and calls for future detailed studies into the relevant growth factors and receptors.
Pigment changes in the ocular fundus have been reported in a number of DBS patients,1 but never investigated in detail. We recently reported the presence of abnormally large RPE melanosomes in globally megalin-deficient mice.13 In the present study, using RPE-specific megalin-deficient mice we investigated the ultrastructure and the formation of these macromelanosomes in the postnatal period. We observed no apparent differences in size or shape of RPE melanosomes at PND5, consistent with the onset of the Cre recombinase at day PND10.17,18 However, our data also showed that striking abnormalities in melanosome size and shape developed in the megalin-deficient mice in only 4 days, between the onset of Cre recombinase expression at day PND10 and ultrastructural analyses at day PND14. Our detailed electron tomography analyses of the abnormal melanosomes, showing a single membrane encompassing multiple melanin-rich cores, suggest that the formation of macromelanosomes in megalin-deficient RPE cells is caused by fusion events between multiple melanosomes.
The RPE is a nonproliferative epithelium and RPE melanosome biogenesis is believed to be largely concluded before birth in mice.35 However, our data suggested that megalin-deficiency introduces a postnatal plasticity to RPE melanosomes. RPE melanosome biogenesis is particularly difficult to study due to a lack of good simple in vitro models. This is mainly because primary RPE cells in culture and immortalized RPE cell lines generally do not display melanosome de novo biogenesis although pigmentation has been observed in RPE cells derived from induced pluripotent stem cells. Thus, our data contributes valuable and rare insight into RPE melanosome biology and suggests that the mature melanosome population may not be as stable as previously thought, requiring megalin-related trafficking activity to maintain melanosome number and morphology.
RPE macromelanosomes have been reported in other genetic disorders including ocular albinism type 1 (OA1)36 and Chediak-Higashi syndrome (CHS).37,38 Defects in the OA1 gene interfere with the stop signal for melanosome growth resulting in abnormally large but otherwise round and elliptical melanosomes.39 In contrast, ultrastructural investigations of RPE melanosomes in patients,40 cats,37 and mice38 with CHS display striking similarities in size and shape irregularities to the abnormal melanosomes in the megalin-deficient RPE cells. Melanosomes are considered lysosome-related organelles and share a common biogenesis pathway with lysosomes.41 Interestingly, it is defects of the lysosomal trafficking regulator (LYST) that result in Chediak-Higashi syndrome and accumulating evidence suggests that LYST is involved in regulation of lysosomal vesicle fission and/or fusion.42,43
Ultrastructural investigations of proximal tubular cells from megalin-deficient mice have similarly revealed significant abnormalities of the endolysosomal system44,45 indicating that the melanosome abnormalities observed in the megalin-deficient RPE cells may be due to disturbances in the melanosome biogenesis pathway. However, detailed cellular studies of megalin-deficient RPE cells are needed to verify this.
Previous mouse models of megalin-deficiency have been based on embryonic deletion of megalin, compromising either survival rates and/or embryonic eye development. Our novel, RPE-specific mouse model has provided the opportunity to study the postnatal role of megalin in ocular morphology. Importantly, it has enabled us to investigate the pigment changes observed in DBS patients and to conclude that lack of megalin in the RPE significantly contributes to the high myopia consistently observed in DBS patients.
In conclusion, postnatal RPE-specific ablation of megalin in mice produces a rapidly developing megaophthalmos phenotype with retinal thinning, RPE macromelanosomes, and compromised vision. Our detailed ultrastructural investigations of RPE melanosomes furthermore provided valuable and rare basic cell biological insight into postnatal RPE melanosome plasticity.
Supplementary Material
Acknowledgments
The authors thank Hanne Sidelmann, Pia Kamuk Nielsen for their technical assistance, and Inger Blenker Kristoffersen as well as Alun R. Barnard, PhD, at Nuffield Laboratory of Ophthalmology, University of Oxford for help improving the manuscript.
Supported by the Velux Foundation, Lundbeck Foundation, Synoptikfonden, Oejenforeningen, The Danish Medical Research Council, the NOVO-Nordisk Foundation, the Wellcome Trust (093445), the NIH (EY015240), Research to Prevent Blindness, and the FM Kirby Foundation.
Disclosure: T. Storm, None; T. Burgoyne, None; J.L. Dunaief, None; E.I. Christensen, None; C. Futter, None; R. Nielsen, None
References
- 1.Pober BR, Longoni M, Noonan KM. A review of Donnai-Barrow and facio-oculo-acoustico-renal (DB/FOAR) syndrome: clinical features and differential diagnosis. Birth Defects Res A Clin Mol Teratol. 2009;85:76–81. doi: 10.1002/bdra.20534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kantarci S, Al-Gazali L, Hill RS, et al. Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes. Nat Genet. 2007;39:957–959. doi: 10.1038/ng2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel N, Hejkal T, Katz A, Margalit E. Ocular manifestations of Donnai-Barrow syndrome. J Child Neurol. 2007;22:462–464. doi: 10.1177/0883073807301933. [DOI] [PubMed] [Google Scholar]
- 4.Kantarci S, Ragge NK, Thomas NS, et al. Donnai-Barrow syndrome (DBS/FOAR) in a child with a homozygous LRP2 mutation due to complete chromosome 2 paternal isodisomy. Am J Med Genet A. 2008;146A:1842–1847. doi: 10.1002/ajmg.a.32381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schrauwen I, Sommen M, Claes C, et al. Broadening the phenotype of LRP2 mutations: a new mutation in LRP2 causes a predominantly ocular phenotype suggestive of Stickler syndrome. Clin Genet. 2013;86:282–286. doi: 10.1111/cge.12265. [DOI] [PubMed] [Google Scholar]
- 6.Khalifa O, Al-Sahlawi Z, Imtiaz F, et al. Variable expression pattern in Donnai-Barrow syndrome: report of two novel LRP2 mutations and review of the literature. Eur J Med Genet. 2015;58:293–299. doi: 10.1016/j.ejmg.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 7.Christensen EI, Birn H, Storm T, Weyer K, Nielsen R. Endocytic receptors in the renal proximal tubule. Physiology (Bethesda) 2012;27:223–236. doi: 10.1152/physiol.00022.2012. [DOI] [PubMed] [Google Scholar]
- 8.Chatelet F, Brianti E, Ronco P, Roland J, Verroust P. Ultrastructural localization by monoclonal antibodies of brush border antigens expressed by glomeruli. II. Extrarenal distribution. Am J Pathol. 1989;122:512–519. [PMC free article] [PubMed] [Google Scholar]
- 9.Chun JT, Wang L, Pasinetti GM, Finch CE, Zlokovic BV. Glycoprotein 330/megalin (LRP-2) has low prevalence as mRNA and protein in brain microvessels and choroid plexus. Exp Neurol. 1999;157;:194–201. doi: 10.1006/exnr.1999.7052. [DOI] [PubMed] [Google Scholar]
- 10.Lundgren S, Carling T, Hjälm G, et al. Tissue distribution of human gp330/megalin, a putative Ca(2+)-sensing protein. J Histochem Cytochem. 1997;45;:383–392. doi: 10.1177/002215549704500306. [DOI] [PubMed] [Google Scholar]
- 11.Storm T, Christensen EI, Christensen JN, et al. Megalin is predominantly observed in vesicular structures in first and third trimester cytotrophoblasts of the human placenta. J Histochem Cytochem. 2016;64:769–784. doi: 10.1369/0022155416672210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng G, Bachinsky DR, Stamenkovic I, et al. Organ distribution in rats of two members of the low-density lipoprotein receptor gene family, gp330 and LRP/alpha 2MR, and the receptor-associated protein (RAP) J Histochem Cytochem. 1994;42:531–542. doi: 10.1177/42.4.7510321. [DOI] [PubMed] [Google Scholar]
- 13.Storm T, Heegaard S, Christensen EI, Nielsen R. Megalin-deficiency causes high myopia, retinal pigment epithelium-macromelanosomes and abnormal development of the ciliary body in mice. Cell Tissue Res. 2014;358:99–107. doi: 10.1007/s00441-014-1919-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veth KN, Willer JR, Collery RF, et al. Mutations in zebrafish lrp2 result in adult-onset ocular pathogenesis that models myopia and other risk factors for glaucoma. PLoS Genet. 2011;7:e1001310. doi: 10.1371/journal.pgen.1001310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cases O, Joseph A, Obry A, et al. Foxg1-Cre mediated Lrp2 inactivation in the developing mouse neural retina, ciliary and retinal pigment epithelia models congenital high myopia. PLoS One. 2015;10:e0129518. doi: 10.1371/journal.pone.0129518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christ A, Christa A, Klippert J, et al. LRP2 acts as SHH clearance receptor to protect the retinal margin from mitogenic stimuli. Developmental Cell. 2015;35:36–48. doi: 10.1016/j.devcel.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Iacovelli J, Zhao C, Wolkow N, et al. Generation of Cre transgenic mice with postnatal RPE-specific ocular expression. Invest Ophthalmol Vis Sci. 2011;52:1378–1383. doi: 10.1167/iovs.10-6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao C, Yasumura D, Li X, et al. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J Clin Invest. 2011;121:369–383. doi: 10.1172/JCI44303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leheste JR, Melsen F, Wellner M, et al. Hypocalcemia and osteopathy in mice with kidney-specific megalin gene defect. FASEB J. 2003;17:247–249. doi: 10.1096/fj.02-0578fje. [DOI] [PubMed] [Google Scholar]
- 20.Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 21.Moestrup SK, Nielsen S, Andreasen P, et al. Epithelial glycoprotein-330 mediates endocytosis of plasminogen activator-plasminogen activator inhibitor type-1 complexes. J Biol Chem. 1993;268:16564–16570. [PubMed] [Google Scholar]
- 22.Sparrow JR, Hicks D, Hamel CP. The retinal pigment epithelium in health and disease. Curr Mol Med. 2010;10:802–823. doi: 10.2174/156652410793937813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Detrick B, Hooks JJ. Immune regulation in the retina. Immunol Res. 2010;47:53–161. doi: 10.1007/s12026-009-8146-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Wildsoet CF. RPE. and choroid mechanisms underlying ocular growth and myopia. Prog Mol Biol Transl Sci. 2015;134:221–240. doi: 10.1016/bs.pmbts.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zidan HE, Rezk NA, Fouda SM, Mattout HK. Association of insulin-like growth factor-1 gene polymorphisms with different types of myopia in Egyptian patients. Genet Test Mol Biomarkers. 2016;20:291–296. doi: 10.1089/gtmb.2015.0280. [DOI] [PubMed] [Google Scholar]
- 26.Zhuang W, Yang P, Li Z, et al. Association of insulin-like growth factor-1 polymorphisms with high myopia in the Chinese population. Mol Vis. 2012;18:634–644. [PMC free article] [PubMed] [Google Scholar]
- 27.Bolós M, Fernandez S, Torres-Aleman I. Oral administration of a GSK3 inhibitor increases brain insulin-like growth factor I levels. J Biol Chem. 2010;285:17693–17700. doi: 10.1074/jbc.M109.096594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carro E, Spuch C, Trejo JL, Antequera D, Torres-Aleman I. Choroid plexus megalin is involved in neuroprotection by serum insulin-like growth factor I. J Neurosci. 2005;25:10884–10893. doi: 10.1523/JNEUROSCI.2909-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roepstorff K, Grandal MV, Henriksen L, et al. Differential effects of egfr ligands on endocytic sorting of the receptor. Traffic. 2009;10:1115–1127. doi: 10.1111/j.1600-0854.2009.00943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choudhry Z, Rikani AA, Choudhry AM, et al. Sonic hedgehog signalling pathway: a complex network. Ann Neurosci. 2014;21:28–31. doi: 10.5214/ans.0972.7531.210109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang XM, Yang XJ. Temporal and spatial effects of sonic hedgehog signaling in chick eye morphogenesis. Dev Biol. 2001;233:271–290. doi: 10.1006/dbio.2000.0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCarthy RA, Barth JL, Chintalapudi MR, Knaak C, Argraves WS. Megalin functions as an endocytic sonic hedgehog receptor. J Biol Chem. 2002;277:25660–25667. doi: 10.1074/jbc.M201933200. [DOI] [PubMed] [Google Scholar]
- 33.Qian YS, Chu RY, Hu M, Hoffman MR. Sonic hedgehog expression and its role in form-deprivation myopia in mice. Curr Eye Res. 2009;34:623–635. doi: 10.1080/02713680903003492. [DOI] [PubMed] [Google Scholar]
- 34.Chen M, Qian Y, Dai J, Chu R. The sonic hedgehog signaling pathway induces myopic development by activating matrix metalloproteinase (MMP)-2 in guinea pigs. PLoS One. 2014;9:e96952. doi: 10.1371/journal.pone.0096952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lopes VS, Wasmeier C, Seabra MC, Futter CE. Melanosome maturation defect in rab38-deficient retinal pigment epithelium results in instability of immature melanosomes during transient melanogenesis. Mol Biol Cell. 2007;18:3914–3927. doi: 10.1091/mbc.E07-03-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Young A, Powelson EB, Whitney IE, et al. Involvement of OA1, an intracellular GPCR, and Gαi3, its binding protein, in melanosomal biogenesis and optic pathway formation. Invest Ophthalmol Vis Sci. 2008;49:3245–3252. doi: 10.1167/iovs.08-1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collier LL, King EJ, Prieur DJ. Aberrant melanosome development in the retinal pigmented epithelium of cats with Chediak-Higashi syndrome. Exp Eye Res. 1998;41:305–311. doi: 10.1016/s0014-4835(85)80021-x. [DOI] [PubMed] [Google Scholar]
- 38.Robison WG, Jr, Kuwabara T, Cogan DG. Lysosomes and melanin granules of the retinal pigment epithelium in a mouse model of the Chediak-Higashi syndrome. Invest Ophthalmol Vis Sci. 1975;14:312–317. [PubMed] [Google Scholar]
- 39.Incerti B, Cortese K, Pizzigoni A, et al. Oa1 knock-out: new insights on the pathogenesis of ocular albinism type 1. Hum Mol Genet. 2000;9:2781–2788. doi: 10.1093/hmg/9.19.2781. [DOI] [PubMed] [Google Scholar]
- 40.Valenzuela R, Morningstar WA. The ocular pigmentary disturbance of human Chédiak-Higashi syndrome: a comparative light- and electron-microscopic study and review of the literature. Am J Clin Pathol. 1981;75:591–596. doi: 10.1093/ajcp/75.4.591. [DOI] [PubMed] [Google Scholar]
- 41.Wasmeier C, Hume AN, Bolasco G, Seabra MC. Melanosomes at a glance. J Cell Sci. 2008;121:3995–3999. doi: 10.1242/jcs.040667. [DOI] [PubMed] [Google Scholar]
- 42.Falkenstein K, De Lozanne A. Dictyostelium LvsB has a regulatory role in endosomal vesicle fusion. J Cell Sci. 2014;127:4356–4367. doi: 10.1242/jcs.138123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ji X, Chang B, Naggert JK, Nishina PM. Lysosomal Trafficking Regulator (LYST) Adv Exp Med Biol. 2016;854:745–750. doi: 10.1007/978-3-319-17121-0_99. [DOI] [PubMed] [Google Scholar]
- 44.Willnow TE, Hilpert J, Armstrong SA, et al. Defective forebrain development in mice lacking gp330/megalin. Proc Natl Acad Sci U S A. 1996;93:8460–8464. doi: 10.1073/pnas.93.16.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Christensen EI, Willnow TE. Essential role of megalin in renal proximal tubule for vitamin homeostasis. J Am Soc Nephrol. 1999;10:2224–2236. doi: 10.1681/ASN.V10102224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.