

ABSTRACT
Cell-free DNA (cfDNA) has been a research hotspot in molecular tumor profiling. In advanced gastric cancer patients, malignant pleural effusion (MPE) and ascites provide a wealth of tumor cells that can be investigated. Here we conducted next-generation sequencing (NGS) on matched cfDNA from plasma, MPE and ascites from a stage-IV gastric cancer patient to identify potential therapeutic targets. In all three samples, we detected an amplification in the cellular-mesenchymal to epithelial transition factor (MET) gene, a truncation mutation in SMAD3 (p.R368X), and four ataxia telangiectasia-mutated gene (ATM) variants, including a missense mutation (p.E2351A), an in-frame deletion (p.NPAVIM2353delinsK), a frame-shift deletion (p.D1758fs) and an ATM- BPI fold containing family B member 1 (BPIFB1) gene fusion. In contrast, we detected amplification of TEK only in malignant ascites. The patient was subjected to Crizotinib to counter MET amplification. Our study demonstrates high accordance in mutational spectra of matched cfDNA from plasma, MPE and ascites, and suggests that it is feasible to utilize these tumor sources in clinical decision-making.
Keywords: CfDNA, mutation accordance, next-generation sequencing, MET amplification, crizotinib, malignant pleural effusion, ascites
Introduction
Gastric cancer (GC) is an important threat to health worldwide, especially in China, being the second most common cancer and the second leading cause of cancer death.1 Despite a decline in incidence and mortality, the outlook of metastatic gastric cancer cases remains poor. The median survival usually does not exceed one year when treated with systematic chemotherapy in metastatic settings.2,3 The past decades have seen development of targeted therapeutics, while a few of them have been approved in GCs. These agents include trastuzumab for first-line treatment in human epidermal growth factor receptor 2 (HER-2) expressing GCs, ramucirumab as second-line treatment, and apatinib for Chinese patients subsets as third-line treatment.4 Unmet clinical need for GC patients refractory to two or more lines chemotherapy has underpinned the investigation of new and effective targeted agents. Aberrant signaling of hepatocyte growth factor (HGF)/mesenchymal-epithelial transition factor (MET) pathway has been proved to enhance tumorigenicity, invasion and metastasis in gastrointestinal tumors. The knowledge of HGF/MET pathway has led to clinical implementation of monoclonal antibodies against HGF or MET and tyrosine kinase inhibitors (TKIs). Crizotinib is approved for patients with anaplastic lymphoma kinase (ALK)-positive and proto-oncogene tyrosine-protein kinase (ROS1)-positive metastatic non-small-cell lung carcinoma (NSCLC) and is being evaluated in a pilot study in patients with MET positive gastric adenocarcinoma as a third-line treatment.5
Deep-sequencing techniques, such as NGS, has been developed to comprehensively and precisely characterize the genomic landscape so as to find therapeutic targets. Pectasides and colleagues have recently demonstrated the extensive genomic heterogeneity within the primary tumor (PT) and between the PT and disseminated disease in gastric and esophageal adenocarcinomas (GEA).6 The results challenged the using of PT biopsies to guide targeted therapy. CfDNA is easy to get serially and is shed from overall tumor lesions including micrometastatics, making it able to uncover targetable genetic events not detected in PT profiling. Thereby, it’s suggested that cfDNA can potentially allow more effective targeted therapy selection. In addition to plasma, MPE and ascites also provide a pool of tumor components for translational research. Here, we performed NGS on matched cfDNA from plasma, MPE and ascites collected from a stage-IV gastric cancer patient. We discovered mutational accordance among the three samples, as well as one clinically actionable aberration in this patient (MET amplification).
Results
Clinicopathologic characteristics of the patient
Our subject was a 62-year-old male, diagnosed with stage-IV gastric cancer (GC) in May 2016. The patient had an Eastern Cooperative Oncology Group (ECOG) performance status of 2 and an abdominal pain score of 6 by numerical rating scale (NRS). Pathological testing of specimens from endoscopy confirmed the presence of moderately and poorly differentiated gastric adenocarcinoma. Computed tomography (CT) scans revealed dissemination of cancer to the peritoneum, pleura, and liver. The patient was then treated with 2 cycles of chemotherapy (intravenous/intraperitoneal docetaxel and oral S-1). However, CT imaging in June 2016 showed no clinical benefit of chemotherapy as growing pleural effusion was observed and other lesions remained stable (Figure 1). The patient experienced gastrointestinal hemorrhage after 2 cycles of chemotherapy. Considering intolerant to intravenous treatment, the patient continued once intraperitoneal docetaxel and once intrapleural cisplatin respectively.
Figure 1.
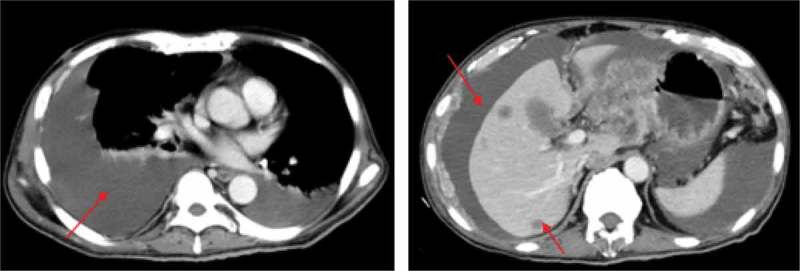
CT imaging after 2 cycles of intravenous chemotherapy. Red arrows indicate the metastatic pleural effusions (left panel), ascites and liver lesions (right panel).
Comparison of mutation patterns in three samples
To provide targeted therapeutic options for the patient, we implemented NGS of cfDNA from plasma, MPE and ascites (referred to as samples 1, 2, and 3 hereinafter), using a gene panel that covers entire exons in 416 cancer-relevant genes (Table S1). Genetic alterations in the coding sequence of ATM, MET, SMAD3, and TEK genes were detected in the present study. These alterations were present in all three samples, except for an ascites-specific TEK gene amplification. This indicated there was a rate of high accordance among samples in mutational spectra (Table 1 and Figure 2).
Table 1.
Genetic aberrations identified in three samples.
| Gene | Gene.ID | AA Change | Chr. start | Chr. end | Plasma | MPE | Ascites | COSMIC |
|---|---|---|---|---|---|---|---|---|
| ATM | ATM:NM_000051.3:exon48 | p.E2351A (c.A7052C) | chr11:108198448 | chr11:108198448 | 8% | 5% | 24% | - |
| ATM | ATM:NM_000051.3:exon48 | p.NPAVIM2353delinsK | chr11:108198452 | chr11:108198466 | 8% | 5% | 24% | - |
| ATM | ATM:NM_000051.3:exon35 | p.D1758fs | chr11:108172469 | chr11:108172479 | 6% | 5% | 24% | - |
| ATM-BPIFB1 | gene fusion | 14% | 6% | 20% | - | |||
| MET | gene amplification | 2 copies | 1.8copies | 2.4copies | 3.27% | |||
| SMAD3 | SMAD3:NM_005902.3:exon8 | p.R368X (c.C1102T) | chr15:67479795 | chr15:67479795 | 22% | 9% | 67% | - |
| TEK | gene amplification | - | - | 2.5copies | 0.82% |
Figure 2.
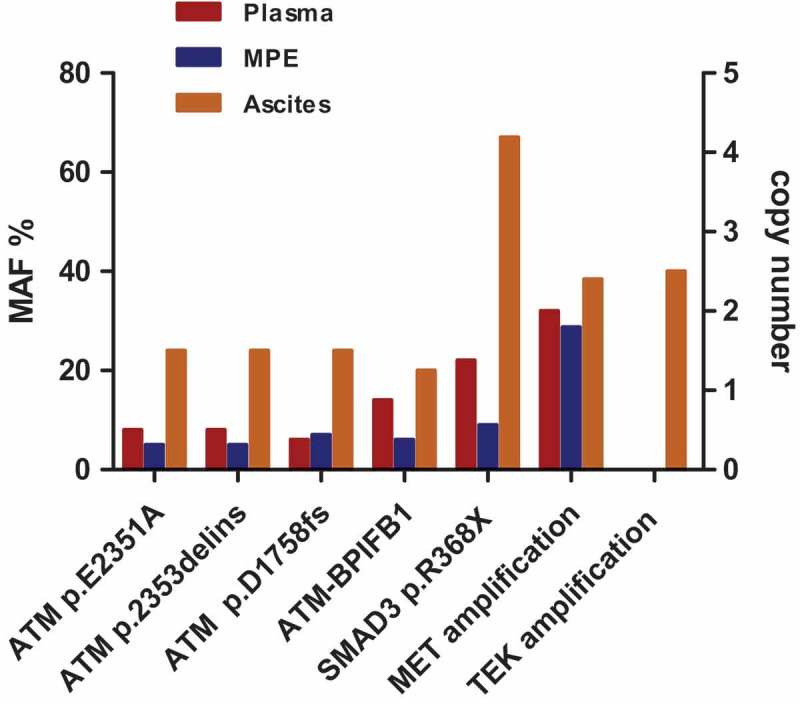
Mutation profiles are highly accordant between cfDNA from plasma, malignant pleural effusion, and ascites. Except for the TEK gene amplification, genetic alterations identified in the present study were all shared among three tumor samples.
Four genetic variants of the ATM gene were found in our study, none of which had been previously reported in the COSMIC database (Figure S1a, Figure 3a-b). A missense mutation (p.E2351A) and an in-frame deletion (p.NPAVIM2353delinsK), both located in exon 48, were detected concurrently. Consequently, these two variants exhibited the same mutant allele frequencies (MAF) in tumor samples: 8%, 5%, and 24% in samples 1, 2, and 3, respectively. The MAF of the ATM frame-shift deletion (p.D1758fs) was 6%, 7%, and 24% in samples 1, 2, and 3, respectively. In addition to these SNVs and INDELs in ATM, we also detected an ATM-BPIFB1 gene fusion composed of a breakage in intron 23 of ATM gene and intron 13 of BPIFB1 gene. This gene fusion was found in all three samples, with a 14%, 6%, and 20% MAF in samples 1, 2, and 3, respectively.
Figure 3.

Mutation distribution within the ATM gene and SMAD3 gene in gastric adenocarcinoma, as reported in the COSMIC database. (a,b) No substitution variants at amino acid site 2351 and no frame-shift or in-frame deletions at amino acid site 1758 or 2353 of ATM gene have been reported. Panel (b) shows the region from position 2161 to 2422, shown in (a), in finer detail. (c) No nonsense substitutions at amino acid site 368 of SMAD3 have been reported. The grey bands show mutations already reported in COSMIC. The black arrows indicate amino acid sites of mutations detected in our study. (d) Representative FISH images showing the presence of MET gene amplification. The right panel shows the cell in dotted box from the left. (red signal MET, green signal CEP7).
The most noticeable genetic abnormality identified in all three samples was a MET amplification with 2-, 1.8-, and 2.4-fold relative copy number increase in samples 1, 2, and 3, respectively. In the COSMIC database, MET amplification could be detected in 3.27% (16/489) gastric adenocarcinoma samples. Fluorescence in situ hybridization (FISH) analysis further validated the presence of MET amplification (Figure 3d). All three samples also contained a truncation mutation located in exon 8 of the SMAD3 gene (p.R368X), with a MAF of 22%, 9% and 67% in samples 1, 2, and 3, respectively. According to the COSMIC database, SMAD3 was mutated in 0.92% (5/542) gastric adenocarcinomas, while the truncation mutation identified in our study has not yet been reported (Figure S1b, Figure 3c). Unlike the genetic aberrations we describe above, TEK amplification was only detected in the malignant ascites sample, with 2.5-fold copy number increase. TEK amplification is reported at a low frequency of 0.82% (4/489) in gastric adenocarcinoma in the COSMIC database.
Clinical implications of genetic abnormalities
Of all the mutations we found, the MET amplification we detected was the optimal clinically actionable variant. ATM mutations can be targeted by inhibitors of Poly (ADP-ribose) polymerase (PARP), ATM and rad3-related (ATR), checkpoint kinase (Chk1/2).7 Clinical trials targeting ATM mutations have been conducted in a variety of tumor types, including two studies with Olaparib and one with AZD6738 in gastric cancer.8 However, ATM mutations identified here did not provide viable therapeutic opportunities for the patient. Therefore, the patient commenced monotherapy with Crizotinib (250 mg twice daily) to target MET amplification beginning in July 2017. Unfortunately, the patient died in August 2017.
Discussion
Plasma, MPE and ascites are superior specimens for molecular analysis when tumor tissues are not available. In addition to mirror molecular profiles of primary tumors, these samples could harbor genetic events necessary for their respective metastatic process.9 The heterogeneity in different distant metastases in GEA have not been clearly stated. Nevertheless, by simultaneously sequencing cfDNA from plasma, MPE, and ascites developed in one advanced stage GC patient, our study addressed the mutational accordance in these sample types, except TEK amplification in ascites.
TEK is reported to favor tumor migration by enhancing tumor cells adhesion to vascular endothelial cells, which indicates TEK amplification probably participate in peritoneum dissemination in this patient, which might explain this moderate discordance between three samples.10 Moreover, given that the ascites had the highest MAF of all other alterations detected, it could harbor more tumor DNA components in cfDNA than other two samples. Due to the relatively lower concentrations of tumor-derived DNA, TEK amplification in plasma and MPE probably did not reach our limit to be detected.
We found MET amplification as an actionable alteration in our study. Although preclinical and phase I-II studies of MET pathway inhibitors showed promising results in MET positive GCs or gastro-esophageal cancers (GECs), the outcomes of three available phase III trials were disappointing (RILOMET-1, RILOMET-2 and METGastric). Rilotumumab was ineffective in MET positive patients regarding overall survival (OS) or progression-free survival (PFS), neither in any patient subgroups stratified by demographics or biomarkers like MET staining intensity/extensity, MET amplification or serum HGF levels.11 Likewise, onartuzumab did not significantly improve OS, PFS or overall response rate (ORR).12 There are some explanations to the failures above. First, ligand blocking approaches are predicted to be ineffective in MET amplification or MET mutation settings, which signal primarily via ligand independent mechanisms and are most likely to benefit from anti-MET therapies. Second, rilotumumab still did not improve survivals in patients with high HGF expression. It is hypothesized that multiple oncogenic pathways are involved in tumor cells and isolated MET inhibition is not sufficient to control tumor growth. HGF overexpressed by tumor cells and stroma may form autocrine and paracrine loops and consequently onartuzumab will not fundamentally affect tumor behavior. Third, crosstalk between MET and other signaling pathways including epidermal growth factor receptor (EGFR) and HER family may lead to poorly response and resistance to MET inhibition.13 Furthermore, anti-MET class effect toxicities, namely oedema and hypoalbuminaemia, should be distinguished from clinical disease progression. Finally, the optimal MET assays, scoring and positive criteria are urgent to be defined and adequately selected. These evidence suggest that MET expression determined by immunohistochemistry is unable to select patient subgroups most likely benefited from anti-HGF/MET treatments. More accurate biomarkers or biomarker combinations are essential for further development of MET pathway inhibitors in GCs.5
MET amplification is predicted with the most evidence to benefit from TKIs therapy. Our team has reported a good response to Crizotinib in a MET-amplified GC patient.14 Nevertheless, the patient in this study was subjected to Crizotinib at a terminal stage with multiple metastatic deposits and lethal complications. The treatment lasted only for one month and we didn’t get any follow-up clinical inspections to evaluate the responses. Earlier targeted interventions should be performed in advanced stage patients. Whether the other alterations identified have a role in treatment effect is also needed to be considered. There is no report about the correlation between ATM mutations nor TEK amplification and anti-MET response. SMAD3 is an important transcription factor in the transforming growth factor-β (TGF-β) pathway.15 The SMAD3 mutation here introduces a stop codon. The truncated portions include part of MH2 region and C-terminal SSXS motif responsive to activated TGF-β receptor type I (TGFBR1) (Figure S1b).16 Therefore, this mutation could be functionally deleterious as interaction of SMAD3 with TGF-β receptors and other transcription factors get impaired. TGF-β has been recently shown to exert an inhibitory effect on MET phosphorylation in glioblastoma.17 The SMAD protein also negatively regulates MET at basal levels, which indicates SMAD3 mutation here has probably involved in ineffective responses to Crizotinib in this patient.
There are also limits of our study. We reported mutational accordance in cfDNA from plasma, MPE, and ascites in only one GC patient. Further validation in more GC patients and large cohorts of patients based on characteristics like prognosis are required before to try general conclusions regarding the sharing of molecular features between these samples and their use in clinical settings.18 In summary, NGS of cfDNA from plasma, MPE, and ascites demonstrated high accordance of mutational spectra between three samples and directed targeted therapy in one gastric cancer patient.
Materials and methods
Ethical compliance
Patient information and clinical samples were obtained from The Comprehensive Cancer Centre of Drum Tower Hospital. The patient has given written consent for specimen collection and the following genetic testing. Sample collection and preparation protocols were approved by the Drum Tower Hospital Ethics Committee.
Sample collection
5–10 mL peripheral blood was collected in an EDTA-coated tube (BD Biosciences, NJ, USA). Plasma was extracted within 2 hours of blood collection. Matched malignant pleural effusion and ascites were collected through catheter drainage from the patient. Samples were sent to the core facility of Nanjing Geneseeq Technology Inc. (Nanjing, China) for DNA extraction and genetic testing.
Targeted NGS and data processing
DNA extraction, sequencing library preparation, and targeted capture enrichment were carried out following the methods as previously described with modifications.19 In brief, genomic DNA from whole blood were extracted using the DNeasy Blood & Tissue kit (Qiagen, Shanghai, China) according to the manufacturer’s protocols. Plasma sample was centrifuged at high speed to remove any cell debris. To prepare cfDNA from MPE and ascites, we first removed cells from the MPE and ascites by low speed centrifugation, followed by high speed centrifugation to remove any debris. The resultant supernatant was then subjected to cfDNA extraction using Qiagen QIAamp Circulating Nucleic Acid Kit (Qiagen, Shanghai, China). Sequencing libraries were prepared using the KAPA Hyper Prep kit (KAPA Biosystems, MA, USA) according to manufacturer’s suggestions for different sample types. In brief, 6.08 – 200 ng (median: 70.5 ng) of cfDNA or 1 μg of fragmented genomic DNA underwent end-repairing, A-tailing and ligation with indexed adapters sequentially, followed by size selection using Agencourt AMPure XP beads (Beckman Coulter, FL, USA). Hybridization-based target enrichment was carried out with GeneseeqOneTM pan-cancer gene panel (416 cancer-relevant genes), and xGen Lockdown Hybridization and Wash Reagents Kit (Integrated DNA Technologies). Captured libraries by Dynabeads M-270 (Life Technologies, MA, USA) were amplified in KAPA HiFi HotStart ReadyMix (KAPA Biosystems, MA, USA) and quantified by qPCR using the KAPA Library Quantification kit (KAPA Biosystems, MA, USA) for sequencing.
The libraries were paired-end sequenced on Illumina HiSeq4000 NGS platforms (Illumina, CA, USA) according to the manufacturer’s instructions. The mean coverage depth was > 100× for the whole blood control samples. For cfDNA samples, the original targeted sequencing depth was > 3000 × . Trimmomatic was used for FASTQ file quality control (below 15 or N bases were removed). Reads were then mapped to the reference Human Genome (hg19) using Burrows-Wheeler Aligner (BWA-mem, v0.7.12) (https://github.com/lh3/bwa/tree/master/bwakit). Local realignment around the indels and base quality score recalibration was applied with the Genome Analysis Toolkit (GATK 3.4.0) (https://software.broadinstitute.org/gatk/), which was also applied to detect germline mutations. VarScan2 was employed for somatic mutation detection. Somatic variant calls with at least 0.2% mutant allele frequency (MAF) and with at least 3 supporting-reads from both directions were retained. Common SNPs were filtered out using dbSNP (v137) and the 1000 Genomes database, followed by annotation using ANNOVAR. Genomic fusions were identified by FACTERA with default parameters. Copy number variations (CNVs) were detected using ADTEx (http://adtex.sourceforge.net) with default parameters. Somatic CNVs were identified using paired normal/tumor samples for each exon with the cut-off of 0.65 for copy number loss and 1.50 for copy number gain.
Fluorescence in situ hybridization (FISH) analysis
FISH analysis was performed on tumor cells prepared from MPE. A MET/CEP7 FISH probe (Vysis MET SpectrumRed FISH probe kit and CEP7 Spectrum Green probe; Abbott Molecular, Abbot Park, IL, USA) was used to identify MET amplifications according to the manufacturer’s instructions. FISH analysis was performed using an Olympus BX61 epifluorescence microscope (Olympus, NY, USA). At least 60 tumor nuclei were counted for each case. Images were captured using a charge-coupled device (CCD) camera and merged using dedicated software (CytoVision, Santa Clara, CA, USA).
Comparison with public database
The latest version of the COSMIC databasev82 was searched as a comprehensive library of somatic mutations in human cancer. We compared our genetic alterations to the gastric adenocarcinoma data reported in the public COSMIC database. Relevant data and figures have been imported from the COSMIC database.
Funding Statement
This study was funded by the National Key Research and Development Program of China (grant number 2017YFC1308900, 2018ZX09301048-003) and the National Natural Science Foundation of China (grant number 81572329); the National Key Research and Development Program of China [2017YFC1308900 2018ZX09301048-003].
Acknowledgments
We thank Xiaohong Pu for her efforts and help in performing FISH analysis.
Disclosure of Interest
The authors declare that they have no conflict of interest.
Supplementary Material
Supplemental data for this article can be accessed here.
References
- 1.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J.. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 2.Narita Y, Muro K.. Challenges in molecular targeted therapy for gastric cancer: considerations for efficacy and safety. Expert Opin Drug Saf. 2017;16:319–327. doi: 10.1080/14740338.2017.1273348. [DOI] [PubMed] [Google Scholar]
- 3.Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. The Lancet. 2016;388:2654–2664. doi: 10.1016/S0140-6736(16)30354-3. [DOI] [PubMed] [Google Scholar]
- 4.Roviello G, Ravelli A, Fiaschi AI, Cappelletti MR, Gobbi A, Senti C, Zanotti L, Polom K, Reynolds AR, Fox SB, et al. Apatinib for the treatment of gastric cancer. Expert Rev Gastroenterol Hepatol. 2016;10:887–892. doi: 10.1080/17474124.2016.1209407. [DOI] [PubMed] [Google Scholar]
- 5.Bradley CA, Salto-Tellez M, Laurent-Puig P, Bardelli A, Rolfo C, Tabernero J, Khawaja HA, Lawler M, Johnston PG, Van Schaeybroeck S, et al. Targeting c-MET in gastrointestinal tumours: rationale, opportunities and challenges. Nat Rev Clin Oncol. 2017;14:562–576. doi: 10.1038/nrclinonc.2017.40. [DOI] [PubMed] [Google Scholar]
- 6.Pectasides E, Stachler MD, Derks S, Liu Y, Maron S, Islam M, Alpert L, Kwak H, Kindler H, Polite B, et al. Genomic heterogeneity as a barrier to precision medicine in gastroesophageal adenocarcinoma. Cancer Discov. 2018;8:37–48. doi: 10.1158/2159-8290.CD-17-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: therapeutic implications. Mol Cancer Ther. 2016;15:1781–1791. doi: 10.1158/1535-7163.MCT-15-0945. [DOI] [PubMed] [Google Scholar]
- 8.Bang YJ, Im SA, Lee KW, Cho JY, Song EK, Lee KH, Kim YH, Park JO, Chun HG, Zang DY, et al. Randomized, double-blind phase II trial with prospective classification by ATM protein level to evaluate the efficacy and tolerability of olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer. J Clin Oncol. 2015;33:3858–3865. doi: 10.1200/JCO.2014.60.0320. [DOI] [PubMed] [Google Scholar]
- 9.Lim B, Kim C, Kim JH, Kwon WS, Lee WS, Kim JM, Park JY, Kim HS, Park KH, Kim TS. Genetic alterations and their clinical implications in gastric cancer peritoneal carcinomatosis revealed by whole-exome sequencing of malignant ascites. Oncotarget. 2016;7:8055. doi: 10.18632/oncotarget.6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu D, Martin V, Fueyo J, Lee OH, Xu J, Cortes-Santiago N, Alonso MM, Aldape K, Colman H, Gomez-Manzano C. Tie2/TEK modulates the interaction of glioma and brain tumor stem cells with endothelial cells and promotes an invasive phenotype. Oncotarget. 2010;1:700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Catenacci DVT, Tebbutt NC, Davidenko I, Murad AM, Al-Batran S-E, Ilson DH, Tjulandin S, Gotovkin E, Karaszewska B, Bondarenko I, et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18:1467–1482. doi: 10.1016/S1470-2045(17)30566-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shah MA, Bang Y-J, Lordick F, Alsina M, Chen M, Hack SP, Bruey JM, Smith D, McCaffery I, Shames DS, et al. Effect of fluorouracil, leucovorin, and oxaliplatin with or without onartuzumab in HER2-negative, MET-positive gastroesophageal adenocarcinoma. JAMA Oncol. 2017;3:620. doi: 10.1001/jamaoncol.2016.5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corso S, Ghiso E, Cepero V, Sierra JR, Migliore C, Bertotti A, Trusolino L, Comoglio PM, Giordano S. Activation of HER family members in gastric carcinoma cells mediates resistance to MET inhibition. Mol Cancer. 2010;9:121. doi: 10.1186/1476-4598-9-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du J, Wu X, Tong X, Wang X, Wei J, Yang Y, Chang Z, Mao Y, Shao YW, Liu B. Circulating tumor DNA profiling by next generation sequencing reveals heterogeneity of crizotinib resistance mechanisms in a gastric cancer patient with MET amplification. Oncotarget. 2017;8:26281–26287. doi: 10.18632/oncotarget.15457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Millet C, Zhang YE. Roles of smad3 in TGF-β signaling during carcinogenesis. Crit Rev Eukaryot Gene Expr. 2007;17:281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Derynck R, Zhang Y. Smad-dependent and Smad-independent pathways in TGF-|[beta]| family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 17.Papa E, Weller M, Weiss T, Ventura E, Burghardt I, Szabó E. Negative control of the HGF/c-MET pathway by TGF-β: a new look at the regulation of stemness in glioblastoma. Cell Death Dis. 2017;8:3210. doi: 10.1038/s41419-017-0051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lan YT, Chen MH, Fang WL, Hsieh CC, Lin CH, Jhang FY, Yang SH, Lin JK, Chen WS, Jiang JK. Clinical relevance of cell-free DNA in gastrointestinal tract malignancy. Oncotarget. 2016;8:3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shu Y, Wu X, Tong X, Wang X, Chang Z, Mao Y, Chen X, Sun J, Wang Z, Hong Z. Circulating tumor DNA mutation profiling by targeted next generation sequencing provides guidance for personalized treatments in multiple cancer types. Sci Rep. 2017;7:583. doi: 10.1038/s41598-017-00520-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.