

ABSTRACT
Background: miR-155 was up-regulated in natural killer/T-cell lymphoma (NKTCL), an aggressive malignancy, and correlated with disease progression. However, minimal is known on biological activities and underlying mechanisms of miR-155 in NKTCL. In this study, we examined BRG1, a potential target of miR-155, and focused on the miR-155/BRG1 signaling in regulating lymphangiogenesis of NKTCL.
Methods: The expression of miR-155, BRG1, VEGFC, and VEGFD was compared between two NKTCL cell lines and normal NK cells. The critical role of miR-155 and STAT3 was assessed using miR-155 inhibitor and STAT3 inhibitor S31-201, respectively. Two biological phenotypes, apoptosis and pro-lymphangiogenesis, were examined in vitro by flow cytometry and lymphatic tube formation, respectively, and in vivo using an NKTCL xenograft model.
Results: The miR-155 level negatively correlated with BRG1, but positively with VEGFC in normal NK as well as two NKTCL cell lines. Targeting miR-155 in NKTCL cells significantly boosted BRG1 expression and decreased the activated STAT3 or VEGFC level, leading to enhanced apoptosis and reduced lymphangiogenesis. STAT3 acted downstream of BRG1 and essentially regulated miR-155-mediated up-regulation of VEGFC and pro-lymphangiogenesis. In vivo, targeting miR-155 inhibited primary xenograft growth as well as tumor-associated lymphangiogenesis.
Conclusions: By inhibiting BRG1 expression, miR-155 activated STAT3/VEGFC signaling and promoted lymphangiogenesis. In addition, miR-155 also controlled the viability of NKTCL cells. Therefore, targeting miR-155 provides a novel therapy for NKTCL.
Keywords: natural killer/T-cell lymphoma, miR-155, BRG1, lymphangiogenesis, VEGFC
Introduction
Natural killer/T-cell lymphoma (NKTCL) is an aggressive malignancy with putative origins from both NK cells and T cells, as represented by negative expression for CD3, positive for cytoplasmic CD3ϵ, CD56 and germline configuration of T cell receptor (TCR) gene, and rearrangement of TCR gene.1 NKTCL presents a geographic preference in Asia and South American countries, with the incidence of approximately 5% and 3%, respectively, significantly higher than that in North America or Europe (approximately 0.3%).2 Almost all NKTCLs develop from extra-nodal anatomic sites, including approximately 80% from the upper aerodigestive tract (also known nasal NKTCLs) and 20% from non-nasal sites such as skin, salivary gland, testis, and gastrointestinal tract.1 Interestingly, a great number of non-nasal NKTCLs are identified as disseminated disease from occult nasal NKTCLs,3,4 presenting more aggressive behaviors in clinic and suggestive of early dissemination during the development of NKTCL. The pathogenesis of NKTCL is associated with the invariable infection of Epstein-Barr virus (EBV), although the underlying mechanisms are not well understood. Genetic aberrations, from the loss of chromosome 6q that harbors many tumor suppressor genes including PRMD1, FOXO3, and PTPRK,5,6 inactivating mutations in tumor suppressor genes, such as TP53,7 to activating mutations in proto-oncogenes, such as JAK3,8 STAT3 and STAT5,9,10 as well as epigenetic modulations of gene expressions,11,12 collectively contribute to the development of NKTCLs. Furthermore, the general expression profile of microRNAs (miRNAs) is significantly altered in NKTCL,13,14 suggestive of their significance in the pathogenesis and treatment of NKTCL.
miRNAs are small (19 to 22 nucleotides) non-coding RNA molecules that control gene expressions on the post-transcriptional level and critically regulate many biological processes and disease development including cancers.15 Studies over the past 10 years identified a repertoire of miRNAs that were aberrantly expressed and functionally important for the differentiation of T cells as well as the development of T-cell malignancies.14 Among these miRNAs, miRNA-155 was up-regulated in NKTCL cell lines when compared to normal NK cells, and its level in serum was higher in NKTCL patients than in healthy individuals and associated with stable or progressive disease instead of disease remission.16,17 Therefore, it is suggested that miRNA-155 was a molecular biomarker for NKTCLs.17 However, minimal is known on the mechanisms by which miRNA-155 promotes the development of NKTCLs.
Lymphangiogenesis provides essential conduits for the dissemination of cancer cells to the draining lymph nodes, which is a most critical biomarker for assessing tumor staging, selecting treatment options, and predicting patient prognosis.18 Current understanding suggests that lymphangiogenesis is controlled by a number of lymphangiogenic growth factors, including the best characterized vascular endothelial growth factor C (VEGFC) and VEGFD. By acting on VEGF receptor 3 (VEGFR3), the VEGFR specific for lymphatic endothelium, VEGFC/VEGFD is not only sufficient to stimulate,19,20 but also functionally essential for tumor lymphangiogenesis and lymph node metastasis.21 In addition, upstream molecules and signaling cascades regulating these critical lymphangiogenic growth factors are also under intensive investigation, since the revelation of controlling mechanisms may yield novel therapeutic targets that will benefit cancer treatment.
Brahma-related gene 1 (BRG1) is a catalytic subunit in the SWI/SNF chromatin-remodeling complex, through which BRG1 regulates a variety of biological processes from gene expression to cell differentiation.22 By targeting multiple lymphatic genes, BRG1 sufficiently and essentially controls the formation of lymphatic system in developing mouse embryos.23 Considering the significance of lymphangiogenesis in the early dissemination of cancer cells, BRG1 may thus regulate cancer metastasis. Consistently, the loss or mutation of BRG1 was a frequent event in some cancers, such as lung cancers,24,25 pancreatic cancer,26 and skin cancer,27 supporting its role as a tumor suppressor. In colorectal cancers, loss of BRG1 stimulated cancer migration and invasion and was associated with increased lymphatic metastasis.28 Further study suggested that BRG1 inhibited the activation of STAT3/VEGFC signaling and thus controlled lymphangiogenesis.29 In human leukemia and lymphoma cell lines, miR-155 suppressed the expression of BRG1.30 However, the status of BRG1 in NKTCL is not well characterized, nor is the potential roles and mechanisms of miR-155/BRG1 signaling in the progression of NKTCL.
In this study, we hypothesized that miR-155, by inhibiting BRG1, activated STAT3/VEGFC signaling, and thus promoted lymphangiogenesis and lymph node metastasis. To test our hypothesis, we explored the potential regulations between miR-155, BRG1, STAT3, VEGFC and lymphangiogenesis in two NKTCL cell lines SNK-6 and YTS in vitro, and assessed the significance of such regulations in vivo. This is the first study revealing the significance of miR-155/BRG1/STAT3/VEGFC axis in controlling lymphangiogenesis and cancer metastasis, and targeting this axis as a potential therapeutic strategy for NKTCL.
Materials and methods
Cell culture and treatments
This study was approved by Ethics Committee of the First Affiliated Hospital of Zhengzhou University (Zhengzhou, China). The human normal NK cells were isolated from the peripheral blood mononuclear cells of healthy volunteers using the EasySep Human NK Cell Isolation Kit (STEMCELL, Vancouver, Canada) according to the manufacturer’s instructions. The human NKTCL cell line SNK-6 was a gift from Professor Norio Shimizu (Chiba University, Chiba, Japan) and cultured in RPMI-1640 (Gibco, Carlsbad, CA, USA) medium containing 10% human AB serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin, and 1000 U/ml interleukin (IL)-2 (Sigma-Aldrich, St. Louis, MA, USA). Another human NKTCL cell line YTS was purchased from Sigma-Aldrich and cultured in IDMEM medium containing 5% fetal bovine serum and 2 mM L-Glutamine (Sigma-Aldrich). The primary human lung lymphatic endothelial cells (HLECs) were purchased from Lonza (Basel, Switzerland) and cultured in EBM2 basal medium supplemented with EGM-2 bullet kit (Lonza). All cells were maintained in an incubator at 37°C in humidified atmosphere of 5% CO2.
For the treatment with miR-155 inhibitor, cells growing in log-phase were transfected through electroporation with miRNA-155 inhibitor or negative control (NC; GenePharma, Shanghai, China).
To knock down the endogenous VEGFR3 level, siRNA specifically targeting human VEGFR3 (siVEGFR3) or control siRNA targeting no known human or mouse gene (siNC) (GenePharma) was transfected into cells using Lipofectamine 2000 according to the manufacturer’s instructions.
Extraction of total RNA and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from cells using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. cDNA was then synthesized using oligo dT as the primer with AMV reverse transcriptase (Invitrogen). RT-PCR was performed in the ABI PRISM 7300 Fast Real-Time PCR System (Ambion, Austin, TX, USA) using the following primers:
miR-155 forward primer 5′-CTCAGACTCGGTTAATGCTAATCGTGATAGG-3′, reverse primer 5′-GCTGTGGCAGTGGAAGCGTGATTTATT-3′;
BRG1 forward primer 5ʹ- CCTCTCTCAACGCTGTCCAACTG-3ʹ, reverse primer 5ʹ- ATCTTGGCGAGGATGTGCTTGTCTT-3ʹ;
VEGFC forward primer: 5ʹ-CACGAGCTACCTCAGCAAGA-3ʹ, reverse primer 5ʹ-GCTGCCTGACACTGTGGTA-3ʹ;
VEGFD forward primer 5ʹ-CCTGAAGAAGATCGCTGTTC-3ʹ, reverse primer 5ʹ-GAGAGCTGGTTCCTGGAGAT-3ʹ;
GAPDH (internal control) forward primer 5ʹ-TGGAGAATGAGAGGTGGGATG-3ʹ, reverse primer 5ʹ-GAGCTTCACGTTCTTGTATCTGT-3ʹ.
Each reaction was set up in triplicate and the relative expression levels of target genes were calculated as a ratio to that of the internal control using 2−ΔΔCt method.31
Western blotting
The cells or tumor tissues were collected and lysed using cell lysis buffer (Beyotime, China). Equal amount of total proteins from each sample were separated on SDS-PAGE gel and blotted onto a polyvinylidene difluoride (PVDF) membrane. The target protein was probed with one of the following primary antibodies (all from Cell Signaling, Danvers, MA, USA) at 4°C overnight: anti-BRG1, anti-VEGFC, anti-VEGFD, anti-p-STAT3, anti-STAT3, anti-LYVE1, or anti-GAPDH (internal control). After incubation with horseradish peroxidase-conjugated secondary antibodies at room temperature for 2 h, the signal was developed using the ECL system (Thermo Fisher Scientific, Waltman, MA, USA) according to the manufacturer’s instructions. The signal density was analyzed using NIH Image J software and the relative protein level was calculated as the density ratio of the target protein to that of GAPDH (internal control).
Luciferase reporter assay
The 3ʹ-UTR sequences of human BRG1 gene containing the wild-type (WT) or a mutated sequence (MUT) for the miR-155-binding site was cloned into psiCHECK-2 luciferase reporter plasmid and transfected into SNK-6 or YTS cells using Lipofectamine 2000 according to the manufacturer’s instructions. Upon transfecting the cells with NC or miR-155 mimics for 36 h, luciferase activity was detected using the Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer’s instructions.
Enzyme-linked immunosorbent assay (ELISA)
Upon treating SNK-6 or YTS cells with NC, S31-201, or miR-155 inhibitor for 48 h, the cells were washed three times with DMEM and cultured in serum-free DMEM for a further 24 h. The conditioned medium (CM) was collected and centrifuged at 2000 × g for 10 min to remove any cell debris. The secretion of VEGFC in CM was measured using human VEGFC ELISA kit (LSBio, Seattle, WA, USA) according to the manufacturer’s instructions.
Apoptosis assay by flow cytometry
To detect cellular apoptosis, SNK-6 or YTS cells were seeded into 6-well plate (3.5 × 105 cells/well) and allowed to grow for 24 h. Upon transfected with miR-155 inhibitor or NC for 48 h, cells were stained with Annexin V-FITC solution (2 µL in 100 µL binding buffer; BD Biosciences, San Jose, CA, USA) on ice for 10 min, followed by propidium iodide (PI; 2 µL in 100 µL binding buffer; BD Biosciences) at room temperature for 3 min. The cells were then analyzed by flow cytometer (BD Biosciencs).
Assay for in vitro lymphangiogenesis
To assess in vitro lymangiogenesis, we first collected conditioned medium (CM) from NKTCLs. Briefly, 2 × 105 cells were seeded into 10-cm tissue culture plate. After overnight, cells were transfected with miR-155 inhibitor or NC for 48 h. After three washes with DMEM, cells were cultured in serum-free DMEM for a further 24 h. The CM was collected and centrifuged at 2000 × g for 10 min to remove any cell debris. Then we coated 24-well plate with Matrigel (Corning, USA). Upon gel solidification, HLECs (1 × 105 cells/well) were seeded on top of the Matrigel in triplicate, transfected with siVEGFR3 or control siRNA (siNC) (Genepharma) for 48 h using Lipofectatimne 2000, treated with the mixture of conditioned medium: EBM2 medium (volume ratio 2:1), and incubated at 37°C, 5% CO2 for 6 h. Each well was imaged under Olympus DP71 microscope (Olympus, Tokyo, Japan) at× 100 magnification and the branching points was quantified and presented as a score using Image J software.
Mouse xenografts
The animal procedures were approved by the Institutional Animal Care and Use Committee of the First Affiliated Hospital of Zhengzhou University. Male immunodeficient nude mice (7–8 weeks old) were purchased from SJA Laboratory Animal Co. (Hunan, China) and housed in specific pathogen-free facility at room temperature of (22 ± 1)°C on a 12/12-h light/dark cycle, with access to food and water ad libitum. On day 0, all mice were randomly divided into two groups (N = 7/group) and subcutaneously injected with miR-155-inhibitor- or NC-transfected SNK-6 cells (2 × 106 cells/mouse) into the right inguinal area. The tumor length (L) and width (W) was measured every five days and the tumor volume (V) was calculated as V = 0.5 × L× W.2. On day 25, all mice were sacrificed. The primary tumor was isolated from each mouse, with the tumor weight and the expression of BRG1, VEGFC and LYVE-1 in tumor tissues measured.
Immunohistochemistry
The mouse xenograft tumor tissues were fixed in 10% neutral formalin and embedded in paraffin. Serial sections of 4 µm in thickness were prepared. For immunohistochemical detection of BRG1 and LYVE-1, respectively, tissue sections were de-paraffinized in xylene and rehydrated in a series of diluted alcohol. Antigen retrieval was performed in boiling 10 mM citrate buffer (pH 6.0) for 10 min. After blocking endogenous peroxidase activity with 0.3% H2O2 in PBS for 10 min and non-specific binding with 5% normal goat serum for 1 h at room temperature, the tissue sections were incubated with either anti-BRG1 (Cell Signaling) or anti-LYVE-1 (R&D, Minneapolis, MN, USA) antibody at 4 ºC overnight. Upon three washes in 0.15M NaCl containing 0.1% v/v Triton-X-100, pH 7.6 (TBST) buffer, the sections were incubated with biotinylated secondary antibody (Vector Labs, Burlingame, CA, USA) at room temperature for 30 min, washed three times in TBST, and then incubated in Vectastain ABC-HRP solution (Vector Labs) at room temperature for 30 min, according to the manufacturer’s instructions. The signal of a target protein was developed using Diaminobenzidine (DAB) substrate (Vector Labs) and the slides were counterstained with hematoxylin. All stained sections were imaged under a light microscope (Zeiss, Jena, Germany).
Statistical analysis
All data were analyzed by SPSS 13.0 software and presented as mean ± SD. Statistical evaluation was performed using Student’s t test (two-tailed) between two groups or one-way analysis of variance (ANOVA) followed by Tukey post hoc test for multiple comparisons. P ≤ 0.05 was considered statistically significant.
Results
miR-155, BRG1, VEGFC and VEGFD were differentially expressed in NKTCL cells vs. in normal NK cells
A previous study showed that miR-155 was up-regulated not only in NKTCL cell lines, but also in the serum of NKTCL patients, and its level was correlated with disease progression.17 However, minimal is known on the status of BRG1 in NKTCLs, or its association with miR-155 or VEGFC/VEGFD. Therefore, we first profiled the expression of these four molecules in two NKTCL cell lines, SNK-6 and YTS. We used normal NK cells as the control. By RT-qPCR analysis, we showed that in either SNK-6 or YTS cells, the steady-state mRNA level of miR-155, VEGFC, or VEGFD was significantly higher, while that of BRG1 significantly lower than in normal NK cells (Figure 1(a)). Consistently, the protein levels of BRG1, VEGFC, and VEGFD presented the same trend of changes as their mRNA levels in all three cell populations examined (Figures 1(b and c)). The data showed that the expression level of BRG1 negatively correlated with that of miR-155, VEGFC, or VEGFD, and the level of miR-155 positively correlated with that of VEGFC or VEGFD in normal NK cells as well as in NKTCL cell lines. Considering the significance of VEGFC and VEGFD in regulating the formation of lymphatic vessels, this finding supported the potential involvement of miR-155 and BRG1 in lymphangiogenesis.
Figure 1.
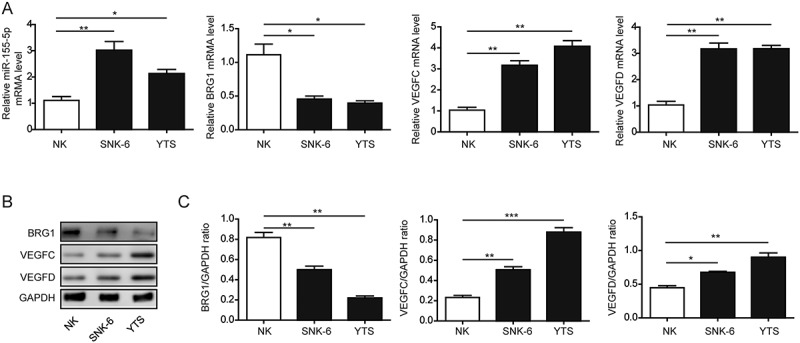
miR-155, BRG1, VEGFC and VEGFD were differentially expressed in NKCL cells vs. in normal NK cells. A. The expression of miR-155, BRG1, VEGFC, and VEGFD on the steady-state mRNA level from indicated cells was examined by RT-qPCR and presented as a ratio relative to that of the internal control (GAPDH). B. and C. The expression of BRG1, VEGFC, and VEGFD on the protein level from indicated cells was examined by Western blotting. The representative Western blotting image was shown in B and the quantification of relative protein levels to that of the internal control (GAPDH) shown in C. *P < 0.05; **P < 0.01; ***P < 0.001.
miR-155 directly inhibited the expression of BRG1 and up-regulated the subsequent VEGFC, but not VEGFD expression in NKTCLs
miR-155 negatively regulated BRG1 expression in human leukemia cell lines.30 To assess whether miR-155 directly inhibited BRG1 transcription, we cloned the 3-UTR sequence of BRG1 that contained the potential miR-155 binding site (WT) or a mutated sequence (MUT) into a luciferase reporter construct. As shown in Figure 2(a), miR-155 potently inhibited the luciferase activity from the WT but not from the MUT construct, suggesting that miR-155 can directly and specifically inhibit the transcription of BRG1. Since BRG1 may target the expression of VEGFC by inhibiting the activation of STAT3,29 we proposed that miR-155, by inhibiting the transcription of BRG1, elevated the expressions of VEGFC. We treated both SNK-6 and YTS cells with miR-155 inhibitor or NC. As shown in Figure 2(b), the miR-155 inhibitor significantly reduced the level of endogenous miR-155 in both cells lines, as expected. Concomitant with the reduction of miR-155, both the mRNA and protein levels of BRG1 was significantly elevated, VEGFC down-regulated, while VEGFD not altered in both SNK-6 and YTS cells treated with miR-155 inhibitor, when compared to cells treated with NC (Figures 2(b-d)), supporting that endogenous miR-155 controlled the expressions of both BRG1 and VEGFC, but not that of VEGFD. Consistently, by ELISA analysis, we found that miR-155 inhibitor drastically reduced the secretion of VEGFC into the CM from SNK-6 or YTS cells, when compared to NC-treated cells (Figure 2(e)). To examine the potential involvement of STAT3 signaling, we measured the p-STAT3 level in both cells (Figures 2(f and g)). The p-STAT3 level was reduced in response to miR-155 inhibitor in both SNK-6 and YTS cells, while the total STAT3 level was not altered, indicating that targeting miR-155, by releasing the inhibition on BRG1, enabled BRG1-mediated inactivation of STAT3 and down-regulation of VEGFC.
Figure 2.
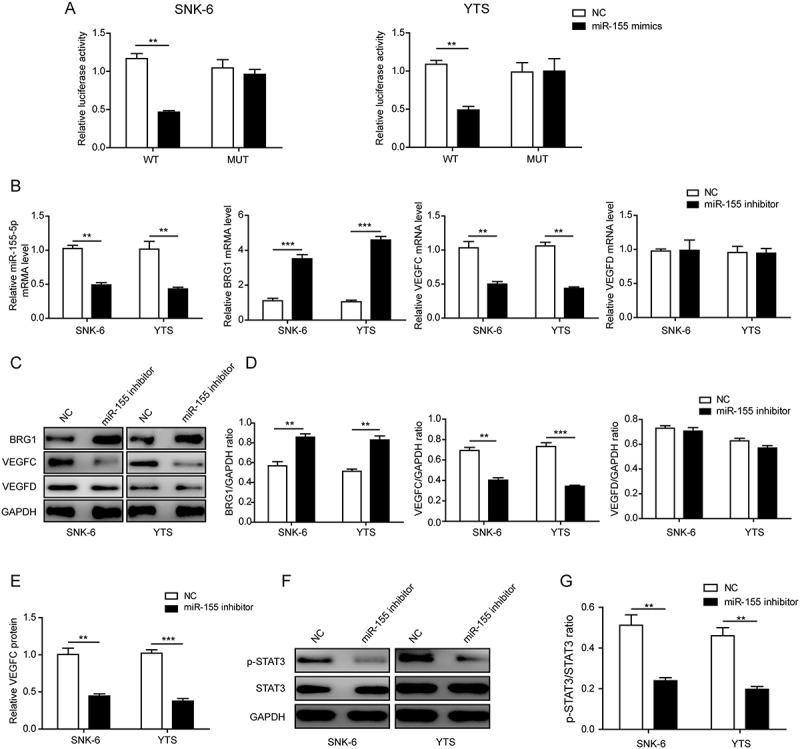
miR-155 directly inhibited the expression of BRG1 and up-regulated the subsequent VEGFC, but not VEGFD expression in NKTCL cells. A. The 3ʹ-UTR sequences of human BRG1 gene containing the wild-type (WT) or a mutated sequence (MUT) for the miR-155-binding site was cloned into the luciferase reporter construct and transfected into SNK-6 (left panel) or YTS (right panel) cells. The cells were then treated with either miR-155 mimics or negative control (NC), and the luciferase activity was measured. B. SNK-6 or YTS cells were transfected with either miR-155 inhibitor or NC. The expression of miR-155, BRG1, VEGFC, and VEGFD on the steady-state mRNA level was examined by RT-qPCR and presented as a ratio relative to that of the internal control (GAPDH). C. and D. The expression of BRG1, VEGFC, and VEGFD on the protein level from indicated cells was examined by Western blotting. The representative Western blotting image was shown in C and the quantification of relative protein levels to that of the internal control (GAPDH) shown in D. E. The secretion of VEGFC into the conditioned medium (CM) from SNK-6 or YTS cells upon treatment with miR-155 inhibitor or NC was measured by ELISA. The relative VEGFC level in CM from NC-treated cells was arbitrally defined as 1. F. and G. The expression of p-STAT3 and STAT3 from indicated cells was examined by Western bloting. The representative Western bloting image was shown in F and the quantification of relative protein levels to that of the internal control (GAPDH) shown in G. **P < 0.01; ***P < 0.001.
Targeting miR-155 promoted the apoptosis and reduced the pro-lymphangiogenic capacity of NKTCLs
BRG1 is well demonstrated for regulating the viability of cancer cells.32,33 By targeting STAT3/VEGFC signaling, BRG1 also regulated lymphangiogenesis of colorectal cancer.29 Since miR-155 could directly target the expression of BRG1 in NKTCLs (Figure 2), we assessed the functional significance of miR-155 in the survival and pro-lymphangiogenic capacity of NKTCLs. By staining the cells with Annexin V and PI, we showed that miR-155 inhibitor significantly boosted the apoptosis of both SNK-6 and YTS (Figures 3(a and b)). Furthermore, CM from both NKTCL cell lines following the treatment with miR-155 inhibitor significantly reduced the tube formation of HLECs, either alone or in conjunction with siVEGFR3, an essential VEGF receptor that mediated the pro-lymphangiogenic activities of VEGFC and VEGFD,34(Figures 3(c and d)). Moreover, concomitant applying miR-155 inhibitor and siVEGFR3 presented a synergistic effect in suppressing lymphangiogenesis than each treatment alone. Taken together, the data suggested that targeting miR-155, by up-regulating BRG1, promoted the apoptosis and reduces the pro-lymphangiogenic capacity of NKTCLs.
Figure 3.
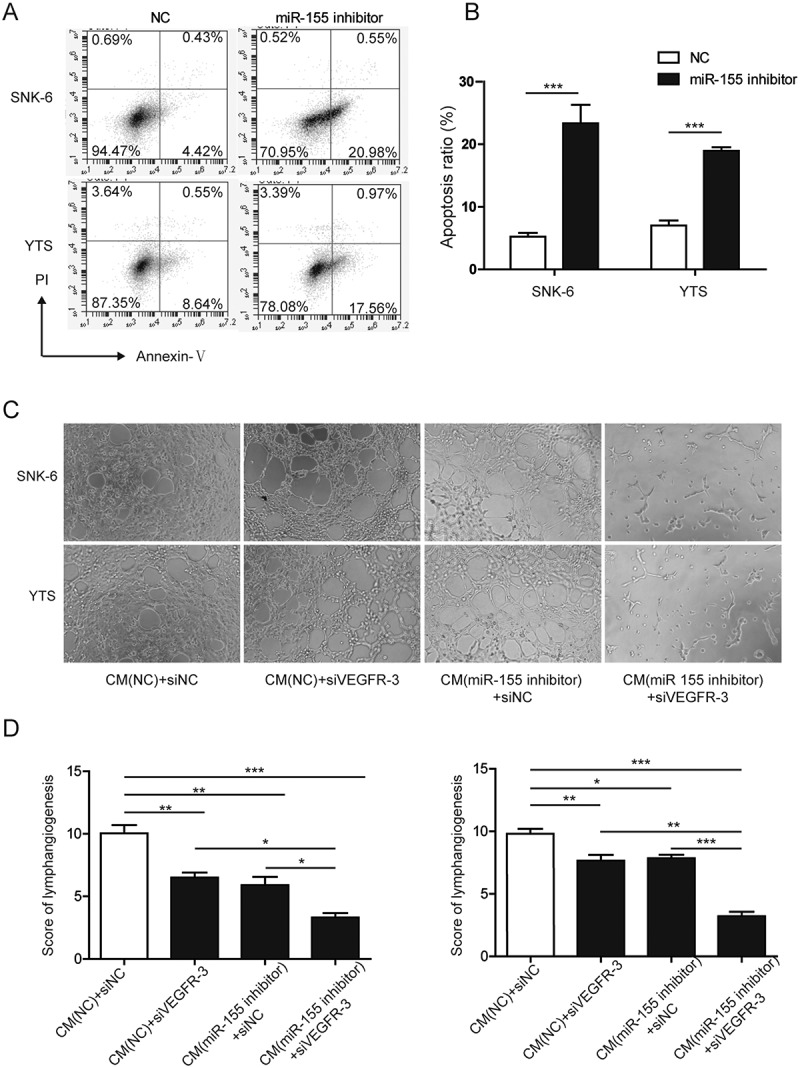
Targeting miR-155 promoted the apoptosis and reduced the pro-lymphangiogenic capacity of NKTCLs. A. and B. SNK-6 or YTS cells were transfected with either miR-155 inhibitor or NC. At 48 h after the transfection, cellular apoptosis was examined by flow cytometry following dual staining with Annexin V and propidium iodide (PI). The representative flow images were shown in A and the quantification of % of AnnexinV+ apoptotic cells shown in B. C. CM was collected from cells treated with NC or miR-155 inhibitor and applied to human lymphatic endothelial cells (HLECs) transfected with siVEGFR3 or control siRNA (siNC). The lymphatic tube formation was imaged under light microscopy. D. The lymphangiogenesis was scored and compared between indicated groups. *P < 0.05; **P < 0.01; ***P < 0.001.
STAT3 was essential for mir-155-induced VEGFC expression and lymphangiogenesis
To assess the significance of STAT3 signaling in miR-155-induced VEGFC expression and the subsequent effect on lymphangiogenesis, we treated both SNK-6 and YTS cells with S31-201, a specific STAT3 inhibitor. As shown in Figures 4(a and b), S31-201 significantly reduced the level of p-STAT3 but not that of total STAT3, indicating its biological activity. Concomitantly, the BRG1 level in cells treated with S31-201 was not changed when compared to cells treated with NC and was significantly lower than in those treated with miR-155 inhibitor, suggesting that STAT3 was a downstream, while miR-155 was an upstream signaling molecule of BRG1. In contrast, the VEGFC level was significantly reduced by S31-201, to a similar level as reduced by miR-155 inhibitor. The effect of S31-201 on VEGFC expression was also determined by ELISA (Figure 4(c)), which showed that both S31-201 and miR-155 inhibitor reduced the secretion of VEGFC into CM to comparable levels, significantly lower than VEGFC in CM from NC-treated cells. Correspondingly, CM from S31-201-treated NKTCLs behaved similarly to CM from miR-155-inhibitor-treated cells, significantly reducing the in vitro tube formation of HLECs, when compared to CM from NC-treated cells (Figures 4(d and e)), supporting that STAT3 functioned downstream of BRG1 and mediated the regulation of miR-155/BRG1 on VEGFC.
Figure 4.
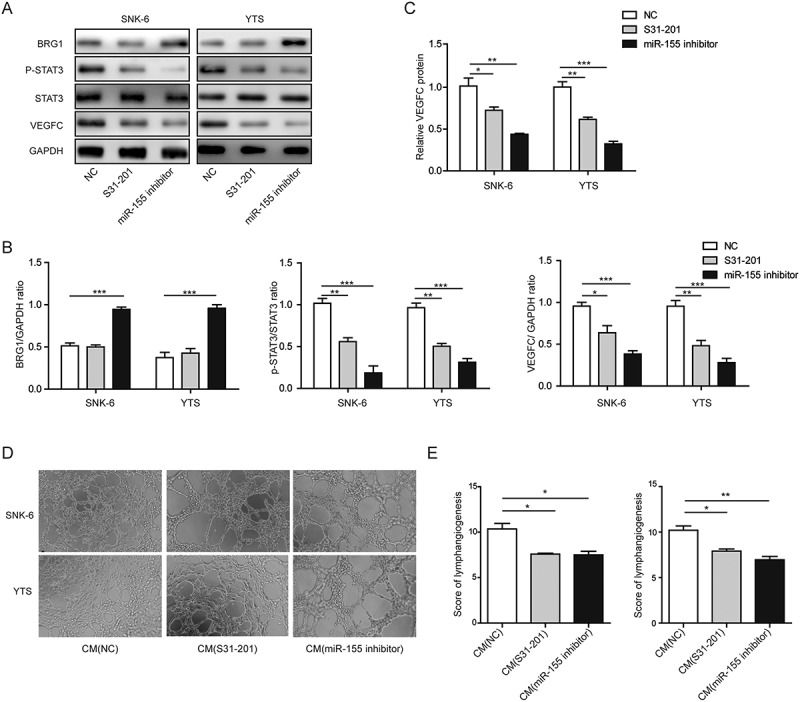
STAT3 was essential for miR-155-induced VEGFC expression and lymphangiogenesis. SNK-6 or YTS cells were treated with NC, S31-201, or miR-155 inhibitor. A. and B. The expression of BRG1, p-STAT3, STAT3, and VEGFC was examined by Western blotting. The representative Western blotting image was shown in A and the quantification of relative protein levels to that of the internal control (GAPDH) shown in B. C. The secretion of VEGFC into CM from SNK-6 or YTS cells treated as indicated was measured by ELISA. The relative VEGFC level in CM from NC-treated cells was arbitrally defined as 1. D. CM was collected from indicated cells and applied to HLECs. The lymphatic tube formation was imaged under light microscopy. E. The lymphangiogenesis was scored and compared between indicated groups. *P < 0.05; **P < 0.01; ***P < 0.001.
Targeting miR-155 inhibited the growth of NKTCL xenografts as well as tumor-associated lymphangiogenesis in vivo
Lastly, we examined whether the in vitro significance of miR-155/BRG1/STAT3/VEGFC signaling cascade may translate in vivo. For this purpose, we established the NKTCL xenografts using SNK-6 cells transfected with either NC or miR-155-inhibitor. We found that the growth of primary tumors from cells treated with miR-155 inhibitor was significantly suppressed (Figure 5(a)), both in tumor volume (Figure 5(b)) and tumor weight (Figure 5(c)), when compared to tumors from NC-treated cells. When analyzing the xenografts for BRG1, VEGFC, and LYVE1 (a marker for lymphatic endothelial cells) by Western blotting, we found that compared to tumors from NC-treated cells, those from miR-155-inhibitor treated cells were associated with significant up-regulation of BRG1 and down-regulation of VEGFC and LYVE1 (Figures 5(d and e)). Consistently, immunohistochemistry revealed that BRG1 was significantly up-regulated, while LYVE-1+ lymphatic vessels significantly reduced in xenografts from SNK-6 cells transfected with miR-155 inhibitor, when compared to xenografts from NC-transfected cells (Figures 5(f and g)), supporting that miR-155 inhibitor inhibited tumor-associated lymphangiogenesis in vivo by inducing the expression of BRG1.
Figure 5.
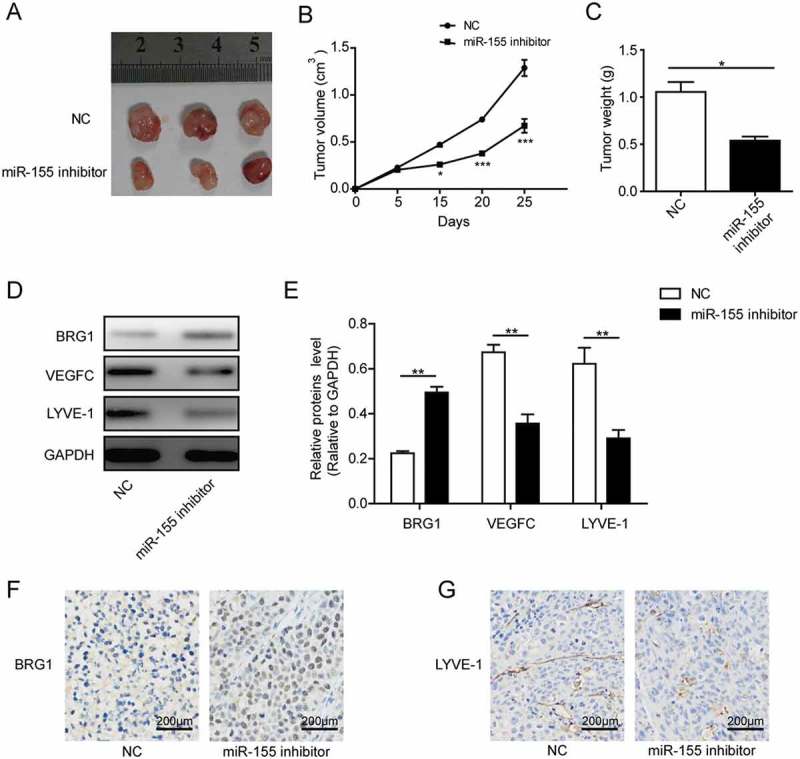
Targeting miR-155 inhibited the growth of NKTCL xenografts as well as tumor-associated lymphangiogenesis in vivo. On day 0, SNK-6 cells transfected with either NC or miR-155 inhibitor was subcutaneously injected into nude mice. On day 25 all mice were sacrificed and the xenografts were isolated from each mouse. A. Picture of three representative xenografts from each group. B. The growth curve of xenografts from each group. C. Upon the sacrifice of mice, the weights of xenograft tumors were measured and compared between the two groups. D. and E. The expression of BRG1, VEGFC, and LYVE1 from xenograft tumors was examined by Western blotting. The representative Western blotting image was shown in D and the quantification of relative protein levels to that of the internal control (GAPDH) shown in E. F. and G. The expression of BRG1 (F) and LYVE-1 (G) between xenografts from cells transfected with NC- and miR-155-inhibitor was examined by immunohistochemistry (brown signals). The tissues were counterstained with hematoxylin (blue-purple signals). *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
In this study, using two different NKTCL cell lines as the model system, we showed for the first time that: 1) miR-155 negatively correlated with BRG1, but positively with VEGFC and VEGFD in NKTCL cells; 2) miR-155 released BRG1-mediated apoptosis and suppression of STAT3/VEGFC signaling in NKTCL cells, and thus contributed to cancer cell survival as well as cancer-induced lymphangiogenesis; 3) targeting miR-155 not only induced apoptosis and dampened the pro-lymphangiogenic capacity of NKTCL cells in vitro, but also reduced tumor growth in vivo. Data from this study supported that BRG1 was a tumor suppressor and targeting its upstream regulator miR-155 might become a novel therapy for NKTCL.
As an important catalytic subunit for the SWI/SNF complex and critical regulatory for gene transcription, DNA repair, cell proliferation and differentiation, the findings that BRG1 was lost or mutated in multiple tumor cell lines, together with its functional phenotypes in inducing cell cycle arrest or apoptosis, supported the notion that BRG1 was a tumor suppressor.24,35 However, later studies suggested that the link of BRG1 to cancer was context-dependent.36 When introduced into breast cancer cell line that contained BRG1 mutation, BRG1 up-regulated multiple cyclin-dependent kinase inhibitors and induced growth arrest,37 supporting its role as a tumor suppressor. In contrast, other studies in breast cancer cells showed that BRG1 knockdown promoted apoptosis or reduced cell growth.38,39 More importantly, sequencing analysis on 507 primary breast tumors revealed none mutations in BRG1 gene.40 Expression analysis showed that BRG1 was up-regulated in breast tumors and correlated with poor survival of patients,38,41 pointing to the oncogenic activities of BRG1. In this study, we focused on two NKTCL cell lines and showed that BRG1 was down-regulated in these cells as compared to normal NK cells. The up-regulation of BRG1 following the treatment of miR-155 inhibitor was associated with an increase in apoptosis and reduction of pro-lymphangiogenesis. Together, the data supported the suppressive activity of BRG1 in NKTCL.
Although the genetic status (mutation or deletion) of BRG1 in the NKTCL cell lines or clinical NKTCL samples are not known, which should be comprehensively addressed in future studies. Our finding that the reduction of miR-155 by its inhibitor resulted in the up-regulation of BRG1 suggested that miR-155 was an essential regulator of BRG1 expression in the two NKTCL cell lines. Therefore, in addition to genomic deletion and genetic mutation, post-transcriptional regulation by miRNAs may also control BRG1 expression in cancer cells. Consistently, although mutations in BRG1 gene were frequently detected in cancer cell lines, they were not common events in human cancer tissues,42 suggesting other mechanisms led to BRG1 silence in multiple human cancers. In addition to NKTCL cells, Cuadros et al. reported the negative correlation between miR-155 and BRG1 in different lymphoma cells.30
As either a tumor suppressor or an oncogene, BRG1 presented pleotropic phenotypes mostly through the association and expressional regulation of different target genes, such as: RB family proteins,43-45 p21,46 and p53,47 for viability and cell cycle progression; p53,47 BRCA1,48 and FANCA49 for DNA repair; CD4450 and E-cadherin for cell adhesion; multiple genes in T-cell development and other immune functions51; miR-550a-5p/RNF43/Wnt/β-catenin signaling28 and miR-148b52 for cancer metastasis. Recently, Zhu et al. showed that by inhibiting the activation of STAT3 and subsequently the expression of VEGFC, BRG1 controlled lymphangiogenesis in colorectal carcinoma, both in vitro and in vivo.29 Consistent with Zhu’s study, here we showed that the expression of BRG1 negatively correlated with that of VEGFC in both normal NK cells and NKTCL cell lines, with BRG1 high and VEGFC low in normal NK cells, while BRG1 low and VEGFC high in NKTCL cell lines. Furthermore, we identified miR-155 as a direct upstream regulator of BRG1 by inhibiting BGR1 expression. When targeting miR-155 with the inhibitor, we detected the restoration of BRG1 expression and the down-regulation of VEGFC. We also established the critical role of STAT3 in miR-155/BRG1/VEGFC signaling, that is, controlling the expression of VEGFC downstream of BRG1. Consistent with the role of VEGFC in lymphangiogenesis, we observed that targeting miR-155 potently reduced the pro-lymphangiogenic capacity of NKTCLs. Interestingly, although we detected the negative correlation between BRG1 and another critical lymphangiogenic factor, VEGFD in both normal NK and NKTCL cells, targeting miR-155 failed to alter VEGFD level, suggesting that the cancer cells deploy distinct mechanisms to up-regulate VEGFC and VEGFD expression. This finding supported the importance of lymphangiogenesis contributing to the malignancy of NKTCL.
In addition to inhibiting the pro-lymphangiogenic capacity of NKTCLs, miR-155 inhibitor significantly induced apoptosis of cancer cells. Consistently, increasing studies support the therapeutic potential of targeting miR-155 in cancerous diseases, such as lung cancer,53 multiple myeloma,54 and breast cancer,55 as well as non-cancerous diseases, including amyotrophic lateral sclerosis56 and systemic lupus erythematosus.57 Considering that BRG1 regulated many downstream genes involved in viability, it was possible that the pro-apoptotic effect of miR-155 inhibitor was mediated through BRG1, but may also be through other downstream targets of miR-155, as revealed by cumulative evidence in the cancer field.58 Future studies should be carried out to address the significance of BRG1 in multiple phenotypes of miR-155 in NKTCL.
In this study, we not only characterized the miR-155/BRG1/STAT3/VEGFC signaling in pro-lymphangiogenic activity of NKTCL cell lines in vitro, but also examined its in vivo importance using a xenograft model. We showed that the tumor growth from miR-155-inhibitor-treated NKTCL cells was significantly suppressed compared to NC-treated cells. Furthermore, when examining equal amounts of xenograft tumor mass from both groups by Western blotting, we detected potent up-regulation of BRG1, and significant reductions of VEGFC and LYVE-1, a biomarker for lymphatic endothelial cells and tumor-associated lymphangiogenesis,59 from miR-155-inhibitor-treated than from NC-treated xenografts. Consistently, immunohistochemistry revealed a marked increase of BRG1 as well as a decrease of LYVE1+ lymphatic vessels in the former xenografts than in the latter, supporting that targeting miR-155, by up-regulating BRG1 and reducing VEGFC, was sufficient to inhibit lymphangiogenesis.
In summary, we identified the miR-155/BRG1/STAT3/VEGFC signaling as a novel mechanism for regulating lymphangiogenesis of NKTCL cells. In addition, we showed that miR-155 regulated the survival of NKTCL cells, and cancer cells utilize other mechanisms other than miR-155/BRG1/STAT3/VEGFC signaling to up-regulate VEGFD expression. Therefore, targeting miR-155 may provide an effective therapy for targeting multiple phenotypes of NKTCL. When combining therapies targeting miR-155/BRG1/STAT3/VEGFC signaling with those targeting VEGFD expression, more robust inhibition of lymphangiogenesis and thus further suppression of lymph node metastasis could be achieved.
Funding Statement
This work was supported by the National Natural Science Foundation of China (NSFC) [No. 81570203].
Disclosure of conflict of interest
The authors declare no competing financial interests.
References
- 1.Tse E, Kwong YL.. The diagnosis and management of NK/T-cell lymphomas. J Hematol Oncol. 2017;10: 85. doi: 10.1186/s13045-017-0452-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perry AM, Diebold J, Nathwani BN, MacLennan KA, Müller-Hermelink HK, Bast M, Boilesen E, Armitage JO, Weisenburger DD. Non-Hodgkin lymphoma in the developing world: review of 4539 cases from the International non-hodgkin lymphoma classification project. Haematologica. 2016;101: 1244–1250. doi: 10.3324/haematol.2016.148809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Au W-Y, Weisenburger DD, Intragumtornchai T, Nakamura S, Kim W-S, Sng I, Vose J, Armitage JO, Liang R. Clinical differences between nasal and extranasal natural killer/T-cell lymphoma: a study of 136 cases from the international peripheral T-Cell Lymphoma Project. Blood. 2009;113: 3931–3937. doi: 10.1182/blood-2008-10-185256. [DOI] [PubMed] [Google Scholar]
- 4.Tse E, Leung R, Khong PL, Lau WH, Kwong YL. Non-nasal natural killer cell lymphoma: not non-nasal after all. Ann Hematol. 2009;88: 185–187. doi: 10.1007/s00277-008-0562-0. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y-W, Guo T, Shen L, Wong K-Y, Tao Q, Choi WWL, Au-Yeung RKH, Chan Y-P, Wong MLY, Tang JCO, et al. Receptor-type tyrosine-protein phosphatase κ directly targets STAT3 activation for tumor suppression in nasal NK/T-cell lymphoma Blood. 2015;125: 1589–1600. doi: 10.1182/blood-2014-07-588970. [DOI] [PubMed] [Google Scholar]
- 6.Karube K, Nakagawa M, Tsuzuki S, Takeuchi I, Honma K, Nakashima Y, Shimizu N, Ko Y-H, Morishima Y, Ohshima K, et al. Identification of FOXO3 and PRDM1 as tumor-suppressor gene candidates in NK-cell neoplasms by genomic and functional analyses. Blood. 2011;118: 3195–3204. doi: 10.1182/blood-2011-04-346890. [DOI] [PubMed] [Google Scholar]
- 7.Jiang L, Gu Z-H, Yan Z-X, Zhao X, Xie -Y-Y, Zhang Z-G, Pan C-M, Hu Y, Cai C-P, Dong Y, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet. 2015;47: 1061–1066. doi: 10.1038/ng.3358. [DOI] [PubMed] [Google Scholar]
- 8.Koo GC, Tan SY, Tang T, Poon SL, Allen GE, Tan L, Chong SC, Ong WS, Tay K, Tao M, et al. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov. 2012;2: 591–597. doi: 10.1158/2159-8290.CD-12-0028. [DOI] [PubMed] [Google Scholar]
- 9.Küçük C, Jiang B, Hu X, Zhang W, Chan JKC, Xiao W, Lack N, Alkan C, Williams JC, Avery KN, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from γδ-T or NK cells. Nat Commun. 2015;6: 6025. doi: 10.1038/ncomms7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee S, Park HY, Kang SY, Kim SJ, Hwang J, Lee S, Kwak SH, Park KS, Yoo HY, Kim WS, et al. Genetic alterations of JAK/STAT cascade and histone modification in extranodal NK/T-cell lymphoma nasal type. Oncotarget. 2015;6: 17764–17776. doi: 10.18632/oncotarget.3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Küçük C, Hu X, Jiang B, Klinkebiel D, Geng H, Gong Q, Bouska A, Iqbal J, Gaulard P, McKeithan TW, et al. Global promoter methylation analysis reveals novel candidate tumor suppressor genes in natural killer cell lymphoma. Clin Cancer Res. 2015;21: 1699–1711. doi: 10.1158/1078-0432.CCR-14-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siu LL, Chan JK, Wong KF, Kwong YL. Specific patterns of gene methylation in natural killer cell lymphomas: p73 is consistently involved. Am J Pathol. 160;2002: 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Motsch N, Alles J, Imig J, Zhu J, Barth S, Reineke T, Tinguely M, Cogliatti S, Dueck A, Meister G, et al. MicroRNA profiling of Epstein-Barr virus-associated NK/T-cell lymphomas by deep sequencing. PLoS One. 2012;7: e42193. doi: 10.1371/journal.pone.0042193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saki N, Abroun S, Soleimani M, Hajizamani S, Shahjahani M, Kast RE, Mortazavi Y. Involvement of MicroRNA in T-Cell differentiation and malignancy. Int J Hematol Oncol Stem Cell Res. 9;2015: 33–49. [PMC free article] [PubMed] [Google Scholar]
- 15.Iorio MV, Croce CM. MicroRNAs in cancer: small molecules with a huge impact. J Clin Oncol. 2009;27: 5848–5856. doi: 10.1200/JCO.2009.24.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamanaka Y, Tagawa H, Takahashi N, Watanabe A, Guo Y-M, Iwamoto K, Yamashita J, Saitoh H, Kameoka Y, Shimizu N, et al. Aberrant overexpression of microRNAs activate AKT signaling via down-regulation of tumor suppressors in natural killer-cell lymphoma/leukemia. Blood. 2009;114: 3265–3275. doi: 10.1182/blood-2009-06-222794. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Ji W, Huang R, Li L, Wang X, Li L, Fu X, Sun Z, Li Z, Chen Q, et al. MicroRNA-155 is a potential molecular marker of natural killer/T-cell lymphoma. Oncotarget. 2016;7: 53808–53819. doi: 10.18632/oncotarget.10780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christiansen A, Detmar M. Lymphangiogenesis and cancer. Genes Cancer. 2011;2: 1146–1158. doi: 10.1177/1947601911423028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karpanen T, Egeblad M, Karkkainen MJ, Kubo H, Ylä-Herttuala S, Jäättelä M, Alitalo K. Vascular endothelial growth factor C promotes tumor lymphangiogenesis and intralymphatic tumor growth. Cancer Res. 61;2001: 1786–1790. [PubMed] [Google Scholar]
- 20.Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, Riccardi L, Alitalo K, Claffey K, Detmar M. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001;7: 192–198. doi: 10.1038/84643. [DOI] [PubMed] [Google Scholar]
- 21.Lin J, Lalani AS, Harding TC, Gonzalez M, Wu -W-W, Luan B, Tu GH, Koprivnikar K, VanRoey MJ, He Y, et al. Inhibition of lymphogenous metastasis using adeno-associated virus-mediated gene transfer of a soluble VEGFR-3 decoy receptor. Cancer Res. 2005;65: 6901–6909. doi: 10.1158/0008-5472.CAN-05-0408. [DOI] [PubMed] [Google Scholar]
- 22.De La Serna IL, Ohkawa Y, Imbalzano AN. Chromatin remodelling in mammalian differentiation: lessons from ATP-dependent remodellers. Nat Rev Genet. 2006;7: 461–473. doi: 10.1038/nrg1882. [DOI] [PubMed] [Google Scholar]
- 23.Singh AP, Foley J, Tandon A, Phadke D, Karimi Kinyamu H, Archer TK. A role for BRG1 in the regulation of genes required for development of the lymphatic system. Oncotarget. 2017;8: 54925–54938. doi: 10.18632/oncotarget.18976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J, Sanchez-Cespedes M. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat. 2008;29: 617–622. doi: 10.1002/humu.20730. [DOI] [PubMed] [Google Scholar]
- 25.Orvis T, Hepperla A, Walter V, Song S, Simon J, Parker J, Wilkerson MD, Desai N, Major MB, Hayes DN, et al. BRG1/SMARCA4 inactivation promotes non-small cell lung cancer aggressiveness by altering chromatin organization. Cancer Res. 2014;74: 6486–6498. doi: 10.1158/0008-5472.CAN-14-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Von Figura G, Fukuda A, Roy N, Liku ME, Morris Iv JP, Kim GE, Russ HA, Firpo MA, Mulvihill SJ, Dawson DW, et al. The chromatin regulator Brg1 suppresses formation of intraductal papillary mucinous neoplasm and pancreatic ductal adenocarcinoma. Nat Cell Biol. 2014;16: 255–267. doi: 10.1038/ncb2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bock VL, Lyons JG, Huang XX, Jones AM, McDonald LA, Scolyer RA, Moloney FJ, Barnetson RS, Halliday GM. BRM and BRG1 subunits of the SWI/SNF chromatin remodelling complex are downregulated upon progression of benign skin lesions into invasive tumours. Br J Dermatol. 2011;164: 1221–1227. doi: 10.1111/j.1365-2133.2011.10267.x. [DOI] [PubMed] [Google Scholar]
- 28.Wang G, Fu Y, Yang X, Luo X, Wang J, Gong J, Hu J. Brg-1 targeting of novel miR550a-5p/RNF43/Wnt signaling axis regulates colorectal cancer metastasis. Oncogene. 2016;35: 651–661. doi: 10.1038/onc.2015.124. [DOI] [PubMed] [Google Scholar]
- 29.Zhu X, Sun L, Lan J, Xu L, Zhang M, Luo X, Gong J, Wang G, Yuan X, Hu J, et al. BRG1 targeting STAT3/VEGFC signaling regulates lymphangiogenesis in colorectal cancer. Oncotarget. 2016;7: 36501–36509. doi: 10.18632/oncotarget.9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cuadros M, Sánchez-Martín V, Herrera A, Baliñas C, Martín-Padrón J, Boyero L, Peinado P, Medina PP. BRG1 regulation by miR-155 in human leukemia and lymphoma cell lines. Clin Transl Oncol. 2017;19: 1010–1017. doi: 10.1007/s12094-017-1633-2. [DOI] [PubMed] [Google Scholar]
- 31.Rao X, Huang X, Zhou Z, Lin X. An improvement of the 2^(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat Bioinforma Biomath. 3;2013: 71–85. [PMC free article] [PubMed] [Google Scholar]
- 32.Liu K, Luo Y, Lin FT, Lin WC. TopBP1 recruits Brg1/Brm to repress E2F1-induced apoptosis, a novel pRb-independent and E2F1-specific control for cell survival. Genes Dev. 2004;18: 673–686. doi: 10.1101/gad.1180204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Q, Madany P, Dobson JR, Schnabl JM, Sharma S, Smith TC, Van Wijnen AJ, Stein JL, Lian JB, Stein GS, et al. The BRG1 chromatin remodeling enzyme links cancer cell metabolism and proliferation. Oncotarget. 2016;7: 38270–38281. doi: 10.18632/oncotarget.9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsumoto M, Roufail S, Inder R, Caesar C, Karnezis T, Shayan R, Farnsworth RH, Sato T, Achen MG, Mann GB, et al. Signaling for lymphangiogenesis via VEGFR-3 is required for the early events of metastasis. Clin Exp Metastasis. 2013;30: 819–832. doi: 10.1007/s10585-013-9581-x. [DOI] [PubMed] [Google Scholar]
- 35.Medina PP, Sanchez-Cespedes M. Involvement of the chromatin-remodeling factor BRG1/SMARCA4 in human cancer. Epigenetics. 3;2008: 64–68. [DOI] [PubMed] [Google Scholar]
- 36.Wu Q, Lian JB, Stein JL, Stein GS, Nickerson JA, Imbalzano AN. The BRG1 ATPase of human SWI/SNF chromatin remodeling enzymes as a driver of cancer. Epigenomics. 2017;9: 919–931. doi: 10.2217/epi-2017-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hendricks KB, Shanahan F, Lees E. Role for BRG1 in cell cycle control and tumor suppression. Mol Cell Biol. 24;2004: 362–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bai J, Mei P, Zhang C, Chen F, Li C, Pan Z, Liu H, Zheng J, El-Maarri O. BRG1 is a prognostic marker and potential therapeutic target in human breast cancer. PLoS One. 2013;8: e59772. doi: 10.1371/journal.pone.0059772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Q, Madany P, Akech J, Dobson JR, Douthwright S, Browne G, Colby JL, Winter GE, Bradner JE, Pratap J, et al. The SWI/SNF ATPases are required for triple negative breast cancer cell proliferation. J Cell Physiol. 2015;230: 2683–2694. doi: 10.1002/jcp.24991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cancer Genome Atlas N Comprehensive molecular portraits of human breast tumours. Nature. 2012;490: 61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Do S-I, Yoon G, Kim H-S, Kim K, Lee H, Do I-G, Kim D-H, Chae SW, Sohn JH. Increased Brahma-related Gene 1 expression predicts distant metastasis and shorter survival in patients with invasive ductal carcinoma of the breast. Anticancer Res. 2016;36: 4873–4882. doi: 10.21873/anticanres.11051. [DOI] [PubMed] [Google Scholar]
- 42.Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene. 2009;28: 1653–1668. doi: 10.1038/onc.2009.4. [DOI] [PubMed] [Google Scholar]
- 43.Bartlett C, Orvis TJ, Rosson GS, Weissman BE. BRG1 mutations found in human cancer cell lines inactivate Rb-mediated cell-cycle arrest. J Cell Physiol. 2011;226: 1989–1997. doi: 10.1002/jcp.22533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dahiya A, Gavin MR, Luo RX, Dean DC. Role of the LXCXE binding site in Rb function. Mol Cell Biol. 20;2000: 6799–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strobeck MW, Knudsen KE, Fribourg AF, DeCristofaro MF, Weissman BE, Imbalzano AN, Knudsen ES. BRG-1 is required for RB-mediated cell cycle arrest. Proc Natl Acad Sci U S A. 97;2000: 7748–7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kang H, Cui K, Zhao K. BRG1 controls the activity of the retinoblastoma protein via regulation of p21CIP1/WAF1/SDI. Mol Cell Biol. 24;2004: 1188–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang M, Gu C, Qi T, Tang W, Wang L, Wang S, Zeng X. BAF53 interacts with p53 and functions in p53-mediated p21-gene transcription. J Biochem. 2007;142: 613–620. doi: 10.1093/jb/mvm176. [DOI] [PubMed] [Google Scholar]
- 48.Bochar DA, Wang L, Beniya H, Kinev A, Xue Y, Lane WS, Wang W, Kashanchi F, Shiekhattar R. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell. 102;2000: 257–265. [DOI] [PubMed] [Google Scholar]
- 49.Otsuki T, Furukawa Y, Ikeda K, Endo H, Yamashita T, Shinohara A, Iwamatsu A, Ozawa K, Liu JM. Fanconi anemia protein, FANCA, associates with BRG1, a component of the human SWI/SNF complex. Hum Mol Genet. 10;2001: 2651–2660. [DOI] [PubMed] [Google Scholar]
- 50.Strobeck MW, DeCristofaro MF, Banine F, Weissman BE, Sherman LS, Knudsen ES. The BRG-1 subunit of the SWI/SNF complex regulates CD44 expression. J Biol Chem. 2001;276: 9273–9278. doi: 10.1074/jbc.M009747200. [DOI] [PubMed] [Google Scholar]
- 51.Gebuhr TC, Kovalev GI, Bultman S, Godfrey V, Su L, Magnuson T. The role of Brg1, a catalytic subunit of mammalian chromatin-remodeling complexes, in T cell development. J Exp Med. 2003;198: 1937–1949. doi: 10.1084/jem.20030714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou Z, Su Y, Fa X. Restoration of BRG1 inhibits proliferation and metastasis of lung cancer by regulating tumor suppressor miR-148b. Onco Targets Ther. 2015;8: 3603–3612. doi: 10.2147/OTT.S95500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van Roosbroeck K, Fanini F, Setoyama T, Ivan C, Rodriguez-Aguayo C, Fuentes-Mattei E, Xiao L, Vannini I, Redis RS, D’Abundo L, et al. Combining Anti-Mir-155 with chemotherapy for the treatment of lung cancers. Clin Cancer Res. 2017;23: 2891–2904. doi: 10.1158/1078-0432.CCR-16-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feng M, Luo X, Gu C, Fei J. Seed targeting with tiny anti-miR-155 inhibits malignant progression of multiple myeloma cells. J Drug Target. 2015;23: 59–66. doi: 10.3109/1061186X.2014.951653. [DOI] [PubMed] [Google Scholar]
- 55.Zuo J, Yu Y, Zhu M, Jing W, Yu M, Chai H, Liang C, Tu J. Inhibition of miR-155, a therapeutic target for breast cancer, prevented in cancer stem cell formation. Cancer Biomark. 2018;21: 383–392. doi: 10.3233/CBM-170642. [DOI] [PubMed] [Google Scholar]
- 56.Butovsky O, Jedrychowski MP, Cialic R, Krasemann S, Murugaiyan G, Fanek Z, Greco DJ, Wu PM, Doykan CE, Kiner O, et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol. 2015;77: 75–99. doi: 10.1002/ana.24304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xin Q, Li J, Dang J, Bian X, Shan S, Yuan J, Qian Y, Liu Z, Liu G, Yuan Q, et al. miR-155 deficiency ameliorates autoimmune inflammation of systemic lupus erythematosus by targeting S1pr1 in Faslpr/lpr Mice. J Immunol. 2015;194: 5437–5445. doi: 10.4049/jimmunol.1403028. [DOI] [PubMed] [Google Scholar]
- 58.Higgs G, Slack F. The multiple roles of microRNA-155 in oncogenesis. J Clin Bioinforma. 2013;3: 17. doi: 10.1186/2043-9113-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jackson DG, Prevo R, Clasper S, Banerji S. LYVE-1, the lymphatic system and tumor lymphangiogenesis. Trends Immunol. 22;2001: 317–321. [DOI] [PubMed] [Google Scholar]