

ABSTRACT
Characterizing the functions of essential cell cycle control genes requires tight and rapid inducible gene inactivation. Drawbacks of current conditional depletion approaches include slow responses and incomplete depletion. We demonstrated that by integrating the tetracycline-controlled promoter system and the auxin-inducible degron (AID) system together, AID-tagged proteins can be downregulated more efficiently than the individual technology alone. When used in conjunction with CRISPR-Cas9-mediated disruption of the endogenous locus, this system facilitates the analysis of essential genes by allowing rapid and tight conditional depletion, as we have demonstrated using several cell cycle-regulatory genes including cyclin A, CDK2, and TRIP13. The vectors constructed in this study allow expression of AID-fusion proteins under the control of tetracycline-controlled promoters and should be useful in studies requiring rapid and tight suppression of gene expression in mammalian cells.
KEYWORDS: Inducible degron, knockout, methods
Introduction
Several loss-of-function strategies have been developed for understanding gene functions in mammalian cells. While direct gene disruption represents the most effective approach in silencing a gene, it is generally irreversible and cannot be used for essential genes. To study the functions of essential genes, tactics involving conditional inactivation or depletion are generally required. For example, RNA interference (RNAi) can be used to downregulate gene expression at the level of mRNA stability and/or translation. But caveats including off-target effects and slow responses limit the use of RNAi in many studies.
A widely used system for downregulating gene expression in mammalian cells involves placing the gene of interest under tetracycline-controlled promoters (Tet-Off and Tet-On), which are based on the Tet repressor protein (TetR) and tet operator (tetO) DNA elements that control the Tn10-encoded tetracycline resistance operon in Escherichia coli [1]. In the Tet-Off system, TetR is fused to the transcription activation domain of VP16 from herpes simplex viruses, resulting in a tetracycline-controlled transcriptional activator (tTA). Tetracycline response element (TRE) is constructed by fusing seven tetO sequences to a minimal TATA-box containing a eukaryotic promoter derived from the CMV immediate early gene. In the absence of tetracycline, tTA dimers bind TRE and activate the expression of the downstream transgene. Binding of tetracycline or derivatives such as doxycycline (Dox) induces a conformational change in the TetR domain of tTA that prevents tetO binding, thus turning off gene expression. By contrast, the TetR variants used in Tet-On systems allow binding to tetO in the presence but not the absence of Dox [2].
Drawbacks of the tetracycline-controlled systems include “leakiness” of the promoters as well as relatively slow responses. An alternative approach that address some of these limitations is to induce conditional depletion by means of an inducible degradation signal, or degron [3]. A system involving the use of auxin-inducible degron (AID) has been adopted in mammalian cells to provide temporal and quantitative control of protein expression. In plants, the phytohormone auxin (indole-3-acetic acid; IAA) induces degradation of a family of transcriptional repressors by mediating the interaction of the AID domain in the target protein with the substrate recognition domain of an F-box protein called TIR1 [4]. Together with cullin, RBX1, SKP1, TIR1 forms a SCF-type ubiquitin ligase (E3), targeting the AID-containing proteins for ubiquitylation and subsequent proteasome degradation. The system plays important roles in various processes including apical dominance, flowering, ethylene biosynthesis, root growth, and fruit development [5].
In mammalian cells, ectopically expressed TIR1 can form an E3 ligase with the remaining SCF components from endogenous proteins and degrade AID-tagged proteins [6]. Likewise, the AID system has also been applied to other organisms including fission yeast [7], Drosophila [8,9], and C. elegans [10]. It has also been used successfully for conditional depletion of both non-essential and essential genes. In these studies, the endogenous genes are targeted by different approaches including siRNA against the UTR [11], the use of previously established knockout DT40 cells [12], CRISPR–Cas9 against the UTR–ORF boundary [13], or tagging AID into the endogenous locus using CRISPR–Cas9-mediated gene editing [14].
A limitation of the AID system is that rapid degradation of AID-fusion proteins requires the expression of relatively high levels of TIR1, which is not readily attainable in many mammalian cell lines. Moreover, residue levels of AID-fusion proteins could often be detected even after prolonged incubation with IAA. We believe that integrating the tetracycline-controlled system and AID system together could complement the limitations of the two systems. The combined system controlled gene expression at both transcriptional and post-translational levels, allowing protein expression to be controlled both rapidly and tightly. When used in conjunction with CRISPR-Cas9-mediated disruption of the endogenous locus, this system facilitates the analysis of essential genes by allowing rapid and tight conditional depletion.
Results
Figure 1 summarises the concept of this conditional deficiency system, in which AID-tagged proteins are degraded rapidly in response to plant auxin (or indole-3-acetic acid, IAA) in cells expressing the ubiquitin ligase SCFTIR1 [6]. The construct is also placed under the control of a Tet-Off promoter, allowing the transcription of the AID-tagged gene to be turned off in the presence of doxycycline (Dox) in Tet-Off cell lines.
Figure 1.
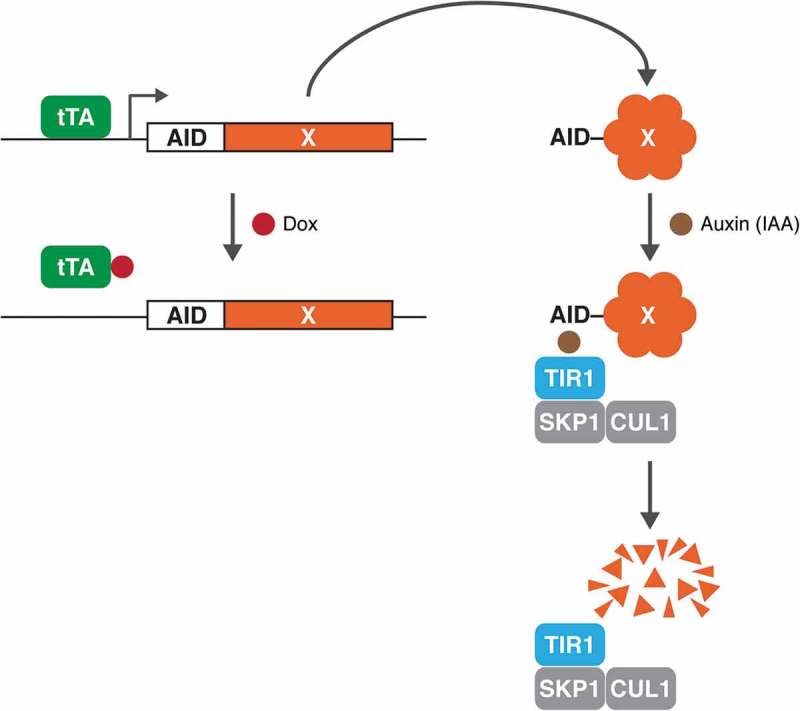
Combining the tetracycline-controlled transcriptional system and the auxin-inducible degron (AID) system. The transcription of AID-tagged genes (X) is placed under the control of a Tet-Off promoter. In Tet-Off cell lines, the tetracycline-controlled transcriptional activator (tTA) binds to the tetracycline response element in the promoter and activates the transcription of AID-tagged gene in the absence of doxycycline (Dox). Addition of Dox turns off the transcription of the promoter. In response to IAA, AID-fusion protein is rapidly targeted for degradation in cells expressing the ubiquitin ligase SCFTIR1.
As many components of the cell cycle engine are essential genes, we first tested the approach by targeting key components of the cell cycle engine. Cyclin-dependent kinase 2 (CDK2) is the catalytic partner of cyclin A and cyclin E and is essential for S phase progression [15]. HeLa cells expressing tTA tetracycline transactivator were transfected with plasmids expressing CRISPR-Cas9 against CDK2, AID-CDK2, and TIR1. Individual clones were then analysed with immunoblotting. Clones containing no detectable endogenous CDK2 (CDK2KO) and expressing AID-CDK2 were isolated (Figure 2(a)). Genome sequencing confirmed that the endogenous CDK2 locus contained base deletion resulting in gene disruption (Figure S1A). The AID-CDK2 was expressed at a similar level as the endogenous CDK2 in parental cells. Treatment of the CDK2KO:AID-CDK2 cells with IAA triggered the destruction of AID-CDK2 (Figure 2(b), compare lanes 1 and 6).
Figure 2.
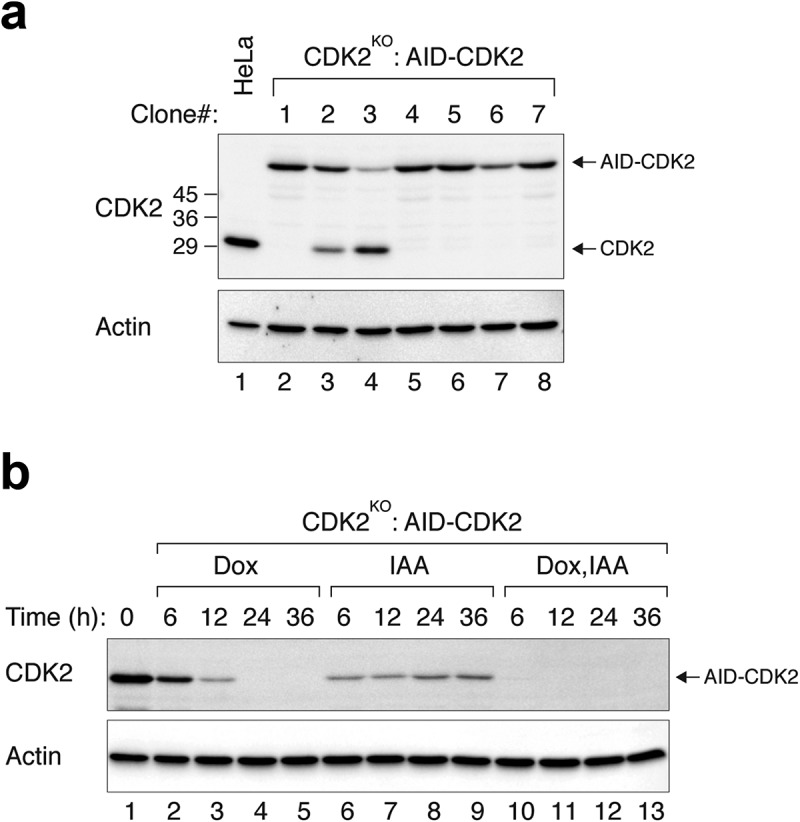
Efficient depletion of CDK2 using IAA and Dox. (a) CDK2KO:AID-CDK2 cells. CDK2KO cells expressing AID-CDK2 were generated as described in Materials and Methods. After growing in the absence of IAA and Dox, individual clones were isolated. Lysates were prepared and the expression of CDK2 was detected with immunoblotting. Equal loading of lysates was confirmed by immunoblotting for actin. The positions of molecular size standards (in kDa) are indicated on the left. Control HeLa cells were included in the analysis to indicate the position and abundance of endogenous CDK2. (b) CDK2KO cells expressing AID-CDK2 were grown in the presence of IAA and/or Dox and harvested at the indicated time. Lysates were prepared and the expression of CDK2 was detected using immunoblotting. Equal loading of lysates was confirmed by actin analysis.
Although addition of IAA could trigger the AID-CDK2 destruction, it was unable to completely remove AID-CDK2 (Figure 2(b), lanes 6–9). As the AID-CDK2 was under the control of a Tet-Off promoter, the expression of AID-CDK2 could be suppressed with Dox. As expected, the kinetics of Dox-mediated decrease of AID-CDK2 was slower than that induced with IAA (Figure 2(b)). However, Dox was able to suppress the expression of AID-CDK2 more completely than IAA. By combining both IAA and Dox, AID-CDK2 was depleted more efficiently than the individual chemicals alone.
We next used a similar strategy to generate conditional depletion cell lines for cyclin A. Cyclin A is an activating subunit for both CDK1 and CDK2 and is involved in controlling both S phase and mitosis [16]. Genome sequencing confirmed that the endogenous cyclin A locus contained indels resulting in gene disruption (Figure S1B). In cell lines lacking endogenous cyclin A and expressing AID-cyclin A, AID-cyclin A could be rapidly degraded in the presence of IAA (Figure 3(a)). Consistent with above, degradation remained incomplete even after prolonged (36 h) incubation with IAA (Figure 3(a,b)). Likewise, residual amount of AID-cyclin A was detected up to 12 h after incubation with Dox (Figure 3(b)). RT-PCR analysis revealed that the mRNA of AID-cyclin A was reduced relatively rapidly in the presence of Dox (similar levels were present between 6 h and 12 h) (Figure S2), confirming the lag between mRNA reduction and the disappearance of the protein. By contrast, AID-cyclin A protein was depleted more rapidly and completely in the presence of Dox and IAA together.
Figure 3.
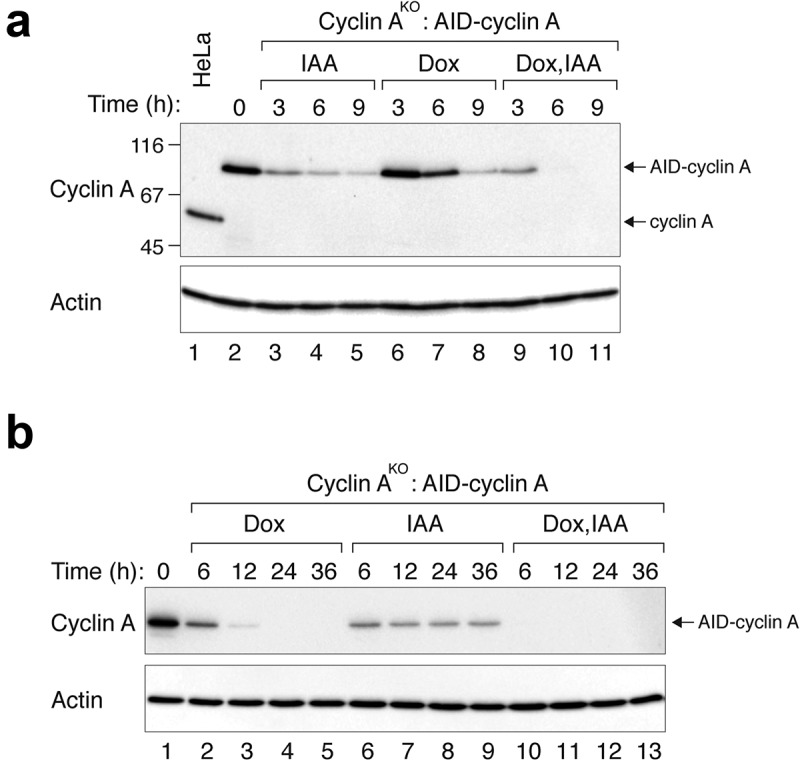
Efficient depletion of cyclin A using IAA and Dox. (a) Cyclin AKO cells expressing AID-cyclin A were generated as described in Materials and Methods. The cells were grown in the presence of IAA and/or Dox and harvested at the indicated time. Lysates were prepared and the expression of cyclin A was detected using immunoblotting. Control HeLa cells were included in the analysis to indicate the position and abundance of endogenous cyclin A. The positions of molecular size standards (in kDa) are indicated on the left. (b) Cyclin AKO cells expressing AID-cyclin A were grown in the presence of IAA and/or Dox and harvested at the indicated time. Lysates were prepared and the expression of cyclin A was detected using immunoblotting.
As cyclin A–CDK2 activity fluctuates during the cell cycle, we further analysed another protein that does not normally vary during the cell cycle. TRIP13 is an AAA+-ATPase that functions in various processes including the silencing of the spindle-assembly checkpoint. It is involved in converting the key checkpoint component MAD2 from the active C-MAD2 conformation to the inactive O-MAD2 conformation [17]. TRIP13KO cells expressing AID-TRIP13 were incubated with IAA, Dox, or both IAA and Dox together. As with other proteins we tested, addition of IAA and Dox together promoted more rapid and complete depletion of AID-TRIP13 compared to individual chemicals alone (Figure 4(a)). To further analyse the functional consequences of TRIP13 depletion, cells were first incubated with IAA and/or Dox for 24 h before challenged with nocodazole. TRIP13 is essential for the cells to be trapped in mitosis by spindle poisons such as nocodazole [13]. We found that treatment with either IAA or Dox individually did not affect the ability of nocodazole to activate the spindle-assembly checkpoint (as indicated by the phosphorylation of histone H3Ser10) (Figure 4(b)). By contrast, the accumulation of histone H3Ser10 phosphorylation was impaired after the cells were treated with IAA and Dox together, indicating that the loss of TRIP13’s functions occurred only after a significant depletion of TRIP13 using both IAA and Dox. In agreement with this, flow cytometry analysis revealed that the nocodazole-treated cells displayed 8N DNA contents (indicating DNA re-replication) only in the presence of both IAA and Dox together (Figure 4(c)).
Figure 4.
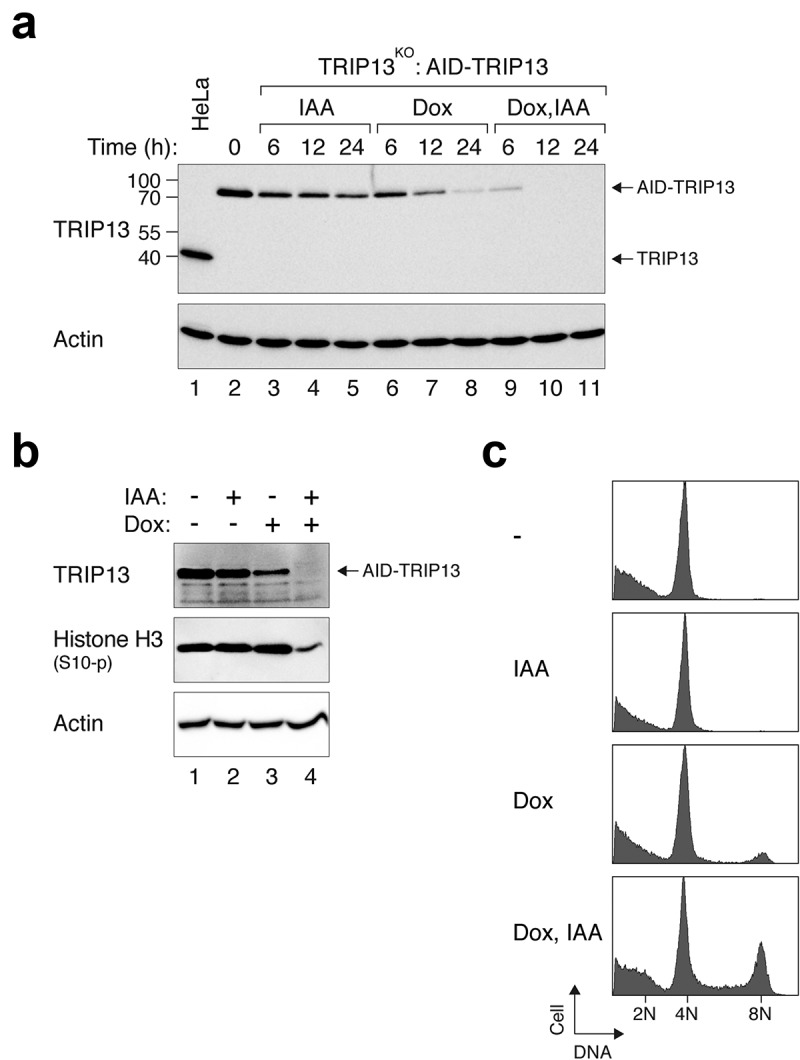
Efficient depletion of TRIP13 using IAA and Dox. (a) Conditional depletion of TRIP13. TRIP13KO cells expressing AID-TRIP13 were grown in the presence of IAA and/or Dox and harvested at the indicated time. Lysates were prepared and the expression of TRIP13 was detected using immunoblotting. Control HeLa cells were included in the analysis to indicate the position and abundance of endogenous TRIP13. The positions of molecular size standards (in kDa) are indicated on the left. (b) Depletion of TRIP13 results in defective spindle-assembly checkpoint. TRIP13KO cells expressing AID-TRIP13 were grown in the presence of IAA and/or Dox for 24 h. The cells were then incubated with nocodazole for 16 h. Lysates were prepared and analysed with immunoblotting. (c) TRIP13KO cells expressing AID-TRIP13 were incubated with IAA and/or Dox and nocodazole as described in panel B. The cells were fixed and analysed with flow cytometry. The positions of 2N, 4N, and 8N DNA contents are indicated.
From the above results, we believed that combining the tetracycline-controlled system and AID system together could be useful for the analysis of many genes. To facilitate others to use the system, we have designed two new vectors that enables simple subcloning to produce AID-fusion proteins under the control of tetracycline-controlled promoters (Figure 5(a,b)). We constructed both a conventional plasmid for transfection (pUHD-AID) and another for retroviral infection (pRevTRE-AID). The pUHD-AID vector can be used for expressing AID-tagged proteins in Tet-Off cell lines. The pRevTRE-AID plasmid can be used to produce retroviruses for transducing into Tet-Off cell lines to express AID-tagged proteins. The plasmids carry either a puromycin-resistant gene (pUHD-AID) or a hygromycin-resistant gene (pRevTRE-AID) to allow antibiotic selection of stable integration in mammalian cells. These constructs should be useful in studies requiring rapid and tight suppression of gene expression in mammalian cells.
Figure 5.

New tetracycline- and AID-controlled vectors. (a) Cloning sites of pUHD-AID. Part of the AID tag is shown (the AID tag is 228 amino acid long (~25 kDa)). The AID tag is followed by a 5xGA linker and multiple cloning sites. Only unique restriction enzyme sites are shown (except the two BamHI sites). *: termination codon. A puromycin-resistant gene is present in the construct for antibiotic selection in mammalian cells. The complete sequence of pUHD-AID can be found in the Supplemental information. (b) Cloning sites of pRevTRE-AID. The AID tag is followed by a 5xGA linker and multiple cloning sites. Only unique restriction enzyme sites are shown. *: termination codon. A hygromycin-resistant gene is expressed from this construct to allow antibiotic selection in mammalian cells. The complete sequence of pRevTRE-AID can be found in the Supplemental information. (c) Workflow for conditional gene inactivation using pUHD-AID or pRevTRE-AID. (1) Silence mutations (changing 3–4 bases) can be introduced into the cDNA to render the cDNA resistant to the CRISPR-Cas9. Alternatively, CRISPR-Cas9 that targets the endogenous gene but not the cDNA (e.g. against a intron-exon boundary) can be used. (2) Cloning sites are shown in panels A and B. (3–4) After cells are transfected with DNA or infected with retroviruses, they are selected with puromycin (pUHD-AID) or hygromycin B (pRevTRE-AID). (5) The cells are then transfected with CRISPR-Cas9 against the gene of interest. Various selection or sorting methods can be used to enrich the transfected cells. (6) Although the entire workflow can be initiated using cells stably expressing TIR1, we found that putting TIR1 into the cells at this stage generated more clones that could effectively respond to IAA. (7) Single clones are isolated and analysed for clones lacking the expression of the endogenous gene of interest, but expressing the AID-tagged version (ideally at a level similar to the endogenous protein) which can be eliminated in the presence of Dox and IAA.
Discussion
Several previous studies had demonstrated that AID-tagged proteins can be degraded relatively rapidly in the presence of IAA. For example, in DLD-1 and RPE-1 cells, 90% of the substrates can be depleted around 30–120 min [11]. Likewise, several AID-fusion proteins can be degraded more rapidly in chicken DT40 cells than we showed in this study [12,18]. It is likely that factors including the nature of the protein and the cell lines used could affect the efficiency of depleting AID-fusion proteins. Furthermore, in both yeast [6] and mammalian cells [14], rapid degradation of AID-fusion proteins requires TIR1 to be expressed at a relatively high level. But a limitation is that a high level of TIR1 induces the destruction of some AID-fusion proteins even before the addition of IAA [3] (our unpublished data). The possibility that this could be in part caused by contaminating auxin in the serum of the culture medium has been suggested [3]. To prevent premature degradation of AID-fusion proteins, strategies including putting the TIR1 under the control of tetracycline-regulated promoters have been used [14]. However, this could defeat the purpose of using the AID system as the induction of TIR1 was relatively slow.
In this study, we found that combining the tetracycline-controlled system and AID system together facilitated faster and tighter control of protein expression than the individual technology alone. Similar approaches also worked well in yeast cells [19]. The properties of the combined system are particularly useful for analysing the effects of the inactivation of essential genes in mammalian cells, when rapid and tight suppression of gene expression is required. In the past, this can be achieved using technologies such as RNAi. For example, we have previously designed a vector system for expressing shRNA to knockdown the endogenous gene while rescuing the cells using a shRNA-resistant cDNA under the control of an inducible promoter [20]. Although this and similar approaches can achieve conditional inactivation of essential genes, the incomplete knockdown by shRNAs and the relatively slow responses of inducible promoters are major drawbacks. The combined tetracycline-controlled system, AID system, and CRISPR-Cas9-mediated gene disruption described here overcomes many of these limitations. The workflow of generating cells expressing for conditional gene inactivation is summarised in Figure 5(c). Advantages of this approach include that the tetracycline-controlled AID-tagged gene does not needed to integrate into the endogenous loci reduces the time required to generate the conditional deficiency cell lines. This is particularly useful for studies involving cancer cell lines because these cells frequently contain multiple gene copies. Moreover, as this is already a rescue system by design, the effects after the AID-tagged gene is turned off can be confidently attributed to the functions of the gene rather than off-target effects of the CRISPR-Cas9.
Experimental procedures
DNA constructs
Cyclin A CRISPR-Cas9 targeting CTCCCGGGAGCAGTGATGTT was prepared by ligating the annealing product of 5´-CACCGCTCCCGGGAGCAGTGATGTT-3´ and 5´-AAACAACATCACTGCTCCCGGGAGC-3´ to BbsI-cut pX330 [21] (obtained from Addgene, Cambridge, MA, USA). AID-cyclin A was not targeted by the CRISPR-Cas9 because the targeting region spanned the UTR and the ORF of cyclin A. To generate the AID-cyclin A retrovirus construct, the XhoI-NcoI fragment from pUHD-AID and the NcoI-BamHI from FLAG-cyclin A in pUHD-P3 [22] were ligated into a modified pRevTRE2 (lacking an XhoI site) (Ma & Poon, 2018).
CDK2 CRISPR-Cas9 targeting CAGAGGCCACTCACGTGTCC was prepared by ligating the annealing product of 5´-CACCGCAGAGGCCACTCACGTGTCC-3´ and 5´-AAACGGACACGTGAGTGGCCTCTGC-3´ to BbsI-cut pX330. AID-CDK2 retrovirus construct was generated as described for AID-cyclin A except that the NcoI–BamHI fragment containing CDK2 [23] was used as an insert.
TRIP13 CRISPR-Cas9 and AID-TRIP13 constructs were generated as described previously [17]. As the PAM sequence of TRIP13 CRISPR-Cas9 spanned the UTR and the ORF, the AID-TRIP13 construct was not targeted by the CRISPR-Cas9.
TIR1-myc retrovirus construct was generated as described previously [17].
New AID constructs
pUHD-AID was generated by first mutating an internal NcoI site in the AID cDNA with a double PCR procedure using 5´-AAGCTAGCATGGGCAGTGTCGAG-3´, 5´-GTTTGCCCATCGTAAAAGAGCTG-3´ and 5´-TCAGCTCTTTTACGATGGGCAAAC-3´, 5´-CACCATGGTGAATAGGATATCGGCA-3´ using pMK106 [6] as the template. The PCR product was digested with NheI and NcoI and ligated to the NheI (partial)- and NcoI-cut pUHD-P3/PUR [24].
To facilitate the cloning of AID-fusion constructs into retroviral vectors, several commonly used restriction enzyme sites in pRevTRE2 (Clontech, Palo Alto, CA, USA) were first removed. To remove one of the two XhoI sites, the XhoI–XhoI fragment pRevTRE2 was removed before ligate with the XhoI–SalI fragment from pRevTRE2 to generate pRevTRE2.1. An NcoI site in the hygromycin-resistant gene in pRevTRE2.1 was mutated by ligating the PvuI-digested PCR product (primers: 5´-CTTCGGTCCTCCGATCGTT-3´ and 5´-TCGATCGCATCCATAGCCTCCG-3´; template: pRevTRE2.1) into the PvuI-digested pRevTRE2.1. To destroy the EcoRI site downstream of the ampicillin-resistant gene, an EcoRI-mutated intermediate plasmid were generated by ligating the MfeI–EcoRI-digested PCR product (primers: 5´-CAACAATTGATACCAGATCACCG-3´ and 5´-TCGATCGCATCCATAGCCTCCG-3´; template: pRevTRE2.1) into EcoRI-digested pRevTRE2.1. The two EcoRI sites downstream of the 5´-LTR were destroyed by ligating the MfeI–ClaI-cut double PCR product (primers of the first PCR: 5´-CGCAATTGCGATCTGATCAGC-3´ and 5´-TCTCGCTGAACTCCCCAATG-3´; 5´-CATTGGGGAGTTCAGCGAGA-3´ and 5´-ACCTACAGGTGGGGTCTTTCATTCCC-3´; template: NcoI-mutated pRevTRE2.1; primers for the second PCR: 5´-CATTGGGGAGTTCAGCGAGA-3´ and 5´-ACCTACAGGTGGGGTCTTTCATTCCC-3´) into the EcoRI–ClaI-cut EcoRI-mutated intermediate. The resulting plasmid was pRevTRE2.2. pRevTRE-AID was generated by putting the TRE mini-CMV (generated by using primers: 5´-CGTATGTCGACGCCCTTTCGTCT-3´ and 5´-CCGATCAATAGATCTTATCATGTCTG-3´; template: modified pUHD-P3 (lacking an XhoI site) [25]; then cut with SalI–NheI) and AID (generated by PCR using primers: 5´-AAGCTAGCATGGGCAGTGTCGAG-3´ and 5´-CCGATCAATAGATCTTATCATGTCTG-3´; template: pUHD-AID; then cut with NheI–EcoRI) into the XhoI–EcoRI-digested pRevTRE2.2. Both pUHD-AID and pRevTRE-AID will be deposited to Addgene.
Cell culture
The HeLa used in this study was a clone that expressed the tTA tetracycline transactivator [23]. Cells were transfected with plasmids using a calcium phosphate precipitation method [26]. Retroviruses were produced by co-transfection of cDNA expressing retrovirus constructs and VSV-G plasmids into Phoenix-gp cells [27] in the presence of 5 µg/ml of polybrene (Sigma-Aldrich, St. Louis, MO, USA). Cells were treated with the following reagents at the indicated final concentration: blasticidin (5 µg/ml) (Life Technologies, Carlsbad, CA, USA), doxycycline hydrochloride (Dox) (2 µg/ml), hygromycin B (200 µg/ml) (Life Technologies), indole-3-acetic acid (IAA) (50 µg/ml) (Sigma-Aldrich), nocodazole (0.33 µM) (Enzo Life Sciences, Farmingdale, NY, USA), and puromycin (0.6 µg/ml) (Sigma-Aldrich).
Conditional depletion cell lines
AID-cyclin A in cyclin AKO cells were generated by first infecting HeLa cells with retroviruses expressing AID-cyclin A and grown in medium containing hygromycin B for 2 wk. The cells were then co-transfected with cyclin A CRISPR-Cas9 in pX330 and a plasmid expressing blasticidin-resistant gene (a gift from Tim Hunt, Cancer Research UK). After enriching the transfected cells with blasticidin selection for 36 h, the cells were infected with retroviruses expressing TIR1-myc. The cells were selected with puromycin and the single cell-derived colonies were obtained by limiting dilution in 96-well plates. AID-CDK2 in CDK2KO cells were generated the same way except that AID and CRIPSPR-Cas9 constructs specific for CDK2 were used. TRIP13KO cells were generated as previously described [17]. AID-TRIP13 in TRIP13KO cells were generated by infecting HeLa cells with retroviruses expressing AID-TRIP13, followed by TRIP13 CRIPSR-Cas9-mediated knockout and TIR1 infection.
Indel analysis
Insertions and deletions at the knockout loci were quantified by PCR amplification of the edited regions with the pair of primers 5´-GGCCGCGAGCGCTTTCATTGGTCCATTTC-3´ and 5´-TGACTGGCAGTGAACCTGAAAGAGCACTGG-3´ (for CCNA2) or 5´-TGGGAGGCGGCAACATTGTTTCAAGTTGGC-3´ and 5´-AGATCTCTCGGATGGCAGTACTGGGCACAC-3´ (for CDK2). The PCR products were sequenced (primers: 5´-GGCCGCGAGCGCTTTCATTGGTCCATTTC-3´ for CCNA2 and 5´-TGGGAGGCGGCAACATTGTTTCAAGTTGGC-3´ for CDK2) and analysed using the Tracking of Indels by Decomposition (TIDE) method [28]. TRIP13 indel analysis was performed as described previously [17].
Quantitative real-time PCR
Total RNA was extracted using NucleoSpin RNA kit (Macherey-Nagel GmbH & Co. KG, Düren, Germany). Reverse transcription was carried out with High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). RT-PCR was performed using Roche LightCycler 480 Instrument II in the presence of SYBR-green (Roche, IN, USA). Primers for qPCR were as follows: 5´-GGGAAATCGTGCGTGACATT-3´ and 5´-GGAACCGCTCATTGCCAAT-3´ (actin), 5´-TCCAAGGAGAAGAGTGCTTGTC-3´ and 5´-TTTGGCAGGAAACCATCACG-3´ (AID). The mRNA expression was normalized to that of actin. Fold-change of sample normalized to control was calculated by 2–∆∆Ct method.
Flow cytometry analysis
Flow cytometry analysis after propidium iodide staining was performed as described previously [29] except that a Coulter Epics XL flow cytometer was used (Beckman Coulter, Brea CA, USA).
Antibodies and immunological methods
Antibodies against beta-actin (Sigma-Aldrich), phospho-histone H3Ser10, and TRIP13 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were obtained from the indicated suppliers. Antibodies against CDK2 (AN4.3) and cyclin A (AT10) were gifts from Tim Hunt (Cancer Research UK). Immunoblotting was performed by separating the samples on SDS-PAGE before transferring to Immobilon PVDF membrane (Merck Millipore, Darmstadt, Germany). The membrane was blocked with TBST (10 mM Tris-HCI pH 8.0, 150 mM NaCl, 0.05% Tween-20) containing 4% dry milk or 1% BSA for 30 min at 25°C. The membrane was incubated with primary antibodies in TBST 2% dry milk or 1% BSA overnight at 4°C. After washing with TBST, the membrane was incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulin (Thermo Fisher Scientific, Waltham, MA, USA) at 25°C for 2 h. The membrane was then washed three times with TBST, developed using ECL chemiluminescence [30], and detected using a ChemiDoc Touch imaging system (Bio-Rad, Hercules, CA, USA).
Funding Statement
This work was supported by the Health and Medical Research Fund [04153526];Innovation and Technology Commission - Hong Kong [ITCPD/17-9];Research Grants Council, University Grants Committee [T12-704/16-R];Research Grants Council, University Grants Committee [16100017];Research Grants Council, University Grants Committee [16100417].
Acknowledgments
We thank Chun Wing Fung and Ziqi Xu for technical support. This work was supported in part by grants from the Research Grants Council (16100017 to H.T.M., 16100417 and T12-704/16-R to R.Y.C.P.), Health and Medical Research Fund (04153526 to H.T.M.), and Innovation and Technology Commission (ITCPD/17-9 to R.Y.C.P.).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Gossen M, Bujard H.. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992;89:5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gossen M, Freundlieb S, Bender G, et al. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268:1766–1769. [DOI] [PubMed] [Google Scholar]
- [3].Natsume T, Kanemaki MT.. Conditional degrons for controlling protein expression at the protein level. Annu Rev Genet. 2017;51:83–102. [DOI] [PubMed] [Google Scholar]
- [4].Hayashi K, Neve J, Hirose M, et al. Rational design of an auxin antagonist of the SCF(TIR1) auxin receptor complex. ACS Chem Biol. 2012;7:590–598. [DOI] [PubMed] [Google Scholar]
- [5].Delker C, Raschke A, Quint M. Auxin dynamics: the dazzling complexity of a small molecule’s message. Planta. 2008;227:929–941. [DOI] [PubMed] [Google Scholar]
- [6].Nishimura K, Fukagawa T, Takisawa H, et al. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. 2009;6:917–922. [DOI] [PubMed] [Google Scholar]
- [7].Kanke M, Nishimura K, Kanemaki M, et al. Auxin-inducible protein depletion system in fission yeast. BMC Cell Biol. 2011;12:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Trost M, Blattner AC, Lehner CF. Regulated protein depletion by the auxin-inducible degradation system in Drosophila melanogaster. Fly (Austin). 2016;10:35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bence M, Jankovics F, Lukácsovich T, et al. Combining the auxin-inducible degradation system with CRISPR/Cas9-based genome editing for the conditional depletion of endogenous Drosophila melanogaster proteins. FEBS J. 2017;284:1056–1069. [DOI] [PubMed] [Google Scholar]
- [10].Zhang L, Ward JD, Cheng Z, et al. The auxin-inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans. Development. 2015;142:4374–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Holland AJ, Fachinetti D, Han JS, et al. Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc Natl Acad Sci U S A. 2012;109:E3350–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wood L, Booth DG, Vargiu G, et al. Auxin/AID versus conventional knockouts: distinguishing the roles of CENP-T/W in mitotic kinetochore assembly and stability. Open Biol. 2016;6:150230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ma HT, Poon RYC. TRIP13 functions in the establishment of the spindle assembly checkpoint by replenishing O-MAD2. Cell Rep. 2018;22:1439–1450. [DOI] [PubMed] [Google Scholar]
- [14].Natsume T, Kiyomitsu T, Saga Y, et al. Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep. 2016;15:210–218. [DOI] [PubMed] [Google Scholar]
- [15].Woo RA, Poon RY. Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle. 2003;2:316–324. [PubMed] [Google Scholar]
- [16].Yam CH, Fung TK, Poon RY. Cyclin A in cell cycle control and cancer. Cell Mol Life Sci. 2002;59:1317–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ma HT, Poon RY. TRIP13 regulates both the activation and inactivation of the spindle-assembly checkpoint. Cell Rep. 2016;14:1086–1099. [DOI] [PubMed] [Google Scholar]
- [18].Samejima K, Ogawa H, Ageichik AV, et al. Auxin-induced rapid degradation of inhibitor of caspase-activated DNase (ICAD) induces apoptotic DNA fragmentation, caspase activation, and cell death: a cell suicide module. J Biol Chem. 2014;289:31617–31623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tanaka S, Miyazawa-Onami M, Iida T, et al. iAID: an improved auxin-inducible degron system for the construction of a ‘tight’ conditional mutant in the budding yeast Saccharomyces cerevisiae. Yeast. 2015;32:567–581. [DOI] [PubMed] [Google Scholar]
- [20].Ma HT, On KF, Tsang YH, et al. An inducible system for expression and validation of the specificity of short hairpin RNA in mammalian cells. Nucleic Acids Res. 2007;35:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cong L, Ran FA, Cox D, et al. Mu.genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fung TK, Yam CH, Poon RY. The N-terminal regulatory domain of cyclin A contains redundant ubiquitination targeting sequences and acceptor sites. Cell Cycle. 2005;4:1411–1420. [DOI] [PubMed] [Google Scholar]
- [23].Yam CH, Siu WY, Lau A, et al. Degradation of cyclin A does not require its phosphorylation by CDC2 and cyclin-dependent kinase 2. J Biol Chem. 2000;275:3158–3167. [DOI] [PubMed] [Google Scholar]
- [24].Ma HT, Tsang YH, Marxer M, et al. Cyclin A2-cyclin-dependent kinase 2 cooperates with the PLK1-SCFbeta-TrCP1-EMI1-anaphase-promoting complex/cyclosome axis to promote genome reduplication in the absence of mitosis. Mol Cell Biol. 2009;29:6500–6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Huang S, Tang R, Poon RY. BCL-W is a regulator of microtubule inhibitor-induced mitotic cell death. Oncotarget. 2016;7:38718–38730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kingston RE, Chen CA, Rose JK. Calcium phosphate transfection. Curr Protoc Mol Biol 2003. Chapter 9: Unit9.1. 63:9–1. [DOI] [PubMed] [Google Scholar]
- [27].Pear WS, Nolan GP, Scott ML, et al. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci U S A. 1993;90:8392–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Brinkman EK, Chen T, Amendola M, et al. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Siu WY, Arooz T, Poon RY. Differential responses of proliferating versus quiescent cells to adriamycin. Exp Cell Res. 1999;250:131–141. [DOI] [PubMed] [Google Scholar]
- [30].Haan C, Behrmann I. A cost effective non-commercial ECL-solution for Western blot detections yielding strong signals and low background. J Immunol Methods. 2007;318:11–19. [DOI] [PubMed] [Google Scholar]