

ABSTRACT
Prostate cancer (PC) is the most commonly diagnosed cancer in men and is the second leading cause of male cancer-related death in North America. Metabolic adaptations in malignant PC cells play a key role in fueling the growth and progression of the disease. Unfortunately, little is known regarding these changes in cellular metabolism. Here, we demonstrate that centromere protein F (CENPF), a protein associated with the centromere-kinetochore complex and chromosomal segregation during mitosis, is mechanically linked to altered metabolism and progression in PC. Using the CRISPR-Cas9 system, we silenced the gene for CENPF in human PC3 cells. These cells were found to have reduced levels of epithelial-mesenchymal transition markers and inhibited cell proliferation, migration, and invasion. Silencing of CENPF also simultaneously improved sensitivity to anoikis-induced apoptosis. Mass spectrometry analysis of tyrosine phosphorylated proteins from CENPF knockout (CENPFKO) and control cells revealed that CENPF silencing increased inactive forms of pyruvate kinase M2, a rate limiting enzyme needed for an irreversible reaction in glycolysis. Furthermore, CENPFKO cells had reduced global bio-energetic capacity, acetyl-CoA production, histone acetylation, and lipid metabolism, suggesting that CENPF is a critical regulator of cancer metabolism, potentially through its effects on mitochondrial functioning. Additional quantitative immunohistochemistry and imaging analyzes on a series of PC tumor microarrays demonstrated that CENPF expression is significantly increased in higher-risk PC patients. Based on these findings, we suggest the CENPF may be an important regulator of PC metabolism through its role in the mitochondria.
KEYWORDS: CENP-F, prostate cancer, mitochondria
Introduction
Prostate cancer (PC) remains a major public health concern; it is the most common malignant neoplasia among men and is the 6th leading cause of cancer-related male mortality worldwide [1]. In the U.S., there are more than 200,000 newly diagnosed cases and nearly 40,000 deaths from PC annually. The current standard methods of diagnosing and monitoring PC involve testing the levels of prostate specific antigen (PSA) and digital rectal screening. However, recent studies have reported that these procedures contribute little to no reduction in overall PC mortality [2,3].
Proliferating PC cells generate the energy required to support accelerated cell division by modulating its metabolism. These cells exhibit increased glucose uptake and lactate production [4,5]. Rapidly dividing cells are able to convert glucose into lactate, regardless of oxygen availability, a phenomenon known as aerobic glycolysis or the Warburg effect [2]. The Warburg effect has been demonstrated to be a key contributor of tumorigenesis and can be a target for cancer therapy [6]. Many oncogenes and/or tumor suppressor genes have been reported to be involved in the metabolic switch towards aerobic glycolysis. For instance, oncogenes AKT [7], c-Myc [8], Ras [9], and HIF-1α [10] promote the Warburg effect; whereas tumor suppressors p53 and PTEN inhibit it [11,12]. Epithelial-mesenchymal transition (EMT) also plays a pivotal role in the development of metastatic castration-resistant prostate cancer (mCRPC) [13] and chemoresistant PC [14]. However, current understandings of the dynamic metabolic changes underlying EMT are limited.
Centromere protein F (CENPF), which is located on chromosome 1q41, encodes for a protein that acts as part of the centromere-kinetochore complex and is a component of the nuclear matrix during G2 of interphase [15]. As a regulator of chromosome segregation, CENPF is expressed in a cell cycle-dependent manner; it gradually accumulates during the cell cycle, reaches peak levels in the G2/M phase, and then degrades upon completion of mitosis [16]. CENPF is upregulated in PC and plays an important part in malignant progression [17,18]. Interestingly, CENPF also significantly enhances chemotherapeutic sensitivity [19]. It was recently observed that CENPF and forkhead box M1 (FOXM1) cooperate together, acting as synergistic master regulators of malignancy in PC [20]. Furthermore, COUP transcription factor 2 (COUP-TFII) may be promoting metastasis in PC through the CENPF signaling pathway [21].
In this study, we aimed to determine the functional role of CENPF and its underlying mechanism in PC progression. Our experimental results showed that silencing CENPF expression significantly inhibited EMT, cell proliferation, and colony formation, while also enhancing global phosphorylation and sensitivity to anoikis-induced apoptosis. Additional proteomics and computational analysis demonstrated that CENPF may regulate cancer cell metabolism through the phosphorylation of pyruvate kinase M2 (PKM2). Moreover, Seahorse Respirometry revealed that CENPF loss leads to a general decrease in metabolic activity in PC cells, which is characterized by reduced respiration and glycolysis. In summary, we propose CENPF as a novel target protein, providing a potential effective paradigm in curing PC.
Materials and methods
Cell culture and cell lines
Human prostate cancer cell lines PC3, DU145, and 22RV1 were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and were cultured in 10% fetal bovine serum (FBS)/Dulbecco’s Modified Eagle medium (DMEM) or RPM1640 medium. Cells were maintained in humidified incubators set at 37°C with 5% CO2. A CENPF-knockout PC3 cell line (CENPFKO) was constructed using the CRISPR/Cas9 system by ALSTEM, LLC (Richmond, CA). The combination of two gRNAs (GTTTCAGCTTGACAGTCTCG and CATTATTGACAGAGAAGTGC) targeted the deletion of an 85 bp fragment in the exon1-intron1 junction, with 32 bp of this deletion fragment in the exon1 and 54 bp deleted in intron. For transient transfection of cells with a CENPF-expressing construct or siRNA CENPF (ThermoScientific), we followed the previously established protocol using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions [22]. All cell lines were confirmed and tested for mycoplasma every 6 months during the experimental period.
Antibodies and reagents
The following antibodies were used: β-Actin (A1978) from Sigma, E-Cadherin (610182), Phosphotyrosine (610000) from BD Transduction Laboratories, ACSS2 (PA5-52059) from ThermoFisher Scientific, ALDH7A1 (ab154218), CENPF (ab5), and OXPHOS (ab110413) from Abcam, and Tom40 (sc365467) from Santa Cruz Biotechnology, phospho-mTOR (2971), phospho-β-catenin (9564), phospho-p38 MAPK (9216), phospho-PAK1 (2601), phospho-NF-κB (3033), TCF8/ZEB1 (3396), phospho-PKM2 (3827), PKM2 (4053), Snail (3879), N-cadherin (13116), MMP2 (13132), MMP9 (13667), Slug (9585), phospho-p70 S6 Kinase (9206), phospho-Src Family (2101), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (4370), β-Catenin (8480), Cleaved-PARP (9541), and Fatty acid and Lipid Metabolism Antibody Kit (8335), Acetyl-Histone Antibody Kit (9933), Tight Junction Antibody Kit (8683) and HRP-conjugated secondary antibodies (7074, 7076) from Cell Signaling Technology.
Proliferation assay
Cell proliferation was analyzed using trypan blue staining. Cells were seeded onto 6-well culture plates (2x102 cells/well) and cultured for 24 hrs. Each well culture was maintained and incubated for 2 d. The medium was replaced every day. The results are presented as the percentage of viable cells relative to that of the control.
Two-dimensional (2D) and three-dimensional (3D) colony formation assay
The growth capability of CENPFKO and control cells was examined using 2D colony formation assays. Approximately 500 controls and CENPFKO cells were seeded onto separate 15 cm culture plates. After incubation at 37°C for 14 d, the cells were washed with PBS twice, fixed with methanol and then stained with 0.1% crystal violet. The number of colonies containing >30 cells was counted under the microscope. Anchorage-independent growth was measured through a 3D colonization assay. Cells were seeded at 1 × 104 in 3 ml 0.35% agar in DMEM/FBS, overlaid on 2 ml of 0.7% agar in DMEM/FBS, in six-well plates. Plates were incubated for up to 14 d and cells were fed every 3–4 d. After two weeks, the plates were stained with MTT and the number of colonies that developed within each well was counted and visualized under a microscope. Colonies that were comprised of more than 10 cells were scored as positive. Experiments were run in triplicate for each cell line and data are representative of three independent trials.
Wound-healing assay
For the wound-healing migration assay, CENPFKO and control cells were seeded on 6-well plates at a density of 1 × 105 cells/well in culture medium. After 24 hrs post seeding, the confluent monolayer of the culture was scratched with a fine pipette tip and migration capacity was visualized under a microscope. The rate of wound closure was observed for an additional 24 hrs during incubation.
Anoikis assays
Cells (5 × 105) were plated onto poly-HEMA-coated six-well plates (Corning, Tewksbury, MA, USA) in growth medium to prohibit attachment. After 24, 48 and 72 hrs in suspension, cells were transferred onto regular cell culture plates in growth medium supplemented with FBS (1%) to aid attachment. Cells were incubated for 0, 1, 2, and 3 hrs prior to the MTT assay (no. G4100, Promega, Madison, WI, USA). Absorbance was read using a microplate reader (Tecan, Männedorf, Switzerland). Experiments were repeated three times.
Western blot analysis
Cells were lysed with RIPA buffer (20 mM Tris, 150 mM NaCl, 1% Nonidet, P-40, 0.1 mM EDTA) (Pierce, ThermoFisher) supplemented with a phosphatase inhibitor cocktail (ThermoFisher). The protein concentration of each sample was measured using the Bradford Protein Kit Assay, according to the manufacturer’s instructions (Pierce, Thermofisher). Equal amounts of protein extract were separated via SDS-PAGE and transferred onto a PVDF membrane. The membranes were then blocked with 5% bovine serum albumin or 5% nonfat milk in TBST buffer [2.42 g/L Tris–HCl, 8 g/L NaCl, and 1 mL/L Tween 20 (pH 7.6)] and incubated overnight at 4°C with specific primary antibodies in TBST. The membranes were then incubated with secondary antibodies conjugated with horseradish peroxidase. β-actin was used as an internal control.
Immunohistochemistry (IHC) analysis
Two independent commercial PC tissue microarrays (TMAs) were purchased from US Biomax (Derwood, MD). Detailed information was not available except sex, TMN, grade, and stages. TMAs were immunestained with mAb CENPF, according to the manufacturer’s recommended protocol. Immunostaining was considered positive when more than 10% of all tumor cells were immunoreactive. For quantification, the TMAs were scored for IHC intensity by two independent investigators. Tumors that showed no positive staining were given a score of 0, those with weak staining were given a score of 1, and tumors with strong IHC staining were given a score of 2.
Acetate measurement assay
The acetate colorimetric assay kit (BioVision, Milpitas, CA) was used according to the instructions provided. The reaction was incubated at room temperature for 40 min. Acetate fluorescence (absorbance at 450nm) was measured using a POLARstar Galazy fluorometer in a 96-well plate.
Free fatty acid (FFA) and cholesterol quantification
Quantification of FFAs and cholesterol were determined using the Free Fatty Acid Quantification Kit and Cholesterol Quantification Kit (FFA: MAK044 and Cholesterol: MAK043, Sigma MO, USA). Briefly, cells were lysed in 1% Triton X-100 in chloroform (w/v). The samples were centrifuged at 13,000 × g for 10 min to remove insoluble debris. The organic phase was collected and air dried in a 50°C dry bath for 20 min. Samples were vacuum dried for 30 min to remove traces of chloroform. The dried lipids were resuspended via vortex in fatty acid assay buffer and further quantified, using the manufacturer’s instructions.
Global proteomics
All chemicals used for preparation of liquid chromatography-tanden mass spectrometry (LC-MS/MS) samples were of at least sequencing grade and were purchased from Sigma-Aldrich, unless otherwise stated. Whole protein was extracted from CENPFKO and control cells using 4% SDS-containing buffer. Protein concentration was measured with the Pierce 660nm Assay Kit. From each sample, 60 μg of protein was digested with trypsin, using FASP, and labeled with TMT6plex reagents in parallel. After TMT labeling, peptides were merged, desalted with C18, and fractionated by high-pH reverse phase liquid chromatography (RPLC). Peptides were sequenced and quantified by LC-MS/MS. Database searching and quantification were performed using Proteome Discoverer (v2.1). A standard false discovery rate of 1% was applied to filter peptide-spectrum matches, peptide identifications, and protein identifications. Next, data was normalized in the Proteome Discoverer environment, assuming that the total peptide amount in each different sample was the same. In addition, TMT126-labeled samples wereselected as a reference channel and ratios between samples were computed by Proteome Discoverer [23]. The fold-changes between CENPFKO and parent PC3 cells were determined as the average ratios of the three TMT tags for each group. The significance of the differential abundances between the two groups was computed using Welch’s t-test on the scaled abundances and Benjamini-Hochberg (BH) procedure for multiple testing corrections. Differentially expressed proteins (DEPs) were identified by applying a threshold of BH FDR ≤ 0.01 and ratio ≥ 1.5.
Phosphoproteomics
Proteins were eluted from p-Y conjugated beads and resolved by a quick separation (5–6mm height) in a 12% SDS-PAGE gel. Upon in-gel reduction, alkylation, and trypsin digestion, the tryptic peptides were extracted and reconstituted in 0.2% formic acid. The peptides were then separated in a 15 cm EASY-Spray C18 column and analyzed on a LTQ Orbitrp Elite Mass Spectrometer (Thermo Scientific). Up to 20 collision-induced dissociation (CID) spectra were acquired per survey scan in the rapid CID scan mode. The raw MS/MS spectra were searched against the Uniprot Human Database (released on 01/22/16, including 20,985 sequences) with MaxQuant (v 1.5.2.8) and Andromeda. A stringent 1% false discovery rate was set for peptide and protein identifications [24].
Network analysis
We identified gene ontology biological processes (GOBPs) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways represented by the DEPs using DAVID [25]. The significance of terms was determined at a BH FDR ≤ 0.1. To delineate the associations of enriched biological functions and DEGs, a network model was constructed using HitPredict, a database of experimentally determined protein-protein interactions [26]. The database update version, 04Jul2017, was downloaded and only high confidence interactions among genes involved in the selected terms were parsed for the network construction. Genes involved in the same biological functions were grouped into the same module, each of which was labeled by their corresponding GOBP and KEGG pathway terms. The network was visualized using Cytoscape (v. 3.2.1) [27].
Seahorse respirometry assay
CENPFKO and control PC3 cells were seeded into a 24-well Seahorse culture plate at a density of 50,000 cells/well 24 hrs before the Seahorse assay. The media was then changed to a XF Base Medium (pH 7.4) supplemented with 10 mM glucose, 1mM sodium pyruvate and 1mM sodium glutamine. Cells equilibrated for 1 hour in a non-CO2 incubator at 37°C before assay start. Chemical reagents (Sigma) were used at final concentrations as follows: 1 μM oligomycin – an ATP synthase inhibitor, 1 μM (FCCP) carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone – an uncoupling agent, and a mixture of 0.5 μM antimycin A – a cytochrome C reductase inhibitor and 0.5 μM rotenone – a complex I inhibitor. Oxygen consumption rate (OCR) and extracellular acidification rate was monitored during the duration of the assay run. Results were normalized to protein concentrations determined by a BCA assay (Thermo Scientific).
Statistical analysis
Statistical analyses were performed using a two-tailed Student’s t-test. For all experiments with error bars, the standard deviation (SD) was calculated to indicate the variation within each experiment, and values represent mean ± SD. Differences were considered significant if p < 0.05.
Results
CENPF is associated with aggressive PC
To evaluate whether CENPF expression is associated with cancer progression, we performed two independent IHC imaging analyses using PC TMAs, as described in Methods section. Using commercial PC TMAs, we found that CENPF was overexpressed in approximately 70% of call cases, with an upward trend in tumors of higher grades (III-IV) (Figure 1(a)). IHC analyses using 2 different commercial TMAs revealed that CENPF was expressed in approximately 60% and 50% of all cases, respectively. We also found moderate staining in the normal prostate tissues and hyperplasia. CENPF expression was considerably higher in PC tissue compared to adjacent normal prostate tissue from the same patient. Levels of CENPF expression were also positively correlated to pathological stage and grade (Figure 2(b)). This observation was consistent with previous literature demonstrating CENPF as part of a signature that distinguishes biochemical recurrence and advanced cancer [28]. Collectively, the expression levels of CENPF in PC tissue were significantly higher than that of normal prostate tissue. These results point to the notion that CENPF expression may be potentially associated with PC progression.
Figure 1.
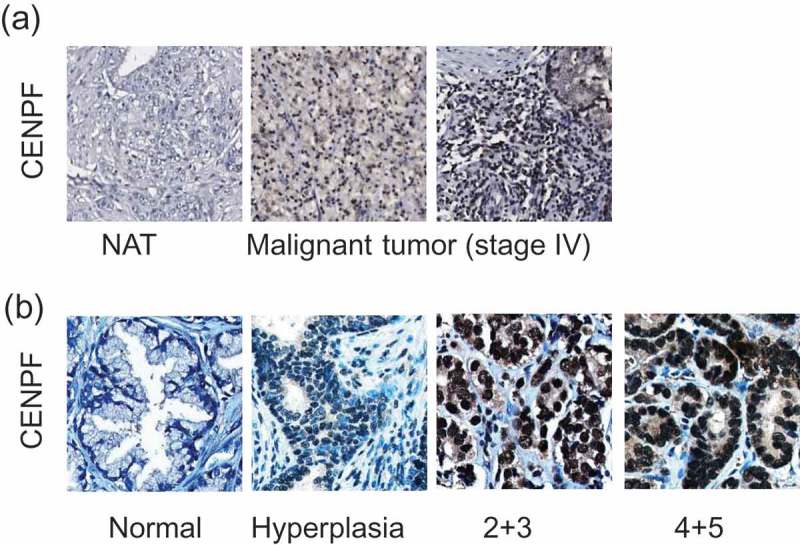
CENPF expression is correlated with PC progression. Two independent tissue microarrays (TMAs) were used. TMA slides were stained with CENPF specific antibody (brown) and counterstained with hematoxylin (blue). (a) The IHC images represent adjacent normal prostate tissues (NAT) or tumors from PC patients with different stages, as described in figures. (b) Representative IHC images show the differential CENPF protein levels.
Figure 2.
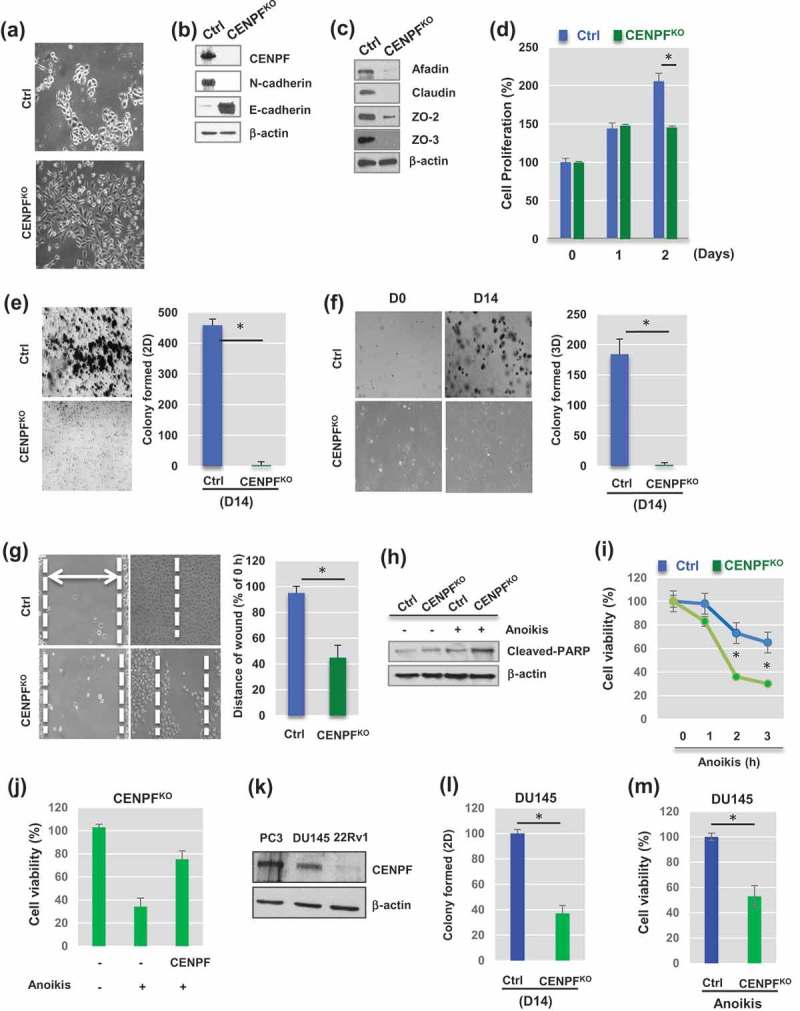
Knockout of CENPF reduces Epithelial-Mesenchymal transition (EMT) and slows proliferation of prostate cancer cells. (a) Morphological changes between parental PC3 cells (Ctrl) and CENPF – knockout PC3 cells (CENPFKO) were observed. Representative images were shown. (b) Western blot analysis demonstrated well-known EMT markers in Ctrl and CENPFKO cells. (c) Cell junction markers were assessed by Western blot analysis. (d) Cell proliferation was determined by trypan blue staining and compared in Ctrl and CENPFKO cells at 0, 24 and 48hrs. (e-f) Gene knockout of CENPF reduced colony formation ability in 2D (e) or 3D (f) settings. (g) Wound-healing assay showed the slower migration of CENPFKO cells, compared to Ctrl. Knockout in prostate cancer cell line. (h-i) Anoikis-induced cell apoptosis was enhanced when CENPF was downregulated. (h) Western blot analysis showed that cleaved form of PARK, an apoptotic marker, was increased in CENPF KO. (i) Levels of cell viability were measured by MTT assay after anoikis for 0, 1, 2, or 3h. All experiments were carried out in triplicate. (j) CENPF overexpression reversed the effects of CENPF knockout on cell viability in response to anoikis. (k) The protein expression of CENPF was compared in PC3, DU145, and 22RV1 PC cells. (l-m) Downregulation of CENPF reduced colony formation (l) and decreased cell viability in response anoikis (m) in DU145 cells. For all Western blot analysis, β-actin was used as the loading control. Data are representative of at least three different experiments and are expressed as the means ± SD.
CENPF loss reversed EMT in PC3 cells
To elucidate the role of CENPF in PC cells, CENPF was stably knocked-out in PC3 cells using the CRISPR/Cas9 system. These CENPF-knockout (CENPFKO) cells were then carefully characterized in the laboratory. There were some noted morphological alterations in the CENPFKO cells, compared to the control PC3 cell line (Figure 2(a)). Western blot analysis demonstrated that CENPF expression was lost in the CENPFKO cells (Figure 2(b)). Consistent with morphological changes, we found that protein expression of mesenchymal cell markers, including N-cadherin (Figure 2(b)), snail, slug, matrix metalloproteinase-2 (MMP2), MMP9, β-catenin (Supplementary Figure 1), was significantly reduced in CENPFKO cells, compared to control cells [29,30]. E-cadherin, an epithelial marker, dramatically increased in CENPFKO cells (Figure 2(b)). In addition, tight junction proteins (e.g. afadin, claudin, and zonula occludens (ZO), such as ZO-2 and ZO-3), which are involved in cell-cell contact regions [31], were also greatly reduced in CENPFKO cells. This was consistent with the morphological changes observed (Figure 2(c)).
Effects of CENPF silencing on the biological outcomes of PC cells
We next sought to determine if CENPF plays any major roles in controlling cell proliferation, migration, invasion, and colony formation capability. In-vitro wound healing, migration, and Matrigel invasion assays were conducted on both PC3 CENPFKO and control cells. Starting from day 2, the cell proliferation rate of CENPFKO cells was significantly reduced compared to controls (Figure 2(d)).
We further assessed the ability of the cells to form colonies in the 2-dimensonal (2D) and 3-dimensional (3D) planes. These colony formation assays revealed that CENPFKO cells formed very few colonies even after 2 wk of incubation (2D, p < 0.0001) (Figure 2(e)). Anchorage-independent cell proliferation was measured with the 3D colony formation assay, and, compared to control cells, CENPFKO cells showed very low colony formation capacity (3D, p < 0.003) (Figure 2(f)).
For the cell wound healing assay, a scratch was inflicted on cell cultures once they reached 90% confluence. We found that the wound healing capability of CENPFKO cells was significantly delayed than the control, suggesting that the migratory ability of CENPFKO cells may be impaired (Figure 2(g)). Knockout of CENPF also increased PC3 cell death by anoikis, which is apoptosis that is induced when cells are unable to attach to the extracellular matrix (ECM) [32]. Expression levels of cleaved PARP (c-PARP), an apoptotic marker, were higher in CENPFKO cells compared to controls (Figure 2(h)). This was consistent with the cell viability assay results (Figure 2(i)). Furthermore, a rescue experiment using a CENPF construct showed that the effects of CENPF knockout could be diminished by inducing overexpression of CENPF (Figure 2(j)). In addition, we found that CENPF plays the same roles in DU145 PC cells, whose CENPF expression levels were as redundant as PC3 cells (Figure 2(k)). Colony formation capability (Figure 2(l)) and viability (Figure 2(m)) were both significantly decreased when CENPF was knocked-down in DU145 cells. Collectively, these results indicate that CENPF inhibition reverses EMT and suppresses the proliferative characteristics of PC cells, while simultaneously enhancing anoikis-induced apoptosis.
CENPF silencing alters the whole proteome to promote signaling and glucose metabolism
Since there have been no previous proteomics studies showing the biological function of CENPF in the context of PC, we were prompted to understand the whole proteome alterations caused by CENPF silencing. In order to understand the mechanisms underlying CENPF function, we performed mass spectrometry-based proteomics analysis, as described in the Methods. Whole-cell lysates in biological triplicates were digested with trypsin. Using LC-MS/MS, we determined which proteins were differentially expressed following CENPF knockout. In total, 549 DEPs were identified (adjusted p-value based on BH procedure ≤ 0.01 and ratio ≥ 1.5) (Supplementary Table 1), of which 130 were upregulated and 419 were downregulated, as shown in the volcano plot (Figure 3(a)). We next conducted a functional gene enrichment analysis of DEPs using the DAVID software to understand the most perturbed pathways based on previously reported GOBPs or KEGGs. Significantly enriched GOBPs included “negative regulation of microtubule polymerization” and “canonical glycolysis” (Figure 3(b)). CENPF silencing significantly enriched KEGG terms for “glycolysis/gluconeogenesis” and various metabolic pathways (Figure 3(c)). To further understand the changes in relationships among biological networks, network analysis was performed. Phosphorylation signaling transduction pathways, various metabolisms (carbohydrate, lipid, amino acids), adhesion, actin cytoskeleton, and endocytosis pathways were clearly marked as being highly enriched for by DEPs in CENPFKO cells (Figure 3(d)). DEPs linked to fatty acid degradation were enriched as well (Figure 3(d)). Figure 3(e) shows the top 10 enriched pathways and the DEPs that belong to each term.
Figure 3.
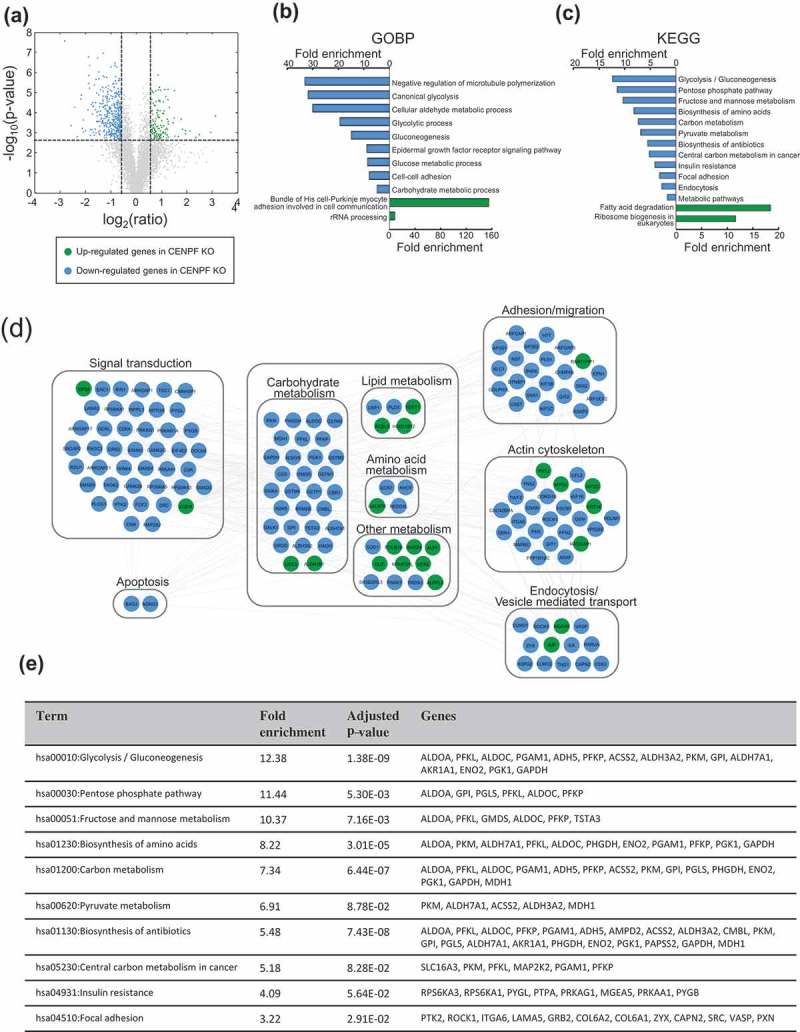
Silencing of CENPF perturbed proteome. (a) Unbiased global proteomics analysis was performed using CENPFKO cells and Ctrl. The volcano plot shows the differentially expressed proteins. Green, upregulated; Blue, downregulated proteins in CENPFKO cells, compared to Ctrl (b-c) Functional enrichment analysis of DEPs was performed using DAVID. GOBP- (b) or KEGG-based analysis (c), (d) Network modeling suggested activation of phosphotyrosine signaling in PC cells. (e) The list of DEPs who belong to the 10 most-enriched pathways.
Silencing of CENPF impeded epigenetic modulation of histone markers and lipid synthesis in PC3 cells
We further defined the biological roles of CENPF in PC3 cells. Fatty acid and lipid metabolism play critical roles in energy maintenance and cellular nutrition in cancer, particularly in PC [33,34]. Biosynthesis of fatty acids utilizes glucose and exploits a pathway that is mainly controlled by an enzyme, fatty acid synthase (FASN). Prior studies have presented evidence showing that FASN is also associated with cell growth, survival, and drug resistance in PC [33]. To test the hypothetical link between CENPF levels and metabolic shifts in PC, cholesterol and free fatty acid levels were quantified in CENPFKO and control PC3 cells. We found significant reduction in both cholesterol and free fatty acid levels in the CENPFKO cells (Figure 4(a,b)). We further measured the expression levels of genes related to fatty acid and lipid synthesis in both cell lines. Expression levels of long-chain fatty-acid-coenzyme A ligase (ACSL1) [35], FASN [36], acetyl-CoA carboxylase (ACC), and phosphorylation levels of ACC (p-ACC) [37] were significantly decreased in CENPFKO cells (Figure 4(c)). This data demonstrated that knocking-out CENPF resulted in decreased expression of lipid metabolism-associated proteins, as well as intracellular concentrations of FFAs and cholesterol.
Figure 4.
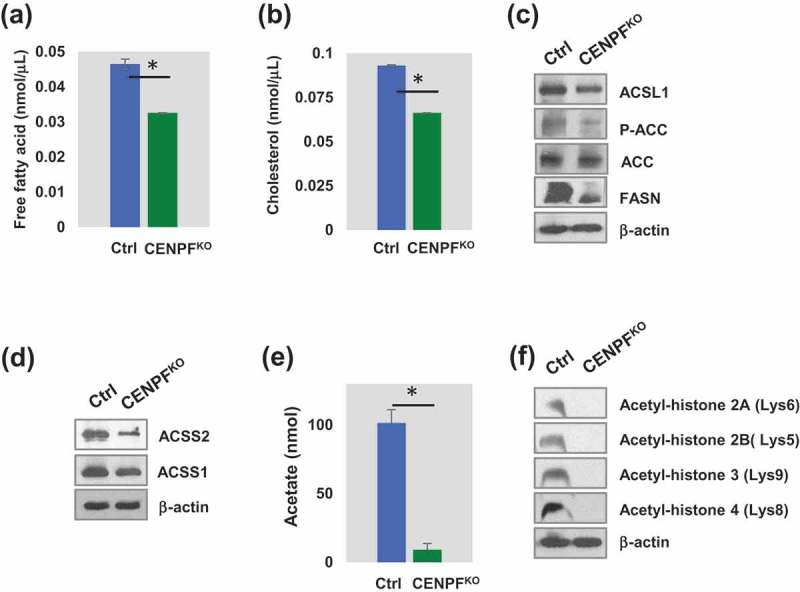
CENPF involved in epigenetic regulation of histone and lipid metabolism. (a) The histogram showed free fatty acid level. (b) Cholesterol level between CENPFKO and Ctrl. (c) Western blot analysis showing long-chain fatty-acid-coenzyme A ligase (ACSL1), acetyl-CoA carboxylase (ACC), phosphorylation level of ACC (p-ACC) and fatty acid synthase (FASN). (d) Both ACSS1 and ACSS2 expression are reduced in CENPFKO. (E) CENPFKO decreased the synthesis of acetate to Ctrl. (f) Western blot analysis of key acetylated histone markers (H2A, H2B, H3, and H4) between CENPFKO and Ctrl. The data represent the mean ± standard deviation. A two-tailed Student’s t-test was used to calculate statistical significance. β-actin was used as the loading control.
In addition, we found that expression levels of acyl-coenzyme A synthetase short-chain family members 1 and 2 (ACSS1 and ACSS2), which are key regulatory proteins for lipid synthesis and energy generation, were also downregulated when CENPF was silenced (Figure 4(d)). Western blot analysis of CENPFKO cells showed that ACSS2 expression greatly decreased, while ACSS1 was modestly downregulated, compared to controls (Figure 4(d)). Interestingly, ACSS2 expression is positively correlated with PC progression [38–40]. ACSS2 has been reported to mediate acetyl modification of histone proteins (histone acetylation) and gene regulation through the generation of acetyl-CoA [41]. Thus, we speculated that CENPF is required for regulating acetate metabolism and contributes to epigenetic modification through its effects on histones via ACSS1 and/or ACSS2 (Figure 4(e)). Our acetate measurements, using a commercial kit, showed that acetate levels in CENPFKO cells were almost completely diminished. We also evaluated for expression of acetylated histones in CENPFKO and control cells. Western blot analysis found that expression levels of key acetylated histones, including H2A, H2B, H3 and H4, were significantly reduced in CENPFKO cells (Figure 4(f)).
CENPFKO cells have a distinct tyrosine phosphorylation profile
We next sought to profile the phosphorylation events that occur when CENPF is knocked-out. Just to get a general idea of the phosphorylation statuses of specific signaling pathways modulated by CENPF silencing, a series of western blot analyses were carried out on both CENPFKO PC3 cells and controls. We found that CENPF is required for maintaining various phosphorylation events that are important for PC progression. Compared to control cells, the phosphorylation levels of Src (Tyr416), β-catenin (Ser45), p21 activated kinase 1 (PAK1) (Thr423), and NF-κB (Ser536) were elevated in CENPFKO cells (Figure 5(a)). In contrast, phosphorylation of Akt (Ser473), extracellular signal-regulated kinase 1/2 (ERK1/2) (Thr202/Tyr204), S6Kinase (Thr389), mTOR (Ser2448), and p38MAPK (Thr180/Tyr182) decreased in CENPFKO cells compared to controls. However, the total protein expression levels remained unchanged (data were not shown) (Figure 5(a)). Interestingly, western blot analysis with anti-phospho-tyrosine antibodies revealed that CENPFKO cells have stronger p-Tyr intensity with distinct phosphorylation patterns (Figure 5(b)), signifying that subduing CENPF increased or changed patterns of tyrosine phosphorylation.
Figure 5.
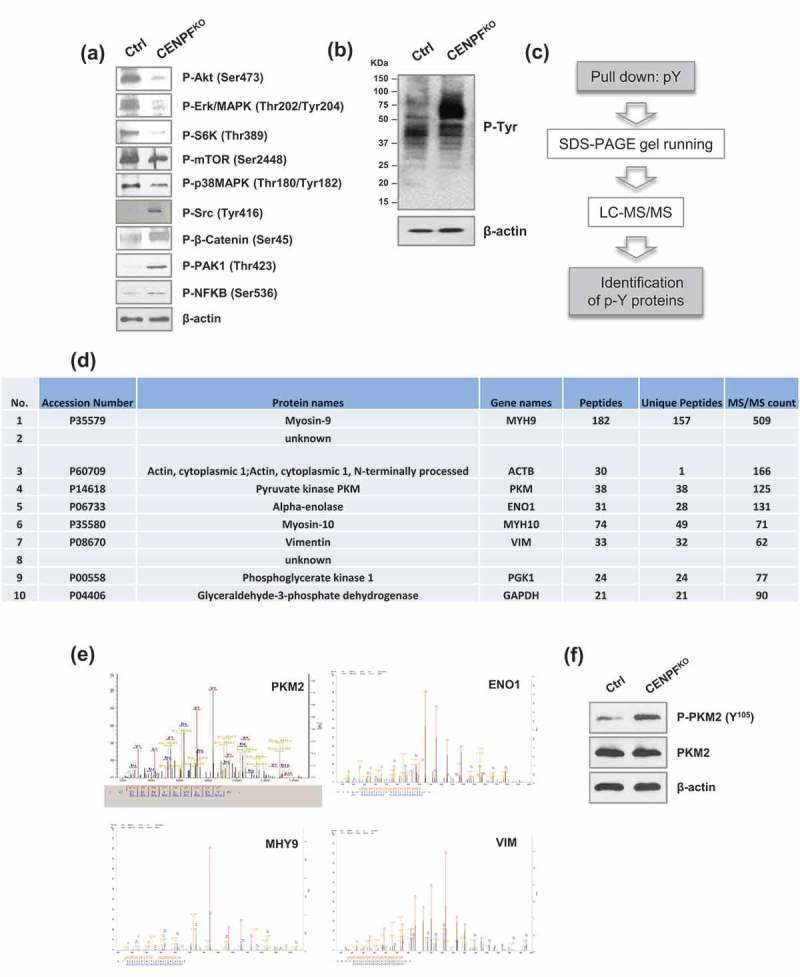
CENPF reduce metabolism in prostate cancer cells. (a) Western blot analysis of key signal transduction proteins. (b) Western blot analysis of phospho-tyrosine (pY) signaling was performed in CENPFKO cells and Ctrl. (c) A workflow including phospho-tyrosine-enrichment by protein pull-down and LC–MS/MS illustrates the experimental design to identify the tyrosine phosphorylated proteins specifically enriched in CENPFKO cells. (d) The 10 top proteins identified from B. (e) Representative MS spectrum of PKM2, ENO1, MHY9, and VIM, which were identified in this study. (f) Phosphorylation of PKM2 at Y105 and non-phosphorylated form of PKM2 were assessed by western blot analysis in CENPFKO cells and Ctrl. β-actin was used as the loading control.
Tyrosine phosphorylation levels of pyruvate kinase M2 (PKM2) were decreased in CENPFKO cells
Although phosphorylation of tyrosine sites constitutes less than a few percentage of all phosphorylation events, we next attempted to specifically focus on defining tyrosine phosphorylation events. This was because previous findings have indicated that phospho-tyrosine (pTyr) based phosphoproteomics can be utilized as a tool for measuring the critical events needed for activating key kinases. In turn, this allows for better recognizing perturbation of downstream signaling pathways [42,43]. To further define tyrosine-phosphorylated proteins in CENPFKO cells and controls, proteomics profiling and bioinformatics approaches were applied. Tyrosine-phosphorylated proteins were enriched for target protein identification via mass spectrometry. This enrichment was done using p-Tyr, an anti-phospho-tyrosine antibody coupled to agarose beads. The coupled proteomic identification was then carried out using LC-MS/MS (Figure 5(c)). This procedure resulted in a list of candidate proteins that may be biologically important for PC progression. Figure 5(d) shows the top 10 proteins, which included myosin-9, actin, PKM, alpha-enolase, myosin-10, vimentin, phosphoglycerate kinase 1, glyceraldehyde-3-phosphate dehydrogenase, and 2 additional proteins without annotations.
PKM2 is known as a tumor-specific isoform of pyruvate kinase (PK), the rate-limiting enzyme during glycolysis, which catalyzes the production of pyruvate and adenosine 5'-triphosphate (ATP) from phosphoenolpyruvate (PEP) and adenosine 5'-diphosphate (ADP) [44]. The detected tandem mass spectrum of PKM2 and other proteins (e.g. Enolase 1 (ENO1), myosin, heavy chain 9 (MHY9), Vimentin (VIM)) are shown in Figure 5(e). Additional western blot analysis confirmed that phosphorylation levels of PKM2 at site Y105 significantly increased in CENPFKO cells, while expression of total PKM2 levels remained unchanged (Figure 5(f)). Given prior findings demonstrating that PKM2 activity is crucial for aerobic glycolysis and that phosphorylation of the Y105 site may inactivate PKM2 [45], our data suggests that knockout of CENPF may be inactivating PKM2 via Y105 site phosphorylation and this may be associated with the biological outcomes observed.
Both mitochondrial respiration and glycolysis are downregulated in CENPFKO cells
Given that CENPF silencing leads to PKM2 inactivation, we hypothesized that CENPF can regulate cancer-associated metabolism, such as aerobic glycolysis and mitochondrial respiration. To determine the effect of CENPF knockout on PC energy shifts, we used the Seahorse XFe24 Analyzer [46]. Mitochondrial respiration was profiled by measuring the oxygen consumption rate (OCR), which can determine the changes in levels of oxidative phosphorylation (OXPHOS)-dependent ATP generation. Compared to the control PC3 cells, CENPFKO cells had a considerable decrease in OCR (Figure 6(a)). Both the basal and maximum oxygen consumption rates were significantly reduced; approximately 35% and 50%, respectively, of the control rates were observed in CENPFKO cells (Figure 6(b,c)). Extracellular acidification rate (ECAR), which is an indicator of glycolysis, also decreased in CENPFKO cells (Figure 6(d)). Collectively, these experimental results suggest that biogenesis was significantly reduced in CENPFKO cells and that these cells were metabolically quiescent, compared to controls.
Figure 6.
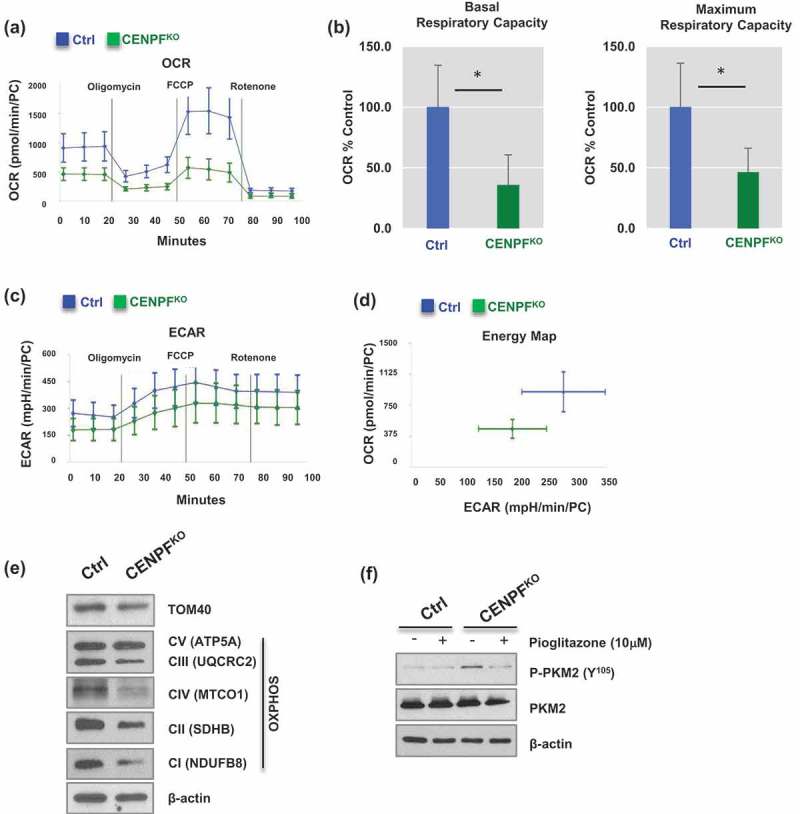
Silencing of CENPF decreased mitochondrial oxidative phosphorylation. The mitochondrial biogenetic activity was determined for CENPFKO and Ctrl in real-time using the Seahorse extracellular flux analyzer. (a) The oxygen consumption rate (OCR) curves in CENPFKO cells and Ctrl treated with oligomycin, FCCP and rotenone/antimycin A (n = 6 independent experiments). The OCR curves were determined by using a Seahorse XF24 Analyzer. (b) Basal and maximum respiratory capacity normalized to the cell numbers in CENPFKO cells and Ctrl. (c) Extracellular acidification rate (ECAR) was determined. (d) An energy map showing difference between CENPFKO cells and Ctrl. (e) Western blot analysis was performed to measure the levels of TAM20 and OXPHOS proteins. (f) Western blot data showing phospho-PKM2 (Y105) and PKM2 expression in CENPFKO cells and Ctrl in presence or absence of pioglitazone. Both cells were treated with 10μM pioglitazone for 24 h before sample preparation. The graphs shown here are representative examples of three independent experiments. The data represent the mean ± standard deviation. A two-tailed Student’s t-test was used to calculate statistical significance.
We next examined whether CENPF regulates mitochondrial quantity. Mitochondrial metabolic respiration-associated proteins, including TOM40 (a central component of the translocase of outer membrane (TOM) receptor complex in mitochondria) and a series of mitochondrial oxidative phosphorylation (OXPHOS) proteins [47], were measured via western blot analysis. The expression levels of TOM 40, C I subunit (NDUFB8), C II subunit (SDHB), C III core protein 2 (UQCRC2), C IV subunit (I MTCO1), and C V alpha subunit (ATP5A) decreased in CENPFKO cells (Figure 6(e)). These findings suggest that CENPF abrogation results in attenuated metabolism evidenced by impaired mitochondrial ATP production and glycolysis. Furthermore, we tested whether the increase in PKM2 phosphorylation can be reversed when mitochondrial biogenesis is boosted by pioglitazone, a medication that is widely used for diabetes [48,49]. Western blot analysis suggested that pioglitazone treatment significantly decreased CENPF knockout-induced tyrosine phosphorylation of PKM2 (Figure 6(f)).
Discussion
Since the discovery of evidence presenting that perturbed metabolic pathways, such as oxidative phosphorylation, glycolysis, and fatty acid biosynthesis, can suppress cancer cell growth, cancer metabolism has become an emerging field [50–52]. In this study, a series of unbiased proteomics and consecutive functional experiments revealed that knocking-out CENPF impaired mitochondrial functioning and signaling activation that is required for PC growth and metastasis. We herein demonstrated that CENPF knock-out can reverse EMT and suppress metabolic rates by reducing OXPHOS and glycolysis, possibly through its effects on PKM2.
High levels of PKM2 are expressed in a variety of human tumors, including lung, breast, and colon cancer [53], and are reported to promote the Warburg effect [44]. PKM2 exists as a low-activity dimeric or high-activity tetrameric form, and cancer cells predominantly express the low-activity dimeric form with a reduced ability to convert phosphoenolpyruvate to pyruvate [54]. Phosphorylation of PKM2 at Tyr105 is reported to disrupt its active tetrameric form [45,55] and reduce its catalytic activity. PKM2 phosphorylation at Tyr105 is known to impair PKM2’s enzymatic activity and ability to catalyze the formation of ATP from ADP using a phosphate group from 2-phosphoenolpyruvate (PEP).
Our histone acetylation data suggested that metabolic reprogramming influences histone acetylation levels in PC (Figure 4(a)). Previous studies have demonstrated the correlation between histone acetylation levels and clinical outcomes, such as recurrence and patient survival, in various cancer types [56]. Other studies have shown that elevated nuclear levels of acetylated histone 2A.Z or decreased global histone acetylation in PC was correlated to poorer prognoses [52,57,58]. Although it is well-accepted that histone acetylation has important roles in gene regulation and DNA repair, the biological significance of global histone acetylation levels and its underlying effects on metabolic rewiring (e.g. how histone modification regulates PC progression and treatment response) remain elusive.
Since nuclear acetylation events are highly dependent on the availability of acetyl-CoA, we decided to survey acetate levels in our control and CENPFKO PC3 cell lines. An acetate assay showed decreased acetate levels in CENPFKO cells, which was consistent with their histone acetylation levels (Figure 4(b)). Lipogenic enzymes, such as ACCS, are the primary enzymatic sources of acetyl-CoA outside of the mitochondria. Several studies have demonstrated that ACLY may also be present in the nucleus and plays a crucial role in regulating histone acetylation. However, our study did not provide any evidence showing CENPF affecting ACLY’s role in histone acetylation.
In summary, our study demonstrates that CENPF, a centromere protein, is required for PC progression through its effects on signaling, glucose metabolism, and epigenetic regulation. Improved understanding of whether a perturbed metabolism precedes changes in epigenetic regulation or vice versa could provide clues on the cross talk that occurs between oncogenic metabolic reprogramming and the epigenome. The finding that the PKM2–mediated glucose metabolism pathway is a key player in maintaining mitochondrial function represents potential therapeutic strategies against PC. Future functional studies assessing the causative role of histone acetylation with altered metabolism by CENPF silencing are needed to further grasp how CENPF regulates mitochondrial functioning during metabolic and epigenetic changes in PC progression.
Funding Statement
This work was supported by the Centers for Disease Control and Prevention [1U01DP006079]; Foundation for the National Institutes of Health [1U01DK103260];U.S. Department of Defense [W81XWH-15-1-0415]. The authors acknowledge support from National Institutes of Health grants (1U01DK103260, 1R01DK100974, U24 DK097154, NIH NCATS UCLA CTSI UL1TR000124), Department of Defense grants (W81XWH-15-1-0415), Centers for Disease Controls and Prevention (1U01DP006079), IMAGINE NO IC Research Grant, the Steven Spielberg Discovery Fund in Prostate Cancer Research Career Development Award, the U.S.- Egypt Science and Technology Joint Fund (to J.K.), and Florida Department of Health, Bankhead- Coley Cancer Research Program (5BC03) (to M.K.). J.K. is former recipient of Interstitial Cystitis Association Pilot Grant, a Fishbein Family IC Research Grant, New York Academy of Medicine,and Boston Children’s Hospital Faculty Development. The funders had no role in the design, datacollection and analysis, decision to publish or preparation of the manuscript. In addition, this article is derived from the Subject Data funded in whole or part by National Academies of Sciences, Engineering, and Medicine (NAS) and The United States Agency for International Development (USAID). Any opinions, findings, conclusions, or recommendations expressed in this article are those of the authors alone, and do not necessarily reflect the views of USAID or NAS.
Acknowledgments
We would like to thank Drs. Wei Yang at the Biomarker Discovery Platform Core (Cedars-Sinai Medical Center) for guidance with proteomics studies and assistance with liquid chromatography-mass spectrometry (LC-MS/MS). The authors acknowledge support from National Institutes of Health grants (1U01DK103260, 1R01DK100974, U24 DK097154, NIH NCATS UCLA CTSI UL1TR000124), Department of Defense grants (W81XWH-15-1-0415), Centers for Disease Controls and Prevention (1U01DP006079), IMAGINE NO IC Research Grant, the Steven Spielberg Discovery Fund in Prostate Cancer Research Career Development Award, the U.S.-Egypt Science and Technology Joint Fund (to J.K.), and Florida Department of Health, Bankhead-Coley Cancer Research Program (5BC03) (to M.K.). J.K. is former recipient of Interstitial Cystitis Association Pilot Grant, a Fishbein Family IC Research Grant, New York Academy of Medicine, and Boston Children’s Hospital Faculty Development. The funders had no role in the design, data collection and analysis, decision to publish or preparation of the manuscript. In addition, this article is derived from the Subject Data funded in whole or part by National Academies of Sciences, Engineering, and Medicine (NAS) and The United States Agency for International Development (USAID). Any opinions, findings, conclusions, or recommendations expressed in this article are those of the authors alone, and do not necessarily reflect the views of USAID or NAS.
Author contributions
JK and HLK designed the study. MS, AY, PL, AA, BZ, and HP carried out all biochemical experiments and mitochondrial metabolomics analysis. ML performed bioinformatics analysis. MS and MK participated in interpretation of data and writing of the manuscript. MK and AA provided valuable insights on study design and data interpretation. JK wrote the manuscript. All authors discussed the results and commented on the manuscript at all stages.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Siegel RL, Miller KD, Jemal A.. Cancer statistics, 2016. CA Cancer J Clin. 2016. Jan-Feb;66(1):7–30. PubMed PMID: 26742998. [DOI] [PubMed] [Google Scholar]
- [2].Schroder FH, Hugosson J, Roobol MJ, et al. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med. 2009. March 26;360(13):1320–1328. PubMed PMID: 19297566. [DOI] [PubMed] [Google Scholar]
- [3].Andriole GL, Crawford ED, Grubb RL 3rd, et al. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med. 2009. March 26;360(13):1310–1319. PubMed PMID: 19297565; PubMed Central PMCID: PMC2944770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cutruzzola F, Giardina G, Marani M, et al. Glucose metabolism in the progression of prostate cancer. Front Physiol. 2017;8:97 PubMed PMID: 28270771; PubMed Central PMCID: PMC5318430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mitsuzuka K, Arai Y.. Metabolic changes in patients with prostate cancer during androgen deprivation therapy. Int J Urol. 2017. October 20 PubMed PMID: 29052905 DOI: 10.1111/iju.13473 [DOI] [PubMed] [Google Scholar]
- [6].Levine AJ, Puzio-Kuter AM.. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010. December 03;330(6009):1340–1344. PubMed PMID: 21127244. [DOI] [PubMed] [Google Scholar]
- [7].Elstrom RL, Bauer DE, Buzzai M, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004. June 1;64(11):3892–3899. PubMed PMID: 15172999. [DOI] [PubMed] [Google Scholar]
- [8].Miller DM, Thomas SD, Islam A, et al. c-Myc and cancer metabolism. Clin Cancer Res. 2012. October 15;18(20):5546–5553. PubMed PMID: 23071356; PubMed Central PMCID: PMCPMC3505847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Smith B, Schafer XL, Ambeskovic A, et al. Addiction to coupling of the Warburg effect with glutamine catabolism in cancer cells. Cell Rep. 2016. October 11;17(3):821–836. PubMed PMID: 27732857; PubMed Central PMCID: PMCPMC5108179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Courtnay R, Ngo DC, Malik N, et al. Cancer metabolism and the Warburg effect: the role of HIF-1 and PI3K. Mol Biol Rep. 2015. April;42(4):841–851. PubMed PMID: 25689954. [DOI] [PubMed] [Google Scholar]
- [11].Dang CV, Kim JW, Gao P, et al. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008. January;8(1):51–56. PubMed PMID: 18046334. [DOI] [PubMed] [Google Scholar]
- [12].Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009. October;9(10):691–700. PubMed PMID: 19759539. [DOI] [PubMed] [Google Scholar]
- [13].Matuszak EA, Kyprianou N. Androgen regulation of epithelial-mesenchymal transition in prostate tumorigenesis. Expert Rev Endocrinol Metab. 2011. May;6(3):469–482. PubMed PMID: 23667383; PubMed Central PMCID: PMC3648215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bitting RL, Schaeffer D, Somarelli JA, et al. The role of epithelial plasticity in prostate cancer dissemination and treatment resistance. Cancer Metastasis Rev. 2014. September;33(2–3):441–468. PubMed PMID: 24414193; PubMed Central PMCID: PMC4230790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Varis A, Salmela AL, Kallio MJ. Cenp-F (mitosin) is more than a mitotic marker. Chromosoma. 2006. August;115(4):288–295. PubMed PMID: 16565862. [DOI] [PubMed] [Google Scholar]
- [16].Liao H, Winkfein RJ, Mack G, et al. CENP-F is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosis. J Cell Biol. 1995. August;130(3):507–518. PubMed PMID: 7542657; PubMed Central PMCID: PMC2120529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Testa JR, Zhou JY, Bell DW, et al. Chromosomal localization of the genes encoding the kinetochore proteins CENPE and CENPF to human chromosomes 4q24–>q25 and 1q32–>q41, respectively, by fluorescence in situ hybridization. Genomics. 1994. October;23(3):691–693. PubMed PMID: 7851898. [DOI] [PubMed] [Google Scholar]
- [18].Li P, You S, Nguyen C, et al. Genes involved in prostate cancer progression determine MRI visibility. Theranostics. 2018;8(7):1752–1765. PubMed PMID: 29556354; PubMed Central PMCID: PMC5858498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mitrofanova A, Aytes A, Zou M, et al. Predicting drug response in human prostate cancer from preclinical analysis of in vivo mouse models. Cell Rep. 2015. September 29;12(12):2060–2071. PubMed PMID: 26387954; PubMed Central PMCID: PMC4591242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Aytes A, Mitrofanova A, Lefebvre C, et al. Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer Cell. 2014. May 12;25(5):638–651. PubMed PMID: 24823640; PubMed Central PMCID: PMC4051317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lin SC, Kao CY, Lee HJ, et al. Dysregulation of miRNAs-COUP-TFII-FOXM1-CENPF axis contributes to the metastasis of prostate cancer. Nat Commun. 2016. April 25;7:11418 PubMed PMID: 27108958; PubMed Central PMCID: PMC4848536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jung JH, You S, Oh JW, et al. Integrated proteomic and phosphoproteomic analyses of cisplatin-sensitive and resistant bladder cancer cells reveal CDK2 network as a key therapeutic target. Cancer Lett. 2018. November 28;437:1–12. PubMed PMID: 30145203; PubMed Central PMCID: PMCPMC6181132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rinas A, Espino JA, Jones LM. An efficient quantitation strategy for hydroxyl radical-mediated protein footprinting using Proteome discoverer. Anal Bioanal Chem. 2016. April;408(11):3021–3031. PubMed PMID: 26873216. [DOI] [PubMed] [Google Scholar]
- [24].Morley S, You S, Pollan S, et al. Regulation of microtubule dynamics by DIAPH3 influences amoeboid tumor cell mechanics and sensitivity to taxanes. Sci Rep. 2015. July 16;5:12136 PubMed PMID: 26179371; PubMed Central PMCID: PMC4503992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. PubMed PMID: 19131956. [DOI] [PubMed] [Google Scholar]
- [26].Patil A, Nakai K, Nakamura H. HitPredict: a database of quality assessed protein-protein interactions in nine species. Nucleic Acids Res. 2011. January 39 (Database issue):D744–9. PubMed PMID: 20947562; PubMed Central PMCID: PMC3013773 DOI: 10.1093/nar/gkq897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003. November;13(11):2498–2504. PubMed PMID: 14597658; PubMed Central PMCID: PMC403769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Horning AM, Wang Y, Lin CK, et al. Single-cell RNA-seq reveals a subpopulation of prostate cancer cells with enhanced cell-cycle-related transcription and attenuated androgen response. Cancer Res. 2018. February 15;78(4):853–864. PubMed PMID: 29233929; PubMed Central PMCID: PMCPMC5983359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sun Y, Wang BE, Leong KG, et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res. 2012. January 15;72(2):527–536. PubMed PMID: 22108827. [DOI] [PubMed] [Google Scholar]
- [30].Gao F, Al-Azayzih A, Somanath PR. Discrete functions of GSK3alpha and GSK3beta isoforms in prostate tumor growth and micrometastasis. Oncotarget. 2015. March 20;6(8):5947–5962. PubMed PMID: 25714023; PubMed Central PMCID: PMCPMC4467413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Landers KA, Samaratunga H, Teng L, et al. Identification of claudin-4 as a marker highly overexpressed in both primary and metastatic prostate cancer. Br J Cancer. 2008. August 5;99(3):491–501. PubMed PMID: 18648369; PubMed Central PMCID: PMCPMC2527792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sakamoto S, Kyprianou N. Targeting anoikis resistance in prostate cancer metastasis. Mol Aspects Med. 2010. April;31(2):205–214. PubMed PMID: 20153362; PubMed Central PMCID: PMCPMC2988681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Suburu J, Chen YQ. Lipids and prostate cancer. Prostaglandins Other Lipid Mediat. 2012. May;98(1–2):1–10. PubMed PMID: 22503963; PubMed Central PMCID: PMC3348998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Itkonen HM, Brown M, Urbanucci A, et al. Lipid degradation promotes prostate cancer cell survival. Oncotarget. 2017. June 13;8(24):38264–38275. PubMed PMID: 28415728; PubMed Central PMCID: PMC5503531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gao Y, Islam MS, Tian J, et al. Inactivation of ATP citrate lyase by Cucurbitacin B: A bioactive compound from cucumber, inhibits prostate cancer growth. Cancer Lett. 2014. July 10;349(1):15–25. PubMed PMID: 24690568. [DOI] [PubMed] [Google Scholar]
- [36].Huang M, Koizumi A, Narita S, et al. Diet-induced alteration of fatty acid synthase in prostate cancer progression. Oncogenesis. 2016. February 15;5:e195 PubMed PMID: 26878389; PubMed Central PMCID: PMC5154344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Park HU, Suy S, Danner M, et al. AMP-activated protein kinase promotes human prostate cancer cell growth and survival. Mol Cancer Ther. 2009. April;8(4):733–741. PubMed PMID: 19372545; PubMed Central PMCID: PMCPMC2775041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mews P, Donahue G, Drake AM, et al. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature. 2017. June 15;546(7658):381–386. PubMed PMID: 28562591; PubMed Central PMCID: PMC5505514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shi L, Tu BP. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol. 2015. April;33:125–131. PubMed PMID: 25703630; PubMed Central PMCID: PMC4380630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schug ZT, Vande Voorde J, Gottlieb E. The metabolic fate of acetate in cancer. Nat Rev Cancer. 2016. November;16(11):708–717. PubMed PMID: 27562461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gao X, Lin SH, Ren F, et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat Commun. 2016. June 30;7:11960 PubMed PMID: 27357947; PubMed Central PMCID: PMCPMC4931325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Drake JM, Graham NA, Stoyanova T, et al. Oncogene-specific activation of tyrosine kinase networks during prostate cancer progression. Proc Natl Acad Sci U S A. 2012. January 31;109(5):1643–1648. PubMed PMID: 22307624; PubMed Central PMCID: PMC3277127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Labots M, van der Mijn JC, Beekhof R, et al. Phosphotyrosine-based-phosphoproteomics scaled-down to biopsy level for analysis of individual tumor biology and treatment selection. J Proteomics. 2017. June 6;162:99–107. PubMed PMID: 28442448. [DOI] [PubMed] [Google Scholar]
- [44].Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008. March 13;452(7184):230–233. PubMed PMID: 18337823. [DOI] [PubMed] [Google Scholar]
- [45].Hitosugi T, Kang S, Vander Heiden MG, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009. November 17;2(97):ra73 PubMed PMID: 19920251; PubMed Central PMCID: PMC2812789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wu M, Neilson A, Swift AL, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007. January;292(1):C125–36. PubMed PMID: 16971499. [DOI] [PubMed] [Google Scholar]
- [47].Georgieva E, Ivanova D, Zhelev Z, et al. Mitochondrial dysfunction and redox imbalance as a diagnostic marker of “Free Radical Diseases”. Anticancer Res. 2017. October;37(10):5373–5381. PubMed PMID: 28982845. [DOI] [PubMed] [Google Scholar]
- [48].Schernthaner G, Currie CJ, Schernthaner GH. Do we still need pioglitazone for the treatment of type 2 diabetes? A risk-benefit critique in 2013. Diabetes Care. 2013. August;36(Suppl 2):S155–S161. PubMed PMID: 23882041; PubMed Central PMCID: PMCPMC3920795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Boxall N, Bennett D, Hunger M, et al. Evaluation of exposure to pioglitazone and risk of prostate cancer: a nested case-control study. BMJ Open Diabetes Res Care. 2016;4(1):e000303 PubMed PMID: 28074141; PubMed Central PMCID: PMCPMC5174825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kim WJ, Kim J. Looking to the metabolic landscapes for prostate health monitoring. Prostate Int. 2017. September;5(3):85–88. PubMed PMID: 28828350; PubMed Central PMCID: PMC5551909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lue HW, Podolak J, Kolahi K, et al. Metabolic reprogramming ensures cancer cell survival despite oncogenic signaling blockade. Genes Dev. 2017. November 14 PubMed PMID: 29138276 DOI: 10.1101/gad.305292.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Webber LP, Wagner VP, Curra M, et al. Hypoacetylation of acetyl-histone H3 (H3K9ac) as marker of poor prognosis in oral cancer. Histopathology. 2017. August;71(2):278–286. PubMed PMID: 28326594. [DOI] [PubMed] [Google Scholar]
- [53].Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab. 2012. November;23(11):560–566. PubMed PMID: 22824010; PubMed Central PMCID: PMCPMC3466350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011. July;43(7):969–980. PubMed PMID: 20156581. [DOI] [PubMed] [Google Scholar]
- [55].Christofk HR, Vander Heiden MG, Wu N, et al. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008. March 13;452(7184):181–186. PubMed PMID: 18337815. [DOI] [PubMed] [Google Scholar]
- [56].Chervona Y, Costa M. Histone modifications and cancer: biomarkers of prognosis? Am J Cancer Res. 2012;2(5):589–597. PubMed PMID: 22957310; PubMed Central PMCID: PMC3433108. [PMC free article] [PubMed] [Google Scholar]
- [57].Ellinger J, Schneider AC, Bachmann A, et al. Evaluation of global histone acetylation levels in bladder cancer patients. Anticancer Res. 2016. August;36(8):3961–3964. PubMed PMID: 27466500. [PubMed] [Google Scholar]
- [58].Valdes-Mora F, Gould CM, Colino-Sanguino Y, et al. Acetylated histone variant H2A.Z is involved in the activation of neo-enhancers in prostate cancer. Nat Commun. 2017. November 07;8(1):1346 PubMed PMID: 29116202; PubMed Central PMCID: PMC5676741. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.