

ABSTRACT
Cancers expressing mutant RAS are associated with a weaker response to chemotherapy and a shorter overall patient survival. We have demonstrated that the irreversible inhibitor of ERBB1/2/4, neratinib, inhibits ERBB1/2/4 and causes their internalization and autolysosomal degradation. Fellow-traveler membrane proteins with RTKs, including mutant K-/N-RAS, were also degraded. We discovered that the CDK4/6 inhibitor palbociclib increased autophagosome and then autolysosome levels in a time dependent fashion, did not reduce mTOR activity, and interacted with temsirolimus to kill. Neratinib and palbociclib interacted in a greater than additive manner to increase autophagosome and then autolysosome levels in a time dependent fashion, and to cause tumor cell killing. Killing required the expression of ATM and AMPKα, Beclin1 and ATG5, BAX and BAK and of AIF, but not of caspase 9. In some cells over-expression of BCL-XL was protective whereas in others it was ineffective. The lethality of [neratinib + palbociclib] was modestly enhanced by the PDE5 inhibitor sildenafil and strongly enhanced by the HDAC inhibitor sodium valproate. This was associated with K-RAS degradation and a greater than additive increase in autophagosome and autolysosome levels. Killing by the three-drug combination required ATM and AMPKα, and, to a greater extent, Beclin1 and ATG5. In vivo, [valproate + palbociclib] and [neratinib + valproate + palbociclib] interacted to suppress the growth of a carboplatin/paclitaxel resistant PDX ovarian tumors that express a mutant N-RAS. Our data support performing a future three-drug trial with these agents.
Keywords: autophagy, HDAC, DNA damage
Introduction
In the recent past, two new drugs for breast cancer have been approved by the FDA, palbociclib (IBRANCE®) and neratinib (NERLYNX®). Palbociclib was developed as an inhibitor of CDK4/6 and neratinib as an inhibitor of ERBB1/2/4.1,2 Palbociclib is approved for the treatment of ER+ ERBB2- patients in combination with aromatase or anti-estrogen therapies. Neratinib is used as an adjuvant therapy alongside the anti-ERBB2 antibody Herceptin in ERBB2+ breast cancers.
Our most recent studies with neratinib, alone or in combination with HDAC inhibitors, have demonstrate a unique biochemical action for the drug.3–5 Neratinib was originally developed as an irreversible inhibitor of ERBB2, and was then shown to inhibit ERBB1 and ERBB4.6 We discovered that neratinib was both a kinase inhibitor and that it possessed the ability to cause ERBB1/2/3/4 receptor internalization with their subsequent proteolytic degradation. As a negative control, we also examined the expression of c-MET and surprisingly discovered that this receptor was also down-regulated by neratinib, even though neratinib does not chemically bind to this receptor. The down-regulation of ERBB1 and c-MET could be molecularly differentiated by the fact that for degradation, ERBB1 also required a ubiquitination step whereas c-MET did not. It has been proposed that growth factor receptors and other signal transduction proteins reside in quaternary complexes on the surface of tumor cells, and based on this concept we determined that neratinib, and particularly neratinib combined with an HDAC inhibitor, caused the autolysosomal-dependent degradation of mutant K-RAS and mutant N-RAS.3,7 These data imply that neratinib, in combination with HDAC inhibitors, could be a therapeutic option to explore in tumors expressing ERBB1/2/4 and mutant RAS proteins.
In prior studies we have demonstrated that the pan-CDK inhibitor flavopiridol (Alvocidib) could interact with the ERBB1/2/4 inhibitor lapatinib to kill breast cancer cells.8 Flavopiridol inhibits multiple CDK enzymes, including CDK4/6, at low nanomolar concentrations. We discovered that flavopiridol interacted with lapatinib to rapidly reduce MCL-1 expression and over-expression of MCL-1 or knock down of BAX and BAK suppressed drug combination lethality. Lapatinib-mediated inhibition of ERK1/2 and to a lesser extent AKT facilitated CDK inhibitor -induced suppression of MCL-1 levels. In clinical trials, as a single agent, however, flavopiridol demonstrated weak activity in solid tumors, including breast cancers.9 Yet, as with palbociclib, it is a potent inhibitor of CDK4/6. These findings suggested to us that in addition to inhibiting CDK4/6, palbociclib must have additional targets in cancer cells that mediate its known efficacy.
In our present studies we discovered that palbociclib as a single agent increased the levels of autophagosomes and then autolysosomes in a time dependent fashion and interacted with the mTOR inhibitor temsirolimus to kill. Neratinib and palbociclib interacted in a greater than additive fashion to both increase autophagosome and then autolysosome levels in a time dependent fashion, and to cause tumor cell killing. Killing required the expression of ATM and AMPKα, Beclin1 and ATG5, BAX and BAK and of AIF, but not of caspase 9. The lethality of [neratinib + palbociclib] was strongly enhanced by the HDAC inhibitor sodium valproate in vitro and in vivo.
Results
Low clinically relevant concentrations of palbociclib in a dose-dependent fashion increased the levels of GFP+ autophagosomes followed several hours later by elevated levels of RFP+ autolysosomes (Figure 1A). Although autophagosome levels had been increased, no significant alteration in the activity of mTOR (mTORC1 S2448 phosphorylation) was observed (Figure 1B). As palbociclib appeared to be stimulating autophagy in an mTOR independent fashion, we determined whether an inhibitor of mTORC1 activity, temsirolimus, could enhance palbociclib lethality. In multiple tumor cell types, including breast cancer cells, temsirolimus and palbociclib interacted to enhance the killing of mammary, colon, NSCLC and melanoma cells, but not of stomach cancer cells (Figure 1C).
Figure 1.
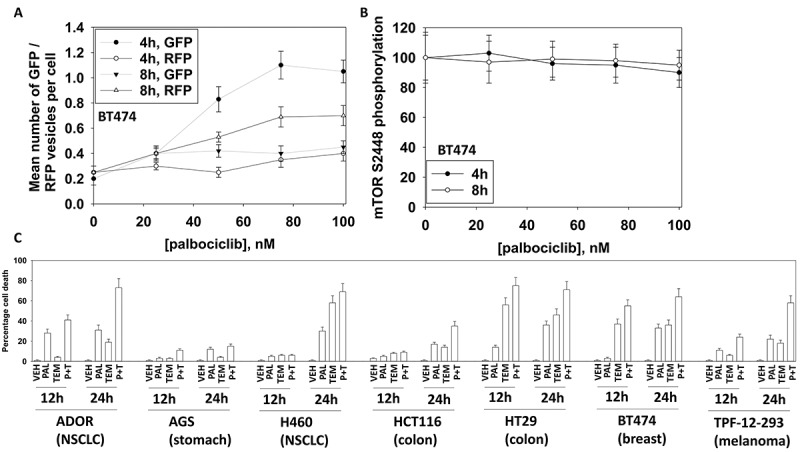
Palbociclib enhances autophagosome and autolysosome levels without inactivating mTOR and interacts with the mTOR inhibitor temsirolimus to kill cancer cells. A. BT474 cells were transfected with a plasmid to express LC3-GFP-RFP. Twenty-four h later cells were treated with vehicle control or with increasing concentrations of palbociclib. Cells were imaged 4h and 8h after the addition of vehicle or palbociclib. At least forty cells per condition were imaged in independent triplicate (n = 3 +/- SD); GFP+ cells indicate autophagosomes; RFP+ cells indicate autolysosomes. B. BT474 cells treated with vehicle control or with increasing concentrations of palbociclib. Cells were fixed 4h and 8h after the addition of vehicle or palbociclib. At least forty cells per condition were imaged in independent triplicate and the intensity ratio of phosphorylated mTOR S2448 levels to total mTOR expression plotted as a percentage of control treatment (n = 3 +/- SD). C. Tumor cells were treated with vehicle control, palbociclib (100 nM), temsirolimus (500 nM) or the drugs in combination for 12h and for 24h. Cells were treated with live/dead reagent and the percentage cell death under each condition determined (n = 3 +/- SD).
Recent studies from our group have demonstrated that the irreversible ERBB1/2/4 inhibitor neratinib can increase autophagosome and autolysosome levels which are responsible for both the down-regulation of RTKs and RAS proteins, as well as causing tumor cell killing.3–5 Thus, we next determined whether the autophagosome-inducing drug palbociclib interacted with the autophagosome-inducing drug neratinib to kill tumor cells. In mammary, colon, NSCLC, sarcoma, pancreatic and renal cancer cells neratinib and palbociclib interacted in an additive to greater than additive fashion to cause tumor cell death after 24h (Figure 2A). Previously we had demonstrated that neratinib could kill afatinib resistant H1975 NSCLC clones.3 The combination of [neratinib + palbociclib] killed both parental wild type H1975 clones and afatinib-resistant H1975 clones after 24h, with the killing of the afatinib-resistant clones being significantly greater than that of the wild type parental clones (Figure 2B).
Figure 2.
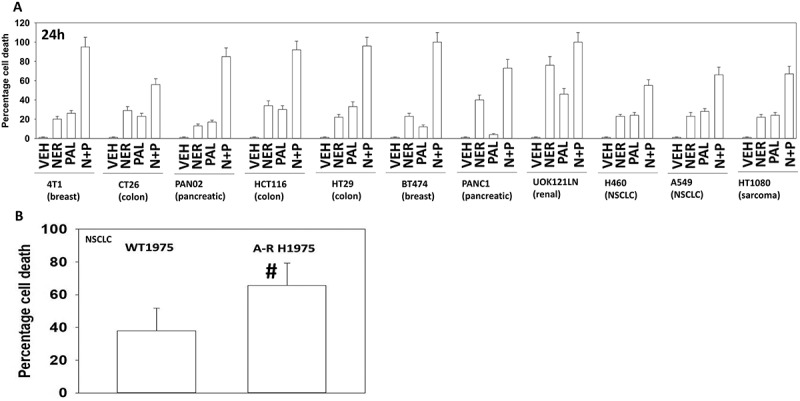
Neratinib and palbociclib interact in a greater than additive fashion to kill cancer cells. A. Cancer cells were treated with vehicle control, neratinib (100 nM), palbociclib (100 nM) or the drug in combination for 24h. Cells were treated with live/dead reagent and the percentage cell death under each condition determined (n = 3 +/- SD). B. Wild type parental and afatinib resistant clones of H1975 NSCLC cells were treated with vehicle control or with [neratinib (100 nM) + palbociclib (100 nM)] for 24h. Cells were treated with live/dead reagent and the percentage cell death under each condition determined (n = 3 +/- SD). # greater cell death an in wild type parental clones.
Based on our killing data, we then performed studies in an agnostic fashion to examine any changes in cell signaling processes caused by [neratinib + palbociclib]. In BT474 (mammary) and Spiky (ovarian) cancer cells the [neratinib + palbociclib] drug combination increased the phosphorylation of eIF2α S51, ATM S1981, AMPKα T172, TSC2 T1496, Raptor S792, ULK-1 S317 and ATG13 S318 (Figure 3A and 3B; Figure S1A). The drug combination reduced the phosphorylation of mTOR S2448 and S2481, ULK-1 S757, ERK1/2, AKT T308 and STAT3 Y705 (Figure 3A and 3B; Figure S1B). Prior studies with neratinib have demonstrated that it can activate an ATM-AMPK-ULK-1-ATG13 pathway and cause mTOR inactivation that leads to elevated autophagosome levels.3 Of note, palbociclib, regardless of LKB-1 expression/functionality, enhanced AMPKα T172 phosphorylation (Figure S1C). In all cell lines tested, neratinib and palbociclib interacted in an additive to greater than additive manner to increase autophagosome levels that were temporally followed by increased levels of autophagosomes (data not shown; see also Figure 6).
Figure 3.
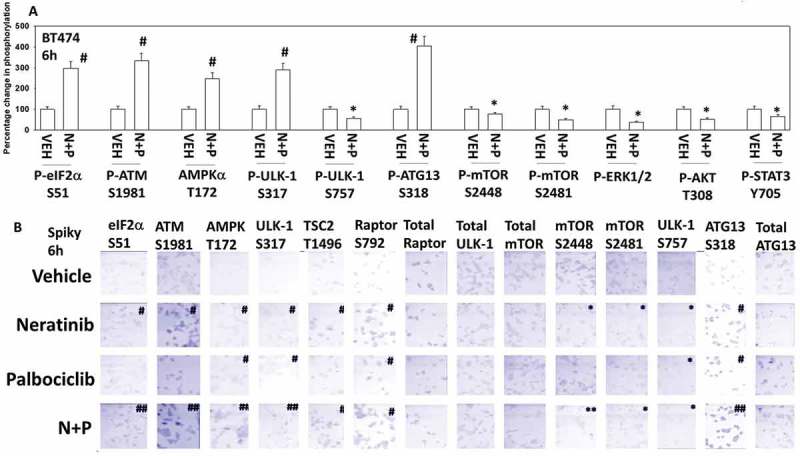
Neratinib and palbociclib interact to activate eIF2α, ATM, AMPK, ULK-1 and inactivate mTOR, ERK1/2, AKT and STAT3. A. BT474 cells were treated with vehicle control or with [neratinib (100 nM) + palbociclib (100 nM)] for 6h. Cells were fixed in place and at least forty cells per condition were imaged in independent triplicate and the intensity ratio of phosphorylated protein levels to total protein expression plotted as a percentage of control treatment (n = 3 +/- SD). # p < 0.05 greater than vehicle control; * p < 0.05 less than vehicle control. B. Spiky ovarian cancer cells were treated with vehicle control, neratinib (100 nM), palbociclib (100 nM) or the drugs in combination for 6h. Cells were fixed in place and at least forty cells per condition were imaged in independent triplicate and the intensity ratio of phosphorylated protein levels to total protein expression plotted as a percentage of control treatment (n = 3 +/- SD). # p < 0.05 greater than vehicle control; * p < 0.05 less than vehicle control.
Figure 6.
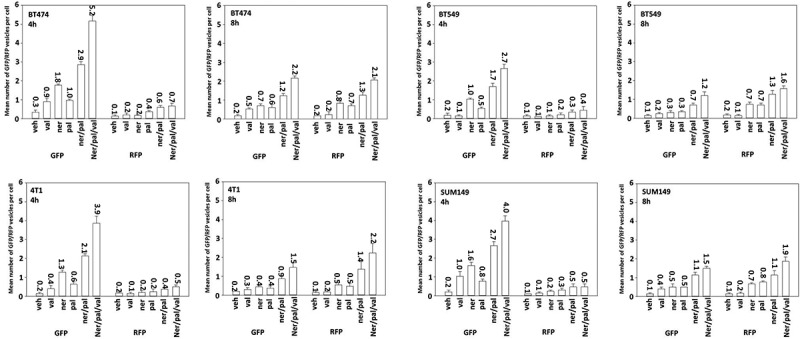
Sodium valproate and [neratinib + palbociclib] interact to stimulate autophagosome and autolysosome production in tumor cells. Mammary carcinoma cells were transfected with a plasmid to express LC3-GFP-RFP. Twenty-four h later cells were treated with vehicle control or with neratinib (100 nM), palbociclib (100 nM), sildenafil (2 μM), sodium valproate (250 μM) or the drugs in combination as indicated in the figure. Cells were imaged 4h and 8h after the addition of vehicle or of other agents. At least forty cells per condition were imaged in independent triplicate (n = 3 +/- SD); GFP+ cells indicate autophagosomes; RFP+ cells indicate autolysosomes.
We next attempted to link changes in cell signaling and biology to the molecular mechanisms by which [neratinib + palbociclib] killed tumor cells. Knock down of ATM, AMPKα, Beclin1 or ATG5 significantly reduced drug combination lethality (Figure 4A). Knock down of Beclin1 or ATG5 also reduced the lethality of palbociclib as a single agent (Figure 4B). The drug combination increased expression of Beclin1 and ATG5 and reduced expression of BCL-XL and MCL-1 (Figure 4C). In breast cancer cells, over-expression of BCL-XL or knock down of BAX, BAK or AIF consistently protected cells. In two of the cell lines, one breast and one ovarian, over-expression of the caspase 8/10 inhibitor c-FLIP-s partially reduced killing and in the same breast line, over-expression of dominant negative caspase 9 also partially reduced killing. In the ovarian cancer cells neither over-expression of BCL-XL nor dominant negative caspase 9 was protective. Collectively these data argue that autophagy plays a key role in mediating tumor cell killing by [neratinib + palbociclib] and that cell execution is more reliant, downstream of the mitochondrion, on AIF than caspase 9.
Figure 4.
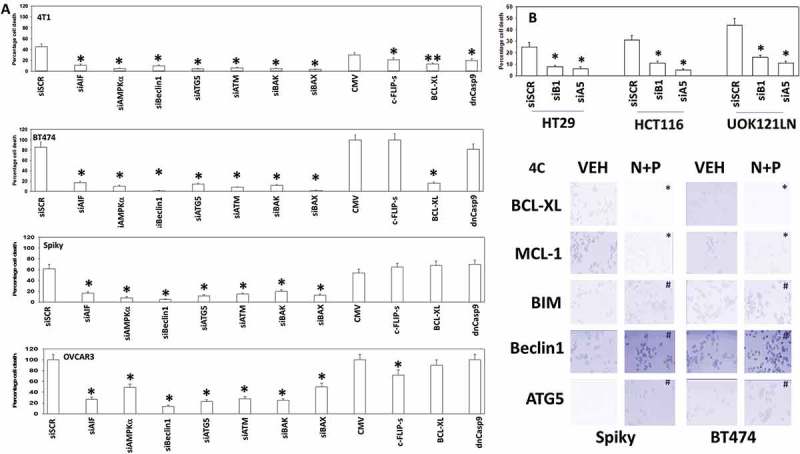
[Neratinib + palbociclib] killing is reduced by knock down of ATM, AMPK, Beclin1, ATG5 and AIF. A. Mammary (BT474, 4T1) and ovarian (Spiky, OVCAR3) cancer cells were transfected with a scrambled siRNA (siSCR) or with validated siRNA molecules to knock down expression of the indicated proteins. In parallel, other portions of cells were transfected to express empty vector (CMV) or to express the indicated proteins. Twenty-four h after transfection cells were treated with vehicle control or with [neratinib (100 nM) + palbociclib (100 nM)] for an additional 24h. Cells were treated with live/dead reagent and the percentage cell death under each condition above that in vehicle control plotted (n = 3 +/- SD). * p < 0.05 less cell death than in the corresponding value in siSCR/CMV treated cells. B. Tumor cells as indicated were transfected with a scrambled siRNA control or with siRNA molecules to knock down the expression of Beclin1 (siB1) or ATG5 (siA5). Twenty-four h after transfection cells were treated with vehicle control or with palbociclib (100 nM) for an additional 24h. Cell viability was determined by trypan blue exclusion and the percentage cell death under each condition above that observed in vehicle control plotted (n = 3 +/- SD). * p < 0.05 less cell death than in the corresponding value in siSCR. C. Tumor cells were treated with vehicle control or with [neratinib (100 nM) + palbociclib (100 nM)] for 6h. Cells were fixed in place and at least forty cells per condition were imaged in independent triplicate and the intensity ratio of phosphorylated protein levels to total protein expression determined (n = 3 +/- SD). # p < 0.05 greater than vehicle control; * p < 0.05 less than vehicle control.
In Figure 3 and Figure S1 we determined that the drug combination reduced the activities of ERK1/2, AKT and STAT3. Over-expression of activated AKT or over-expression of activated MEK1, to a greater extent than expression of activated STAT3, protected BT474 mammary tumor cells from [neratinib + palbociclib] (Figure 5A). Expression of activated AKT suppressed the inactivation of mTOR and activation of ULK-1, also as judged by reduced ATG13 phosphorylation (Figure 5B). Activated AKT and activated MEK1 both prevented [neratinib + palbociclib] from enhancing expression of BIM, Beclin1 and ATG5. Activated MEK1, and to a lesser extent activated AKT, however, did not alter the levels of drug-induced eIF2α S51 phosphorylation. i.e. the observed elevated endoplasmic reticulum stress signaling is likely to have less importance in killing tumor cells, per se, than the inactivation of AKT, mTOR and the ERK1/2 MAP kinase pathway. Similar data were also obtained in Spiky ovarian cancer cells (Figure S2).
Figure 5.
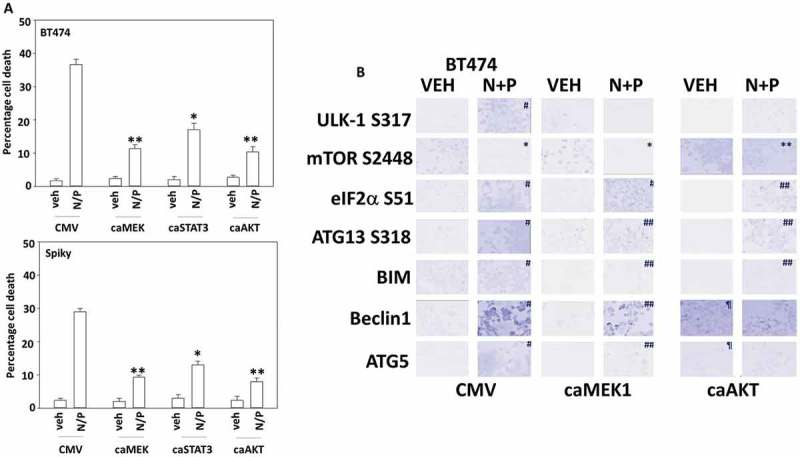
[Neratinib + palbociclib] enhances the expression of BIM, Beclin1 and ATG5 that is suppressed by expression of activated MEK1 or activated AKT. A. Mammary (BT474) and ovarian (Spiky) cancer cells were transfected with empty vector (CMV) or to express the indicated “activated” proteins. Twenty-four h after transfection cells were treated with vehicle control or with [neratinib (100 nM) + palbociclib (100 nM)] for an additional 24h. Cell viability was determined by trypan blue exclusion and the percentage cell death under each condition above that in vehicle control plotted (n = 3 +/- SD). * p < 0.05 less cell death than in the corresponding value in CMV; ** p < 0.05 less than value in activated STAT3 expressing cells. B. BT474 cells were transfected with empty vector (CMV) or to express the indicated “activated” proteins. Twenty-four h after transfection cells were treated with vehicle control or with [neratinib (100 nM) + palbociclib (100 nM)] for an additional 6h. Cells were fixed in place and at least forty cells per condition were imaged in independent triplicate and the intensity ratio of phosphorylated protein levels to total protein expression was determined (n = 3 +/- SD).* p < 0.05 less intensity in the corresponding value in vehicle control; # p < 0.05 greater intensity in the corresponding value in vehicle control; ¶ p < 0.05 greater intensity than corresponding value in CMV transfected cells; ** p < 0.05 lower percentage reduction in intensity than in CMV cells; ## p < 0.05 lower percentage increase in intensity than in CMV cells.
At present there is an open clinical trial combining neratinib and palbociclib in breast cancer patients (NCT03065387). We are about to open a phase I trial in collaboration with Puma Biotechnology combining neratinib with the HDAC inhibitor sodium valproate for all qualifying solid tumor patients. In several prior publications we have shown that both the HDAC inhibitor sodium valproate (Depakote®) and the PDE5 inhibitor sildenafil (Viagra®) can enhance the formation of autophagosomes and autolysosomes caused by other agents, which in turn leads to elevated levels of tumor cell killing.10–12 Thus, we next determined whether valproate or sildenafil could enhance “autophagy” and thus the lethality of [neratinib + palbociclib].
Treatment of multiple mammary and ovarian tumor cell lines with either neratinib or palbociclib increased the numbers of autophagosomes within tumor cells (Figure 6). Both agents combined in a greater than additive fashion to increase autophagosome levels. Sodium valproate in a greater than additive manner further enhanced autolysosome formation. Autophagic flux appeared to be occurring under all treatment conditions as the elevated levels of GFP+ autophagosomes observed at 4 hours were reduced by 8 hours and the numbers of RFP+ autolysosomes observed at 4 hours were negligible but were significantly enhanced by 8 hours. We next determined the impact of sodium valproate and sildenafil on the lethality of [neratinib + palbociclib], note, after 12 hours of incubation. Neratinib and palbociclib interacted in a greater than additive fashion to kill mammary and ovarian tumor cells (Figure 7). As observed for the autophagy biomarkers, sodium valproate in a greater than additive manner further enhanced tumor cell killing by [neratinib + palbociclib]; sildenafil was less effective. As clinical studies are underway combining [neratinib + palbociclib] and [neratinib + valproate], this data would collectively argue for a possible future trial combining the three drugs at the safe RP2D in both trials with neratinib.
Figure 7.
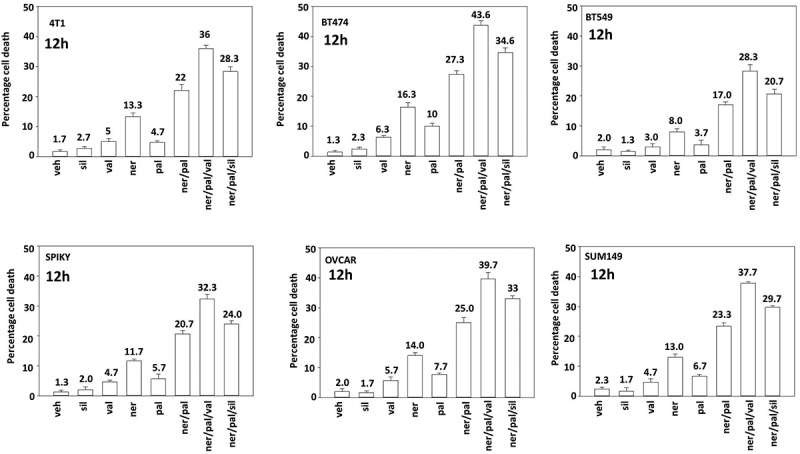
Sodium valproate and [neratinib + palbociclib] interact in a greater than additive fashion to cause tumor cell death. Mammary carcinoma cells (BT474 (ERBB2+), BT549, SUM149, 4T1 (TNBC)) and ovarian cancer cells (Spiky, OVCAR3) were treated with vehicle control or with neratinib (100 nM), palbociclib (100 nM), sildenafil (2 μM), sodium valproate (250 μM) or the drugs in combination as indicated in the figure. Cells were isolated after 12h and cell viability determined by trypan blue exclusion assay. A representative study from 3 is shown (n = 3 +/- SD).
We next performed studies to determine how valproate altered cell signaling processes and enhanced the autophagy and the cell killing response to [neratinib + palbociclib]. Treatment of cells with [neratinib + palbociclib + valproate] resulted in a greater and more prolonged activation of ATM, AMPK and ULK-1; this was associated with greater ATG13 S318 phosphorylation and greater eIF2α S51 phosphorylation (Figure 8A). Treatment of cells with [neratinib + palbociclib + valproate] resulted in a greater and more prolonged inactivation of mTOR, AKT and ERK1/2. The three-drug combination caused a prolonged inactivation of p70 S6K, which was congruent with our mTOR and AKT phosphorylation data. The three-drug combination caused a prolonged reduction in the expression of MCL-1 and of c-FLIP-s, that, as will be presented in Figure 9, is also associated with enhanced death receptor activation. Similar data were observed in CT26 mouse colon cancer cells that express a mutant K-RAS (Figure S3). The three-drug combination significantly reduced K-RAS protein expression within 4h in CT26 cells (Figure 8B, upper). Note, that in [neratinib + palbociclib] treated cells after 4h, the K-RAS protein became punctate in a manner like that observed in the “capping” process of growth factor receptors (Figure 8B, lower; Figure S4). After 8h of incubation, [neratinib + palbociclib] and the three-drug combination had profoundly reduced K-RAS expression.
Figure 8.
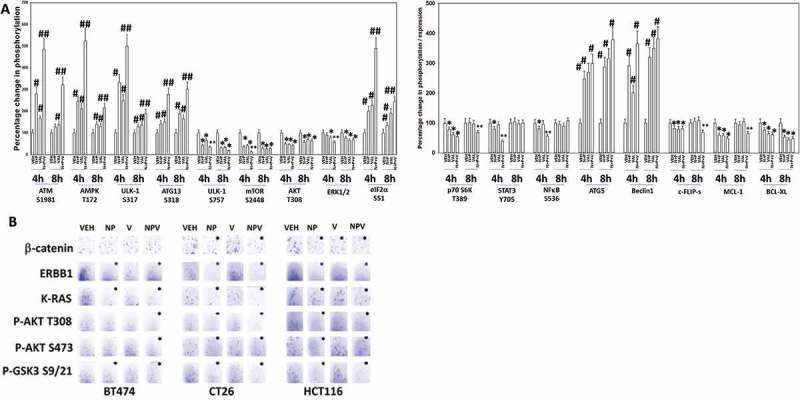
Sodium valproate enhances [neratinib + palbociclib] -induced activation of ATM, AMPK, ULK-1 and eIF2α, and facilitates a greater inactivation of mTOR, AKT, p70 S6K and ERK1/2. A. Spiky ovarian cancer cells were treated with vehicle control, [neratinib (100 nM) + palbociclib (100 nM)], sodium valproate (250 μM) or the drugs in combination for 4h and for 8h. Cells were fixed in place and at least forty cells per condition were imaged in independent triplicate and the intensity ratio of phosphorylated protein levels to total protein expression was determined (n = 3 +/- SD).* p < 0.05 less intensity in the corresponding value in vehicle control; # p < 0.05 greater intensity in the corresponding value in vehicle control; ** p < 0.05 lower intensity than in individual drug-treated cells; ## p < 0.05 greater intensity than in individual drug-treated cells. B. BT474 (human, mammary); HCT116 (human, colorectal); CT26 (mouse, colon) cancer cells that express a mutant K-RAS protein were treated with vehicle control, [neratinib (100 nM) + palbociclib (100 nM)], sodium valproate (250 μM) or the drugs in combination for 4h and for 8h. Cells were fixed in place and at least forty cells per condition were imaged in independent triplicate and the intensity ratio of phosphorylated protein levels to total protein expression was determined (n = 3 +/- SD).* p < 0.05 less intensity in the corresponding value in vehicle control; ** p < 0.05 lower intensity than in individual drug-treated cells.
Figure 9.
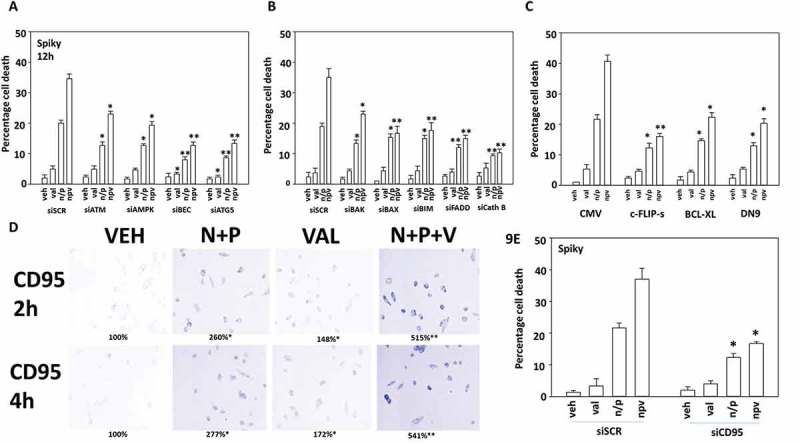
The induction of autophagy and the lysosomal protease cathepsin B play essential roles in the ability of valproate to enhance [neratinib + niraparib] lethality. A.-C. Spiky ovarian carcinoma cells were transfected with a scrambled siRNA (siSCR) or with siRNA molecules to knock down the indicated proteins. In parallel studies, cells were transfected with an empty vector plasmid (CMV) or with plasmids to express the indicated proteins. Twenty-four h after transfection, cells were treated with vehicle control or with neratinib (100 nM), palbociclib (100 nM), sildenafil (2 μM), sodium valproate (250 μM) or the drugs in combination as indicated in the figure. Cells were isolated after 12h and cell viability determined by trypan blue exclusion assay. A representative study from 3 is shown (n = 3 +/- SEM). * p < 0.05 less than corresponding value in siSCR/CMV transfected cells; in Panel A: ** p < 0.05 less than corresponding values in siATM and siAMPK cells; Panel B: ** p < 0.05 less than corresponding value in siBAK cells; Panel C. ** p < 0.05 less than corresponding values in cells expressing BCL-XL or dominant negative caspase 9. D. Spiky cancer cells were treated with vehicle control, [neratinib (100 nM) + palbociclib (100 nM)], valproate (250 μM) or the drugs in combination, as indicated for 4h. Cells were fixed in place and not permeabilized. Immunostaining was performed to detect the cell surface levels of the death receptor CD95 (n = 3 +/- SD) * p < 0.05 greater than corresponding value in vehicle treated cells; ** p < 0.05 greater than corresponding value in [N + P] cells. E. Spiky cells were transfected with a scrambled control siRNA (siSCR) or with an siRNA molecule to knock down the expression of CD95. Twenty-four h after transfection, cells were treated with vehicle control or with neratinib (100 nM), palbociclib (100 nM), sodium valproate (250 μM) or the drugs in combination as indicated in the figure. Cells were isolated after 12h and cell viability determined by trypan blue exclusion assay. A representative study from 3 is shown (n = 3 +/- SEM). * p < 0.05 less than corresponding value in siSCR transfected cells.
Knock down of ATM or AMPKα significantly reduced the lethality of [neratinib + palbociclib] and of [neratinib + palbociclib + valproate] (Figure 9A). However, knock down of either Beclin1 or ATG5 more effectively suppressed three-drug combination lethality than did knock down of ATM or AMPKα. i.e. under scrambled control transfection the true lethality of [neratinib + palbociclib] was enhanced ~ 15% by valproate; under siATM enhanced by ~ 10%; under siAMPKα enhanced by ~ 7%; and for siBeclin1 and siATG5 enhanced by ~ 4%. Thus, the ability of valproate to enhance autophagosome formation is essential for it to also enhance [neratinib + palbociclib] killing. In agreement with autophagy playing a key role in killing, and in particular with autophagic flux, knock down of the lysosomal protease cathepsin B also prevented the two- and three-drug combinations from killing (Figure 9B). Similar protective findings were made for knock down of FADD or over-expression of c-FLIP-s, that together with data from Figure 8 implies that death receptor signaling is also being enhanced by valproate (Figure 9C). As judged by increased cell surface localization, [neratinib + palbociclib] activated CD95 that was significantly enhanced by valproate (Figure 9D). Finally, confirming the importance of death receptor signaling in our system, knock down of CD95 significantly reduced the ability of valproate to enhance [neratinib + palbociclib] lethality (Figure 9E).
Finally, using the Spiky PDX ovarian cancer isolate, we determined the interactions between neratinib, palbociclib and valproate. [Valproate + palbociclib] significantly enhanced the anti-tumor efficacy of neratinib against Spiky ovarian cancer tumors (Figure 10). No apparent toxicities were observed in normal tissues judged by H&E staining (brain, lung, heart, liver, spleen, colon and kidneys). Our findings argue that the HDAC inhibitor sodium valproate represents a cost-effective drug to enhance the efficacy of neratinib and palbociclib.
Figure 10.
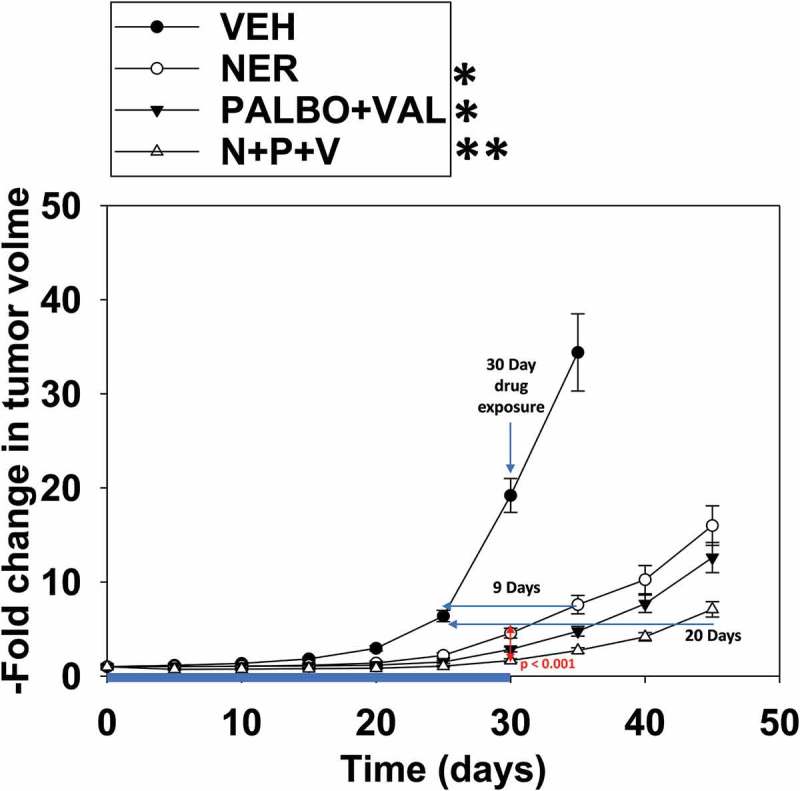
[Valproate + palbociclib] interact to suppress tumor growth that is further enhanced by neratinib. Animals were then segregated into groups with near identical mean volumes and the animals then treated for 30 days with the indicated therapeutic agents: vehicle control (cremophore); neratinib 15 mg/kg QD, [palbociclib 5 mg/kg QD and sodium valproate 50 mg/kg QD]; or the three drugs in combination. Tumor volumes were measured prior to drug administration and every five days after the initiation of therapeutic interventions. (n = 8 mice per group +/-SEM). Before, during and after drug treatment tumors are calipered as indicated in the Figure and tumor volume was assessed up to ~ 40 days later. When the volume of the tumor reached > 1,000 mm3, animals were humanely sacrificed. * p < 0.05 less growth than vehicle control; ** p < 0.05 less growth than neratinib alone; *** p < 0.05 less growth than [valproate + palbociclib].
Discussion
The present studies were performed to determine whether the drugs neratinib and palbociclib, both of which induce autophagosome formation, interacted to kill tumor cells. Our data demonstrated that these drugs interact in a greater than additive fashion to kill a diverse cohort of different tumor cell types. Based on our positive data, and our prior experience with PDE5 inhibitors and HDAC inhibitors both facilitating greater levels of toxic autophagosome levels, we then determined that the cost-effective HDAC inhibitor sodium valproate caused a greater enhancement of [neratinib + palbociclib] killing than did the PDE5 inhibitor sildenafil. In vivo, neratinib, palbociclib and sodium valproate interacted to significantly suppress tumor growth.
Prior studies from this laboratory demonstrated that the NSCLC medication pemetrexed interacted with the liver/kidney cancer mediation sorafenib to promote elevated levels of autophagosome formation, which was causal in tumor cell killing.13 Of note were the observations made in the ER+ breast cancer line MCF7. It is known that reduced Beclin1 function plays an important role in mammary tumorigenesis, and MCF7 cells are known to be haplotype deleted for Beclin1.14–17 Our prior studies demonstrated that paired MCF7 cells made resistant to the pure anti-estrogen fulvestrant had dramatically increased their basal expression levels of Beclin1, and of ATG5-ATG12, and as a result, exhibited greater autophagosome formation and were more sensitive to the [pemetrexed + sorafenib] combination than the parental MCF7 cells.13 Neratinib is approved as an adjuvant therapy in ER- ERBB2+ breast cancer patients, and the BT474 cells used in our studies were obtained from the ATCC and originally grown in bulk using female athymic mice but without any additional estrogen supplementation, and as a result these tumorized BT474 cells express low levels of the estrogen receptor and grow in vitro and in vivo in an estrogen-independent fashion. i.e. they represent a model for both de novo ERBB2+ breast cancer and also for ER+ breast cancers that have evolved from estrogen receptor addiction to requiring ERBB2 signaling as a survival mechanism; the disease sub-types for which neratinib is approved. We have previously published that our tumorized BT474 cells are killed by neratinib as single agents through a mechanism requiring expression of Beclin1 and ATG5.3
Palbociclib is approved for the treatment of ER+ ERBB2 negative breast cancer, and the standard of care for its use in patients includes anti-estrogen or anti-aromatase therapies in combination with the CDK4/6 inhibitor. Our data demonstrated that for palbociclib as a single agent to kill also required the expression of Beclin1 and ATG5, as did the combination with neratinib. The precise target(s) by which palbociclib stimulated autophagosome formation are at present somewhat unclear. Others have observed that palbociclib and other CDK4/6 inhibitors can induce autophagy.18,19 Some have argued that Cyclin D1 can inhibit AMPK activation which may link the CDK4/6 inhibitors to autophagy.20 Knock down of Cyclin D1 enhanced the phosphorylation of AMPKα T172 whereas over-expression of the cyclin prevented metabolic stresses from increasing T172 levels; this was linked to Cyclin D1/CDK4/6 phosphorylating and inactivating LKB-1. Unfortunately, these authors used 5-fold greater concentrations of palbociclib to our own which is above the safe plasma C max concentration, and our ability of palbociclib to enhance AMPKα T172 phosphorylation occurs in cells with very low/no LKB-1 expression such as HT1080 (sarcoma), A549 and H460 (NSCLC) and HCT116 (colon).21 However, others have reported that palbociclib in a CDK4/6-independent fashion activates the AMPK.22 And, that this effect may be due to inhibition of other targets such as CDK9, which has a similar consensus sequence to that of CDK4/6 and ERK1/2.23,24 Inhibition of CDK9 in parallel with elevated eIF2α phosphorylation would be predicted to profoundly reduce the expression of multiple proteins with short half-lives such as the cyto-protective protein MCL-1, that would in turn promote mitochondrial dysfunction and Beclin1 release.
We found that palbociclib does not cause mTOR inactivation, although it did promote a modest amount of ULK-1 S757 dephosphorylation. Thus, palbociclib could be reducing mTOR catalytic activity or modifying mTOR complex formation. In agreement with reference 21, palbociclib activated the AMPK, as judged by elevated T172 phosphorylation, which in turn is directly responsible for the elevated phosphorylation observed in ULK-1 S317, TSC2 T1496 and Raptor S792; and, activation of the AMPK played an essential role in drug combination lethality. In prior studies using neratinib, we have linked enhanced AMPKα T172 phosphorylation to activation of ATM, however palbociclib did not significantly enhance ATM activity. Studies outside of the scope of this manuscript will be required to determine whether palbociclib alters the activities of LKB-1 and calcium-calmodulin dependent kinases that also target AMPKα T172. Collectively, through convergent and independent signaling pathways, neratinib and palbociclib interact to promote autophagosome formation that is essential for tumor cell execution (Figure 11). As breast cancer cells, in part, survive anti-estrogen therapy via increased Beclin1 expression and autophagy, the use of palbociclib alone or in combination with neratinib will circumvent both the autophagy resistance mechanism in parallel to the mechanism of elevated ERBB2 expression and signaling.
Figure 11.
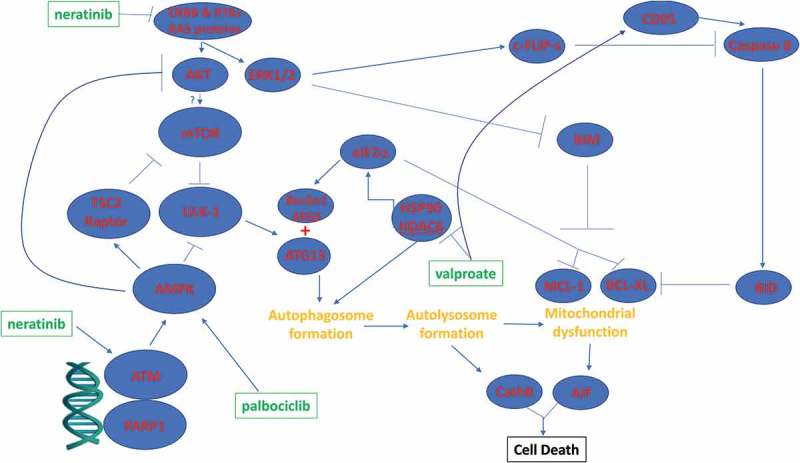
Putative mechanisms by which neratinib, palbociclib and valproate interact to kill tumor cells.
A phase I trial combining neratinib and palbociclib is already underway at M.D. Anderson Cancer Center (NCT03065387). The dose-escalation portion of this study is to find the highest tolerable dose of neratinib in combination with either everolimus, palbociclib, or trametinib that can be given to patients who have advanced cancers; specifically, tumors with a specific mutation in either ERBB1, ERBB2, ERBB3 or ERBB4. Whether such an approach would also be successful in amplified non-mutated ERBB2 is unknown. Based on our present findings, the combination of [neratinib + palbociclib] in ERBB2+ breast cancer may be a novel useful modality. We will soon be opening a phase I trial at Massey Cancer Center in late 2018 in all solid tumor patients combining neratinib with the HDAC inhibitor sodium valproate.
In other pre-clinical and clinical studies, we have used the PDE5 inhibitor sildenafil to promote the lethality of kinase inhibitors (NCT02466802; NCT01817751). Thus, based on our preliminary studies examining the molecular mechanisms by which neratinib and palbociclib interact, we wished to develop a novel three drug combination that would further enhance tumor cell killing. We determined whether either valproate or sildenafil, agents that also enhance autophagosome formation, could increase the efficacy of [neratinib + palbociclib].12,25 Although both valproate and sildenafil enhanced [neratinib + palbociclib] lethality in a greater than additive fashion, at the clinically relevant concentrations used, valproate was superior to sildenafil. The addition of valproate to [neratinib + palbociclib] enhanced the formation of autophagosomes and autolysosomes in a greater than additive fashion which correlated with the enhanced levels of tumor cell killing. Knock down of ATG5 or Beclin1 profoundly reduced the levels of tumor cell death induced by [neratinib + palbociclib] and by [neratinib + palbociclib + valproate]. This also correlated with a strong protective effect in cells with reduced expression of the lysosomal protease cathepsin B. In tumor cells exposed to [neratinib + palbociclib], expression of the caspase 8 inhibitor c-FLIP-s modestly reduced the levels of tumor cell killing whereas in cells exposed to the three-drug combination both expression of c-FLIP-s or knock down of FADD exhibited strong protective effects. Cells treated with the three-drug combination exhibited elevated plasma membrane levels of the death receptor CD95, which is indicative of receptor activation. Thus, the addition of valproate to [neratinib + palbociclib] both increases the levels of toxic autophagic flux with cathepsin B activation, and in parallel promotes death receptor activation that through caspases 8/10 also leads to tumor cell death (Figure 11).
The safety of any drug or drug combination is initially determined in a phase I trial. The dose-limiting toxicity of palbociclib is neutropenia; of neratinib is colon dysfunction/diarrhea; for valproate liver toxicity. Thus, none of the proposed three drugs in our combination has an overlapping primary dose-limiting toxicity with either of the other drugs. Once the [neratinib + palbociclib] and [neratinib + valproate] trials are completed, with their own defined toxicities, it may be possible with careful scheduling to safely combine all three agents.
The mechanisms by which valproate can enhance the lethality of other agents have previously been explored in detail by our group. Valproate can stimulate endoplasmic reticulum stress via inhibition of HDAC6 that causes inactivation of HSP90, with subsequent elevated levels of misfolded proteins. Of note, HSP90 stabilizes LKB-1, arguing that LKB-1 is unlikely to be our palbociclib regulated kinase.26,27 Knock down of eIF2α reduced both [neratinib + palbociclib] and [neratinib + palbociclib + valproate] -induced cell killing (Booth and Dent, unpublished observations). In the presence of other agents which induce autophagy, such as neratinib, this also causes the lysosomal-dependent degradation of HDAC6 as well as other cellular HDACs, profoundly altering cellular biology. In tumors, this alteration of biology can be observed up to two weeks after cessation of drug treatments.5 For example, neratinib and valproate, through down-regulation of HDAC function and expression, alters the protein expression of immunogenic biomarkers. In the present studies we discovered that [neratinib + palbociclib] also could rapidly decrease the expression of PD-L1, PD-L2, IDO-1 and increase the levels of MHCA (Figure S5). These events were associated with the extracellular release of the immunogenic protein HMGB1. Whether [neratinib + palbociclib] can be utilized as an approach to enhance tumor cell immunogenicity and enhance the actions of immune checkpoint inhibitory antibodies will require studies beyond the scope of this manuscript.
The studies in this manuscript utilized the cheap generic low-efficacy HDAC inhibitor sodium valproate. This was based on the fore-knowledge that drug expenses can often make the performance of drug-combination clinical trials cost-prohibitive. We have performed studies with other FDA-approved HDAC inhibitors including vorinostat and etinostat, as well as the clinically relevant HDAC inhibitor AR42, and demonstrated that these agents too also enhance [neratinib + palbociclib] activity. However, use of those FDA agents would likely make the translation of [neratinib + palbociclib + HDAC inhibitor] too expensive to implement as a clinical trial. It is hoped, at the completion of the [neratinib + palbociclib] and [neratinib + valproate] trials that a new clinical study can be initiated combining [neratinib + palbociclib + valproate].
Materials and methods
Materials
Palbociclib, valproate and sildenafil were from Selleckchem (Houston, TX). Neratinib was supplied by Puma Biotechnology Inc. (Los Angeles, CA). Trypsin-EDTA, DMEM, RPMI, penicillin-streptomycin were purchased from GIBCOBRL (GIBCOBRL Life Technologies, Grand Island, NY). Tumor cells were purchased from the ATCC and were not further validated beyond that claimed by ATCC. Cells were re-purchased every ~ 6 months. Spiky ovarian cancer cells, an established PDX model, were kindly provided by Dr. Karen Paz (Champions Oncology, NJ). Commercially available validated short hairpin RNA molecules to knock down RNA/protein levels were from Qiagen (Valencia, CA) (Figure 12). See references 10–13.
Figure 12.
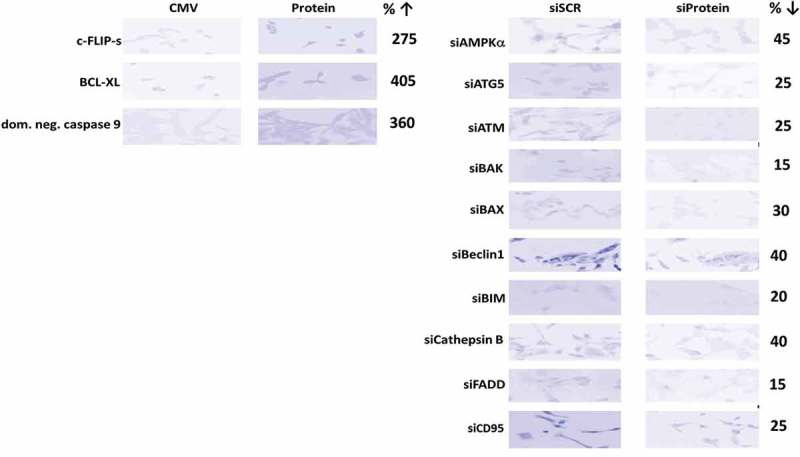
Representative images of proteins knocked down or over-expressed in the present studies. The percentage of protein expression remaining after knock-down is presented.
Methods
Culture and in vitro exposure of cells to drugs. All cell lines were cultured at 37°C (5% (v/v CO2) in vitro using RPMI supplemented with dialyzed 5% (v/v) fetal calf serum and 1% (v/v) Non-essential amino acids. The safe achievable plasma Cmax for neratinib is ~ 150 nM and for palbociclib it is ~ 0.5 μM. Neratinib and palbociclib were both used, in vitro, at 100 nM.
Transfection of cells with siRNA or with plasmids. See references 10–13.
Detection of cell viability, protein expression and protein phosphorylation by immuno-fluorescence using a Hermes WiScan wide-field microscope. http://www.idea-bio.com/. See references 10–13.
Detection of cell death by Trypan Blue assay. See references 10–13.
Assessment of autophagy: See references 10–13.
Animal Studies. Studies were performed per USDA regulations under VCU IACUC protocol AD20008. Spiky ovarian carcinoma cells (2 x 106) were implanted into rear flanks of female NRG mice. Tumors were permitted to form until the mean tumor volume was ~ 40 mm3. Animals were then segregated into groups with near identical mean volumes and the animals then treated for 30 days with the indicated therapeutic agents: vehicle control (cremophore); neratinib 15 mg/kg QD, [palbociclib 5 mg/kg QD and sodium valproate 50 mg/kg QD]; or the three drugs in combination. Tumor volumes were measured prior to drug administration and every five days after the initiation of therapeutic interventions. (n = 8 mice per group ± SEM). Before, during and after drug treatment tumors are calipered as indicated in the Figure and tumor volume was assessed up to 20–45 days later. When the volume of the tumor reached > 1,000 mm3, animals were humanely sacrificed.
Data analysis. Comparison of the effects of various treatments (performed in triplicate three times) was using one-way analysis of variance and a two tailed Student’s t-test. Differences with a p-value of < 0.05 were considered statistically significant. Experiments shown are the means of multiple individual points from multiple experiments (± SEM).
Funding Statement
This work was supported by the HHS | NIH | National Cancer Institute (NCI) [R01 CA192613].
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Support for the present study was funded from philanthropic funding from Massey Cancer Center, the Universal Inc. Chair in Signal Transduction Research and PHS R01-CA192613 (PD). Thanks to Dr. H.F. Young and the Betts family fund for support in the purchase of the Hermes Wiscan instrument. The authors have no conflicts of interest to report.
Supplementary Material
Supplemental data for this article can be accessed Supplemental Material.
Abbreviations
ERK extracellular regulated kinase
PI3K phosphatidyl inositol 3 kinase
ca constitutively active
dn dominant negative
ER endoplasmic reticulum
AIF apoptosis inducing factor
AMPK AMP-dependent protein kinase
mTOR mammalian target of rapamycin
JAK Janus Kinase
STAT Signal Transducers and Activators of Transcription
MAPK mitogen activated protein kinase
PTEN phosphatase and tensin homologue on chromosome ten
ROS reactive oxygen species
CMV empty vector plasmid or virus
si small interfering
SCR scrambled
PDE phospho-diesterase
IP immunoprecipitation
VEH vehicle
NER neratinib
PAL palbociclib
VAL sodium valproate
SIL sildenafil
HDAC histone deacetylase
CDK cyclin dependent kinase
TEM temsirolimus
References
- 1.Zardavas D, Pondé N, Tryfonidis K.. CDK4/6 blockade in breast cancer: current experience and future perspectives. Expert Opin Investig Drugs. 2017;26(12):1357–1372. doi: 10.1080/13543784.2017.1389896. [DOI] [PubMed] [Google Scholar]
- 2.Echavarria I, López-Tarruella S, Márquez-Rodas I, Jerez Y, Martin M. Neratinib for the treatment of HER2-positive early stage breast cancer. Expert Rev Anticancer Ther. 2017;17:669–679. doi: 10.1080/14737140.2017.1338954. [DOI] [PubMed] [Google Scholar]
- 3.Booth L, Roberts JL, Poklepovic A, Avogadri-Connors F, Cutler RE, Lalani AS, Dent P. HDAC inhibitors enhance neratinib activity and when combined enhance the actions of an anti-PD-1 immunomodulatory antibody in vivo. Oncotarget. 2017;8:90262–90277. doi: 10.18632/oncotarget.21660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Booth L, Roberts JL, Poklepovic A, Kirkwood J, Sander C, Avogadri-Connors F, Cutler RE Jr., Lalani AS, Dent P. The levels of mutant K-RAS and mutant N-RAS are rapidly reduced in a Beclin1/ATG5 -dependent fashion by the irreversible ERBB1/2/4 inhibitor neratinib. Cancer Biol Ther. 2018;19:132–137. doi: 10.1080/15384047.2017.1394556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Booth L, Roberts JL, Rais R, Kirkwood J, Avogadri-Connors F, Cutler RE Jr., Lalani AS, Poklepovic A, Dent P. [Neratinib + Valproate] exposure permanently reduces ERBB1 and RAS expression in 4T1 mammary tumors and enhances M1 macrophage infiltration. Oncotarget. 2017;9:6062–6074. doi: 10.18632/oncotarget.23681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rabindran SK, Discafani CM, Rosfjord EC, Baxter M, Floyd MB, Golas J, Hallett WA, Johnson BD, Nilakantan R, Overbeek E, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64:3958–3965. doi: 10.1158/0008-5472.CAN-03-2868. [DOI] [PubMed] [Google Scholar]
- 7.Needham SR, Zanetti-Domingues LC, Hirsch M, Rolfe DJ, Tynan CJ, Roberts SK, Martin-Fernandez ML, Clarke DT. Structure-function relationships and supramolecular organization of the EGFR (epidermal growth factor receptor) on the cell surface. Biochem Soc Trans. 2014;42:114–119. doi: 10.1042/BST20130236. [DOI] [PubMed] [Google Scholar]
- 8.Mitchell C, Yacoub A, Hossein H, Martin AP, Bareford MD, Eulitt P, Yang C, Nephew KP, Dent P. Inhibition of MCL-1 in breast cancer cells promotes cell death in vitro and in vivo. Cancer Biol Ther. 2010;10:903–917. doi: 10.4161/cbt.10.9.13273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan AR, Yang X, Berman A, Zhai S, Sparreboom A, Parr AL, Chow C, Brahim JS, Steinberg SM, Figg WD, et al. Phase I trial of the cyclin-dependent kinase inhibitor flavopiridol in combination with docetaxel in patients with metastatic breast cancer. Clin Cancer Res. 2004;10:5038–5047. doi: 10.1158/1078-0432.CCR-04-0025. [DOI] [PubMed] [Google Scholar]
- 10.Tavallai M, Hamed HA, Roberts JL, Cruickshanks N, Chuckalovcak J, Poklepovic A, Booth L, Dent P. Nexavar/Stivarga and viagra interact to kill tumor cells. J Cell Physiol. 2015;230:2281–2298. doi: 10.1002/jcp.24961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Booth L, Albers T, Roberts JL, Tavallai M, Poklepovic A, Lebedyeva IO, Dent P. Multi-kinase inhibitors interact with sildenafil and ERBB1/2/4 inhibitors to kill tumor cells in vitro and in vivo. Oncotarget. 2016;7:40398–40417. doi: 10.18632/oncotarget.9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Booth L, Roberts JL, Poklepovic A, Gordon S, Dent P. PDE5 inhibitors enhance the lethality of pemetrexed through inhibition of multiple chaperone proteins and via the actions of cyclic GMP and nitric oxide. Oncotarget. 2017;8:1449–1468. doi: 10.18632/oncotarget.13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bareford MD, Park MA, Yacoub A, Hamed HA, Tang Y, Cruickshanks N, Eulitt P, Hubbard N, Tye G, Burow ME, et al. Sorafenib enhances pemetrexed cytotoxicity through an autophagy-dependent mechanism in cancer cells. Cancer Res. 2011;71:4955–4967. doi: 10.1158/0008-5472.CAN-11-0898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Motyl T, Gajkowska B, Zarzyńska J, Gajewska M, Lamparska-Przybysz M. Apoptosis and autophagy in mammary gland remodeling and breast cancer chemotherapy. J Physiol Pharmacol. 2006;57(S7):17–32. [PubMed] [Google Scholar]
- 15.Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ. 2010;17:268–277. doi: 10.1038/cdd.2009.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z, Chen B, Wu Y, Jin F, Xia Y, Liu X. Genetic and epigenetic silencing of the beclin 1 gene in sporadic breast tumors. BMC Cancer. 2010;10:98–110. doi: 10.1186/1471-2407-10-663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valenzuela CA, Vargas L, Martinez V, Bravo S, Brown NE. Palbociclib-induced autophagy and senescence in gastric cancer cells. Exp Cell Res. 2017;360:390–396. doi: 10.1016/j.yexcr.2017.09.031. [DOI] [PubMed] [Google Scholar]
- 19.Iriyama N, Hino H, Moriya S, Hiramoto M, Hatta Y, Takei M, Miyazawa K. The cyclin-dependent kinase 4/6 inhibitor, abemaciclib, exerts dose-dependent cytostatic and cytocidal effects and induces autophagy in multiple myeloma cells. Leuk Lymphoma. 2018;59:1439–1450. [DOI] [PubMed] [Google Scholar]
- 20.Casimiro MC, Di Sante G, Di Rocco A, Loro E, Pupo C, Pestell TG, Bisetto S, Velasco-Velázquez MA, Jiao X, Li Z, et al. Cyclin D1 restrains oncogene-induced autophagy by regulating the AMPK-LKB1 signaling axis. Cancer Res. 2017;77:3391–3405. doi: 10.1158/0008-5472.CAN-16-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin L, Chun J, Pan C, Kumar A, Zhang G, Ha Y, Li D, Alesi GN, Kang Y, Zhou L, et al. The PLAG1-GDH1 axis promotes anoikis resistance and tumor metastasis through CamKK2-AMPK signaling in LKB1-deficient lung cancer. Mol Cell. 2018;69:87–99.e7. doi: 10.1016/j.molcel.2017.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsieh FS, Chen YL, Hung MH, Chu PY, Tsai MH, Chen LJ, Hsiao YJ, Shih CT, Chang MJ, Chao TI, et al. Palbociclib induces activation of AMPK and inhibits hepatocellular carcinoma in a CDK4/6-independent manner. Mol Oncol. 2017;11:1035–1049. doi: 10.1002/1878-0261.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sumi NJ, Kuenzi BM, Knezevic CE, Remsing Rix LL, Rix U. Chemoproteomics reveals novel protein and lipid kinase targets of clinical CDK4/6 inhibitors in lung cancer. ACS Chem Biol. 2015;10:2680–2686. doi: 10.1021/acschembio.5b00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shore SM, Byers SA, Dent P, Price DH. Characterization of Cdk9(55) and differential regulation of two Cdk9 isoforms. Gene. 2005;350:51–58. doi: 10.1016/j.gene.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 25.Booth L, Roberts JL, Poklepovic A, Dent P. [pemetrexed + sildenafil], via autophagy-dependent HDAC downregulation, enhances the immunotherapy response of NSCLC cells. Cancer Biol Ther. 2017;18:705–714. doi: 10.1080/15384047.2017.1362511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boudeau J, Deak M, Lawlor MA, Morrice NA, Alessi DR. Heat-shock protein 90 and Cdc37 interact with LKB1 and regulate its stability. Biochem J. 2003;370:849–857. doi: 10.1042/BJ20021813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. PNAS. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.