

ABSTRACT
Maintenance of mitochondrial quality is essential for skeletal muscle function and overall health. Exercise training elicits profound adaptations to mitochondria to improve mitochondrial quality in skeletal muscle. We have recently demonstrated that acute exercise promotes removal of damaged/dysfunctional mitochondria via mitophagy in skeletal muscle during recovery through the Ampk-Ulk1 signaling cascade. In this Extra View, we explore whether Pink1 is stabilized on mitochondria following exercise as the signal for mitophagy. We observed no discernable presence of Pink1 in isolated mitochondria from skeletal muscle at any time point following acute exercise, in contrast to clear evidence of stabilization of Pink1 on mitochondria in HeLa cells following treatment with the uncoupler carbonyl cyanide m-chlorophenyl hydrazone (CCCP). Taken together, we conclude that Pink1 is not involved in exercise-induced mitophagy in skeletal muscle.
KEYWORDS: Mitophagy, skeletal muscle, exercise, Pink1
Introduction
Exercise is well known to promote systemic health and, hence, is an effective intervention for the prevention and treatment of a number of chronic diseases [1]. In particular, regular exercise maintains skeletal muscle health and function, as well as mitigates the loss of skeletal muscle mass with age, known as sarcopenia [2]. Mitochondria are the primary producers of cellular ATP in skeletal muscle required for exercise and other physiological functions [3]. Detriments in the quality of skeletal muscle mitochondria result in tissue dysfunction and are thought to be primary contributors to the development of chronic disease [2,4,5]. Therefore, understanding how mitochondrial quality is regulated and how those processes are influenced by exercise can lead to the development of novel therapies to maintain skeletal muscle function and, thereby, overall health.
Skeletal muscle mitochondria exist as an interconnected reticulum [6,7]. In response to yet unknown signals, damaged, dysfunctional, and/or energetically stressed regions of the reticulum are selectively removed through fission and mitophagy, a highly regulated process of selective autophagy of mitochondria [3,8]. Recently, we have demonstrated that acute exercise induces mitophagy in skeletal muscle during the recovery period, which was dependent upon Ampk-mediated phosphorylation of Ulk1 at Ser555 [4]. By imaging mitophagy via a fluorescent mitochondrial reporter, MitoTimer [9,10], we were able to observe mitophagy in response to a single bout of exercise [4]. However, it is unclear what mechanism(s) may mediate the recognition of regions of the mitochondrial reticulum destined for mitophagy in response to exercise.
Arguably, the best understood means by which damaged regions of the mitochondrial reticulum are differentiated for mitophagy is through PTEN-induced putative kinase 1 (Pink1). In this model, Pink1 is thought to be constantly transported to the mitochondria from the cytosol and, when the mitochondrial reticulum is healthy, Pink1 is imported, cleaved, and subsequently degraded in the mitochondria [11,12]. However, when mitochondrial quality is compromized (e.g. a decline in membrane potential, accumulation in misfolded proteins, and/or damage to mtDNA), Pink1 is stabilized on the outer mitochondrial membrane (OMM) [13–18]. Once stabilized, Pink1 recruits the E3 ubiquitin ligase, Parkin, which initiates a cascade of events leading to the degradation of the affected region(s) of the reticulum [15,18–20]. However it has not been demonstrated whether stabilization of Pink1 on the mitochondria occurs in skeletal muscle in response to exercise.
Results
To determine if Pink1 stabilization is involved in mitophagy in skeletal muscle following acute exercise, we isolated mitochondria-enriched fractions from gastrocnemius muscle (GA) at five time points post-exercise, using mitochondria-enriched fractions from GA of sedentary mice as controls. Interestingly, we did not detect any Pink1 in mitochondrial fractions but an abundant prescence in the corresponding cytosolic fractions (Figure 1(a)). On the contrary, treatment of HeLa cells with the mitochondria uncoupler carbonyl cyanide m-chlorophenyl hydrazone (CCCP) at 10 µM, a concentration shown to collapse mitochondrial membrane potential resulting in Pink1 stablization [18], resulted in a marked increase of Pink1 in mitochondria-enriched fractions (Figure 1(b)) and co-localization with the mitochondria reticulum (Figure 1(d,e)) using the same Pink1 antibody used in isolated GA mitochondria fractions (Figure 1(a,c)). Collectively, these data suggest that Pink1 is not stabilized on the OMM in skeletal muscle following acute exercise in vivo, thus Pink1 stabilization may not be required for exercise-induced mitophagy in skeletal muscle.
Figure 1.
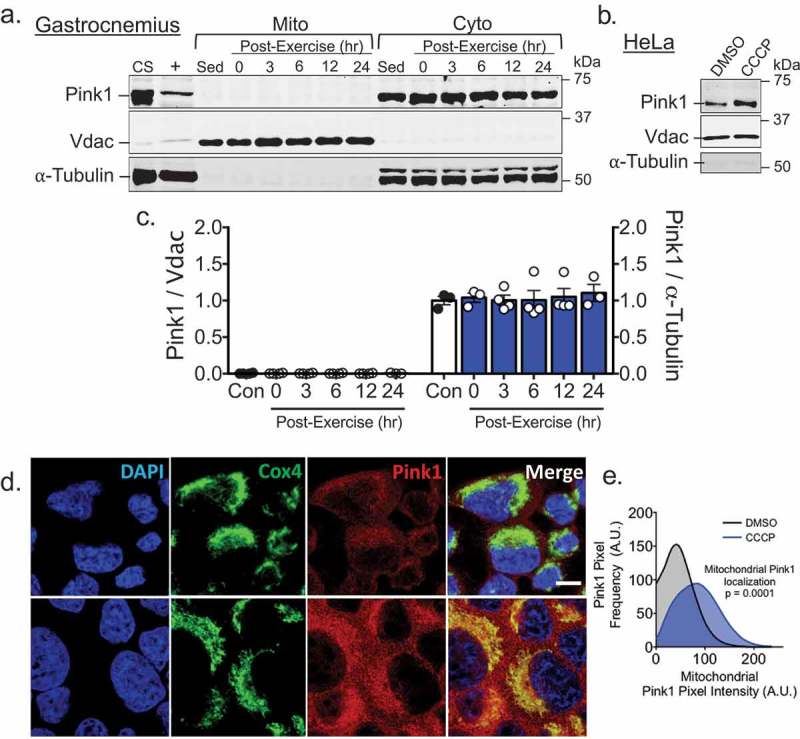
Pink1 is not present in mitochondria fraction from skeletal muscle at any point following acute exercise but is in HeLa cells following incubation with CCCP. A) Western blot analysis was performed for mitochondrial or cytosolic fractions from mouse gastrocnemius mucles (GA) at 0, 3, 6, 12 or 24 hrs following an acute bout of treadmill running with sedentary control mice as control. CS, i.e. common standard, represents mixed tissue homogenate of skeletal muscle, heart and liver from sedentary control mice. + represents whole cell lysate of HeLa cell treated for 8 hrs with 1 µM CCCP, Vdac (control for mitochondria), and α-tubulin (control for cytosol); B) Western blot image of Pink1, Vdac and α-tubulin in isolated mitochondria fraction from HeLa cells treated with 10 µM of CCCP for 1 hr; C) Quantification of Pink1 in both mitochondrial and cytosolic fractions of GA presented as mean ± standard error of the mean. n = 3–4 per time point; D) Immunofluorescent staining of Pink1 and CoxIV in HeLa cells 1 hr following exposure to 10 µM of CCCP. Scale bar = 30 µM; and E) Quantification of Pink1 positive pixels on mitochondria. n = 8 images per three independent experiments.
Discussion
Endurance exercise requires a profound increase in mitochondrial metabolism in contracting skeletal muscle to meet energetic demands. While it has been known for many decades that exercise training results in the expansion of the mitochondrial reticulum in skeletal muscle through biogenesis [21] in order to better meet energetic demands, the impact of exercise on mitochondrial breakdown is not as well understood. Recently, we demonstrated that an acute bout of endurance exercise (i.e. treadmill running) promotes removal and degradation of, presumably dysfunctional, regions of the mitochondrial reticulum through mitophagy [4]. Given the established role of Pink1 stabilization on areas of the reticulum that are ultimately degraded through mitophagy in some model systems [13–15,18], we hypothesized that this same mechanism is activated following acute exercise.
We have previously demonstrated that the final step in mitophagy, the incorporation of mitochondria into autolysosomes, occurs approximately six hours into the recovery period with the necessary and sufficient Ampk-Ulk1 signaling events occurring during and/or immediately following exercise [4]. However, when we examined the presence of Pink1 in the same isolated mitochondrial fractions from GA muscle immediately, 3, 6, 12, and 24 hrs following a single bout of treadmill running [4], we observed no detectable presence of Pink1, though Pink1 was readily apparent in the corresponding cytosolic fractions. These data in skeletal muscle post-exercise were in contrast to the readily detectable presence of Pink1 in isolated mitochondrial fractions and co-localization on mitochondria following CCCP treatment in HeLa cells. Therefore, we conclude that, although Pink1 is known to orchestrate the localization of the mitophagic machinery to specific regions of the mitochondrial reticulum in response to certain stresses in some cell backgrounds [13–18], it does not appear to be involved in exercise-induced mitophagy in skeletal muscle (Figure 2).
Figure 2.
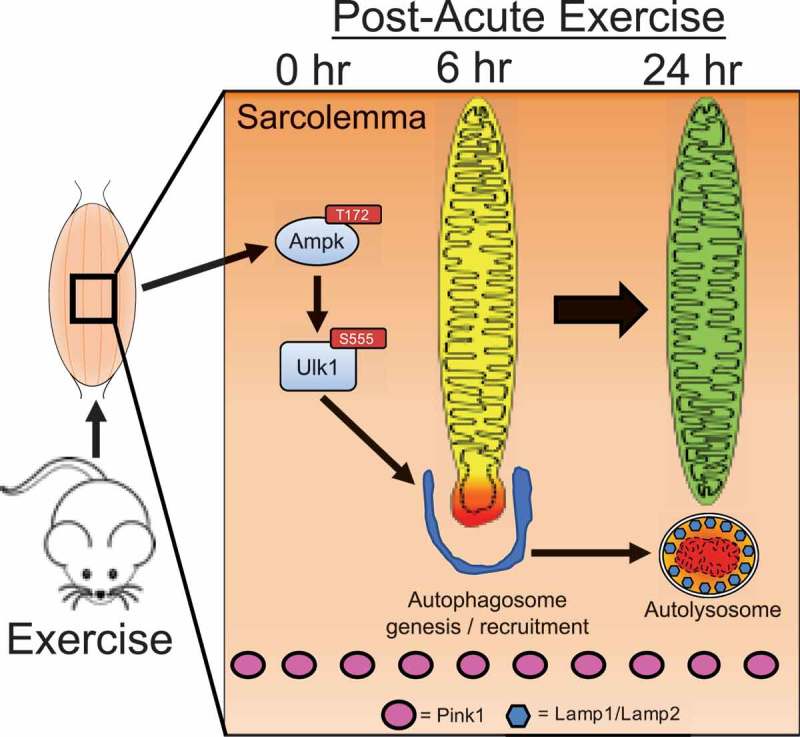
Schematic presentation of timeline for mechanisms regulating acute exercise-induced mitophagy in skeletal muscle.
The stabilization of Pink1 on the OMM has been primarily described in response to a loss in mitochondrial membrane potential, accumulation in misfolded proteins, and mtDNA damage [13–18]. However, skeletal muscle mitochondria are more resistant to a collapse in membrane potential following acute exercise [22], suggesting that Pink1 should not be expected to be stabilized on the OMM in response to exercise. There is also little reason to suspect exercise results in any accumulation in misfolded proteins. Exercise is a potent inducer of the unfolded protein response (UPR) [23–27], suggesting, in a non-diseased state, that proper folding of nascent proteins should occur following acute exercise. Furthermore, while mtDNA can be damaged by excessive generation of reactive oxygen species (ROS) [28], ROS-induced mtDNA mutations are not observed in response to acute exercise [29]. Furthermore, endurance exercise training protects mtDNA from ROS-induced damage [30]. Taken together, in a non-diseased condition, the appropriate stressor may be lacking for Pink1 stabilization on OMM in response to acute exercise to initiate mitophagy within the current context.
Indeed, it is becoming increasing clear that there exists a certain specificity for mechanisms that recognize regions of the mitochondrial reticulum for mitophagy. The mitochondrial protein FUN domain containing 1 (Fundc1) has been shown to orchestrate the recruitment of Ulk1 and Lc3 to the mitochondrial reticulum only in response to hypoxia [31–36]. There is evidence that the Pink1 substrate, Parkin, is enriched in mitochondrial fractions following exhaustive treadmill running in mice [37,38]. In these studies, the exercise protocol used was cumulatively more intense with shorter durations compared to the protocol used in our current investigation. Therefore, it may still be possible that acute exercise can result in the stabilization of Pink1 on mitochondria given a robust enough energetic stress (i.e. exhaustive exercise), but it is unclear whether such a result would be beneficial for optimal adaptation to exercise training. Taken together, it may be that the mechanism(s) of mitophagy initiation are stress-dependent.
While our data do not support a role for Pink1-mediated mitophagy in exercise-induced mitochondrial quality control in the non-diseased state, Pink1 may very well be important for maintenance of mitochondrial quality in other contexts. Sensitivity to mitochondrial transition pore opening, which causes detriments in mitochondrial membrane potential, is increased in skeletal muscle from elderly humans and is accompanied by a reduction in content of the Pink1 substrate Parkin [39]. Loss in mitochondrial membrane potential has also been linked to the development of skeletal muscle insulin resistance [40]. Therefore, the functional importance of Pink1 in skeletal muscle for maintenance of mitochondrial quality may be better appreciated within the context of chronic disease.
In conclusion, we have recently demonstrated that acute exercise promotes mitophagy, which is dependent on activation of Ulk1 through Ampk-dependent phosphorylation at Ser555. However, distinguishing specific regions of the reticulum for mitophagy in response to exercise does not appear to be mediated by a stabilization of Pink1 on mitochondria. It seems likely that a different mechanism may be responsible for the recognition of regions of the mitochondrial reticulum in skeletal muscle that are unable to maintain the energetic output required during prolonged exercise, thus triggering their removal via mitophagy for the maintenance of mitochondrial quality.
Methods
Animals
All experimental procedures were approved by the University of Virginia, Institutional Animal Care and Use Committee. As in our previous publication [4], male C57BL/6J mice (10–12 weeks old) were obtained commercially (Jackson Laboratories) for post-exercise time course experiments.
Acute treadmill running
C57BL/6 mice were acclimatized to the treadmill and acute treadmill running was performed as described in our previous publication [4].
Isolation of mitochondria from skeletal muscle
Skeletal muscle mitochondrial and cytosolic fractions were isolated via differential centrifugation as described in our previous publication [4]. Mitochondria fractions from HeLa cells were isolated using the same procedure from cells grown on two 100 mm plates and combined for centrifugation.
Cell culture and reagents
HeLa cells were cultured in DMEM with 10% FBS. For fractionation experiment, HeLa cells were treated with either 10 µM CCCP or DMSO (control) for 1 hr. For immunohistochemistry, HeLa cells were fixed in 4% paraformaldehyde/PBS, permeabilized in 0.3% Triton/PBS, blocked in 5% NGS/PBS, and stained with primary antibodies. Primary antibodies used were Pink1 (Santa Cruz #33796) and CoxIV (CST #11967) at a concentration of 1:50. Secondary antibodies FITC and Cy5 were used as well as DAPI for detection of nuclei. Slides were imaged at X100 magnification under a confocal microscope (Olympus Fluoview FV1000) with identical pre-determined acquisition parameters for all samples to ensure no saturation of the signals.
Western blot
Immunoblotting procedures and antibody catalog numbers are reported in our previous publication [4].
Statistical Analyses
Data are presented as the mean ± SEM. Time course experiments are analyzed via one-way ANOVA. IHC Pink1 l colocalization on mitochondria was performed using Image J by designating mitochondria ROI’s and averaging individual cell Pink1 histograms across each condition and analyzed via two-way ANOVA. Statistical significance was established a priori as p < 0.05.
Funding Statement
This work was supported by National Institutes of Health (R01-AR050429) to Z.Y., National Institutes of Health (K99-AG057825) and American Diabetes Association post-doctoral fellowship (1-16-PDF-030) to J.C.D., American Heart Association post-doctoral fellowship (14POST20450061) to R.C.L., National Institutes of Health (T32 HL007284-37) and American Heart Association (114PRE20380254) to R.J.W; National Institute of Arthritis and Musculoskeletal and Skin Diseases [R01-AR050429]; National Institute on Aging [K99-AG057825].
Acknowledgments
The authors thank David Kashatus for providing the whole cell lysate of CCCP treated HeLa cells used in Figure 1. We also thank members of the Yan Lab for critical feedback and discussion.
Disclosure statement
The authors have no conflicts of interest.
References
- [1].Warburton DER, Nicol CW, Bredin SSD.. Health benefits of physical activity : the evidence. Can Medial Assoc J. 2006;174:801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cartee GD, Hepple RT, Bamman MM, et al. Exercise promotes healthy aging of skeletal muscle. Cell Metab. 2016;23:1034–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Drake JC, Wilson RJ, Yan Z. Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. Faseb J. 2015;30:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Laker RC, Drake JC, Wilson RJ, et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun. 2017;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Call JA, Wilson RJ, Laker RC, et al. Ulk1-mediated autophagy plays an essential role in mitochondrial remodeling and functional regeneration of skeletal muscle. Am J Physiol Cell Physiol. 2017;312:C724–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Glancy B, Hartnell LM, Malide D, et al. Mitochondrial reticulum for cellular energy distribution in muscle. Nature. 2015;523:617–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kirkwood SP, Munn EA, Brooks GA. Mitochondrial reticulum in limb skeletal muscle. Am J Physiol. 1986;251:C395–402. [DOI] [PubMed] [Google Scholar]
- [8].Mishra P, Varuzhanyan G, Pham AH, et al. Mitochondrial dynamics is a distinguishing feature of skeletal muscle fiber types and regulates organellar compartmentalization. Cell Metab. 2015;22:1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Laker RC, Xu P, Ryall KA, et al. A novel mitotimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J Biol Chem. 2014;289:12005–12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wilson RJ, Drake JC, Cui D, et al. Conditional MitoTimer reporter mice for assessment of mitochondrial structure, oxidative stress, and mitophagy. Mitochondrion. 2017;S1567-7249(17)30196–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Greene AW, Grenier K, Aguileta MA, et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy. 2013;9:1758–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Aerts L, Craessaerts K, De Strooper B, et al. PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J Biol Chem. 2015;290:2798–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Okatsu K, Oka T, Iguchi M, et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun. 2012;3:1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kondapalli C, Kazlauskaite A, Zhang N, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Suen D, Narendra DP, Tanaka A, et al. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci U S A. 2010;107:11835–11840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy. 2013;9:1750–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Matsuda N, Sato S, Shiba K, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ordureau A, Heo J-M, Duda DM, et al. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc Natl Acad Sci. 2015;112:6637–6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Koyano F, Okatsu K, Kosako H, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. [DOI] [PubMed] [Google Scholar]
- [21].Hollozy J. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem. 1967;242:2278–2282. [PubMed] [Google Scholar]
- [22].Fernström M, Tonkonogi M, Sahlin K. Effects of acute and chronic endurance exercise on mitochondrial uncoupling in human skeletal muscle. J Physiol. 2004;554:755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Smolka MB, Zoppi CC, Alves AA, et al. HSP72 as a complementary protection against oxidative stress induced by exercise in the soleus muscle of rats. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1539–45. [DOI] [PubMed] [Google Scholar]
- [24].Pierce A, Wei R, Halade D, et al. A Novel mouse model of enhanced proteostasis: full-length human heat shock factor 1 transgenic mice. Biochem Biophys Res Commun. 2010;402:59–65. [DOI] [PubMed] [Google Scholar]
- [25].Wu J, Ruas JL, Estall JL, et al. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1α/ATF6α complex. Cell Metab. 2011;13:160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Memme JM, Oliveira AN, Hood DA. Chronology of UPR activation in skeletal muscle adaptations to chronic contractile activity. Am J Physiol Cell Physiol. 2016;310:C1024–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mattson JP, Ross CR, Kilgore JLON, et al. Induction of mitochondrial stress proteins following treadmill running. Physiology. 2000;32:365–369. [DOI] [PubMed] [Google Scholar]
- [28].Mattiazzi M, Vijayvergiya C, Gajewski CD, et al. The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum Mol Genet. 2004;13:869–879. [DOI] [PubMed] [Google Scholar]
- [29].Jafari A, Hosseinpourfaizi MA, Houshmand M, et al. Effect of aerobic exercise training on mtDNA deletion in soleus muscle of trained and untrained Wistar rats. Br J Sports Med. 2005;39:517–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Safdar A, Little JP, Stokl AJ, et al. Exercise increases mitochondrial PGC-1α content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J Biol Chem. 2011;286:10605–10617. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [31].Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. [DOI] [PubMed] [Google Scholar]
- [32].Lv M, Wang C, Li F, et al. Structural insights into the recognition of phosphorylated FUNDC1 by LC3B in mitophagy. Protein Cell. 2017;8:25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wu W, Tian W, Hu Z, et al. ULK 1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014;15:566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kuang Y, Ma K, Zhou C, et al. Structural basis for the phosphorylation of FUNDC1 LIR as a molecular switch of mitophagy. Autophagy. 2016;12:2363–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chen G, Han Z, Feng D, et al. Article a regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54:362–377. [DOI] [PubMed] [Google Scholar]
- [36].Chen Z, Liu L, Cheng Q, et al. Mitochondrial E 3 ligase MARCH 5 regulates FUNDC 1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017;18:495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chen CCW, Erlich AT, Crilly MJ, et al Parkin is required for exercise-induced mitophagy in muscle: impact of aging. Am J Physiol Endocrinol Metab. 2018;315:E404–E415. [DOI] [PubMed] [Google Scholar]
- [38].Chen CW, Erlich AT, Hood DA. Role of Parkin and endurance training on mitochondrial turnover in. Skelet Muscle. 2018;8:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gouspillou G, Sgarioto N, Kapchinsky S, et al. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. Faseb J. 2014;28:1621–1633. [DOI] [PubMed] [Google Scholar]
- [40].Taddeo EP, Laker RC, Breen DS, et al. Opening of the mitochondrial permeability transition pore links mitochondrial dysfunction to insulin resistance in skeletal muscle. Mol Metab. 2014;3:124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]