

ABSTRACT
Targeting the early steps of the glycolysis pathway in cancers is a well-established therapeutic strategy; however, the doses required to elicit a therapeutic effect on the cancer can be toxic to the patient. Consequently, numerous preclinical and clinical studies have combined glycolytic blockade with other therapies. However, most of these other therapies do not specifically target cancer cells, and thus adversely affect normal tissue. Here we first show that a diverse number of cancer models – spontaneous, patient-derived xenografted tumor samples, and xenografted human cancer cells – can be efficiently targeted by 2-deoxy-D-Glucose (2DG), a well-known glycolytic inhibitor. Next, we tested the cancer-cell specificity of a therapeutic compound using the MEC1 cell line, a chronic lymphocytic leukemia (CLL) cell line that expresses activation induced cytidine deaminase (AID). We show that MEC1 cells, are susceptible to 4,4ʹ-Diisothiocyano-2,2ʹ-stilbenedisulfonic acid (DIDS), a specific RAD51 inhibitor. We then combine 2DG and DIDS, each at a lower dose and demonstrate that this combination is more efficacious than fludarabine, the current standard- of- care treatment for CLL. This suggests that the therapeutic blockade of glycolysis together with the therapeutic inhibition of RAD51-dependent homologous recombination can be a potentially beneficial combination for targeting AID positive cancer cells with minimal adverse effects on normal tissue.
Implications: Combination therapy targeting glycolysis and specific RAD51 function shows increased efficacy as compared to standard of care treatments in leukemias.
Keywords: RAD51, glycolysis, AID, cancer, xenograft
Introduction
Glycolysis is a major ATP-producing pathway in mammalian cells, and can lead to either lactate fermentation or pyruvate oxidation, with lactate fermentation yielding significantly fewer ATP molecules per molecule of glucose metabolized.1 Cancer cells are characterized by a high rate of glycolysis as compared to normal cells, leading to excessive lactate fermentation despite inefficient ATP production, a phenomenon termed the Warburg Effect.2–4 This effect was initially thought to be a cause of neoplasticity, but now is considered a key feature of neoplastic cells in numerous types of cancers.5,6 Because of this increased reliance on glycolysis compared to non-neoplastic cells, glycolytic inhibitors have been considered an attractive means of targeting cancer.7 However, because glycolysis is a universal metabolic pathway, its blockade by inhibitors such as 2-deoxy-glucose (2DG) at doses that are efficacious can yield adverse side effects.8–10 Therefore, glycolytic inhibitors have been tested at low doses in combination with other cytotoxic therapies.10 While this combination approach has been shown to be successful in targeting a number of malignancies, most compounds used in combination with glycolytic inhibitors are not tumor-specific and can therefore damage non-malignant cells.10 In addition, such compounds can also potentially introduce DNA damage that can lead to mutations or breaks, potentially resulting in neomalignancies.10 Consequently, there is increasing consideration of the synergistic potential of using glycolytic inhibitors in combination with cytotoxic chemotherapies administered at doses lower than those used when such chemotherapies are used alone. More ideally, glycolytic inhibitors could be therapeutically conjoined with chemotherapeutic agents that more specifically target the cancer while sparing normal tissues.
Activation Induced Cytidine Deaminase (AID, AICDA) is an enzyme that initiates the formation of double-strand breaks (DSBs) in the heavy chain locus of the immunoglobulin heavy chain locus, a mechanism that leads to antibody isotype class switching in B-cells.11,12 In addition, AID is also responsible for somatic hypermutation in the V(D)J locus of the immunoglobulin genes.13 However, we and others have shown that AID generates collateral non-immunoglobulin DNA breaks throughout the genome and that these breaks can be repaired by XRCC2- and RAD51-dependent homologous recombination.14–16
A number of B-cell malignancies expresses functional AID.17,18 In addition, a number of studies have shown that non-B-cell malignancies also express this enzyme.19–23 It has been hypothesized that the presence of AID assists in enhancing the generation of somatic mutations of the cancer genome, resulting in increased survival and/or propagation of the cancer.18,22,24,25 We previously identified a small-molecule RAD51-specific inhibitor, 4,4ʹ-Diisothiocyano-2,2ʹ-stilbenedisulfonic acid (DIDS), that specifically disrupts the repair of AID-induced breaks in primary and neoplastic mouse B-cells and inhibits the growth of AID-positive human CLL cancers ex vivo.17 The effect of DIDS in vivo was strain-dependent: In C57BL/6J mice DIDS significantly reduced the number of post-germinal B-cells; however, in the autoimmune strain NOD/ShiLtDvs, DIDS significantly increased the number of autoregulatory CD73 + B-cells and suppressed Type I diabetes.17,26 These strain-dependent differences in response to DIDS suggest a complex role for RAD51 inhibition in B-cells.
Here we investigate the potential of a glycolytic inhibitor, 2DG, to alleviate tumor burden in spontaneous and patient-derived xenograft (PDX) cancer mouse models. Furthermore, we show that DIDS can reduce tumor burden in xenografted cell lines in vivo. Ultimately, we observe that the efficacy of DIDS in reducing tumor burden in vivo in mice can be enhanced by the effect of 2DG, both used at dosages that lower the risk of adverse effects, indicating that the combination of RAD51 inhibition and glycolytic blockage can be a potentially effective therapy against AID-positive cancers.
Results
2DG alleviates tumor burden in a spontaneous mouse model of lymphomagenesis
SJL/J mice spontaneously develop a hyperplastic disorder involving CD4 + T-cells and B-cells that resembles non-Hodgkin lymphoma and is evident after one year of age.27,28 It is thought that activated CD4+ T-cells secreting interleukin 21 drive B-cells to transformation in this model.29 SJL/J mice deficient in CD8a and thus lacking CD8 + T-cells show significantly accelerated development of B-cell lymphomas, with no change in other aspects of their phenotype.30 Since the growth or maintenance of any tumor requires energy, and highly proliferative cells such as cancer cells depend on numerous modes of ATP production, including glycolysis, to meet their energetic demands, blocking glycolysis in cancer cells at the first steps following cellular glucose intake should, in theory, reduce tumor burden.4,6,7 To test the extent to which inhibition of glycolysis by 2DG can alleviate these spontaneously arising lymphomas, we first aged a cohort of SJL.CD8a-/- female mice to 13 months of age and monitored them for signs of lymphoma development. Visible growth was most evident in cervical lymph nodes and in some cases in spleens (indicated by arrows, Figure 1A). Once tumors were sufficiently large to palpate, mice were placed on water with 6 g/L of dissolved 2DG provided ad libitum. The average mouse consumed approximately 4 ml of water each day; hence, based on the average weight of the mice, each mouse received a dose of 2DG of approximately 900 mg/kg/day. Photographs of the tumors were taken weekly to document disease regression, and the time points, in weeks, at which the tumors visibly shrank or, later, reemerged, were recorded. Via gross observation of the mice, we observed no adverse side effects of this treatment, such as weight loss, lethargy, or lack of grooming, either shortly after treatment initiation, or at any time during the treatment.
Figure 1.
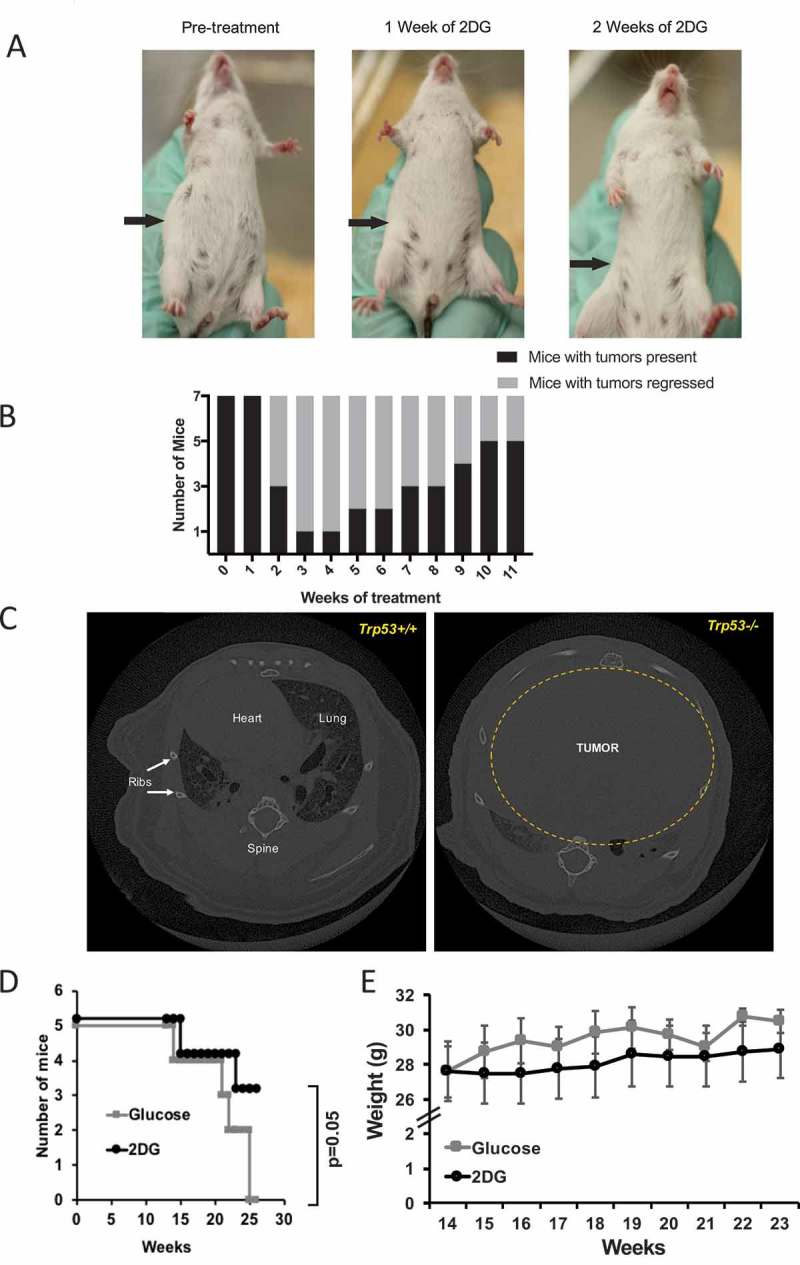
2-deoxy-D-Glucose (2DG) alleviates tumor burden in a spontaneous mouse cancer model. (A) Representative images showing inguinal lymph node tumor reduction on an SJL mouse treated with 2DG dissolved in drinking water (6 g/L). (B) Chart showing the transient effect of 2DG on tumor regression, and subsequent re-emergence (N = 7). (C) Computed tomography (CT) scans of a p53 wildtype (Trp53+/+) and p53 mutant (Trp53-/-) mouse, showing the maximum engulfment of a thymic lymphoma in the chest cavity. (D) Survival curve of Trp53-/- mice treated with2DG (670 mg/kg) or glucose (control) three times per week via intraperitoneal injections. (E) Weights of mice during glucose or 2DG treatment.
Of the seven mice in this study, six showed evidence of tumor regression after two or three weeks of treatment (Figure 1A and B). However, in four of these six, the tumors returned within 5–11 weeks, despite continuation of the treatment. This significant regression, which is similar to what is observed in mouse models of solid cancer treated with 2DG (see ref. 10), suggested that SJL lymphomas are partially responsive to relatively high therapeutic doses of a combination treatment for lymphoid cancers.
We wanted to extend the above findings by testing a more homogeneous and acute spontaneously arising lymphoma. In addition, we wanted to test the extent to which 2DG could affect a purely T-cell lymphoma. To meet all of these criteria, we turned to a classic mouse model of T-cell cancer, the p53-deficient mouse.31 The Trp53 gene codes for the p53 protein, and deficiency of this gene in mice leads to thymic lymphomas as early as 14 weeks of age (Figure 1C; Supplementary Figure 1); because of this phenotype, the Trp53-/- mouse is considered a model of Li-Fraumeni Syndrome {Jacks, 1994 #134}. To test the effect of 2DG on these thymic lymphomas, B6.Trp53-/- mice were treated with either 2DG (200 µL of 2DG at 600 mM in DPBS (670 mg/kg)) or glucose, intraperitoneally (I.P.) three times weekly, starting at 14 weeks of age and continuing for 10 weeks. We observed that mice treated with 2DG were significantly protected (Log rank Mantel Cox test P = .04 and Gehan-Breslow-Wilcoxon test p = .05) from developing neoplasms compared to glucose-treated mice (Figure 1D). Two notable adverse effects were observed with 2DG treatment delivered I.P.: first, upon injection, 2DG-treated mice showed inactivity for 10–60 minutes, and, second, as the experiment progressed, the 2DG-treated mice showed lower weight gain compared to glucose-treated mice, although the difference did not achieve significance (Figure 1E).
Together, based on two different mouse models of spontaneous cancer, 2DG administered orally or I.P. can alleviate both B- and T-cell tumor burdens; however, in the latter case, treatment resulted in notable adverse effects.
Lung tumor PDX models indicate that metabolic differences and not proliferation determine susceptibility to 2DG
We then sought to eventuate the effects of 2DG in human lung carcinoma PDX (patient-derived xenograft) models. While it is understood that 2DG primarily affects glycolysis, studies have shown that 2DG can interfere with other systems, such as the cell cycle, independently of its effects on metabolism.8,32,33 Changes in the cell cycle would affect the proliferation of cancer cells, ultimately influencing tumor growth. To test whether 2DG affects glycolysis independently of proliferation, we tested two PDX human lung carcinomas (TM00244 and TM00921) with similar growth kinetics (Figure 2A) but differing in the use of glycolytic pathways (Table 1; Supplementary Table 1; Supplementary Figure 2). Both of these PDX tumors are from the primary malignancy lung squamous cell carcinoma Grade 2. TM00244 is from a 62-year old white female and TM00921 is from a 66-year old American Indian or Alaskan male. A direct comparison of overall gene expression between the two carcinomas reveals that one carcinoma, TM00244, shows transcripts for alternative metabolic pathways as compared to the TM00921 carcinoma (Table 1; Supplementary Table 1; Supplementary Figure 2). We xenografted tumors from these two carcinomas into immunodeficient NOD.Scid.IL2R gamma null (NSGTM) mice, and, after tumors of a measurable size (> 50mm3) were observed, we treated mice of each model with 200 µL of either 600 mM 2DG or glucose three times weekly via I.P. injection. After 4 weeks of treatment, TM00921-xenografted mice showed a significantly smaller tumor volume with 2DG treatment compared to glucose treatment, whereas TM00244-xenografted mice showed no difference in tumor size between 2DG and glucose-treated mice (Figure 2B). Several genes that allow the utilization of other forms of metabolism or pathways that feed into the glycolysis pathway downstream of hexokinase are overexpressed in the resistant TM00244 tumor as compared to the susceptible TM00921 tumor (Supplementary Table 1), suggesting that inherent differences in metabolic pathway use – i.e., reliance on glycolysis or not – may impact tumor responses to 2DG.
Figure 2.
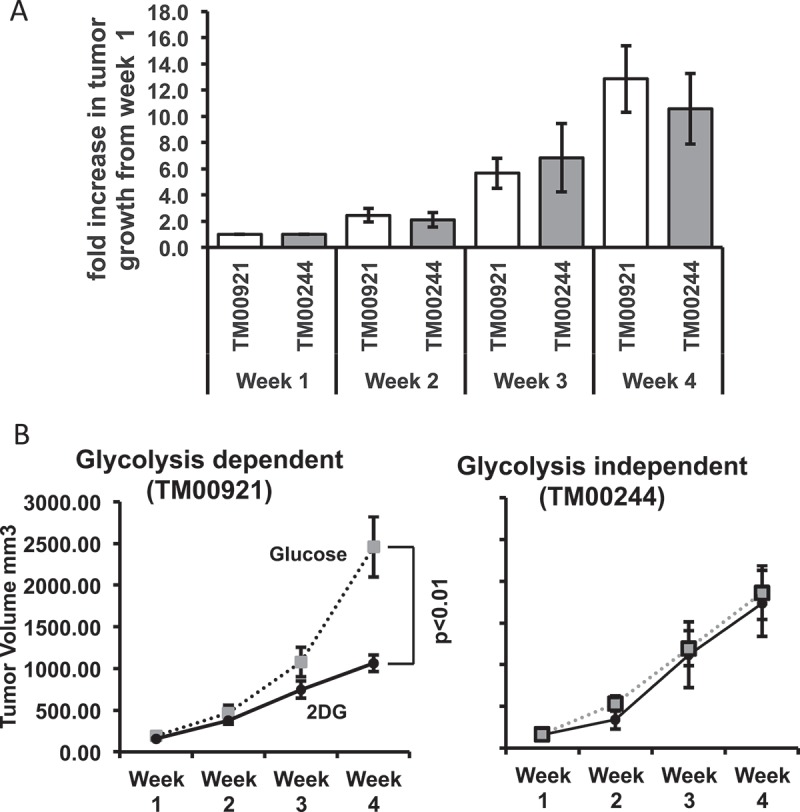
Glycolysis sensitivity rather than proliferation determines susceptibility to 2DG in lung PDX tumor models. (A) Glycolysis-dependent (TM00921) and glycolysis-independent (TM00244) PDX lung tumors were subcutaneously xenografted in NSGTM mice, and tumor growth was measured for 4 weeks. Growth was normalized to the initial size of the tumor for each individual mouse (N = 4–5) and plotted as fold increase. (B) Growth of the two lung tumors in mice treated with either 2DG or glucose (control).
Table 1.
Pathway differences between TM00921 and TM00244 (INGENUITY/KEGG PATHWAY).
| Pathway | Number of genes | p-Value |
|---|---|---|
| Metabolic pathways | 11 | 6.6E-2 |
| Arachidonic acid metabolism | 4 | 3.4E-3 |
| Drug metabolism-cytochrome P450 | 4 | 4.4E-3 |
| Metabolism of xenobiotics by cytochrome P450 | 4 | 5.6E-3 |
| Tyrosine metabolism | 3 | 1.3E-2 |
| Glutathione metabolism | 3 | 2.6E-2 |
| Chemical carcinogenesis | 3 | 5.9E-2 |
| Toll-like receptor signaling pathway | 3 | 9.5E-2 |
| Phenylalanine metabolism | 2 | 8.1E-2 |
Glycolysis-dependent B-cell cancers are sensitive to 2DG
While the above experiments using PDX models give a strong indication that lung tumors relying on glycolysis will be susceptible to 2DG therapy, the heterogeneity of PDX tumors can be a confounding variable, as each PDX tumor contains multiple cell types with different glycolytic demands.34,35 To control for this cellular heterogeneity, and to extend our findings to human B-cell cancers, we screened B-cell cancer cell lines and obtained two lines, CCRF-SB and MEC1, that share similar aerobic respiration rates but differ in their use of glycolysis as a source of energy (Figure 3A), and yet have similar proliferation rates ex vivo (Figure 3B).36,37 These data allow us to hypothesize that MEC1 cells, which are more glycolysis-dependent than are CCRF-SB cells, will be more sensitive to 2DG. To test that hypothesis, the two cell lines were tested for 2DG sensitivity in vivo: immunodeficient NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl/SzJ (NRGTM) mice were xenografted with either MEC1 or CCRF-SB cells, and the mice were then treated with 200 µL of 600 mM 2DG or glucose I.P. three times per week for two weeks. Both MEC1 and CCRF-SB cells migrate to the spleen and bone marrow, and since these mice express CD19 + B-cells, the tumor burden can be measured in these organs by anti-human CD19 staining via flow cytometry after euthanasia at the end of the treatment period. In contrast to CCRF-SB cells, which showed no significant response to 2DG in spleens or bone marrow, MEC1 cells in the bone marrow showed significant reductions in numbers with 2DG treatment (Figure 3C). Interestingly, while MEC1 cells in the bone marrow showed sensitivity to 2DG, those in the spleen did not (Figure 3C).
Figure 3.
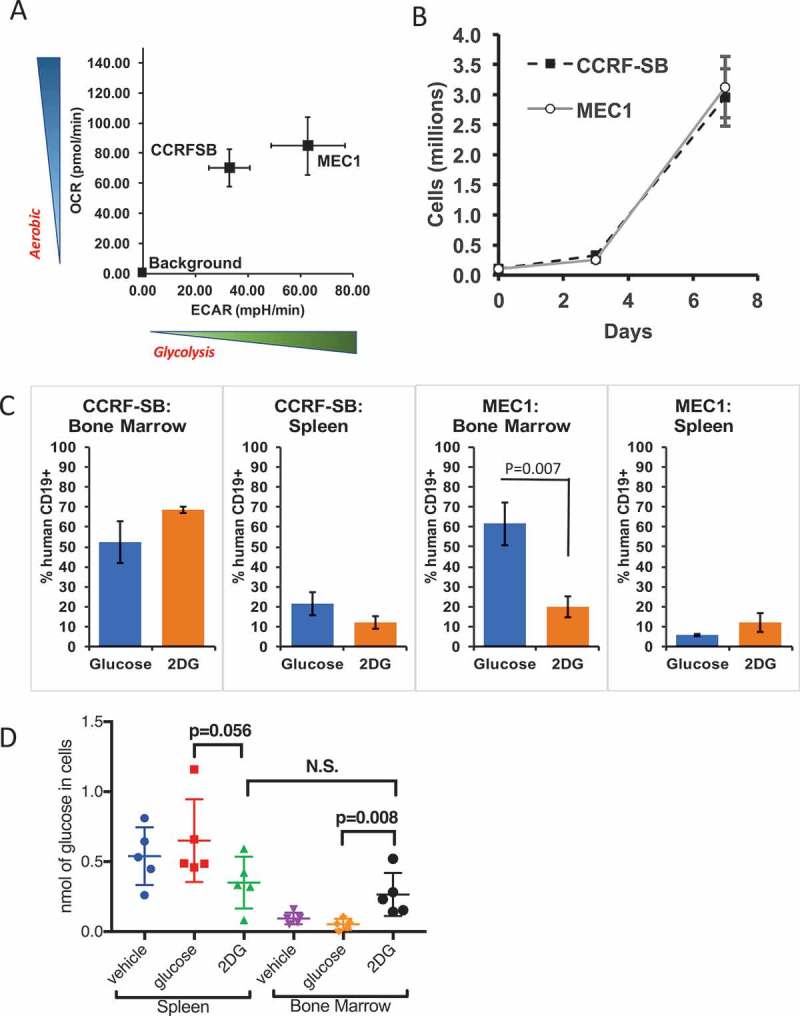
The glycolysis-dependent MEC1 cell line is susceptible to 2DG in vivo. (A) Metabolic comparison of two AID-positive B-cell lines, MEC1 and CCRF-SB with (B) similar growth rates ex vivo (N = 3 in two experiments). (C) Graphs showing bone marrow and spleen tumor burden as measured by human CD19+ cells (N = 4 mice). (D) Comparison of free glucose inside spleen and bone marrow cells of mice treated with 2DG or Glucose (6 g/L) or regular water (vehicle) for 1 week (N = 5 Mice per group).
To further investigate this disparity, we determined the cellular concentrations of free glucose in spleens and bone marrow of NRGTM mice treated for 1 week with 2DG or glucose in drinking water at 6 g/L or water with no additives provided ad libitum. We found only a trending drop (P = .056) of free glucose in spleen cells from 2DG treated compared to glucose treated mice (Figure 3D). In contrast there was a significant increase in free glucose in the bone marrow of 2DG treated compared to glucose or vehicle treated mice (Figure 3D). We hypothesize that bone marrow cells are more proliferative and, due to this increased activity, they take up more 2DG that blocks the glycolysis pathway; this leads to a subsequent increase in the unused glucose levels in the bone marrow cells. This would mean that MEC1 cells that home to the bone marrow as opposed to the spleen are more prolific and are therefore more affected by glycolytic blockade. This result indicates that there are organ-specific differences in the response of MEC1 cells to 2DG. Overall, the results suggest that glycolysis-dependent B-cell cancers are more sensitive than glycolysis-independent B-cell cancers to 2DG, with the proviso that MEC1 cells in different sites may also differ in their sensitivity to 2DG.
The RAD51 inhibitor DIDS reduces the splenic tumor burden of a chronic lymphocytic leukemia (CLL) xenografted cell line
As noted above, AID generates immunoglobulin locus-independent DNA breaks throughout the genome that are repaired by XRCC2- and RAD51-dependent homologous recombination.15,16 It was previously shown that DIDS could specifically target AID-positive neoplastic cell lines in tissue culture.17 Importantly, DIDS also targeted AID-positive, but not AID-negative, human CLL cells from patients ex vivo.17 We wanted to test the extent to which DIDS could target a xenografted AID-positive CLL cell line in vivo. We utilized the glycolytic MEC1 B-cell line, which can be quantified by CD19 expression ex vivo.36 We first confirmed AID positivity of MEC1 B-cells, using reverse transcriptase (RT)-PCR, and then compared AID expression of MEC1 B-cells with that of K562 cells, an AID-deficient cell line, and of CCRF-SB, an AID-positive ALL cell line. To control for the amount of RNA input, the samples were also tested for the control gene Gapdh. Results show that MEC1 B-cells constitutively express AID (Figure 4A). AID was also detected by intracellular staining of AID and flow cytometry (Figure 4B). We then proceeded to test the sensitivity of MEC1 B-cells to DIDS ex vivo. DIDS was titrated from 0 to 0.2 mM, with 2 × 105 MEC1 B-cells. Cytotoxic effects of DIDS on MEC1 B-cells were seen as early as 5 days at 0.05 mM, and a significant effect was observed with doses at 0.1 and 0.2 mM after 5 days (Figure 4C).
Figure 4.
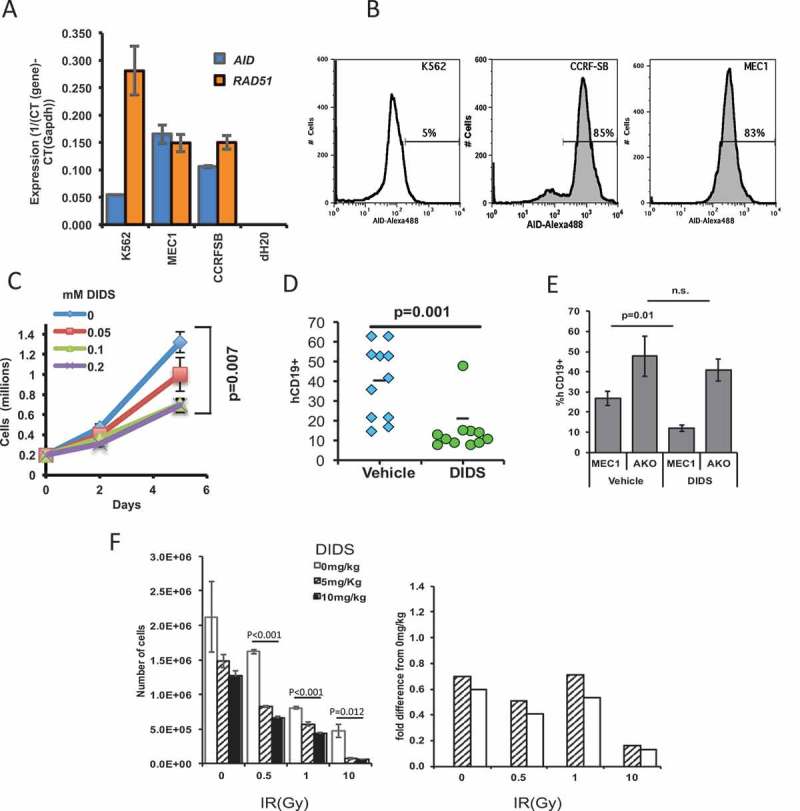
The RAD51-sensitive MEC1 cell line can be targeted by the RAD51 inhibitor DIDS in vivo. (A) Quantitative RT-PCR of AICDA (AID) and RAD51 of MEC1, K562, and CCRF-SB cells. (B) Flow cytometry of intracellular staining of AID in the aforementioned cell lines. (C) Ex vivo growth of MEC1 B-cells in the presence of different doses of DIDS. (D) MEC1 tumor burden in NRG™ mice after two-week treatment with DIDS as measured by hCD19+ cells in the spleen. Each point represents a mouse (data pooled from 3 experiments). (E) Tumor burden of MEC1 cells and MEC1 cells without AID (AKO) after DIDS treatment (N = 5 to 9 mice). (F) In vivo treatment of AID-/- mice with different concentrations of DIDS, followed by treatment of splenocytes with radiation ex vivo, illustrating the capacity of DIDS to work in combination treatment with IR (N = 5 mice).
Because DIDS was effective in reducing numbers of MEC1 B-cells ex vivo, we wanted to test its effectiveness in vivo. To accomplish this, 2 × 107 MEC1 cells were xenografted into immunodeficient NRGTM mice for two weeks, and mice were then treated either with DIDS at 50 mg/kg or with a 0.1 M potassium bicarbonate/PBS vehicle, once per week for an additional two weeks. We used NRGTM mice because they are deficient in CD19 cells, allowing us to quantify MEC1 B-cells by staining spleen cells for human CD19, as the spleen is a homing organ for MEC1 B-cells, all of which are positive for CD19. We observed that, despite the wide range of xenograft capabilities among the tested mice, 10 of 11 mice treated with DIDS showed a significant (p = 0.001) reduction in the number of MEC1 cells in the spleen (Figure 4D).
The therapeutic effect of DIDS on MEC1 cells requires AID
To directly determine if the effect of DIDS was dependent on AID, we generated three CRISPR guides that targeted exon 2 of AID (Supplementary Figure 3A). This exon was targeted because it is the first exon sufficiently large for the efficient design of CRISPR guides. The guides were transfected into MEC1 cells, and the cells were cloned by limiting dilution. One clone (Clone #14) that grew at a rate similar to the parental cell line, MEC1 was divided into four subclones (Clones #14–1, 14–3, 14–14 and 14–15). Upon sequencing, it was discovered that they all had a 25-base pair deletion in exon 2 of one allele and a complete deletion of exon 2 on the other (Supplementary Figure 3A and 3B). Theoretically, this would result in a truncated, non-functional AID, which we termed AID knockout MEC1 (AKO) cells. To test the dependence of DIDS on AID, we treated AKO cells with DIDS and compared their growth with that of parental MEC1 cells. The impact of DIDS on the proliferation of AKO cells was significantly different than its impact on proliferation of parental MEC1 cells (Supplementary Figure 3C). Moreover, unlike the parental MEC1 cells, AKO cells grew at a similar rate regardless of whether the cells were treated with vehicle or DIDS (Supplementary Figure 3C). To test whether AID sufficiency was required for DIDS efficacy in vivo, we xenografted AKO and parental cells in NRGTM mice, and treated the mice with 50 mg/kg of DIDS for two weeks. Compared to parental xenografted MEC1 cells, AKO cells were more resistant to DIDS as measured by the percentages of human CD19 cells (Figure 4E) and by spleen weights (Supplementary Figure 3D). Thus, the therapeutic effect of DIDS in MEC1 xenografts depends on AID.
The results above indicate that DIDS affects AID-initiated breaks in vivo. We next sought to determine whether DIDS could block repair of AID-independent double-strand breaks (frank or single-strand staggered double-strand breaks) in vivo. A common way to generate double-strand breaks is via ionizing radiation (IR).1,38,39 For this, we treated AID-null mice with different concentrations of DIDS (0, 5, or 10 mg/kg) for eight hours, and then harvested splenocytes and treated them with different doses of IR (0, 1, 5, 10 Gy). Eighteen hours after IR, the numbers of live white blood cells (CD45+ Ter119- PI-) were measured by flow cytometry. Results showed that IR alone results in decreased survival of the cells; however, in the presence of DIDS, the effect is significantly greater (Figure 4F). This effect is particularly evident at 10 Gy when the cell data are normalized to the 0 mg/Kg DIDS-treated data, where even a tenth of the dose of DIDS can synergistically enhance the severity of cell ablation (Figure 4F). These data indicate, that while DIDS can block repair of AID-initiated DSBs, it can also block repair of IR-induced DSBs. Importantly, the results indicate that DIDS, or any RAD51 inhibitor, has the potential to be paired with ionizing radiation to enhance the therapeutic effect of the IR alone.
Rapid synergistic effect of glycolytic blockade and RAD51 inhibition on MEC1 cells ex vivo and in vivo
Having found that RAD51 inhibition by DIDS can be improved by adjunct therapy such as radiation, we sought to test whether the combined use of DIDS and 2DG would have a synergistic effect on AID-positive neoplasms, both in vitro and in vivo. We reasoned that RAD51 function requires ATP and that, therefore, a reduction in the pool of ATP should decrease the efficiency of RAD51-dependent repair and enhance the cytotoxic effect of DIDS on rapidly proliferating AID+ neoplastic cells (Supplementary Figure 4).39,40 To test this possibility, we titrated 2DG with DIDS on MEC1 cells, since MEC1 cells are sensitive to both 2DG and DIDS. By day 2 there was a reduction of cell numbers as the concentration of DIDS was increased (Figure 5A), consistent with our earlier results (Figure 4C). However, the addition of 0.2 mM of 2DG, a concentration that does not have an effect on its own (y-axis, Figure 5A), enhanced the cytotoxic effect of DIDS (Figure 5A). From the slopes of the titration, it can be surmised that 2DG has a synergistic effect with DIDS, not only with respect to the lower concentrations of DIDS required to elicit an effect, but also kinetically, since we see the effects by 48 hours, in contrast to 5 days (Figure 5A versus Figure 4C). In order to determine the extent to which this 2DG-mediated synergistic effect holds for any AID-positive cell treated with any RAD51 inhibitor, we used a different RAD51 inhibitor, B02 (Sigma), on MEC1 and on another AID positive cell line, SuDHL. In addition, we also tested DIDS on SuDHL cells. In each experiment, we combined RAD51-inhibitor treatment (B02 or DIDS) with 2DG treatment. Together, results of the two experiments and our earlier experiment treating MEC1 cells with DIDS and 2DG (Figure 5A) show that treatment of either of two AID-positive cell lines with 2DG has a similar synergistic effect on the efficacy of two different RAD51 inhibitors (Figure 5B, Supplementary Figure 4). Importantly, the results also suggest that the combination of 2DG with any RAD51 inhibitor will have a synergistic therapeutic effect on any AID-positive cancer.
Figure 5.
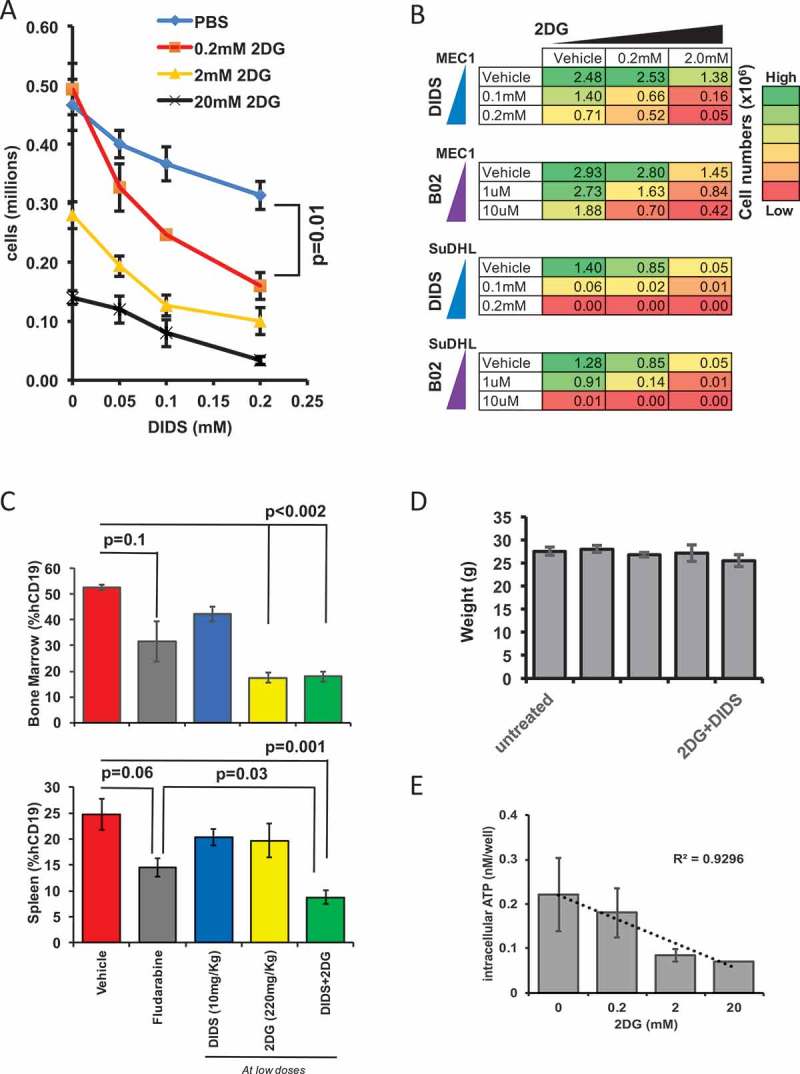
Glycolysis blockade and RAD51 inhibition exhibits a synergistic and enhanced anti-cancer effect on tumor burden. (A) Ex vivo intracellular ATP levels in MEC1 cells incubated with different doses of DIDS and 2DG 48 hours after incubation. (B) Heat map of the proliferation of MEC1 and SuDHL cells treated with 2DG or DIDS ex vivo. The color shows the numbers (in millions) of cells after 5 days of proliferation. Initial number for MEC1 was 1 million cells and SuDHL was 0.5 million cells. (C) In vivo MEC1 tumor burden as measured by the percentage of hCD19+ cells in the spleen and bone marrow after two-week treatment with low doses of DIDS and/or 2DG, with fludarabine, or with glucose (N = 4–13 mice in two experiments). (D) Average weight of the mice in each group at the end of the treatment regimen. (E) Ex vivo intracellular ATP of MEC1 cells incubated in 2DG at varying concentrations for 24 hours (N = 3).
To test whether this synergistic effect at low doses can be recapitulated in vivo, MEC1-xenografted mice were treated with DIDS alone, 2DG alone, or DIDS and 2DG in combination, all at low concentrations, or with a positive or negative control. Mice were treated I.P. three times weekly for two weeks. 1) Mice were dosed I.P. with 10 mg/kg DIDS, a fifth of the dose required to elicit an effect in vivo (Figure 4D); 2) mice were treated I.P. with 220 mg/kg of 2DG or glucose; a third of the dose of 2DG required for an effect (Figure 3C); 3) mice were treated with both DIDS and 2DG, at the same doses used in the first two groups; 4) as a positive control, mice were treated with fludarabine, a compound frequently used to treat CLL, at 35 mg/kg, a dose commonly used to elicit an effect; and 5) as a negative control, mice were treated with vehicle consisting of glucose in PBS.41 Following the 2-week treatment period, bone marrow and spleens were assessed for tumor burden, by determining the percentages of human CD19-positive cells via flow cytometry (Figure 5C).
In the bone marrow (Figure 5C), the combination DIDS/2DG treatment resulted in a significant (p < 0.002) decrease in tumor burden compared to vehicle, and also a decrease compared to treatment with fludarabine, although this decrease was not statistically significant (p = 0.26). Interestingly, in the bone marrow, 2DG alone elicited as much effect as the combination treatment (Figure 5C). In the spleen, the combination treatment produced the best outcome compared to either compound separately or vehicle (p = 0.001), and even performed significantly better than fludarabine (p = 0.03) (Figure 5C). Together, these data suggest that a combination RAD51-inhibitor/glycolysis-inhibitor treatment at lower doses is more effective in treating MEC1 cells in vivo than the current standard of treatment, fludarabine. Furthermore, at these reduced doses of DIDS and 2DG, the adverse effects of these treatments, including change in weight, were not observed, as at the end of the treatment regime the weights of the treated mice were similar to those of the untreated or vehicle-treated mice (Figure 5D). To test for a mechanism of the observed synergy, we exposed MEC1 cells for 24 hours to various concentrations of 2DG and measured intracellular ATP levels. This revealed a dose dependent decrease (R2 = .9296) in intracellular ATP as 2DG doses increase (Figure 5E). The reduced ATP pool should further inhibit ATP dependent RAD51 function and augment the cytotoxic effects of DIDS. Together, these results indicate that 2DG, in combination with the RAD51 inhibitor DIDS, can be administered safely at reduced doses of each compound to produce a cytotoxic effect on the cancer that is more effective compared to treatment with DIDS alone, while eliciting no evidence for adverse effects, which is the best outcome for the treatment of any disease.
Discussion
Glycolysis is a primary energy source for all cells. The concept of blocking glycolysis to prevent the expansion of cancer cells is well established.3,4,6,10 It has been known for decades that a number of cancer-cell types require a high rate of glycolysis compared to normal cells (the Warburg Effect) that favors lactate fermentation rather than aerobic respiration.3,4 However, since both pathways begin with glycolysis, therapeutic targeting of early steps in glycolysis poses a challenge with respect to specificity. Moreover, cancers can adapt to the therapeutic and/or favor the growth of cells whose glycolytic demands are lower than those of typical cancer cells and therefore do not require the targeted pathway.42,43 Therefore, targeting the glycolytic pathway has the potential not only to directly interfere with normal cell metabolism but also to provide selection pressure for tumors to adapt to sources of energy other than glucose. For these reasons, a number of pre-clinical studies and clinical trials combine glycolysis blockade with other chemotherapies.10 In several of these, the total dose of combined chemotherapy required to elicit a favorable therapeutic outcome was significantly less than the dose required for either therapy alone.6,10
Here we first show that 2DG monotherapy administered at high doses can be efficacious in treating two different spontaneous mouse models of lymphoma: the SJL/J model of non-Hodgkin lymphoma and the B6.Trp53-/- model of T-cell lymphoma. Furthermore, we show that high doses of 2DG show greater efficacy in glycolysis-dependent human PDX lung cancers and B-cell line lymphoma xenografts than they do in those that are not strictly reliant on glycolysis10. Together, these findings support the concept that glycolysis-dependent tumors can be targeted by 2DG. However, the high dose of 2DG required to elicit an effect, combined with the transience of the tumor reduction/resistance (Figure 1A, B), suggest that 2DG is not suitable as a stand-alone lymphoma therapy. Synergistic efficacy has been described when glycolysis inhibition by 2DG is combined with conventional, non-specific, anti-cancer therapies that target basic functions such as DNA replication, cell cycle or transcription.10 2DG, even at reduced dosages as is the norm in combination therapies, combined with compounds that more specifically target cancer cells, may result in safer and more effective combination therapies with fewer off-target effects.
Previously, we showed that the RAD51 inhibitor DIDS can specifically target AID-positive but not AID-negative primary or neoplastic cells ex vivo.17 Herein, we show that DIDS has a therapeutic effect in vivo specifically on AID-positive cells. While DIDS itself is not clinically translatable due to its chemical composition, the concept of using a RAD51 inhibitor to specifically target AID-positive and not AID-negative cells – as we have now shown is possible using in vivo xenograft models – allows the clinical development of a RAD51 inhibitor that could be used as a chemotherapeutic targeting the AID-RAD51 pathway. Furthermore, while it can be argued that RAD51-dependent repair is a universal double-strand break repair mechanism, we have not observed adverse effects of a RAD51 inhibitor on any AID-negative cells or any mouse models treated with DIDS, either in the present study or previous studies.17,44,45 The only reported adverse effect of DIDS treatment was in C57BL/6J mice, which showed a reduction in the percentage of AID-positive post-germinal B-cells following DIDS treatment; however, this effect was not observed in NOD mice, where DIDS increased the number of CD73 + B-cells, while suppressing the autoimmune disease.17,26,46,47 Therefore, targeting AID-positive cancers could potentially target mature class-switching B-cells should stable derivatives of DIDS be available for human treatment, and the effect could be potentially overridden by immunoglobulin transfusions in the clinic.
In the current study, we found that in SJL.CD8a-/- mice, even when treated with 2DG for 11 weeks, did not result in observable adverse effects, while B6.Trp53-/- mice showed a transient adverse effect with 2DG administration. We combined the specific inhibition of RAD51, via DIDS, with the generalized anti-glycolytic effect of 2DG, each at a lower dose than is used when the compound is administered alone, with the overall goal of narrowing the specificity of an anti-glycolytic inhibitor such that it impacts only cancer cells. While it is true that this therapy would be limited to AID-positive cancers, this study is proof-of-concept that simultaneous inhibition of glycolysis and RAD51-dependent repair can augment the synthetic lethal effect specifically on AID+ cancers. As published previously, about half of all CLLs express AID, and a number of publications show the expression of AID in non-lymphoid tumors, presenting the possibility that this therapy could potentially be used for non-lymphoid cancers.17,18,20,21,23
The observation that the combination of both glycolytic blockade and RAD51 inhibition leads to enhanced efficacy against tumorigenesis can be explained in two ways: First, ATP is required for multiple processes other than repair. Thus, reducing glycolysis by 2DG (Figure 5E) weakens the cancer cell, regardless of the action of DIDS. This was observed in the bone marrow in the MEC1 cell-xenografted study where there was no difference in the tumor burden with or without DIDS. Second, in the spleen, the presence of DIDS made a significant difference, and the two compounds showed a synergistic effect.
Lastly, we also observe that simultaneous blocking of glycolysis, with 2DG, and inhibition of RAD51, with DIDS, not only has a synergistic effect, but also results in a substantially more rapid effect compared to the use of DIDS alone. This advantage could be translated into a shorter treatment time for chemotherapy, which could lead to less severe adverse effects, lower costs, and a smaller chance that a tumor will develop resistance against the treatment.
In summary, we show that combining glycolytic blockade using 2DG, and RAD51 inhibition using DIDS, each at lower doses compared to solo administered, can synergistically reduce the numbers of AID-positive cancer cells both ex vivo and in vivo. While only AID-positive cancer cells are targeted in this study, our results suggest that this approach could be used with tumors that are not AID-positive, as RAD51 is used to repair fork collapses in rapidly replicating cells as well as to repair double-strand breaks.48,49 Furthermore, while AID has been implicated as a tumor-promoting gene in a number of cancers, a modification of our combination therapy approach could potentially be effective in homologous recombination-negative cancers such as BRCA mutants, in which AID is unlikely to play a tumorigenic role.50 Specifically, patients could be administered recombinant AID and 2DG simultaneously. This could potentially result in a lethal combination in the cancer cell through administration of a clastogenic protein – AID – together with the creation of conditions for reduced levels of homologous recombination via depletion of the ATP pool. This combination could expand application of the therapy concept discussed herein to AID-negative cancers. Taken together, we have provided proof-of-concept that the combination of AID, RAD51 inhibition, and glycolysis blockade can be a beneficial combination for treating cancers while minimizing the potential for adverse effects, due to the cell specificity and kinetics, and the lower doses required to elicit a lethal effect on cancer cells.
Materials and methods
Mice
C57BL/6J (JR664, JAX), B6.129S2-Trp53tm1Tyj/J (B6.Trp53-/-, JR2101, JAX), NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl/SzJ (NRGTM, JR7799, JAX), NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSGTM, JR5557, JAX), and SJL.129S2(B6)-Cd8atm1Mak/1Dcr (SJL.Cd8a-/- JR4023, private strain: Derry Roopenian) mice used in this study were bred and housed at The Jackson Laboratory (Bar Harbor, Maine). Mice were provided with food and water ad libitum and were housed on a 14-hour light, 10-hour dark cycle. All procedures were approved by The Jackson Laboratory Institutional Animal Care and Use Committee (IACUC).
Cells
Human peripheral blood acute B-lymphoblastic leukemia cells (the CCRF-SB cell line (ATCC)); human chronic lymphocytic leukemia cells (the MEC1 cell line (Cat. no. ACC 497, DSMZ)); AID-positive peritoneal effusion B-lymphoblast SUDHL-4 cells (a gift from Cyteir Therapeutics); and K562 myeloid leukemia cells (a gift from Dr. Jennifer Trowbridge, The Jackson Laboratory) were cultured according to manufacturer’s/donor’s recommendations. Cell viability counts were done on the Countess II Automated Cell Counter (Invitrogen) according to manufacturer’s recommendations.
Generation of AID KO MEC1 cell line
AID knockout (AKO) MEC1 cell lines were developed by targeting Exon 2 of AID with the following guide RNAs: CTTGATGAACCGGAGGAAG, GTCCGCTGGGCTAAGGGTC, GTGCTACATCCTTTTCAC. These guides were cloned into the Cas9-EGFP vector, pX330 (Addgene, Cat. no. 66582). These vectors were nucleofected into MEC1 cells using Amaxa Cell Line Nucleofector Kit V using program X-001, and according to manufacturer’s instructions (Lonza, Cat. no. VACA-1003). Nucleofected cells were sorted for GFP positivity and cloned by limited dilution to generate AKO cell lines. Two independent AKO cell lines (14–1 and 14–3) were confirmed for AID nullizygosity using both genomic and transcript PCRs.
Compounds
2-deoxy-D-glucose (Cat. no. D8375), the RAD51 inhibitor B02 (Cat. no. SML0364), and fludarabine phosphate (Cat. no. 1272204) were obtained from Sigma-Aldrich. DIDS was obtained from ChemCruz (Cat. no. sc-203919).
Quantification of AID and RAD51
Total RNA was extracted from tissue samples using the RNeasy Mini Kit according to manufacturer’s protocol (QIAGEN, Cat. no. 74104). In order to detect AID and RAD51 by qPCR, 1% β-mercaptoethanol in Buffer RLT was added to the tissue at a ratio of 100 mg/1.25 mL. Synthesis of cDNA was done using the RT2 First Strand Kit following manufacturer’s protocol (QIAGEN, Cat. no. 330404). 500 ng of RNA was used when available; otherwise, the next-largest amount of RNA for the set of samples was used. For qPCR, oligonucleotides to detect GAPDH transcripts were 5ʹ-GAGTCAACGGATTTGGTCGT-3ʹ (forward) and 5ʹ-TTGATTTTGGAGGGATCTGC-3ʹ (reverse). Oligonucleotides to detect AICDA transcripts were 5ʹ-TTCTTTTCACTGGACTTTGG-3ʹ (forward) and 5ʹ-GACTGAGGTTGGGGTTCC-3ʹ (reverse). Oligonucleotides to detect RAD51 transcripts were 5ʹ-CAACCCATTTCACGGTTAGAGC-3ʹ (forward) and 5ʹ-TTCTTTGGCGCATAGGCAACA-3ʹ (reverse). For each sample, three 25 µL reactions were run of varying cDNA concentrations (10, 5, and 1 µL). The reactions were run on an Applied Biosystem Model 7500 thermocycler using RT2 SYBR Green ROX qPCR Mastermix (QIAGEN, Cat. no. 330520). PCR conditions were 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, 60°C 1 min.
To detect AID by flow cytometry, cells were fixed in 3% Neutral Buffered Formalin (Cat. no. MER 44991 GL, Mercedes Chemicals), 2% sucrose (Cat. no. S8501 Sigma-Aldrich) in DPBS (Cat. no. 14190250, ThermoFisher) at a concentration of one million per mL for 10 minutes in suspension at room temperature. The cells were then washed with DPBS by centrifugation (400 x g x 5 mins) and permeabilized with 0.1% Triton-X-100 (Cat. no. X100, Sigma-Aldrich) in DPBS, and washed once with DPBS. The cells were then incubated with 1 mL of 10% FBS in DPBS for 1 hour, and stained overnight at 4°C with anti-AID antibody (ab93596, Abcam) at 1:100 in 0.1 mL, 10% FBS in DPBS. The cells were washed twice with DPBS, and incubated with a 1:1,000 dilution of Alexa 488 goat anti rabbit IgG (Cat. no. A27016, ThermoFisher) in 0.1 mL of 10% FBS in DPBS for 1 hour at room temperature in the dark. The cells were washed twice with DPBS, and data were acquired using a FACScaliber II and were analyzed by FlowJo Version 8.8.7.
PDX xenografts and treatments
Patient-derived xenograft (PDX) tumors were obtained from the JAX PDX Resource. Tumor fragments were minced separately and xenografted subcutaneously in an NSGTM mouse to establish P1. Upon growth of the tumor fragment, the tumor was harvested, minced, and xenografted in multiple mice to establish P2 mice. All experiments were conducted on P2 or higher mice that arose from engraftment from one of multiple tumor fragments.
Expression analyses of early JAX PDX oncology models were performed utilizing the ThermoFisher Scientific (formerly Affimetrix) GeneChip™ Human Genome U133 Plus 2.0 Array or GeneChip™ Human Gene 1.0 ST Array.51 Arrays from all microarray-assayed models were processed together to generate normalized expression of all genes in the two-microarray platforms. The affyPLM R package was used, performing quantile normalization and no background correction. The data were fitted to a simple model that treats the log intensity as a sum of array effect, probe effect, and residual. The array effect is the equivalent to the median polished value produced by standard RMA analysis. The gene expressions are provided as log2-transformed output (normalized expression).
The differences between the normalized expression for each gene in models TM00244 and TM00921 were calculated to determine which genes displayed the greatest differential expression between the two models. The gene list was then exported into Ingenuity Pathway Analysis software that revealed the effected pathways.
Xenograft studies
Human cancer cell lines, mentioned above, were xenografted into NRGTM mice in the following way: Cells grown in flasks were given fresh media the night before xenograftment and seeded at 106 cells per mL. The next day, the cells were washed twice with DPBS without calcium or magnesium and resuspended to 1 – 2 × 108 cells per mL of DPBS at room temperature and immediately injected via tail vein at 100 µL per mouse. The cells were given 7–10 days to fully xenograft, after which tumor load in the blood was assessed via flow cytometry (FACSCaliber II) for human CD19+ cells (Antibody cat no. 555413, BD Parmingen). Mice with similar tumor loads were assigned to different treatment groups to decrease bias and ensure more even tumor load across treatments. Mice were treated as described in the Results section for two weeks unless otherwise indicated. Upon euthanasia, the spleen and the bone marrow were harvested and stained for human CD19, and the cells were measured by flow cytometry on the FACSCaliber II. Analysis of the flow data was done by Flowjo version 8.8.7 (BD Pharmingen).
Metabolic measurements
Metabolic measurements were done on the Agilent Seahorse XFe96 Analyzer using the Agilent XF cell energy phenotype testing test kit according to the manufacturer’s instructions (Cat. no. 103325–100). Measurements of intracellular ATP were done using a commercially available fluorescence assay from abcam (ab83355) using manufacturer’s recommendations. Measurements of free glucose were done using the Glucose Assay kit from abcam (ab65333) using the colorimetric methods according to manufacturer’s recommendations.
Treatment of the spontaneous tumor models with 2DG
SJL.CD8a-/- female mice were aged and monitored for signs of tumor development weekly after reaching 6 months of age. Tumors were evident by 7 to 12 months of age. Once tumors had achieved clearly visible sizes, the mice were administered ad libitum with 6 g/L of 2DG dissolved in drinking water. This concentration of 2DG in the water supplied the mice with a daily dose of approximately 900 mg/kg of 2DG. Mice were monitored weekly to determine changes in tumor mass by palpation and photographs, and overall tumor size changes, (same, smaller or larger) compared with the preceding week of treatment, was recorded. Treatments lasted for 11 weeks or until the tumors returned and mice became too sick to continue in the study. The treatment of one mouse was carried out to week 16 to determine what, if any, side effects would occur with prolonged 2DG exposure. This tumor never returned and no adverse effects were noted.
Microct imaging and image reconstruction
MicroCT was performed using a high-speed in vivo µCT scanner (Quantum GX, PerkinElmer, Hopkinton, MA, USA). The images were acquired using a High Resolution Scan mode with a 4-min scan time. The X-ray source was set to a current of 88 µA, voltage of 90 kVp, and a 36 mm FOV for a 50 µm voxel size. Animals were anesthetized with 2% isoflurane via nose cone while imaging. Administration of anesthesia helped to minimize motion artifacts during scanning. Animals were recovered in a clean box with pine shavings placed on a 37°C heating pad until fully mobile and then returned to their home cages.
The µCT imaging was visualized via 3D Viewer, existing software within the Quantum GX system. The greyscale image slices were selected on the basis of internal landmarks such as ribs and spinal column so that images were generated in approximately the same location within each animal. These images were saved as JPEG files. Colored images were reconstructed using Image J32 (V1.49) or the PerkinElmer 3D Viewer application. Thresholds were used to visually determine optimal separation of the histogram into bone and soft tissue. In this way, the lungs and tumor could be viewed separately from the bone.
Gene expression analysis
Expression analyses of early JAX PDX oncology models were performed utilizing the ThermoFisher Scientific (formerly Affimetrix) GeneChip™ Human Genome U133 Plus 2.0 Array or the GeneChip™ Human Gene 1.0 ST Array. Arrays from all microarray-assayed models were processed with the AffyPLM R package, using quantile normalization and no background correction, and fitted to a simple model that treats the log intensity as a sum of array effect, probe effect, and residual. The array effect is the ‘summarized expression’ that is equivalent to the median polished value produced by standard RMA analysis. The differences between the normalized expression for each gene in models TM00244 and T
M00921 were calculated to identify the genes that showed the greatest difference in expression between the models. The gene list was then exported into Ingenuity Pathway Analysis software, which revealed the effected pathways.
Statistics
All statistical parameters were calculated using EXCEL version 15.4. Error bars are standard error of the mean (SEM) and p-values are calculated by student t-test, unless indicated otherwise.
Funding Statement
Linda Tallen & David Paul Kane Educational and Research Foundation funds to Muneer Hasham (TALLEN KANE FDN FY 15-18), Lupus Research Alliance and John and Marcia Goldman Foundation funds to Derry Roopenian, and The Jackson Laboratory Principal Investigator to Kevin Mills (TJL DIF FY13 KDM) paid for the personnel, costs, and fees for materials and use of Scientific Services for this study. The fellowships of authors Elise Wolf and Nathan Labrie were funded by JAX Genomic Education and the JAX Summer Student Program, a National Cancer Institute Cancer Education Grant to Dr. Lenny Shultz (5R25CA174584-05), and The Betterment Fund Scholarship Endowment of The Jackson Laboratory.
Acknowledgments
The authors acknowledge the following individuals from The Jackson Laboratory Scientific Services (fee-for-service departments): the Flow Cytometry Service: William Schott and Ted Duffy for their technical support; and the PDX R&D Core: Dr. Kin-Hoe Chow (manager), Amy Lambert, Ed Keniston, and Srijoy Gupta for xenograft studies.
In addition, the authors acknowledge Stephen Sampson for assistance in manuscript preparation, Dr. Michael Wiles for assistance in CRISPr construct generation, Dr. Heidi Kocalis for imaging consultation, and Dr. Ewelina Bolcun-Filas for critical reading of the paper.
Disclosure of Potential Conflicts of Interest
Kevin Mills is an employee of Cyteir Therapeutics.
Grant Sources
See funding sources in the document.
Supplementary material
Supplemental data for this article can be accessed here.
References
- 1.Berg JM, Tymoczko JL, Stryer L.. Biochemistry. 5th ed. New York: (NY): W H Freeman; 2002. [Google Scholar]
- 2.Foley GEL H, Farber S, Uzman B, Adams R.. Studies on human leukemia cells in vitro. The proliferation and spread of neoplastic cells. Baltimore (MD): The Williams & Wilkins Co; 1968. p. 65–97. [Google Scholar]
- 3.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–899. Epub 2004/ 11/02. doi: 10.1038/nrc1478 PubMed PMID: 15516961. [DOI] [PubMed] [Google Scholar]
- 4.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. Epub 2009/ 05/23. doi: 10.1126/science.1160809 PubMed PMID: 19460998; PMCID: PMC2849637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25(34):4633–4646. PubMed PMID: 16892078. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 6.Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66(18):8927–8930. Epub 2006/09/20. doi: 10.1158/0008-5472.CAN-06-1501 PubMed PMID: 16982728. [DOI] [PubMed] [Google Scholar]
- 7.Chen Z, Lu W, Garcia-Prieto C, Huang P. The Warburg effect and its cancer therapeutic implications. J Bioenerg Biomembr. 2007;39(3):267–274. Epub 2007/06/07. doi: 10.1007/s10863-007-9086-x PubMed PMID: 17551814. [DOI] [PubMed] [Google Scholar]
- 8.Muley P, Olinger A, Tummala H. 2-Deoxyglucose induces cell cycle arrest and apoptosisin colorectal cancer cells independent of its glycolysis inhibition. Nutr Cancer. 2015;67(3):514–522. Epub 2015/03/10. doi: 10.1080/01635581.2015.1002626 PubMed PMID: 25751508. [DOI] [PubMed] [Google Scholar]
- 9.Vijayaraghavan R, Kumar D, Dube SN, Singh R, Pandey KS, Bag BC, Kaushik MP, Sekhar K, Dwarakanath BS, Ravindranath T. Acute toxicity and cardio-respiratory effects of 2-deoxy-D-glucose: a promising radio sensitiser. Biomed Environ Sci. 2006;19(2):96–103. PubMed PMID: 16827179. [PubMed] [Google Scholar]
- 10.Zhang D, Li J, Wang F, Hu J, Wang S, Sun Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Letters. 2014;355(2):176–183. PubMed PMID: 25218591. doi: 10.1016/j.canlet.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Stavnezer J, Schrader CE. IgH chain class switch recombination: mechanism and regulation. J Immunology. 2014;193(11):5370–5378. PubMed PMID: 25411432; PMCID: 4447316. doi: 10.4049/jimmunol.1401849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matthews AJ, Zheng S, DiMenna LJ, Chaudhuri J. Regulation of immunoglobulin class-switch recombination: choreography of noncoding transcription, targeted DNA deamination, and long-range DNA repair. Adv Immunol. 2014;122:1–57. doi: 10.1016/B978-0-12-800267-4.00001-8 PubMed PMID: 24507154; PMCID: 4150736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teng G, Papavasiliou FN. Immunoglobulin somatic hypermutation. Annu Rev Genet. 2007;41:107–120. doi: 10.1146/annurev.genet.41.110306.130340 PubMed PMID: 17576170. [DOI] [PubMed] [Google Scholar]
- 14.Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, McBride KM, Klein IA, Stone G, Eisenreich TR, et al. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol Cell. 2009;36(4):631–641. PubMed PMID: 19941823; PMCID: 2805907. doi: 10.1016/j.molcel.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hasham MG, Donghia NM, Coffey E, Maynard J, Snow KJ, Ames J, Wilpan RY, He Y, King BL, Mills KD. Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nat Immunol. 2010;11(9):820–826. PubMed PMID: 20657597; PMCID: 2930818. doi: 10.1038/ni.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hasham MG, Snow KJ, Donghia NM, Branca JA, Lessard MD, Stavnezer J, Shopland LS, Mills KD. Activation-induced cytidine deaminase-initiated off-target DNA breaks are detected and resolved during S phase. J Immunology. 2012;189(5):2374–2382. PubMed PMID: 22826323; PMCID: 3424338. doi: 10.4049/jimmunol.1200414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamont KR, Hasham MG, Donghia NM, Branca J, Chavaree M, Chase B, Breggia A, Hedlund J, Emery I, Cavallo F, et al. Attenuating homologous recombination stimulates an AID-induced antileukemic effect. J Exp Med. 2013;210(5):1021–1033. PubMed PMID: 23589568; PMCID: 3646491. doi: 10.1084/jem.20121258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu X, Shivarov V, Strout MP. The role of activation-induced cytidine deaminase in lymphomagenesis. Curr Opin Hematol. 2012;19(4):292–298. Epub 2012/04/21. doi: 10.1097/MOH.0b013e328353da3a PubMed PMID: 22517589. [DOI] [PubMed] [Google Scholar]
- 19.Matsumoto T, Shimizu T, Nishijima N, Ikeda A, Eso Y, Matsumoto Y, Chiba T, Marusawa H. Hepatic inflammation facilitates transcription-associated mutagenesis via AID activity and enhances liver tumorigenesis. Carcinogenesis. 2015;36(8):904–913. Epub 2015/05/15. doi: 10.1093/carcin/bgv065 PubMed PMID: 25969143. [DOI] [PubMed] [Google Scholar]
- 20.Nonaka T, Toda Y, Hiai H, Uemura M, Nakamura M, Yamamoto N, Asato R, Hattori Y, Bessho K, Minato N, et al. Involvement of activation-induced cytidine deaminase in skin cancer development. J Clin Invest. 2016;126(4):1367–1382. Epub 2016/03/15. doi: 10.1172/JCI81522 PubMed PMID: 26974156; PMCID: PMC4811119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sapoznik S, Bahar-Shany K, Brand H, Pinto Y, Gabay O, Glick-Saar E, Dor C, Zadok O, Barshack I, Zundelevich A, et al. Activation-Induced Cytidine Deaminase Links Ovulation-Induced Inflammation and Serous Carcinogenesis. Neoplasia. 2016;18(2):90–99. Epub 2016/03/05. doi: 10.1016/j.neo.2015.12.003 PubMed PMID: 26936395; PMCID: PMC5005261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimizu T, Marusawa H, Endo Y, Chiba T. Inflammation-mediated genomic instability: roles of activation-induced cytidine deaminase in carcinogenesis. Cancer Sci. 2012;103(7):1201–1206. Epub 2012/ 04/04. doi: 10.1111/j.1349-7006.2012.02293.x PubMed PMID: 22469133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sawai Y, Kodama Y, Shimizu T, Ota Y, Maruno T, Eso Y, Kurita A, Shiokawa M, Tsuji Y, Uza N, et al. Activation-induced cytidine deaminase contributes to pancreatic tumorigenesis by inducing tumor-related gene mutations. Cancer Res. 2015;75(16):3292–3301. Epub 2015/06/27. doi: 10.1158/0008-5472.CAN-14-3028 PubMed PMID: 26113087. [DOI] [PubMed] [Google Scholar]
- 24.Casellas R, Basu U, Yewdell WT, Chaudhuri J, Robbiani DF, Di Noia JM. Mutations, kataegis and translocations in B cells: understanding AID promiscuous activity. Nat Rev Immunol. 2016;16(3):164–176. Epub 2016/ 02/24. doi: 10.1038/nri.2016.2 PubMed PMID: 26898111; PMCID: PMC4871114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451(7180):841–845. PubMed PMID: 18273020. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 26.Ratiu JJ, Racine JJ, Hasham MG, Wang Q, Branca JA, Chapman HD, Zhu J, Donghia N, Philip V, Schott WH, et al. Genetic and small molecule disruption of the AID/RAD51 axis similarly protects nonobese diabetic mice from type 1 diabetes through expansion of regulatory B lymphocytes. J Immunology. 2017;198(11):4255–4267. PubMed PMID: 28461573; PMCID: 5474749. doi: 10.4049/jimmunol.1700024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lacoste-Collin L, Jozan S, Cances-Lauwers V, Pipy B, Gasset G, Caratero C, Courtade-Saidi M. Effect of continuous irradiation with a very low dose of gamma rays on life span and the immune system in SJL mice prone to B-cell lymphoma. Radiat Res. 2007;168(6):725–732. Epub 2007/12/20. doi: 10.1667/RR1007.1 PubMed PMID: 18088184. [DOI] [PubMed] [Google Scholar]
- 28.Thrush GR, Butch AW, Lerman SP. CD8 suppressor cell activity and its effect on CD4 helper cell-dependent growth of SJL/J B-cell lymphomas. Cell Immunol. 1989;122(2):555–562. Epub 1989/09/01 PubMed PMID: 2569938. [DOI] [PubMed] [Google Scholar]
- 29.Jain S, Chen J, Nicolae A, Wang H, Shin DM, Adkins EB, Sproule TJ, Leeth CM, Sakai T, Kovalchuk AL, et al. IL-21-driven neoplasms in SJL mice mimic some key features of human angioimmunoblastic T-cell lymphoma. Am J Pathol. 2015;185(11):3102–3114. Epub 2015/09/13. doi: 10.1016/j.ajpath.2015.07.021 PubMed PMID: 26363366; PMCID: PMC4630166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McPhee CG, Sproule TJ, Shin DM, Bubier JA, Schott WH, Steinbuck MP, Avenesyan L, Morse HC 3rd, Roopenian DC. MHC class I family proteins retard systemic lupus erythematosus autoimmunity and B cell lymphomagenesis. J Immunology. 2011;187(9):4695–4704. Epub 2011/10/04. doi: 10.4049/jimmunol.1101776 PubMed PMID: 21964024; PMCID: PMC3381364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4(1):1–7. Epub 1994/01/01 PubMed PMID: 7922305. [DOI] [PubMed] [Google Scholar]
- 32.Ralser M, Wamelink MM, Struys EA, Joppich C, Krobitsch S, Jakobs C, Lehrach H. A catabolic block does not sufficiently explain how 2-deoxy-D-glucose inhibits cell growth. Proc Natl Acad Sci U S A. 2008;105(46):17807–17811. Epub 2008/11/14. doi: 10.1073/pnas.0803090105 PubMed PMID: 19004802; PMCID: PMC2584745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong D, Xiong L, Liu T, Liu X, Liu X, Chen J, Sun SY, Khuri FR, Zong Y, Zhou Q, et al. The glycolytic inhibitor 2-deoxyglucose activates multiple prosurvival pathways through IGF1R. J Biol Chem. 2009;284(35):23225–23233. Epub 2009/07/04. doi: 10.1074/jbc.M109.005280 PubMed PMID: 19574224; PMCID: PMC2749096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siolas D, Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013;73(17):5315–5319. Epub 2013/ 06/05. doi: 10.1158/0008-5472.CAN-13-1069 PubMed PMID: 23733750; PMCID: PMC3766500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okegawa T, Morimoto M, Nishizawa S, Kitazawa S, Honda K, Araki H, Tamura T, Ando A, Satomi Y, Nutahara K, et al. Intratumor heterogeneity in primary kidney cancer revealed by metabolic profiling of multiple spatially separated samples within tumors. EBioMedicine. 2017;19:31–38. Epub 2017/04/15. doi: 10.1016/j.ebiom.2017.04.009 PubMed PMID: 28408240; PMCID: PMC5440602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stacchini A, Aragno M, Vallario A, Alfarano A, Circosta P, Gottardi D, Faldella A, Rege-Cambrin G, Thunberg U, Nilsson K, et al. MEC1 and MEC2: two new cell lines derived from B-chronic lymphocytic leukaemia in prolymphocytoid transformation. Leuk Res. 1999;23(2):127–136. PubMed PMID: 10071128. [DOI] [PubMed] [Google Scholar]
- 37.Kaplan J, Mastrangelo R, Peterson WD Jr.. Childhood lymphoblastic lymphoma, a cancer of thymus-derived lymphocytes. Cancer Res. 1974;34(3):521–525. Epub 1974/03/01 PubMed PMID: 4590920. [PubMed] [Google Scholar]
- 38.Haber J. Genome instability: DNA repair and recombination. 1st ed. New York: (NY): Garland Science; 2014. December 16 2013 p. 399. [Google Scholar]
- 39.Sung P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science. 1994;265(5176):1241–1243. Epub 1994/ 08/26 PubMed PMID: 8066464. [DOI] [PubMed] [Google Scholar]
- 40.Chi P, Van Komen S, Sehorn MG, Sigurdsson S, Sung P. Roles of ATP binding and ATP hydrolysis in human Rad51 recombinase function. DNA Repair. 2006;5(3):381–391. PubMed PMID: 16388992. doi: 10.1016/j.dnarep.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 41.Skarbnik AP, Faderl S. The role of combined fludarabine, cyclophosphamide and rituximab chemoimmunotherapy in chronic lymphocytic leukemia: current evidence and controversies. Ther Adv Hematol. 2017;8(3):99–105. Epub 2017/ 03/02. doi: 10.1177/2040620716681749 PubMed PMID: 28246553; PMCID: PMC5305006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rebucci M, Michiels C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem Pharmacol. 2013;85(9):1219–1226. PubMed PMID: 23435357. doi: 10.1016/j.bcp.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 43.Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, Keating MJ, Huang P. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005;65(2):613–621. Epub 2005/ 02/08 PubMed PMID: 15695406. [PubMed] [Google Scholar]
- 44.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28(5):739–745. PubMed PMID: 18082599. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 45.Kawabata M, Kawabata T, Nishibori M. Role of recA/RAD51 family proteins in mammals. Acta Med Okayama. 2005;59(1):1–9. doi: 10.18926/AMO/31987 PubMed PMID: 15902993. [DOI] [PubMed] [Google Scholar]
- 46.Bibby MC. Orthotopic models of cancer for preclinical drug evaluation: advantages and disadvantages. Eur J Cancer. 2004;40(6):852–857. Epub 2004/ 05/04. doi: 10.1016/j.ejca.2003.11.021 PubMed PMID: 15120041. [DOI] [PubMed] [Google Scholar]
- 47.Pirmohamed M, Park BK. Genetic susceptibility to adverse drug reactions. Trends Pharmacol Sci. 2001;22(6):298–305. Epub 2001/06/08 PubMed PMID: 11395158. [DOI] [PubMed] [Google Scholar]
- 48.Atkinson J, McGlynn P. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 2009;37(11):3475–3492. Epub 2009/05/02. doi: 10.1093/nar/gkp244 PubMed PMID: 19406929; PMCID: PMC2699526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Caddle LB, Hasham MG, Schott WH, Shirley BJ, Mills KD. Homologous recombination is necessary for normal lymphocyte development. Mol Cell Biol. 2008;28(7):2295–2303. PubMed PMID: 18212067; PMCID: 2268416. doi: 10.1128/MCB.02139-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88(2):265–275. Epub 1997/01/24 PubMed PMID: 9008167. [DOI] [PubMed] [Google Scholar]
- 51.Bolstad BM, Collin F, Brettschneider J, Simpson K, Cope L, Irizarry RA, Speed TP. Quality assessment of affymetrix GeneChip data In: Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S, editors. Bioinformatics and computational biology solutions using R and bioconductor, chapter 3. New York: (NY): Springer; 2005. p. 33–47. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.