

ABSTRACT
Glioblastoma are among the most common forms of cancer affecting the central nervous system, and yet there is currently no effective means of treating them. In the current study, we reported that tousled-like kinase 2 (TLK2) is a key factor in glioblastoma that modulates SRC signaling, thereby driving tumor malignancy. TLK2 is commonly upregulated in glioblastoma, and such upregulation was associated with poor patient outcomes. TLK2 overexpression induced cell growth, migration, invasion, and epithelial-mesenchymal transition, and cell cycle arrest, while TLK2 knockdown had the opposite effect. SRC pathway inhibition by Saracatinib resulted in reduced TLK2-mediated glioblastoma migration, invasion, confirming a key role for SRC signaling in regulating the functions of TLK2. Together, our findings demonstrate that glioblastoma TLK2 overexpression acts as a key driver of tumor malignancy via SRC signaling pathway.
Keywords: TLK2, glioblastoma, SRC, tumor malignancy, cell cycle arrest
Introduction
Glioblastoma (GBM) is the most abundant form of brain tumor in adults, and are a major and increasingly common source of malignancy adversely impacting human healt.1,2 5-year survival rates remain low, even when treated aggressively, and the disease is associated with a 12–15 month median surviva.3,4 While great effort has been put towards understanding how glioblastoma develop and progress the molecular mechanisms governing these processes remain to be fully clarifie.5,6
Tousled-like kinases (TLKs) are a family of serine/threonine kinases located in the nucleus that promote S-phase chromatin assembly and mitotic chromosome segregatio.7,8 There are two genes in the TLK family-TLK1 and TLK.7 Previous studies have primarily focused on TLK1, whereas the role of TLK2 in human cancers is unclea.9 Mutations in TLK2 are associated with an elevated risk of breast cance.10 However, the function of TLK2 in the context of glioblastoma remains to be determined.
In this study, we found that TLK2 expression is increased in glioblastoma. Furthermore, we determined that TLK2 overexpression resulted in tumor growth and metastasis. This provides key insights into the role of TLK2 in glioblastoma progression, offering valuable opportunities for future therapeutic intervention.
Materials and methods
Cell culture
American Type Culture Collection (ATCC) was the source of the U251 and U87 glioblastoma lines. DMEM supplemented with 10% FBS, penicillin, streptomycin, and 1% L-glutamine at was used for all cell culture, in a 5% CO2 incubator at 37°C.
Tissue arrays and immunohistochemistry
Paraffin-embedded human glioblastoma tissue microarrays were obtained from SuperBioChips. They were probed with anti-TLK2 (1:200) in 5% BSA/PBS for 16 hours at 4°C. 3,3-diaminobenzidine was used to identify antibody staining, while hematoxylin (Sigma) served as a counterstain. Two scorers then graded TLK2 levels in a blinded manner based on the percentage of TLK2+ cells (0, 0%; 1, < 25%; 2, 25–50%; 3, 51–75%; and 4, > 75%), and staining intensity (0 – none; 1 – weak; 2 – moderate; and 3 – strong). The staining index formula was: staining intensity × percentage score. A score of > 6 was deemed “high expression”.
Real-time PCR
Real-time PCR was performed as previously stud.11 An RNeasy Mini Kit (Qiagen) was used for sample RNA isolation. cDNA was synthesized with the Transcriptor First Strand cDNA Synthesis Kit (Roche). The comparative threshold cycle (Ct) approach was used for quantifying changes in gene expression, with respective β-actin as a normalizing control.
Cell line engineering
TLK2 cDNA was inserted into the inducible lentiviral pTINDLE vector, which has an inducible promoter and a transactivator. TLK2 shRNA and non-silencing controls were inserted into the pINDUCER lentiviral vector. After packaging, cells were infected with lentiviruses carrying doxycycline (Dox) inducible plasmids, with 8 mg/ml polybrene added to the transfection mixture. Stable shTLK2 cell lines sorted based on GFP-positive cells on a FACSAria (BD Biosciences) flow cytometer. 200 ng/ml Dox induced TLK2 expression in appropriate cells. In overexpression assays, Dox was continuously administered for two weeks prior to downstream assays.
Western blotting
Western blotting was performed as previously stud.12,13 Briefly, RIPA lysis buffer (Sigma-Aldrich) was used for cell lysis, with complete protease inhibitor tablets (Roche) added before use. After running proteins on an SDS-PAGE gel, samples were transferred to a 0.2 mm nitrocellulose membrane. Primary antibodies were used against TLK2, ZO-1 (Abcam), p-SRC, SRC, p-AKT, AKT, p-ERK, ERK, E-Cadherin, Vimentin, p21 (Cell signaling technology), cyclin D1, cyclin B1 (Santa cruz biotechnology).
Cell viability and colony formation
Cells were plated in 6-well plates. Cell viability was assessed with trypan at 0, 24, 48, and 72 hours after plating. To measure anchorage-dependent colony formation, 500 cells were seeded per well and were allowed to incubate for 14 days. Methanol was then used to fix colonies, followed by 0.1% crystal violet (Sigma) staining and counting.
Assays for migration and invasion
24-well plates with porous Transwell filter inserts with an 8 μm pore size were used for migration assays, whereas for invasion assays Matrigel (BD Biosciences) coated filters were used. 2 × 104 cells were added into these inserts, with 600uL media added to the lower chamber. U251 cells were allowed to incubate for 16 hours, while U87 cells were allowed to incubate for 48 hours. Crystal violet was used to stain and count cells that entered the lower chamber, with three fields of view being counted in three independent experiments.
Cell cycle and apoptosis assessment by FACS
70% EtOH was use to fix cells before propidium iodide staining. The Annexin V-FITC apoptosis detection kit was used for apoptosis detection, following the instructions of the manufacturer (Abcam). A FACSCantoll cell analyzer (BD Biosciences) was used for all data acquisition.
Mouse models
3 × 106 U87 cells were injected subcutaneously in the left flank of nude mice, while TLK2 transfected cells were simultaneously injected into right side (n = 6). Tumor volumes were monitored for 21 days. Tumors were then removed, weighed, and assessed by qPCR and IHC. For assays assessing metastatic potential, treated U87 cells (1 × 106/0.2 ml) were injected into the tail veins of nude mice. After 8 weeks, mice were euthanized and the metastatic lung tumors were counted. No toxicity or weight loss was observed in the course of this study.
Statistical analyses
GraphPad Prism V was used for data analyses. Two-sided Fisher exact tests were used to compare TLK2 expression and pathologic features. Kaplan–Meier analyses and the log-rank test were used to assess overall survival. Student’s t-tests were also used. P < 0.05 served as the threshold for statistical significance.
Results
TLK2 is upregulated in glioblastoma and correlated with poor survival
Using tissue microarrays, we measured TLK2 levels in glioblastoma samples and determined that its protein levels were markedly elevated in invasive glioblastoma samples than in either non-invasive samples or normal tissues (Figure 1A). Patients that had experienced glioblastoma recurrence had higher mRNA expression of TLK2 than did those in whom the cancer did not recur (Figure 1B). Similarly, expression of TLK2 was significantly elevated in primary glioblastoma samples from patients experiencing metastasis than those in whom the cancer did not metastasize (Figure 1C). Kaplan–Meier survival analyses revealed that patients with high TLK2 expression has significantly worse survival rates as compared to those with low TLK2 expression (Figure 1D). These results indicate that TLK2 is often upregulated in the context of glioblastoma, and this upregulation is associated with poor disease outcomes for affected patients.
Figure 1.
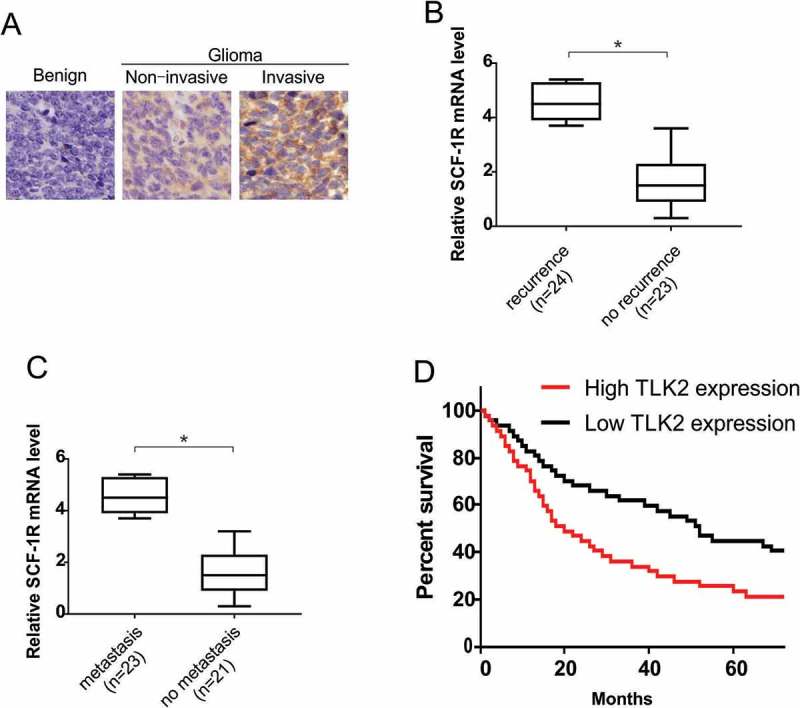
TLK2 overexpression indicates poor prognosis and promotes glioblastoma metastases. (A) Representative images of immunohistochemistry (IHC) staining of TLK2 protein in glioblastoma tissue microarrays. (B) Relative mRNA expression of TLK2 in glioblastoma patient samples with recurrence or without recurrence. (C) Relative mRNA expression of TLK2 in glioblastoma patient samples with metastasis or without metastasis. (D) Kaplane-Meier analysis of overall survival for patients with glioblastoma. *, P < 0.05.
TLK2 regulates glioblastoma cell growth
We next stably overexpressed TLK2 in U87 cells, and stably knocked it down in U251 cells, verify altered protein levels by Western blotting (Figures 2A and 2B). Knockdown of TLK2 was associated with a slight decrease in proliferation (Figures 2C and 2E), while its overexpression was associated with increased glioblastoma cell growth (Figures 2D and 2F). A clonogenic assay revealed that TLK2 silencing resulted in reduced colony generation, while its overexpression was linked to increased clonogenicity (Figures 2G and 2H).
Figure 2.
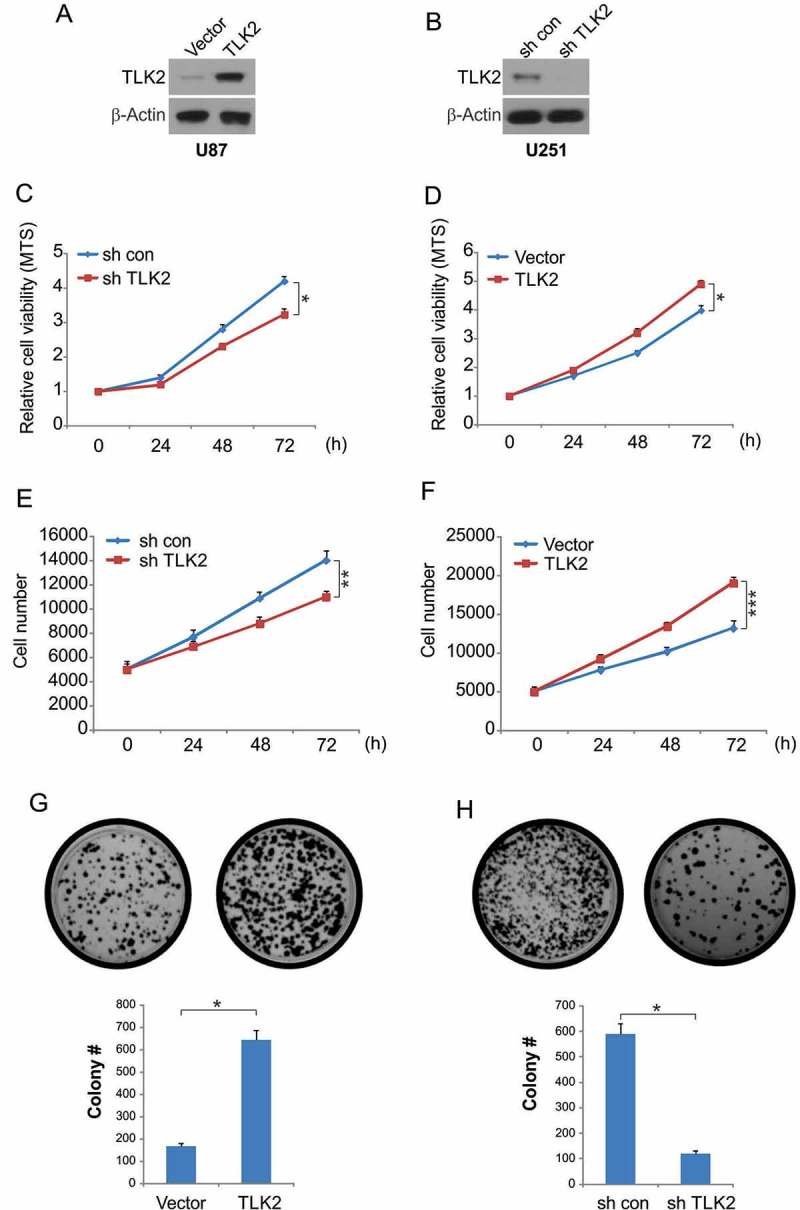
Effect of TLK2 on glioblastoma cells proliferation. (A) Western blotting of TLK2 overexpression in U87 cells. (B) Western blotting of TLK2 knockdown in U251 cells. (C) TLK2 modulated cell viability in vitro. Cell viability of U251 cells was analyzed by trypan blue exclusion assays at different time points. Data were represented as means ± SD from three independent experiments. *, P < 0.05. (D) Cell viability of U87 cells was analyzed by trypan blue exclusion assays at different time points. Data were represented as means ± SD from three independent experiments. *, P < 0.05. (E) U251 cells were tansfected with sh TLK2 for 24 hours, cell number was analyzed at different time points, **, P < 0.01. (F) U87 cells were tansfected with TLK2 for 24 hours, cell number was analyzed at different time points, ***, P < 0.001. (G) Effects of TLK2 on anchorage dependent colony formation of U87 cells. Results were presented as means ± SD from three independent experiments. *, P < 0.05. (H) Effects of TLK2 on anchorage dependent colony formation of U251 cells. Results were presented as means ± SD from three independent experiments. *, P < 0.05.
TLK2 regulates glioblastoma cell malignancy and EMT
We next investigated the association between TLK2 and glioblastoma cell migration and invasion. Silencing of TLK2 was associated with significantly reduced cellular migration (Figure 3A), and invasive potential was similarly reduced (Figure 3B). When we assessed the effects of TLK2 overexpression on these same malignant phenotypes, we found that both migration and invasion were increased upon overexpression of this gene (Figures 3C and 3D). These results indicate that TLK2 expression is linked to the invasive potential of glioblastoma cells. Given the well-documented link between invasive potential and the epithelial-mesenchymal transition (EMT), we used Western blotting to assess epithelial (E-cadherin, ZO-1) and mesenchymal (Vimentin) marker expression. We found that overexpression of TLK2 was associated with lower E-cadherin and ZO-1 levels, whereas vimentin levels were increased. TLK2 knockdown conversely revealed elevated E-cadherin and ZO-1 but decreased vimentin (Figure 3E). This indicates that TLK2 regulates the growth, migration, invasive potential, and EMT of glioblastoma cells.
Figure 3.
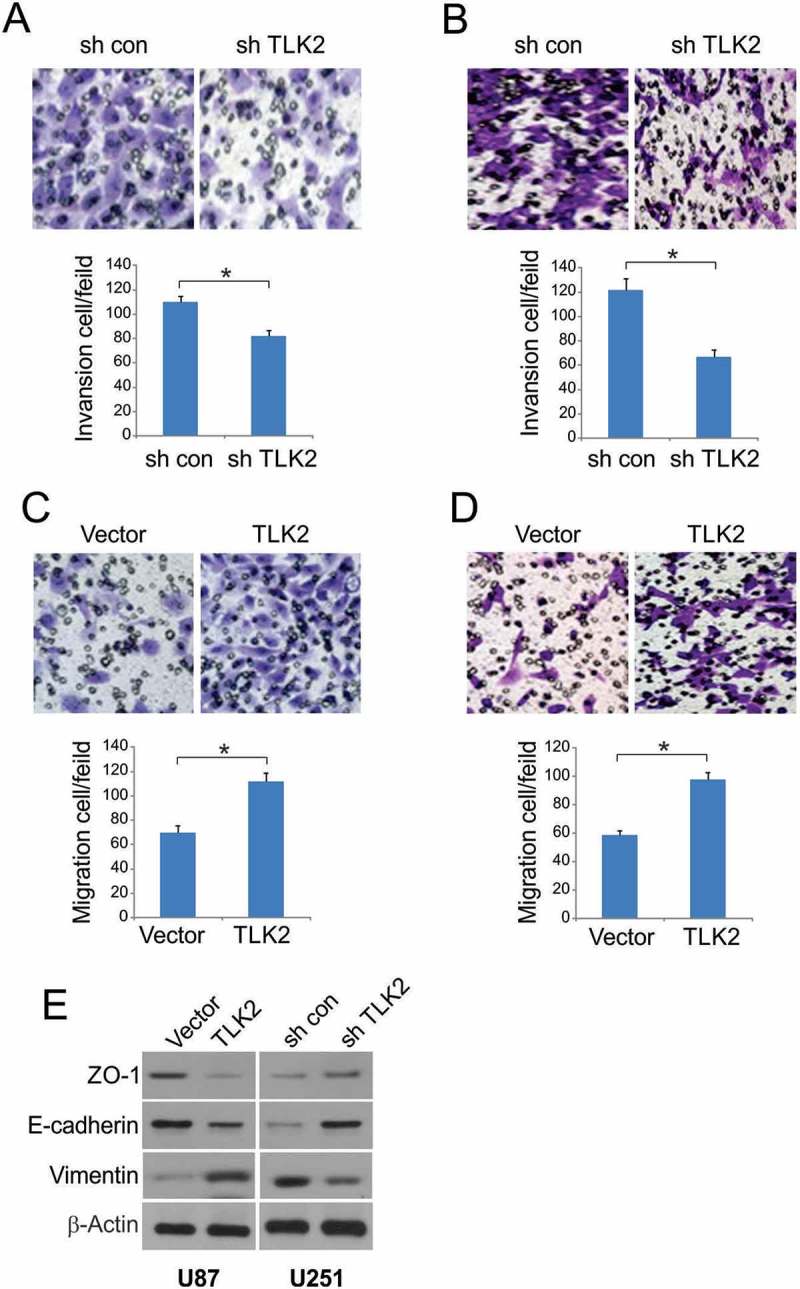
Effect of TLK2 on glioblastoma cells migration and invasion. (A-B) Knockdown TLK2 suppressed cell migration and invasion in U251 cells. (C-D) Overexpression of TLK2 significantly enhanced migration and invasion in U87cells. (E) TLK2 regulated epithelial-mesenchymal transition of glioblastoma cells. Expression of epithelial markers, E-cadherin and ZO-1, and mesenchymal markers, vimentin, were analyzed by Western blotting. All results were represented as means ± SD from three independent experiments. *, P < 0.05.
TLK2 silencing disrupts that G2/M transition and induces apoptosis
To investigate the importance of TLK2 in the context of cell cycle progression at the molecular level, we investigated DNA content in U251 cells after knockdown of TLK2. Through these FACS analyses, we found that the frequency of G2/M phase cells was significantly increased by TLK2 silencing, while the frequency of G0/G1 cells was significantly decreased (Figure 4A). By examining the levels of cell cycle regulatory proteins, we further found that cyclin B1 and cyclin D1, which promote the G2/M transition, were significantly decreased in TLK2 knockdown cells (Figure 4B). TLK2 inhibition was also linked to marked induction of apoptosis in these U251 cells (Figure 4C). TLK2 inhibition thus leads to cell cycle arrest at the G2/M phase, ultimately causing these glioblastoma cells to undergo apoptosis.
Figure 4.
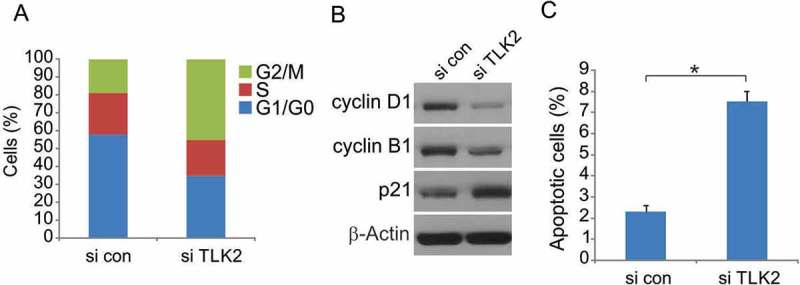
TLK2 inhibition results in impaired G1/S progression and induction of apoptosis in TLK2-amplified glioblastoma cells. (A) Flow cytometry results showing cell cycle changes after TLK2 knockdown in U251 cells. (B) U251 cells were transfected with si control or si TLK2 for 24 hours, indicated protein level were analyzed by Western blotting. (C) Cell apoptosis assessed by Annexin V assay in asynchronized U251 cells following TLK2 knockdown. All results were represented as means ± SD from three independent experiments. *, P < 0.05.
TLK2 enhances SRC signaling
We next assessed changes in cell signaling in responses to TLK2 overxpression, using Western blotting to investigate relevant signaling pathways. We observed no AKT or ERK activation in these TLK2 overexpressing glioblastoma cells (Figure 5A). SRC phosphorylation, however, was significantly increased in these cells. We therefore treated cells with the SRC inhibitor Saracatinib, and found that the increases in cell migration and invasion observed in TLK2 overexpressing cells were significantly ablated upon Saracatinib treatment (Figures 5B and 5C). This suggests that SRC activation downstream of TLK2 is important to glioblastoma cell malignancy in vitro.
Figure 5.
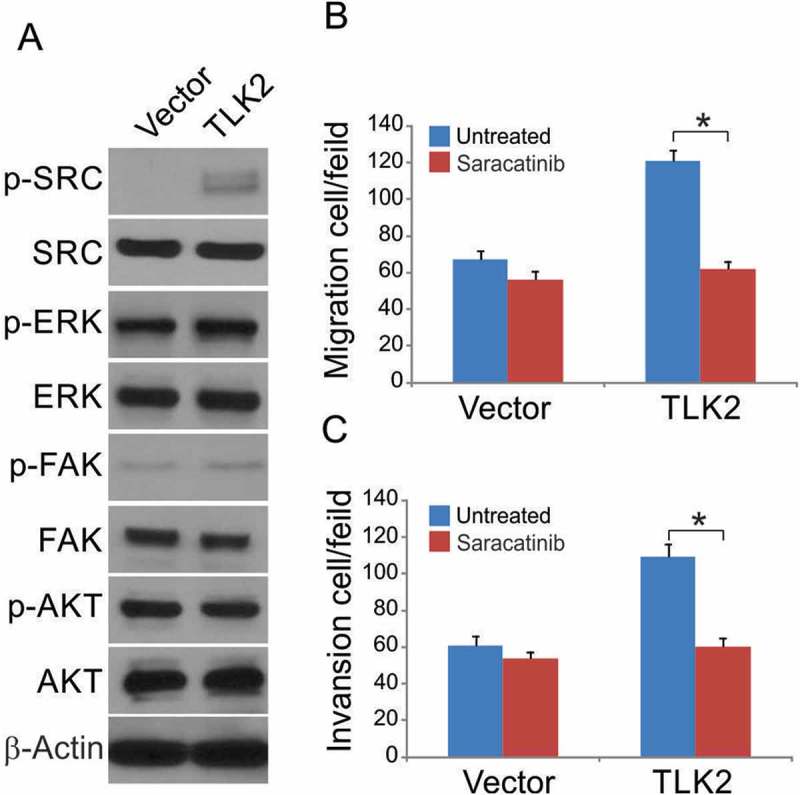
TLK2 enhances SRC signaling in glioblastoma cells. (A) U87 stable transfectants were treated Dox at indicated time points. Indicated proteins were analyzed by Western blotting. (B-C) Cells were treated with Saracatinib (5 mM) or DMSO during the migration and invasion assays. Data were represented as means ± SD from three independent experiments. *, P < 0.05.
TLK2 knockdown inhibits in vivo tumor growth and lung metastases
To better understand the role of TLK2 in glioblastoma in vivo, we used a nude mouse model into which cells with stable TLK2 shRNA (lenti-TLK2) or control shRNA were injected subcutaneously. TLK2 silencing was associated with significantly reduced tumor growth relative to control (Figure 6A). Xenografts that were treated with the TLK2 shRNA lentivirus were confirmed to have a durable decrease in TLK2 expression (Figure 6B). Western blotting also confirmed that p-SRC levels were markedly decreased if mice were injected with shTLK2 tumors, consistent with in vitro results (Figure 6C). Control or lenti-TLK2 cells were also injected intravenously in nude mice, and the occurrence of lung metastases was assessed. We found that TLK2-silenced tumor cells were associated with significantly fewer lung metastases relative to mice injected with control TLK2-competent cells (Figure 6C). TLK2 expression was confirmed to be decreased in these metastatic nodules from mice in the lenti-TLK2 group as compared to the control shRNA group.
Figure 6.
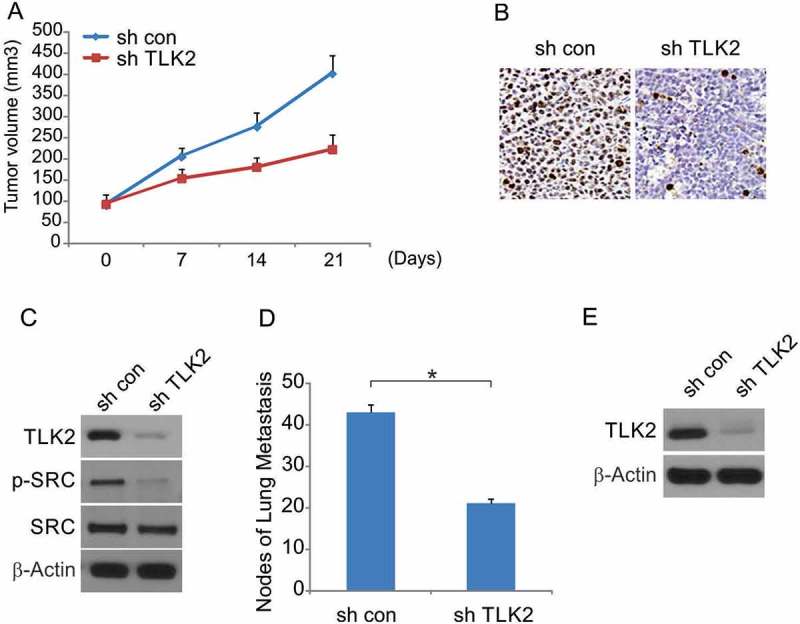
TLK2 enhanced tumor growth in vivo. (A) U87 mock transfectants were subcutaneously injected into left rear flank of nude mice and equal number of TLK2 overexpressed cells injected into right rear flank of each mouse. The size of tumors was measured at indicated time points, and was shown as mean ± SD. Mice were sacrificed at day 21. *, P < 0.05, n = 6. (B) Immunohistochemistry of transplanted tumors. Paraffin-embedded sections were immunostained with TLK2. (C) Analysis of indicated proteins expression in transplanted tumors by Western blotting. (D) Effects of TLK2 on tumor metastasis. The number of metastatic tumors was decreased in the TLK2 knockdown group. (E) Western blotting of TLK2 in metastatic tumors (n = 6). *, P < 0.05.
Discussion
Tlk2 is a homolog of Tousled, first identified in Arabidopsis thalian.14 Humans contain two homologues of Tousled, TLK1 and TLK2, whose activity is cell cycle regulated and peaks during S phas.9,10,15 This research revealed that TLK2 upregulation in glioblastoma is associated with poor patient outcomes and an overall worse prognosis. Consistence with previously stud,10,16 overexpression of TLK2 in U87 cells was associated with increased migration and invasion, while its knockdown had the opposite effect, suggesting a direct role for TLK2 in glioblastoma invasion. We further determined that TLK2 is linked to the SRC signaling axi,17–19 by which it facilitates the increased invasiveness observed in glioblastoma tissues. TLK2 knockdown was found to lead to cell cycle arrest and apoptosis in glioblastoma tissues, suggesting that its expression is vital for tumor cell survival and malignancy.
Additional factors within the tumor microenvironment may further regulate the role of TLK2 in glioblastoma in an endogenous setting, potentially complicating any interpretation of these in vitro finding.20 Further research in autochthonous model systems not reliant on developed cell lines will shed further light on the true relevance of TLK2 to glioblastoma progression in vivo. Ultimately, this study suggests that inhibiting TLK2 may be a viable therapeutic strategy for combating glioblastoma in patients whose tumors exhibit high levels of TLK2 expression. Indeed, in a murine model system, TLK2 silencing was associated with significantly reduced tumor growth and a significant reduction in metastasis. While these in vivo preclinical models fail to recapitulate the true complexities of complex stromal microenvironments and tumor cell heterogeneity, they provide key evidence underscoring the relevance of TLK2 to glioblastoma progression in vivo. Future work with patient-derived xenograft models will provide key insights into the importance of TLK2 in human glioblastoma, allowing for the development of appropriate therapeutic regimens aimed at curing glioblastoma.
In conclusion, this study suggests that TLK2 is a regulator of the SRC signaling pathway in glioblastoma cells, thereby driving malignant phenotypes in these tumors. Further study of TLK2 may offer valuable scientific and therapeutic insights into the biology and treatment of this complex and dangerous tumor type.
Acknowledgments
This research was not supported by any grants from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Punsoni M, Donahue JE, Elinzano HD, Kinsella T.. Updates in molecular pathology of central nervous system gliomas in adults. R I Med J (2013). 2015;98(11):17–19. [PubMed] [Google Scholar]
- 2.Davis ME. Glioblastoma: overview of Disease and Treatment. Clin J Oncol Nurs. 2016;20(5 Suppl):S2–8. doi: 10.1188/16.CJON.S1.2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15:422–442. doi: 10.1038/s41571-018-0003-5. [DOI] [PubMed] [Google Scholar]
- 4.Shergalis A, Bankhead A, Luesakul U, Muangsin N, Neamati N. Current challenges and opportunities in treating glioblastoma. Pharmacol Rev. 2018;70(3):412–445. doi: 10.1124/pr.117.014944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noroxe DS, Poulsen HS, Lassen U. Hallmarks of glioblastoma: a systematic review. ESMO Open. 2016;1(6):e000144. doi: 10.1136/esmoopen-2016-000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi PJ, Tubbs RS, Oskouian RJ. Emerging cellular therapies for glioblastoma multiforme. Cureus. 2018;10(3):e2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carrera P, Moshkin YM, Gronke S, Sillje HHW, Nigg EA, Jackle H, Karch F. Tousled-like kinase functions with the chromatin assembly pathway regulating nuclear divisions. Genes Dev. 2003;17(20):2578–2590. doi: 10.1101/gad.276703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelly R, Davey SK. Tousled-like kinase-dependent phosphorylation of Rad9 plays a role in cell cycle progression and G2/M checkpoint exit. PLoS One. 2013;8(12):e85859. doi: 10.1371/journal.pone.0085859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Segura-Bayona S, Knobel PA, González-Burón H, Youssef SA, Peña-Blanco A, Coyaud É, López-Rovira T, Rein K, Palenzuela L, Colombelli J, et al. Differential requirements for Tousled-like kinases 1 and 2 in mammalian development. Cell Death Differ. 2017;24(11):1872–1885. doi: 10.1038/cdd.2017.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim JA, Anurag M, Veeraraghavan J, Schiff R, Li K, Wang X-S. Amplification of TLK2 induces genomic instability via impairing the G2-M checkpoint. Mol Cancer Res. 2016;14(10):920–927. doi: 10.1158/1541-7786.MCR-16-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tong J, Wang P, Tan S, Chen D, Nikolovska-Coleska Z, Zou F, Yu J, Zhang L. Mcl-1 degradation is required for targeted therapeutics to eradicate colon cancer cells. Cancer Res. 2017;77(9):2512–2521. doi: 10.1158/0008-5472.CAN-16-3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tong J, Tan S, Nikolovska-Coleska Z, Yu J, Zou F, Zhang L. FBW7-dependent Mcl-1 degradation mediates the anticancer effect of Hsp90 inhibitors. Mol Cancer Ther. 2017;16(9):1979–1988. doi: 10.1158/1535-7163.MCT-17-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tong J, Tan S, Zou F, Yu J, Zhang L. FBW7 mutations mediate resistance of colorectal cancer to targeted therapies by blocking Mcl-1 degradation. Oncogene. 2017;36(6):787–796. doi: 10.1038/onc.2016.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Liu J, Xia R, Wang J, Shen J, Cao R, Hong X, Zhu J-K, Gong Z. The protein kinase TOUSLED is required for maintenance of transcriptional gene silencing in Arabidopsis. EMBO Rep. 2007;8(1):77–83. doi: 10.1038/sj.embor.7400852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Z, Gourguechon S, Wang CC. Tousled-like kinase in a microbial eukaryote regulates spindle assembly and S-phase progression by interacting with Aurora kinase and chromatin assembly factors. J Cell Sci. 2007;120(Pt 21):3883–3894. doi: 10.1242/jcs.007955. [DOI] [PubMed] [Google Scholar]
- 16.Kim JA, Tan Y, Wang X, Cao X, Veeraraghavan J, Liang Y, Edwards DP, Huang S, Pan X, Li K, et al. Comprehensive functional analysis of the tousled-like kinase 2 frequently amplified in aggressive luminal breast cancers. Nat Commun. 2016;7:12991. doi: 10.1038/ncomms12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lohman AW, Weilinger NL, Santos SM, Bialecki J, Werner AC, Anderson CL, Thompson RJ. Regulation of pannexin channels in the central nervous system by Src family kinases. Neurosci Lett. 2017; doi: 10.1016/j.neulet.2017.09.019. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Zhuang S. Src family kinases in chronic kidney disease. Am J Physiol Renal Physiol. 2017;313(3):F721–F728. doi: 10.1152/ajprenal.00141.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Q, Zhou Z, Shan L, Zeng H, Hua Y, Cai Z. The importance of Src signaling in sarcoma. Oncol Lett. 2015;10(1):17–22. doi: 10.3892/ol.2015.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu H, Dowdle JA, Khurshid S, Sullivan NJ, Bertos N, Rambani K, Mair M, Daniel P, Wheeler E, Tang X, et al. Discovery of stromal regulatory networks that suppress ras-sensitized epithelial cell proliferation. Dev Cell. 2017;41(4):392–407 e6. doi: 10.1016/j.devcel.2017.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]