

ABSTRACT
Contemporary in vivo and in vitro discovery platform technologies greatly increase the odds of identifying high-affinity monoclonal antibodies (mAbs) towards essentially any desired biologically relevant epitope. Lagging discovery throughput is the ability to select for highly developable mAbs with drug-like properties early in the process. Upstream consideration of developability metrics should reduce the frequency of failures in later development stages. As the field moves towards incorporating biophysical screening assays in parallel to discovery processes, similar approaches should also be used to ensure robust chemical stability. Optimization of chemical stability in the early stages of discovery has the potential to reduce complications in formulation development and improve the potential for successful liquid formulations. However, at present, our knowledge of the chemical stability characteristics of clinical-stage therapeutic mAbs is fragmented and lacks comprehensive comparative assessment. To address this knowledge gap, we produced 131 mAbs with amino acid sequences corresponding to the variable regions of clinical-stage mAbs, subjected these to low and high pH stresses and identified the resulting modifications at amino acid-level resolution via tryptic peptide mapping. Among this large set of mAbs, relatively high frequencies of asparagine deamidation events were observed in CDRs H2 and L1, while CDRs H3, H2 and L1 contained relatively high frequencies of instances of aspartate isomerization.
KEYWORDS: antibody, developability, chemical liability, asparagine deamidation, aspartic acid isomerization, forced degradation, low and high pH stress, tryptic peptide mapping
Introduction
The comparatively high approval rate1,2 and relative ease of engineering for monoclonal antibody- (mAb) based therapeutics have garnered substantial investment in the discovery and development efforts of this seemingly ever-expanding drug class. Still, the overall process of antibody therapeutics development, from lead selection to commercial product approval, is inefficient and thus economically wasteful. Over the past decade, drug developers have become increasingly proactive about early identification and risk assessment to combat attrition. Efforts include the quality-by-design global initiative (where statistical tools are used to ensure reproducible experiments and material productions), discovery- stage screening of materials for biophysical and biochemical properties, and implementing predictive in silico tools to avoid or reduce development liabilities. One strategy now commonly used to avoid late-stage failures is the so-called developability or molecular assessment approach, which aims to optimize biophysical and chemical properties of molecules prior to product development.3,4
The field has recently incorporated parallel screening of biophysical properties in discovery.5 However, in addition to good biophysical properties, mAbs also need to possess sufficient chemical stability to conform to stringent process development parameters.6 For those engineering efforts to be successful, a comprehensive survey of the field is required to determine how clinical-stage mAbs are expected to perform. The predominant chemical modifications of interest for mAbs are oxidation, deamidation and isomerization.4 Oxidative modification of methionine residues with peroxide is a commonly used technique that has recently been applied to the study of samples with variable region sequences sourced from 121 clinical-stage mAbs.7 Unlike the straightforward peroxide oxidation chemistry used to induce methionine oxidation,8 Asn deamidation and Asp isomerization reactions occur through complex and interrelated pathways. These chemical mechanisms are pH- and temperature-dependent and have several competing transition states.9-13 In the case of deamidation, the chemistry is further complicated by competing and alternative mechanisms.13,14
As shown in Figure 1, the aspartate isomerization reaction proceeds through an aspartyl-succinimide (Asu) intermediate. This reaction is accelerated under acidic conditions.15,16 Additionally, at low pH, the Asu intermediate is relatively stable (maximally at pH 4–5) due to a reduced rate of hydrolysis of the 5-membered ring.17-19 Under these acidic conditions, hydrolysis of the Asu intermediate becomes rate-limiting, permitting the accumulation of these species within the sample20 and the subsequent facile detection of an 18 Da mass loss. This mass loss is detected and used as a surrogate for the isomerization modification, which is more difficult to detect via liquid chromatography-mass spectrometry. Conversely, deamidation proceeds predominantly through the Asu-mediated asparagine deamidation pathway, under neutral or basic conditions.9,11 At alkaline pHs, the rate of Asu hydrolysis is much faster than the rate of Asu formation,10,11 nearly eliminating the Asu population16 while simultaneously increasing the rate of deamidation (subsequently observed as a + 1 Da mass shift variant). As both reactions occur through the same reaction intermediate with opposing pH preferences, changing the pH of the reaction is a suitable toggle to control the partitioning of each pathway. For example, deamidation rates at pH 5.5 are approximately 40-fold slower than at pH 8.0,9,11,15,21 while the rate of Asu intermediate formation increases 6-fold over the same pH range.15 Although several studies have been conducted where both products are induced at a single pH,12,22-24 others have preferentially induced isomerization at low pH16,25,26 or deamidation at high pH.26-28 The reaction conditions used in this study employ the latter approach to amplify the observable modified populations with concurrent simplification of isomerization detection.
Figure 1.
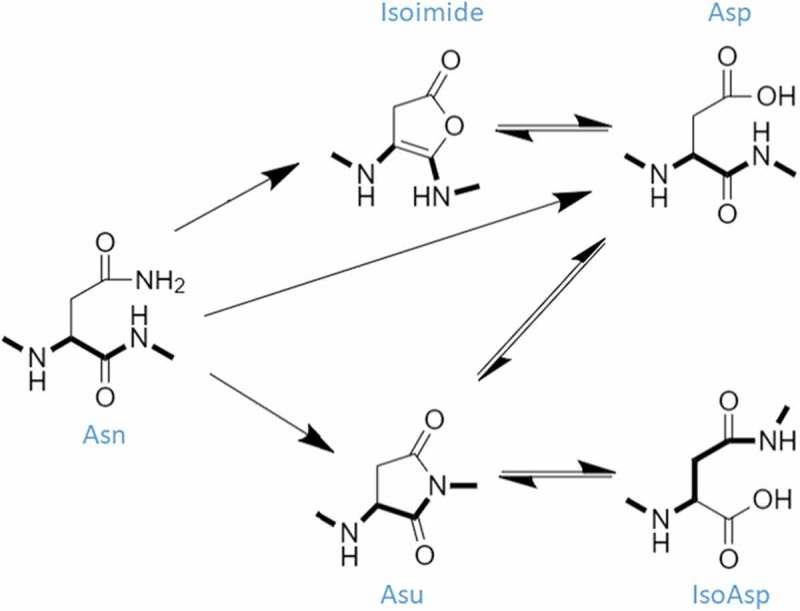
Deamidation and isomerization mechanism. The dominant deamidation pathway at pH 8.5 is Asu-mediated (Asn → Asu ⇌ [IsoAsp ⇌ Asp]). The alternative direct hydrolysis pathway (Asn → Asp) is also operative at this pH. Additionally, there is a minor Isoimide-mediated pathway that may also contribute to deamidation under our experimental conditions (Asn → Isoimide ⇌ Asp). The Asu-mediated (Asp ⇌ Asu ⇌ IsoAsp) isomerization pathway is accelerated at pH 5.5. The peptide backbone is highlighted in bold to illustrate isomerization.
Typically, clinical-stage mAbs are stressed under a combination of heat and low or high pH to accelerate the degradation that is expected to occur during manufacturing processes and long-term storage. Similar accelerated studies may also be carried out during the lead selection stage to identify the most stable candidates for further development, or to identify or confirm degradation sequence liabilities for re-engineering before committing resources to process and product development. With the goal of avoiding unanticipated issues in product development, it is worth noting that optimizing the chemical stability of a mAb will not only improve the shelf-life of the molecule, but ideally also translate into serum stability.29 Therefore, upstream optimization of mAb chemical stability using information from accelerated degradation studies has the potential to expediate process and product development, as well as produce a more homogeneous and stable drug product.
Looking forward, the field appears to be working towards a priori predictions of chemical modifications.7,23,30 Currently, cursory ab initio analysis can be performed at the amino acid sequence level to identify potential degradation sequence liabilities based on motifs of two amino acids, such as NG or DG sequences; however, simple counting of such motifs leads to a high frequency of false positives. For example, in the most extensive study to date, 37 mAbs were stressed at pH 6.0 at 40 °C for two weeks, which resulted in 67% or 21% of samples with expected-labile NG or NS motifs showing detectable levels of deamidation, respectively. For isomerization, the false positive rate was even higher, with only 36% and 13% of samples with DG and DS motifs, respectively, showing detectable level of isomerization.23 With sufficient data, computational and statistical models may be improved to accurately predict chemical stability.
To encourage the general understanding of chemical liabilities in the context of clinical-stage mAbs, and to establish a more comprehensive and transparent (by identifying all molecules by their international non-proprietary names (INNs)) dataset on the subject, we report here our data for accelerated degradation of 131 isotype-matched samples with variable region sequences corresponding to clinical-stage mAbs. Two different stress conditions (pH 5.5 at 40 °C for two weeks and pH 8.5 at 40 °C for one week) were used to identify labile sites for isomerization and deamidation. Through this effort, we observed that complementarity-determining regions (CDRs) H2, H3, and L1 are particularly susceptible to modification, with each containing a single exceptionally labile sequence position or sub-section where deamidation and isomerization motifs are frequently observed to be modified.
Results
All antibodies used in this study were formatted and expressed as IgG1 isotype mAbs, regardless of original drug isotype, with variable regions matching published sequences (see Jain et al.,31 for further details). For ease of exposition, we generally use the INNs (e.g., evolocumab) to refer to the samples, with the understanding that our sample is not identical to the original clinical material. To minimize sample treatment and data acquisition-induced artifacts, the difference between stressed and unstressed samples was used to quantify the amount of observed deamidation or isomerization upon stress. Only degradation events occurring in the CDRs are discussed here due to the low frequency of framework region isomerization and deamidation modifications (for completeness, reported in Table S1). All sequences, INNs, modification quantities, and modified residues are included in Supplemental Tables S2-S5 and in a supplemental workbook.
In this section, deamidation and isomerization sequence liabilities, as well as hot-spots (frequently modified sequence positions) and cold-spots (frequently unmodified sequence positions) are described in the context of “canonical” and “non-canonical” motifs. Canonical motifs are specified using the standard n, n + 1 notation,32 and include motifs that have been previously implicated in CDR deamidation and isomerization events, and/or have otherwise demonstrated modification rates in time-scales aligned with our experiments. Thus, the canonical motifs for isomerization are DG,16,20,22,23,25,29,33,34 DS,10,23,35,36 DD,23,36 DT,23 and DH.37 For deamidation, the canonical motifs are NG,20,22,23,32,34,38 NS,22,23,32,39 NN,23,32 NT,22,23,32,34,40 and NH.32 From these published studies, it is also possible to discern an approximate hierarchy. For example, DG is expected to be more labile than DS, and DS more so than DD or DT. Similarly, for deamidation motifs there is the expectation of decreasing stability in the order NG, NS, NT.
CDR deamidation at high pH
Upon high pH stress (pH 8.5) for 1 week at 40 °C, 39 deamidation sites were identified among these samples (see Figure S1 for example data), with degradation levels ranging from 2.0% (our reporting criterion) to 67.7% (Fig. S2A). Approximately half of these sites (22/39) occurred in light chain (LC) CDRs (Figure 2, Table S2). Of all heavy chain (HC) CDR deamidation events, 64.7% (11/17) occurred in CDRH2 with the highest deamidation level (65.1%, evolocumab) and occurrence (8/11) of modification at an NG motif. However, only 47.0% (8/17) of all observed occurrences of NG motifs within the CDRH2 underwent detectable deamidation upon high pH treatment (Figure 2(b)).
Figure 2.
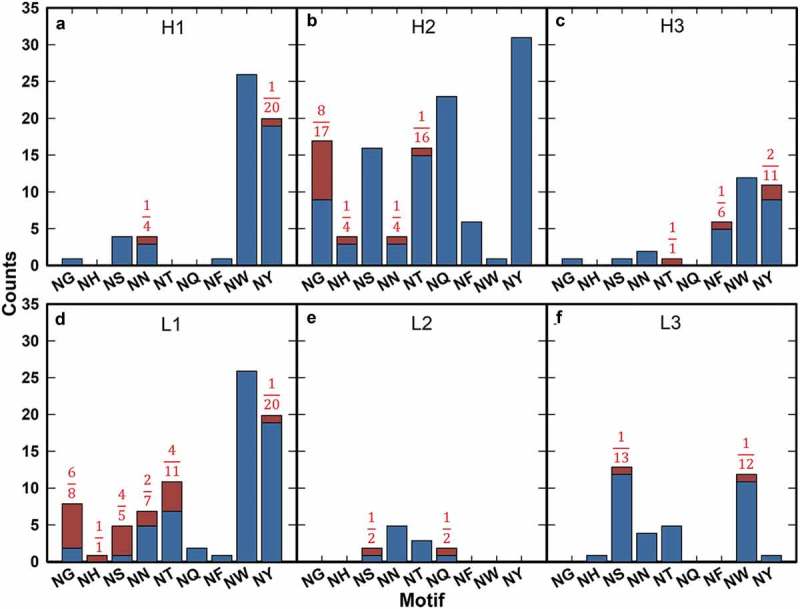
Motif deamidation frequency by CDR under high pH stress. motif occurrences are plotted by unmodified (blue) and modified (red) for CDRH1 (a), CDRH2 (b), CDRH3 (c), CDRL1 (d), CDRL2 (e), and CDRL3 (f). Fractions of total occurrences modified are noted for all motifs when applicable.
In CDRH3, four deamidation events were observed. There is no obvious motif bias within this CDR as three of the four observed modifications (NT, NF, and two instances of NY) occur at non-canonical motifs (Figure 2(c)). Deamidation modifications ranged from 2.6 to 61.7 % upon stress (Fig. S2A). Although these four instances only accounted for 23.5% of all deamidation species in the HC CDRs within this dataset, the average deamidation observed in CDRH3 was higher than observed in CDRH2 (32.4% ± 26.2%, n = 4 versus 14.3% ± 17.8%, n = 11).
Similarly, CDRL1 was frequently deamidated under these conditions. This CDR accounted for 81.8% (18/22) of all observed LC deamidation events, and it had the second highest average percent deamidation of all six CDRs (Figure 2(d), S2A). The observed deamidation events occurred at motifs consistent with previous findings,23 except for a single instance of deamidation occurring at the non-canonical NY motif.
Both deamidation-labile CDRs (H2 and L1) had the highest frequency of canonical motifs compared to all other CDRs. In terms of fractional occurrence (number modified/number of motif instances within a given CDR), all canonical motifs within CDRL1 appeared to be relatively susceptible to deamidation (Figure 2(d)), while NG motifs appeared to be preferentially modified in CDRH2 (Figure 2(b)). The canonical NS motif was observed in relatively high frequencies within these CDRs as well as in CDRL3, but only within CDRL1 did one observe high occurrence of modifications at NS (4/5 or 80% for NS in CDRL1 vs. 6/41 or 14.6% in CDRL3). These results are consistent with previous observations that deamidation of a canonical motif within mAb CDRs is likely dependent on local structure or conformation.21,23
CDR isomerization at low pH
Upon low pH stress (pH 5.5) for 2 weeks at 40 °C, 31 isomerization events (including both IsoAsp or Asu species) were observed (see Figure S3 for example data) with total modification measurements ranging from 2.1 to 44.0% (Fig. S2B, Table S3). Approximately 39% of all observed isomerization (12/31 instances) occurred on the LC (Figure 3). Of all HC isomerization events, 63.1% (12/19) occurred in CDRH3, with an apparent DG motif preference (Figure 3(c)). The DG motif accounted for 58.3% (7/12) of all isomerization events occurring in this CDR with the presence of a DG motif at any position within CDRH3 modified in 53.8% (7/13) of cases.
Figure 3.
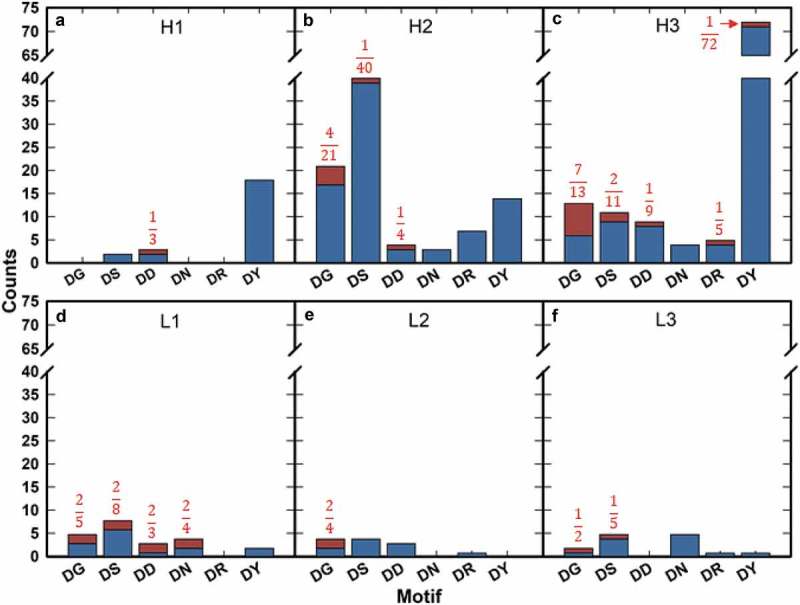
Motif isomerization frequency by CDR under low pH stress. Motif occurrences are plotted by unmodified (blue) and modified (red) for CDRH1 (a), CDRH2 (b), CDRH3 (c), CDRL1 (d), CDRL2 (e), and CDRL3 (f). Fractions of total occurrences modified are noted for all motifs when applicable.
We found that 31.5% (6/19) of all HC isomerization events were located within CDRH2 (Figure 3(b)). Of these 6 isomerization events, 66.7% (4/6) occurred at a DG motif. However, DG at any position within CDRH2 underwent isomerization upon low pH stress only 19.0% of the time (4/21).
Isomerization events in the LC occurred most frequently in CDRL1, accounting for 66.7% (8/12) of all LC modification instances (Figure 3(d)). As with the deamidation sequence liabilities observed in the high pH stress condition, there was no apparent canonical motif bias, with the observed labile motifs being generally consistent with those described previously.
All isomerization-labile CDRs (H2, H3, and L1) had higher frequencies of canonical motifs than all other CDRs. In terms of fractional occurrence, all canonical motifs within CDRL1 appeared to be relatively susceptible to isomerization (Figure 3(d)), while only DG motifs appeared to be modified at high frequency in CDRH3 (Figure 3(c)). In contrast, CDRH2 (Figure 3(b)) showed a low frequency of modification at the, a priori, most labile canonical motifs (DG and DS) compared to all other CDRs except for CDRH1, which contains limited instances of DG and DS motifs. These observations suggest that, as with deamidation, isomerization of canonical motifs is structurally or conformationally dependent.
It is also notable that no Asp isomerization event was observed in canonical motif DT or DH sites within CDRs, of 32 and 6 occurrences, respectively. This observation is consistent with the results reported by Sydow et al.23
Sequence analysis
All mAb variable region sequences are structurally aligned using a modification of the Kabat-Chothia numbering scheme (see Methods).41-43 Comparison of observed modification sites across mAbs in the context of canonical motifs revealed three deamidation and isomerization hot-spots and a single cold-spot within individual CDRs. We define hot- and cold-spots as residue positions with a high frequency of canonical motif occurrence and high and low levels, respectively, of chemical degradation (Table S6). The single cold-spot resides in CDRH2 (H61) where the canonical DS or DD motifs occurred 37 times with zero instances of detectable isomerization. However, it should be noted that under alternative CDR definitions (e.g., IMGT®44) the second CDR of the heavy chain ends at position H58, and thus H61 would be considered part of the framework.
We identified three locations that were frequently susceptible to deamidation and/or isomerization modifications under our stress conditions: H54, H98 and a stretch of amino acids in the CDRL1 “insertion sub-region,” namely positions L30 through L30F. Position H54 contains 50.0% (3/6) and 63.6% (7/11) of all isomerization and deamidation modifications in CDRH2, respectively. Of all canonical motifs at position H54, 35% (7/20) of deamidation motifs and 20% (3/15) of isomerization motifs were modified (Table S6). Similarly, sub-region L30 (as defined above) contained 87.5% (7/8) of all isomerization events, and 88.9% (16/18) of all deamidation events in CDRL1. In this region, 64% (14/22) of canonical deamidation motifs, and 33% (7/21) of isomerization motifs were modified. Lastly, position H98 accounted for 33% (4/12) of all isomerization events and 25% (1/4) of all deamidation events in CDRH3. Considering all canonical motifs at position H98, 100% (1/1) of deamidation motifs and 67% (4/6) of isomerization motifs were modified (Table S6). Notably, occurrences of an Asp (12/131) and Asn (3/131) at H98 were rare within the set.
Although only one cold-spot was identified, there are five positions within the CDRs where Asp and Asn residues occur frequently without modification. In all cases the n + 1 position leads to a non-canonical motif. For instance, a Pro residue occurs at position H52A in 45.0% (59/131) of all sequences. Of the 59 cases, 22 sequences have an Asn residue, and 10 sequences have an Asp residue at the preceding, n, position. There are no instances of isomerization or deamidation at this position because the n + 1 proline prevents Asu formation due to the lack of an N-terminal backbone amide. Less extreme examples can be observed where the n + 1 position is typically a Y, R, or a branched-chain aliphatic (I, L, V) residue. These motif positions include H31 (n + 1 is Y), H101 (n + 1 is Y, I, L, or V), L28 (n + 1 is I or V), and L53 (n + 1 is L or R).
Motif modification may also be modulated by near-in-sequence but non-adjacent residues. In CDRL3, a germline-encoded proline residue (L95) is notably mutated in the two instances of L3 isomerization within the set, cixutumumab and anifrolumab. The absence of this proline in cixutumumab is as generally expected for lambda L3 sequences. Conversely, in anifrolumab, with a kappa L3 sequence, position L95 is mutated to alanine, relative to proline in the germline VK3-20. Of 101 nine-residue long L3 sequences, only six in our set lack a proline at position L95 (Table S7A). Of those six, anifrolumab is the only one that contains a D (L92) at any position within CDRL3; it also shows a relatively high rate of isomerization (8.8% modification upon low pH stress). Likewise, lirilumab has the same L3 length of nine, does not have proline at position L95, and shows low amounts of deamidation (2.1%) at Asn (position 93) upon high pH stress. Meanwhile, four closely related L3 sequences (those in the samples corresponding to ofatumumab, daratumumab, olaratumab, and tabalumab) are identical across the L91-L95 stretch, with proline at L95, and did not undergo deamidation at Asn in position L93 (Table S7B).
Chemical modifications observed under both low and high pH stress
Deamidation modifications were observed at 21 unique sites under low pH stress across 18 mAbs (Table S4). All deamidation events occurring at low pH were also observed under high pH conditions, with the one exception being the Asn at position H52 of urelumab, which showed no observable deamidation (< 2%) under high pH stress (Fig. S4A). The motif at position H52 of urelumab is NH, which may be responsible for the discrepancy. Histidine, with a sidechain pKa of 6.0, has a weak affinity for H+ at pH 5.5 and potentially facilitates the formation of the Asu intermediate by donating a proton to the OH− leaving group.45,46
In contrast to the congruity observed for the deamidation modifications across both pH conditions, a similar comparison of modifications with identical masses but different retention times reveals that only 7 of 17 cases occurring at high pH were also found after low pH stress, while the remaining 10 cases do not have corroborating evidence at low pH (Fig. S4B, Table S5). This observation is quite striking considering previous characterizations of the pH dependence of isomerization rates. For example, the rate of isomerization measured in a model peptide, VYPDGA, increased 10-fold from pH 8 to pH 5,15 suggesting that any isomerization detected at high pH should also be detected at low pH with higher intensity. In several cases our data did not trend as expected, suggesting that there may be alternative chemistries operating at the high pH condition. It was previously reported that several IgG antibody samples showed racemization modifications after being stressed at alkaline pHs and elevated temperatures.19 Since the conversion of L-amino acids to their D-enantiomers does not lead to any mass shift either, our current experimental measurement cannot discriminate between racemization and isomerization (Table S5B). Due to this ambiguity, these 10 exceptional cases are not included in further analysis or discussion of isomerization.
Discussion
Of the 131 isotype-matched clinical-stage-like mAbs surveyed in our study, 63 (48%) were chemically modified under low or high pH stress conditions. A total of 33 mAbs incurred deamidation upon high pH stress (for a total of 39 events), while 28 mAbs underwent isomerization upon low pH stress (31 events). Only 7 of the 63 mAbs were susceptible to both isomerization and deamidation. Notably, 18 out of 131 mAbs were modified by deamidation under low pH stress. In contrast, 17 out of 131 mAbs were modified by either isomerization or racemization under the high pH condition.
As we continue the interpretation of our data, it is important to point out the possibility of false negative results when unmodified and degraded peptides cannot be separated chromatographically (see Methods for details). The only way to avoid such false negative interpretation would be to synthetically generate all possible forms of modified peptides and confirm that the corresponding retention times are sufficiently different from the unmodified peptides. As this is an especially onerous task at the scale of this study, these control experiments were not performed.
While the isomerization reaction requires the Asu intermediate, the deamidation modification may proceed through alternate parallel mechanisms (Figure 1). In addition to a proposed deamidation mechanism that populates an isoimide intermediate that hydrolyses to Asp,14 the direct deamidation mechanism may also contribute to our observed modifications. Under acidic conditions (pH< 4), deamidation will preferentially occur via direct hydrolysis.11,47 Some research even suggests that at neutral and basic pHs, direct hydrolysis may occur at competitive rates to the Asu-mediated mechanism.13 It is important to emphasize that at any pH, all reactions occur simultaneously with partitioning of the reaction pathways dependent on pH, temperature,9,48 acidity of the subsequent (n + 1) backbone amide hydrogen,45,49 and the chemistry and sterics of the local environment.12,13,32 These observations allow us to better understand our experimental conditions, confirm chemical degradation assignments across experimental conditions, and may help to identify other modes of risk factors in the future.
Comparison to historical data
Historical review of the literature reveals that certain sequence motifs are particularly labile to modification under the stress conditions used in this study.17-19 Previously reported canonical motifs are NG, NS, NN, NT, NH and DG, DS, DD, DT, and DH for deamidation and isomerization modification under our conditions, respectively.23,32 Three publications contain the INNs (enabling unambiguous identification of the amino acid sequence) of the mAbs being investigated and study chemical modifications under reaction conditions similar to those used in this study. Sydow et al., is the most comprehensive, with isomerization and deamidation data (resulting post-incubation at pH 6.0 and 40 °C for two weeks) for 37 mAbs, 13 of which are identified by their INN.23 Huang et al., provides high pH stress (pH 10.4 at 25 °C for 7 days) data for the Fc fusion etanercept,50 and Yang et al., published low pH data (pH 6.0 at 40 °C for 14 days) on trastuzumab.40 All other available publications either do not include a molecule’s name, do not have comparable reaction conditions, investigate the constant region, or interrogate the reactions of interest with peptides or antibody fragments (instead of full-length mAbs).
Although the available historical data are limited, the results presented in this work corroborate previous observations by Sydow et al., in that the majority of deamidation and isomerization events are observed at defined canonical motifs at generally the same rank-order of modification incidence.23 Although the percent modification observed for NG, NS, DG and DS motifs tends to be lower (“blue circles”, Figure 4) than as described by Sydow et al. (“red circles”, Figure 4), the observed rank-order in our data is consistent with experiments conducted on penta-peptides.32 The subtle differences observed when compared to the order observed by Sydow et al. are likely a consequence of the disparate sample sizes.23
Figure 4.
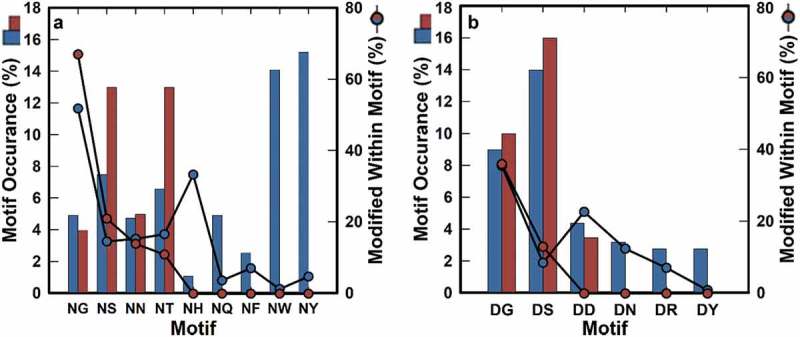
Frequency of liability motifs in all CDRs. Circles show percentages of modified deamidation motifs under high pH stress (a), and isomerization motifs under low pH stress (b) for the current dataset (blue) and the one adapted from Sydow et al.23 (red), respectively. There is no available motif occurrence data for NF, NH, NQ, NW, NY, DN, DR, and DY in the Sydow et al., dataset (red). Percentages shown are the percentages of total motif occurrence (bar graph).
Of the 13 named mAbs evaluated by Sydow et al.,23 12 samples are also within our dataset. Of these 12, nine show no deamidation or isomerization in either dataset. The remaining three mAbs (natalizumab, nimotuzumab, and trastuzumab) all have deamidation consistently identified at the same CDRs and motifs in both studies. Asparagine isomerization of trastuzumab CDRL3 and deamidation of natalizumab CDRH2 were consistently observed in both datasets. Nimotuzumab, while found to be isomerized in both datasets, has the modification assigned to CDRH3 at position H100 (DS) by Sydow et al.,23 but at H100W (DG) in the data presented here. Our data unambiguously assign this modification to the H100W position, suggesting that subtler differences in reagents may be responsible for this discrepancy (Fig. S5). The samples studied by Sydow and colleagues do correspond to actual clinical material for those 12 (approved) antibodies;23 the fact that their results largely agree with those within this study suggest that the differences in material (isotype in some cases, expression host, production, and purification in all cases) are unlikely to affect the kind of data reported here.
Isomerization events at non-canonical motifs
Within this dataset isomerization is rarely observed at non-canonical motifs, occurring twice in CDRH3 and twice in CDRL3 (Table S3). In all cases the modified residue is within a variable length insertion sub-region of the CDR. The two cases in CDRL3 are both DN motifs in the L30 sub-region of gemtuzumab and glembatumumab. Although DN and the related ND motifs have not been found to be liabilities existing within full-length mAbs in the literature (and are therefore not defined as canonical motifs in this manuscript), peptide studies suggest that isomerization51 and deamidation32 at these motifs transpire on comparable timescales to the previously defined canonical motifs.
Peptide studies also indicate that the modified DY and DR motifs within the H3 of fletikumab and olaratumab, respectively (Table 1), are not expected to be modified on the timescale of our experiments. It is unclear if the local sequence contributes to isomerization at these motifs. Previous work on the related deamidation chemistry at the peptide level suggests that the obligate Asu intermediate is not likely to occur when the n + 1 residue is bulky, suggesting that the observations at the DY and DR motifs are expectedly rare and likely the result of favorable conformations or local environment (i.e., near-in-space to a proton donor).32 Interestingly, the fletikumab DY motif is preceded by a histidine residue that, we speculate, may act as a proton donor under favorable conditions, with a chemistry similar to that proposed for a histidine in the n + 1 position.45 In contrast, the DR motif in olaratumab is unlikely to contribute to Asu formation. Arginine’s ionizable group has been previously noted to be incapable of similar direct proton transfer to the preceding amide due to the length of the side chain,32 and is unlikely to donate a proton at a solution pH of 5.5. However, arginine has been shown to facilitate Asu formation of an adjacent NG motif at near neutral and slightly acidic pHs by coordinating buffer anions that subsequently induce Asu cyclization via proton transfer.52 Therefore, if arginine is contributing to the observed modification, it would be highly dependent on conformation and the chemistry of the local environment.
Table 1.
Non-canonical motifs. Instances of non-canonical motifs are found across all CDRs. All deamidation events at non-canonical motifs contain a reverse motif at the modification position (indicated in bold blue). Non-canonical motifs are modified at low pH in only two instances, one of which is not located at a “reverse motif” (olaratumab).
| mAb | Motif | % Modified | Modification | Stress | CDR | CDR Sequence |
|---|---|---|---|---|---|---|
| robatumumab | NF | 45.20 | Deamidation | High pH | H3 | ARLGNFYYGMDV |
| inotuzumab | NY | 20.20 | Deamidation | High pH | H3 | TREGYGNYGAWFAY |
| gemtuzumab | NY | 2.60 | Deamidation | High pH | L1 | RASESLDNYGIRFLT |
| olaratumab | NY | 2.60 | Deamidation | High pH | H3 | ARQSTYYYGSGNYYGWFDR |
| lenzilumab | NY | 2.30 | Deamidation | High pH | H1 | YSFTNYYIH |
| gemtuzumab | NQ | 2.30 | Deamidation | High pH | L2 | AASNQGS |
| lirilumab | NW | 2.10 | Deamidation | High pH | L3 | QQRSNWMYT |
| fletikumab | DY | 10.10 | Isomerization | Low pH | H3 | AREPLWFGESSPHDYYGMDV |
| olaratumab | DR | 6.00 | Isomerization | Low pH | H3 | ARQSTYYYGSGNYYGWFDR |
Deamidation events at non-canonical motifs
The generality of the chemical stability of canonical motifs appears to apply less to CDRH3 deamidation where, of four events observed, only one occurred at a canonical deamidation motif (NT). The other three events occurred at NF (one) and NY (two) motifs (Figure 2(c)). The NF deamidation site is unique within this data set and had not been previously reported to occur within antibody CDRs. By contrast, deamidation occurring at the previously unreported NY motif is not unique to CDRH3, with instances occurring at low frequency in CDRH1 and CDRL1. Interestingly, all observed deamidation motifs in CDRH3 have a preceding glycine residue that is not unique nor required for non-canonical deamidation events within CDRs. Across all CDRs, nearly all non-canonical deamidation motifs have a relatively bulky residue at the n + 1 position (F, Y, W, and to a certain extent, Q). All non-canonical motif modification instances occur at sequences preceded by G (n = 3), D (n = 1), T (n = 1), or S (n = 2) residues, which all lead to putative “reverse” canonical motifs. The implications of this observation are not obvious, as the non-canonical motif deamidation reactions are observed to occur with half-lives over 63 times longer in penta-peptides GGN[F/Y/W/Q]G compared to those observed in the penta-peptide GGNGG.32 Considering that the analogous glutaminyl deamidation via a 6-membered, glutarimide, intermediate53,54 occurs with reaction half-times only 6 times longer than their asparaginyl counterparts,55 and that these reaction products are not observed in this data set, it is unlikely that the observed modifications at non-canonical motifs are occurring in the absence of other local environment factors. If the “reverse” motif is partially responsible for the apparent acceleration of non-canonical motif reaction rates, the n-1 residues may contribute to general local flexibility as to facilitate Asu cyclization. This hypothesis is supported by experiments on degradation rates in unstructured model penta-peptides that demonstrate, at the sequence level, that the preceding residue (particularly G) can modestly accelerate the deamidation reactions.32 The slightly accelerated reaction kinetics observed with a preceding G residue are attributed to the assumed increased local flexibility.32 Presumably the increased local dynamics increases the water occupancy of the Asn sidechain to initiate the reaction, as well as access to the subsequent residue’s backbone amide. Thus, increasing the local flexibility at the n-1 position is consistent with previous observations of structural dependence on deamidation rates,12,21 and is a reasonable explanation for the observed modifications at non-canonical motifs.
Potential racemization under high pH conditions
In the Asp isomerization reaction, the Asu intermediate is first populated, with isomerization occurring because of subsequent hydrolysis of the cyclized intermediate. Because the Asu intermediate can be populated under either high or low pH, it is expected that isomerization-labile residues would be detected in the products of both treatments. However, agreement is not observed in 10 of 17 occurrences. Instead, in these 10 cases, an isomerization modification is detected only at the high pH condition without the corresponding observation at low pH (Fig S4B). Therefore, the observed retention time shift may be the result of a concurrent racemization reaction (Table S5B). It has been previously suggested that racemization is a product of proton abstraction from the Cα of amino acid residues,19 a reaction that will accelerate under high pH conditions. Our observation of this phenomenon in only the high pH stress is consistent with existing racemization data produced with alkaline treatment. The detection of racemization has been previously demonstrated via a shift in retention time using reverse-phase chromatography.19
Because our detection method for Asp isomerization similarly relies on retention time shifts and MS/MS identification of chromatographically distinct but identical masses, in the absence of extensive isotope labeling experiments we cannot discriminate between racemization or isomerization products. Further, because racemization is not limited to Asp residues, and most minimal peptides contain Ser and/or Thr residues, we are unable to determine which chemistry is operative. With the evidence at hand, we can only confidently suggest that one of the presumed isomerization events at high pH, observed in the H3 of mepolizumab, is likely S or T racemization. In this case, we have data that suggests that the only D that could not be isomerized would be at a DP motif. Assuming the isomerization chemistry occurs through an Asu intermediate, the proline would prevent isomerization at this position.32 Therefore, racemization is the most likely explanation for the observed retention time shift.
Structural analysis
We identified five positions where Asp and Asn residues are frequently present yet rarely modified; H52A (n + 1 = P), H31 (n + 1 = Y), H101 (n + 1 = Y, I, L, or V), L28 (n + 1 = I or V), and L53 (n + 1 = L or R). In all cases, the n + 1 position is a residue that constitutes a non-canonical motif, suggesting that the limited occurrence of canonical motifs at these positions are responsible for the local chemical stability. Conversely, cold-spot positions where a canonical motif typically occurs and is not modified are rare, only being observed once within CDRH2 at H61 (DS). At this position, the DS motif occurs 37 times, none of which incur detectable isomerization. This cold-spot is likely the result of the DS motif residing in a generally structured region of the antibody. An analysis of 942 unique antibody structures from the Protein Data Bank (PDB)56 reveals that position H61-H63 are part of a secondary structure turn motif in all but 34 instances, including the 181/185 cases with the canonical DS isomerization motif at H61-H62 (data not shown). Therefore, this region is likely structurally constrained to disfavor the formation of the Asu intermediate.
Turning our attention to the hot-spots, modifications of H54 and H98 typically occur at NG and DG motifs. However, in the CDRL1 “insertion sub-region” (L30 to L30F), motifs DG, DS, DD and DN are all nearly equally modified. Further, 13/18 CDRL1 deamidation events occur at positions L30A-L30F, i.e., in antibodies with 12 amino acid and longer L1s, suggesting that loop flexibility may encourage Asu formation.
Although local structure is known to modulate deamidation and isomerization rates,21,23,30 the ability to predict such local structural effects from sequence alone is not fully developed. For instance, 9.8% deamidation was observed at H54 upon high pH stress of rituximab. The related ocrelizumab molecule, with an identical CDRH2 sequence, accumulates only 3.5% deamidated species under the same condition. A comparison of gemtuzumab and lintuzumab echoes the same results, with only two sequence differences at positions H58 and H65. Similarly, gemtuzumab was found to accumulate 17.3% deamidated species at H54 while the same position was unmodified in lintuzumab, which has a closely related sequence. Of pembrolizumab and ozanezumab (which differ in their CDRH2 sequences at just 3 positions H50, H59, and H65, but have the same NG motif at position H54), only the former shows deamidation at H54 upon stress (16.9%). For all three pairs discussed above, the less chemically stable antibody shows modestly lower thermal stability as assessed by the melting temperature (Tm) of the antigen-binding fragment (Fab).31
An additional example concerns the CDRH2 of tabalumab, where the NH motif shows 4.3% deamidation upon high pH stress; two other samples in the set (patritumab and zanolimumab) have identical sequences across the entire CDRH2 yet show no detectable modification. It should be noted that the apparent N-linked glycosylation motif, NHS, does not lead to actual glycan occupancy in any of the three cases; see McCann et al.,57 for a literature precedent of similar sequences. The Fab Tms previously reported were 64, 71.5 and 80.0 °C, respectively, for tabalumab, patritumab and zanolimumab.31 However, over the entire data set, correlation between Tm and chemical stability (as assessed in this work) does not reach statistical significance (data not shown). These findings suggest that the complex global dynamics of the molecule may result in structural differences in the CDRs that likely contribute to the observed chemical stability. In light of this observation, we suspect that the sequence and structure inputs into recent predictive methods23,30 may be too similar (as currently encoded or examined) to allow for meaningful differentiation between the modified and non-modified instances. For example, referring to the NH example above, preliminary homology models of the three antibodies (tabalumab, patritumab and zanolimumab) followed by analysis as in Yan et al.30 leads to very similar values for the key descriptors (secondary structure, solvent accessibility and Cγ to N distance) at the NH motif, and thus no suggestion that they may differ in their deamidation propensity.
Our data suggest that the absence of proline at L95 in nine-residue long CDRL3s may be a contributing factor to the chemical stability of either isomerization or deamidation within the segment. Of the 49 kappa mAbs with L3 of length 9 in our dataset with at least one N or D within the CDR, 47 have proline at position L95 and no modification is observed for any of them. Modification is only observed in the two exceptional cases listed earlier (anifrolumab and lirilumab, exhibiting isomerization and deamidation, respectively). It had been proposed previously that higher local flexibility,23,30 as reflected in higher B-factor values,58 may correlate with propensity for isomerization or deamidation. To examine the specific effect due to a presence or absence of Pro at L95, we performed an analysis of B-factors using a set of 359 crystal structures of un-complexed kappa antibodies from the PDB having L3 length of 9; n = 337 with Pro and n = 22 without Pro. A significant difference emerges between the sum of average B-factors for the backbone atoms of residues at the L91 to L94 positions: those lacking Pro at L95 have larger B-factor values (data not shown). These effects are independent of the resolution at which the respective sets of structures were solved. Thus, greater L3 loop flexibility for anifrolumab and lirilumab in the absence of the L95 Pro could encourage the formation of the mechanistically common Asu intermediate and ensuing chemical degradation.
Degradation events and stage of development
In our previous work studying biophysical properties of a similar set of clinical-stage antibodies, we noticed a clear trend for molecules in more advanced stage of development having fewer biophysical problems than those in earlier stages (see Figure 3(a) of Xu et al.5). We wondered if such an effect could also occur here. Surprisingly, we found more deamidation events in approved antibodies than in those in clinical Phase 2 or 3 studies (Fig. S6). Reasoning that this may be a function of the time in which development was initiated, and that many approved molecules originated as long as two decades ago, we looked explicitly at a timeline of canonical sequence motifs (using time of INN designation as a surrogate for relative “age” of a given mAb; see Figure S7). An apparent trend emerges revealing that, since the early 2000s, instances of these motifs have been declining, (Fig. S8A). The same analysis applied to DG motifs reveals the opposite trend (Fig. S8A). However, despite these trends, there has been little change in the number of mAbs containing chemical liabilities over time (Fig S8).
Chemical liabilities in a clinical context
This study was designed to characterize the chemical stability of a set of transiently expressed mAb samples with variable regions matching clinical-stage antibodies under commonly applied stressing conditions. Under these conditions, we identified at least one deamidation and/or isomerization site (in the variable region) in 56 of 131 mAbs, with a wide range of observed degradation. Similar approaches are usually applied during late-stage discovery of mAbs for lead selection to identify candidates with the best chemical stability profiles or to identify degradation sites from the leading candidates for re-engineering to de-risk downstream process development.
It was surprising to observe relatively high levels of degradation (under our current stressing conditions) for some mAb samples with variable region matching approved drugs, such as trastuzumab. However, multiple possible degradation variants can complicate process development. The lack of chemical stability may also render liquid formulation challenging, as is evident from Herceptin’s (trastuzumab) lyophilized formulation.
It is worth noting that all the mAb samples in this study were intentionally designed as IgG1 iso-type to compare properties of variable regions in the background of a common constant region. Although the agreement of our data with data collected using commercial material23 suggests that our method is generally non-disruptive, subtle differences may be expected when the same variable region is fused to a different isotype such as IgG2 or IgG4. Additionally, the conditions used in this study are intended to accelerate rates of chemical degradation, which could lead to over estimation of the degradation levels for corresponding commercial materials in a clinical environment.
Conclusion
Rapid assessment strategies in early post-discovery processes to evaluate the chemical stability of therapeutic proteins are necessary to identify and correct particularly labile residues prior to committing time and resources towards accommodating such inherent risks downstream. The wealth of knowledge on this topic for mAbs in advanced stages of development is fragmented and incomplete. In the interest of improving the state of the field, we produced and tested 131 samples corresponding to clinical-stage mAb molecules and subjected them to low and high pH stresses to rapidly induce isomerization and deamidation reactions, respectively. Specifically, CDRs H2, H3, and L1 were found to be particularly labile to deamidation and isomerization modifications. Within those CDRs, just three motif sequence positions (H54, H98, and L30) account for most modifications. In combination with our previously published results on the stability to oxidation of 121 clinical-stage mAbs,7 the chemical liability landscape for clinical-stage mAbs is currently well described.
Materials and methods
mAb production
All 131 antibodies were expressed in HEK293 cells as reported earlier by Jain et al.31 All variable region sequence information for the 131 samples is included in a supplemental workbook. The VH and VL encoding gene fragments (Integrated DNA Technologies, Coralville, IA) were subcloned into heavy and light chain pcDNA 3.4+ vectors (ThermoFisher, Waltham MA). All mAbs were expressed as IgG1 isotype. The corresponding vectors were co-transfected into HEK293 suspension cells. After six days of growth, the cell culture supernatant was harvested by centrifugation and passed over Protein A agarose (MabSelect SuRe™, GE Healthcare Life Sciences, Pittsburgh PA). The bound antibodies were then washed with phosphate-buffered saline and eluted with buffer (200 mM acetic acid (BDH, 3092)/50 mM NaCl (Sigma, S9888) pH3.5) into 1/8th volume 2 M HEPES, pH 8.0. The final products were buffer exchanged into 25 mM HEPES (TEKNOVA, H1035), 150 mM NaCl, and pH 7.3.
Low and high pH treatment
Samples (1 mg/mL) were buffer exchanged at 100 µL volumes using 40K Zeba Spin Desalting Columns (Thermo, 87766). High pH samples were exchanged into 20 mM Tris (Sigma, 93362), 10mM EDTA (Sigma, E6758), pH 8.5. Low pH samples were exchanged into 50 mM sodium acetate (Sigma, S2889), pH 5.5. Low pH and high pH samples were incubated at 40 °C for 2 weeks and 1 week, respectively. Reactions were stored at −80 °C prior to further treatment or analysis.
Tryptic peptide mapping
All mAb samples were digested using trypsin (Pierce 90,058, 5 µg/mL) as previously described with minor modification.7 Briefly, 5 µL of each sample (1 mg/mL) was unfolded and reduced by addition of 8 M urea (JT Baker 4206), 100 mM ammonium bicarbonate (Sigma, 09830), 20 mM DTT at pH 7.8, and incubated at 70 °C for 3 minutes. Reduced samples were subsequently mixed with 100 µL of 5 ng/µL trypsin in 20 mM ammonium bicarbonate, 5 mM DTT at pH 7.8. Low pH stressed samples were digested at 37 °C for 5 minutes and high pH stressed samples were incubated at 37 °C for 20 minutes. Digestions were quenched by the addition of trifluoroacetic acid (final concentration 0.8%). For comparison purposes, unstressed samples were digested side-by-side with stressed samples. All tryptic peptides were eluted on an Acquity Ultra Performance liquid chromatography (UPLC) system (Waters) equipped with a BEH C18 column (Waters, 1.7 µm, 2.1 × 150 mm, 186003687). The flow rate was 0.2 mL/min, and the column temperature was 60 °C. The elution gradient was 0% to 10% solvent B (0.1% formic acid at acetonitrile) in 9 minutes, to 40% B over 60 minutes, and then to 95% B in 2 minutes. Peptides separated on the column were detected by a Q ExactiveTM mass spectrometer (Thermo) in positive electrospray ionization mode. The instrument parameters were set as spray voltage of 3.5 kV, capillary temperature of 320 °C, sheath gas flow rate at 35 and aux gas flow rate at 10 and S-lens RF level at 70. MS spectra were acquired at the scan range of 150–2000 m/z with a resolution of 70,000. Data-dependent MS/MS was performed at a resolution of 17,500, and five most abundant ions from the previous MS scan were selected for higher-energy C-trap dissociation fragmentation (30% normalized collision energy).
Acquired MS data were analyzed using Biopharma Finder software (Thermo Scientific) followed by manual inspection to ensure correct assignment and relative quantification accuracy. A lower limit cut-off of 2% modified material was used to distinguish between modified and unmodified material, due mainly to the signal requirements for verifying the modification. In this assessment, only the variable regions were investigated for chemical stability.
For all deamidation events, site identifications were made using both Biopharma Finder software and manual inspection of MS/MS spectra of deamidated peptides. In one case, the sample corresponding to dacetuzumab, there was insufficient data to determine which one of the two Asn residues were modified in the tryptic peptide (annotated in Table S2). In this case, the assignment was made to the more chemically reactive motif. For isomerization events observed after low pH stress, residue assignments primarily relied on the observation of the Asu intermediates. Including overlapping mis-cleaved peptides in our analysis also helped to exclude alternative potential modification sites, when available. In one case, romosozumab, there is insufficient MS/MS data to determine the exact residue being isomerized, narrowing the options down to two DD motifs (annotated in Table S3). In this case, the assignment produced by BioPharma Finder software was used. For the isomerization modifications observed after high pH stress, Asu species were expectedly not observed. These modification sites were identified by aligning the retention times of modified peptides with isomerized ones observed after low pH stress (Table 5A). If there was no orthogonal corresponding data at low pH conditions, different mis-cleaved peptides were overlapped to provide the minimum fragments containing those modification sites (Table S5B).
Deamidation and isomerization modifications rely on sufficient chromatographic separation for identification. Deamidation modifications, having a mass shift of + 1 Da can be isotopically resolved using the Q Exactive, even when poor chromatographic separation exists. Isomerization modifications do not have this advantage and can be overlooked when chromatographic peak separation is poor. This separation relies on several factors, including peptide hydrophobicity, conformation, sequence position, the specific gradient and column being used. We find that the method used here can sufficiently resolve isomerization (Fig S9) and deamidation (Fig S10) modifications occurring on long peptides, suggesting that our method is generally applicable. We also note that the chromatographic advantage of using a UPLC system over an HPLC system has pronounced resolution differences (Fig S10) that aid in making these identifications possible.
Sequence analysis
Antibody sequences were numbered using a modification of the Kabat-Chothia rules as described previously.42 The rules derive from Kabat’s original sequence-based CDR definitions,59 and incorporate updates that structurally position CDR insertions and deletions outside of the conserved β-sheet framework. Accordingly, the listed sequences for CDRs L1-L3 and H1-H3 span positions 24–34, 50–56, 89–97 and 27–35, 50–65 and 93–102, respectively. For CDRs L1 and L3, insertions are located after position 30 (i.e., 30A..30F) and 95 (i.e., 95A..95D). For CDRs H1 and H2, insertions are located after position 31 (i.e., 31A..31D) and 52 (i.e., 52A..52D). For CDR H3, a custom numbering scheme was used; briefly the original Kabat insertion point at position H99 was moved to H100; in addition, insertions are added to the C-terminal end of H99 as H99A, H99B, etc. and, as appropriate, also added to the N-terminal end of H100 in descending order as H100Z, H100Y, etc. (the approach is similar in spirit to that employed in the IMGT® numbering scheme44). The supplemental workbook provides 131 examples of how the scheme is applied.
Abbreviations
- Ala (A)
Alanine
- Arg (R)
Arginine
- Asn (N)
Asparagine
- Asp (D)
Aspartic Acid
- Asu
Aspartyl-succinimide
- CDR
Complementarity-Determining Region
- EIC
Extracted Ion Chromatography
- Gly (G)
Glycine
- HC
Heavy chain
- His (H)
Histidine
- HPLC
High Performance Liquid Chromatography
- IgG
Immunoglobulin G
- Ile (I)
Isoleucine
- IMGT®
ImMunoGeneTics information system®
- INN
International Non-proprietary Name
- IsoAsp
Isoaspartic Acid
- Leu (L)
Leucine
- mAbs
Monoclonal Antibodies
- Met (M)
Methionine
- Pro (P)
Proline
- QC
Quality Control
- RF
Radio Frequency
- Ser (S)
Serine
- Thr (T)
Threonine
- TPM
Tryptic Peptide Mapping
- Tm
Melting Temperature
- Tyr (Y)
Tyrosine
- UPLC
Ultra Performance Liquid Chromatography
- Val (V)
Valine
Acknowledgments
All the antibodies characterized in this work were produced, purified and subjected to quality control through the combined efforts from the molecular core, high throughput expression and protein analytics departments at Adimab LLC. We appreciate the critical review, discussion, guidance and support provided by Tingwan Sun, Noel Pauli, Anna Wec, Amy Bray, Eric Krauland, K. Dane Wittrup and Tillman Gerngross.
Supplemental material
Supplementary material for this article can be accessed here.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Dimasi JA, Feldman L, Seckler A, Wilson A.. 2010. Trends in risks associated with new drug development : success rates for investigational drugs. Clin Pharmacol Ther. 87:272–277. doi: 10.1038/clpt.2009.295. [DOI] [PubMed] [Google Scholar]
- 2.Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC.. 2005. Monoclonal antibody successes in the clinic. Nat Biotechnol. 23:1073–1078. doi: 10.1038/nbt0905-1073. [DOI] [PubMed] [Google Scholar]
- 3.Zurdo J. 2013. Developability assessment as an early de-risking tool for biopharmaceutical development. Pharm Bioprocess. 1:29–50. doi: 10.4155/pbp.13.3. [DOI] [Google Scholar]
- 4.Jarasch A, Koll H, Regula JT, Bader M, Papadimitriou A, Kettenberger H. Developability assessment during the selection of novel. J Pharm Sci. 2015; 104:1885–1898. [DOI] [PubMed] [Google Scholar]
- 5.Xu Y, Roach W, Sun T, Jain T, Prinz B, Yu T, Torrey J, Thomas J, Wittrup KD, Bobrowicz P, et al. Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system : a FACS-based, high-throughput selection and analytical tool. Protein Eng Des Sel. 2013;26:663–670. doi: 10.1093/protein/gzt047. [DOI] [PubMed] [Google Scholar]
- 6.Wang WEI, Singh S, Zeng DL, King K, Nema S; Al WET . antibody structure, instability, and formulation. J Pharm Sci. 2007; 96:1–26. doi: 10.1002/jps.20727. [DOI] [PubMed] [Google Scholar]
- 7.Yang R, Jain T, Lynaugh H, Nobrega RP, Lu X, Boland T, Burnina I. 2017. Rapid assessment of oxidation via middle-down LCMS correlates with methionine side-chain solvent-accessible surface area for 121 clinical stage monoclonal antibodies. MAbs. 9:646–653. doi: 10.1080/19420862.2017.1290753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bach RD, Su M, Schlegel BH. Oxidation of amines and sulfides with hydrogen peroxide and alkyl hydrogen peroxide. The nature of the oxygen-transfer step. J. Am. Chem. Soc. 1994;116:5379–5391. doi:10.1021/ja00091a049. [Google Scholar]
- 9.Peters B, Trout BL. 2006. Asparagine deamidation : pH-dependent mechanism from density functional theory. Biochemistry. 45:5384–5392. doi: 10.1021/bi052438n. [DOI] [PubMed] [Google Scholar]
- 10.Patel K, Borchardt RT. chemical pathways of peptide degradation. II. Kinetics of deamidation of an asparaginyl residue in a model hexapeptide. Pharm Res. 7;1990:703–711. [DOI] [PubMed] [Google Scholar]
- 11.Bhatt NP, Patel K, Borchardt RT. Chemical pathways of peptide degradation. I. deamidation of adrenocorticotropic hormone. Pharm Res. 7;1990:593–599. [DOI] [PubMed] [Google Scholar]
- 12.Geiger T, Clarke S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. J Biol Chem. 262;1987:785–794. [PubMed] [Google Scholar]
- 13.Catak S, Monard G, Aviyente V, Ruiz-Lopez MF. 2009. Deamidation of asparagine residues : direct hydrolysis versus succinimide-mediated deamidation mechanisms. J Phys Chem. 113:1111–1120. doi: 10.1021/jp808597v. [DOI] [PubMed] [Google Scholar]
- 14.Clarke S. 1987. Propensity for spontaneous succinimide formation from aspartyl and asparaginyl residues in cellular proteins. Int J Pept Protein Res. 30:808–821. doi: 10.1111/j.1399-3011.1987.tb03390.x. [DOI] [PubMed] [Google Scholar]
- 15.Wakankar AA, Borchardt RT. 2006. Formulation considerations for proteins susceptible to asparagine deamidation and aspartate isomerization. J Pharm Sci. 95:2321–2336. doi: 10.1002/jps.20740. [DOI] [PubMed] [Google Scholar]
- 16.Wakankar AA, Borchardt RT, Eigenbrot C, Shia S, Wang YJ, Shire SJ, Liu JL. 2007. Aspartate isomerization in the complementarity-determining regions of two closely related monoclonal antibodies. Biochemistry. 46:1534–1544. doi: 10.1021/bi061500t. [DOI] [PubMed] [Google Scholar]
- 17.Klaene JJ, Ni W, Alfaro JF, Zhou ZS. 2015. Detection and quantification of succinimide in intact protein via hydrazine trapping and chemical derivatization. J Pharm Sci. 103:3033–3042. doi: 10.1002/jps.24074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang HZ, Nichols A, Liu D. 2009. Direct identification and quantification of aspartyl succinimide in an IgG2 mAb by rapigest assisted digestion. Anal Chem. 81:1686–1692. doi: 10.1021/ac802708s. [DOI] [PubMed] [Google Scholar]
- 19.Huang L, Lu X, Gough PC, De Felippis MR. 2010. Identification of racemization sites using deuterium labeling and tandem mass spectrometry. Anal Chem. 82:6363–6369. doi: 10.1021/ac101348w. [DOI] [PubMed] [Google Scholar]
- 20.Yan B, Steen S, Hambly D, Valliere-Douglass J, Bos TIMV, Smallwood S, Yates ZAC, Arroll T, Han Y, Gadgil H, et al. succinimide formation at asn 55 in the complementarity determining region of a recombinant monoclonal antibody IgG1 heavy chain. J Pharm Sci. 2009;98:3509–3521. doi: 10.1002/jps.21655. [DOI] [PubMed] [Google Scholar]
- 21.Capasso S, Salvadori S. 1999. Effect of the three- dimensional structure on the deamidation reaction of ribonuclease A. J Pept Re. 54:377–382. doi: 10.1034/j.1399-3011.1999.00111.x. [DOI] [PubMed] [Google Scholar]
- 22.Diepold K, Bomans K, Wiedmann M, Zimmermann B, Petzold A, Schlothauer T, Mueller R, Moritz B, Stracke JO, Mølhøj M, et al. Simultaneous assessment of asp isomerization and asn deamidation in recombinant antibodies by LC-MS following incubation at elevated temperatures. PLoS ONE. 2012;7(1):e30295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sydow JF, Lipsmeier F, Larraillet V, Hilger M, Mautz B, Mølhøj M, Kuentzer J, Klostermann S, Schoch J, Voelger HR, et al Structure-based prediction of asparagine and aspartate degradation sites in antibody variable regions. PLoS ONE. 2014;9(6):e100736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fukuda M, Takao T. 2012. Quantitative analysis of deamidation and isomerization in β 2- microglobulin by 18 O labeling. Anal Chem. 84:10388–10394. doi: 10.1021/ac302603b. [DOI] [PubMed] [Google Scholar]
- 25.Jr LWD, Qiu D, Cheng K. 2009. Identification and measurement of isoaspartic acid formation in the complementarity determining region of a fully human monoclonal antibody. J Chromatogr B. 877:3841–3849. doi: 10.1016/j.jchromb.2009.09.031. [DOI] [PubMed] [Google Scholar]
- 26.Nowak C, Cheung JK, Dellatore SM, Katiyar A, Bhat R, Sun J, Ponniah G, Neill A, Mason B, Beck A, et al. Forced degradation of recombinant monoclonal antibodies : A practical guide. MAbs. 2017;9:1217–1230. doi: 10.1080/19420862.2017.1368602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hains PG, Truscott RJW. 2018. Age-dependent deamidation of lifelong proteins in the human lens. Invest Ophthalmol Vis Sci. 51:3107–3114. doi: 10.1167/iovs.09-4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu M, Cheetham J, Cauchon N, Ostovic J, Ni W, Ren D, Zhou ZS. 2012. protein isoaspartate methyltransferase-mediated 18O-labeling of isoaspartic acid for mass spectrometry analysis. Anal Chem. 84:1056–1062. doi: 10.1021/ac202652z. [DOI] [PubMed] [Google Scholar]
- 29.Yang N, Tang QM, Hu P, Lewis MJ, Yang N, Tang QM, Hu P, Lewis MJ. Use of in vitro systems to model in vivo degradation of therapeutic monoclonal antibodies. Anal Chem. 2018;90(13):7896–7902. [DOI] [PubMed] [Google Scholar]
- 30.Yan Q, Huang M, Lewis MJ, Hu P. 2018. Structure based prediction of asparagine deamidation propensity in monoclonal antibodies. MAbs. 10:901–912. doi: 10.1080/19420862.2018.1478646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jain T, Sun T, Durand S, Hall A, Rewa N, Nett JH, Sharkey B. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci USA. 114;2017:944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robinson NE, Robinson AB. 2000. Molecular clocks. Pnas. 98:944–949. doi: 10.1073/pnas.98.3.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cacia J, Keck R, Presta LG, Frenz J. 1996. Isomerization of an aspartic acid residue in the complementarity-determining regions of a recombinant antibody to human IgE : identification and effect on binding affinity. Biochemistry. 35:1897–1903. doi: 10.1021/bi951526c. [DOI] [PubMed] [Google Scholar]
- 34.Harris RJ, Kabakoff B, Macchi FD, Shen FJ, Kwong M, Andya JD, Shire SJ, Bjork N, Totpal K, Chen AB. 2001. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J Chromatogr B. 752:233–245. doi: 10.1016/S0378-4347(00)00548-X. [DOI] [PubMed] [Google Scholar]
- 35.Patel CN, Bauer SP, Davies J, Durbin JD, Shiyanova TL, Zhang K, Tang JX. 2016. N+1 engineering of an aspartate isomerization hotspot in the complementarity-determining region of a monoclonal antibody. J Pharm Sci. 105:512–518. doi: 10.1016/S0022-3549(15)00185-9. [DOI] [PubMed] [Google Scholar]
- 36.Sreedhara A, Cordoba A, Zhu Q, Kwong J, Liu J. 2012. Characterization of the isomerization products of aspartate residues at two different sites in a monoclonal antibody. Pharm Res. 29:187–197. doi: 10.1007/s11095-012-0843-0. [DOI] [PubMed] [Google Scholar]
- 37.Rehder DS, Chelius D, Mcauley A, Dillon TM, Xiao G, Crouse-Zeineddini J, Vardanyan L, Perico N, Mukku V, Brems DN, et al. Isomerization of a single aspartyl residue of anti-epidermal growth factor receptor immunoglobulin γ 2 antibody highlights the role avidity plays in antibody activity. Biochemistry. 2008;47:2518–2530. doi: 10.1021/bi7018223. [DOI] [PubMed] [Google Scholar]
- 38.Ouellette D, Chumsae C, Clabbers A, Radziejewski C, Correia I. 2013. Comparison of the in vitro and in vivo stability of a succinimide intermediate observed on a therapeutic IgG1 molecule. MAbs. 5:432–444. doi: 10.4161/mabs.22965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Timm V, Gruber P, Wasiliu M, Lindhofer H, Chelius D. 2010. Identification and characterization of oxidation and deamidation sites in monoclonal rat/mouse hybrid antibodies. J Chromatogr B. 878:777–784. doi: 10.1016/j.jchromb.2010.01.036. [DOI] [PubMed] [Google Scholar]
- 40.Yang Y, Zhao J, Geng S, Hou C, Li X, Lang X, Qiao C, Li YAN, Feng J, Lv M, et al. Improving trastuzumab’s stability profile by removing the two degradation hotspots. J Pharm Sci. 2015;104(6):1960–1970. doi: 10.1002/jps.24435. [DOI] [PubMed] [Google Scholar]
- 41.Chothia C, Lesk AM, Tramontano A, Levitt M, Sith-Gill SJ, Air G, Sheriff S, Padlan EA, Davies D, Tulip WR, et al. Conformations of immunoglobulin hypervariable regions. Nature. 1989;342:877–883. doi: 10.1038/342708a0. [DOI] [PubMed] [Google Scholar]
- 42.Al-Lazikani B, Lesk AM, Chothia C. 1997. Standard conformations for the canonical structures of immunoglobulins. J Mol Biol. 273:927–948. doi: 10.1006/jmbi.1997.1354. [DOI] [PubMed] [Google Scholar]
- 43.T Te W, Kabat EA. An analysis of the sequences of the variable regions of bence jones proteins and myeloma light chains and their implications for antibody complementarity. J Exp Med. 132;1970:18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lefranc M, Giudicelli V, Ginestoux C, Jabado-Michaloud J, Folch G, Bellahcene F, Wu Y, Gemrot E, Brochet X, Lane J, et al. IMGT, the international immunogenetics information system. Nucleic Acids Res. 2009;37:1006–1012. doi: 10.1093/nar/gkn838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brennan TV, Clarke S. Effect of adjacent histidine and cysteine residues on the spontaneous degradation of asparaginyl- and aspartyl-containing peptides. Int J Pept Protein Res. 45;1995:547–553. [DOI] [PubMed] [Google Scholar]
- 46.Pace AL, Wong RL, Zhang YT, Kao Y, Wang YJ. asparagine deamidation dependence on buffer type, pH, and temperature. Protein Anal Chem. 102;2013:1712–1723. [DOI] [PubMed] [Google Scholar]
- 47.Song Y, Schowen RL, Borchardt RT, Topp EM. 2001. Effect of pH on the rate of asparagine deamidation in polymeric formulations : pH- rate profile. J Pharm Sci. 90:141–156. doi:. [DOI] [PubMed] [Google Scholar]
- 48.Oliyai C, Borchardt RT. 1993. Chemical pathways of peptide degradation. IV. Pathways, kinetics, and mechanism of degradation of an aspartyl residue in a model hexapeptide. Pharm Res. 10:95–102. doi: 10.1023/A:1018981231468. [DOI] [PubMed] [Google Scholar]
- 49.Radkiewicz JL, Zipse H, Clarke S, Houk KN. 2001. Neighboring side chain effects on asparaginyl and aspartyl degradation : an ab initio study of the relationship between peptide conformation and backbone NH acidity. J Am Chem Soc. 7:3499–3506. doi: 10.1021/ja0026814. [DOI] [PubMed] [Google Scholar]
- 50.Huang L, Chiang C, Lee Y, Wang T, Fong C, Chen S. 2016. Characterization and comparability of stress-induced oxidation and deamidation on vulnerable sites of etanercept products. J Chromatogr B. 1032:189–197. doi: 10.1016/j.jchromb.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 51.Yi LI, Beckley NIA, Gikanga B, Zhang J, Wang YJ, Chih H, Sharma VK. 2013. Isomerization of asp – asp motif in model peptides and a monoclonal antibody fab fragment. J Pharm S. 102:947–959. doi: 10.1002/jps.23423. [DOI] [PubMed] [Google Scholar]
- 52.Kirikoshi R, Manabe N, Takahashi O. 2017. Succinimide formation from an NGR-containing cyclic peptide : computational evidence for catalytic roles of phosphate buffer and the arginine side chain. Int J Mol Sci. 18:1–15. doi: 10.3390/ijms18020429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Capasso S, Mazzarella L, Sica F, Zagari A. First evidence of spontaneous deamidation of glutamine residue via cyclic lmide to a- and y-glutamic residue under physiological conditions. J Chem Soc Chem Commun. 1991:1667–1668. doi: 10.1039/C39910001667. [DOI] [Google Scholar]
- 54.Wright HT. 1991. Nonenzymatic deamidation of asparaginyl and glutaminyl residues in proteins. Crit Rev Biochem Mol Biol. 26:1–52. doi: 10.3109/10409239109081719. [DOI] [PubMed] [Google Scholar]
- 55.Robinson AB, Rudd CJ Deamidation of glutaminyl and asparaginyl residues in peptides and proteins. Curr Top Cell Regul. 1974;8(0):247–295. [DOI] [PubMed] [Google Scholar]
- 56.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The protein data bank. Nucleic Acids Res. 28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mccann KJ, Ottensmeier CH, Callard A, Radcliffe CM, Harvey DJ, Dwek RA, Rudd PM, Sutton BJ, Hobby P, Stevenson FK. 2008. Remarkable selective glycosylation of the immunoglobulin variable region in follicular lymphoma. Mol Immunol. 45:1567–1572. doi: 10.1016/j.molimm.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 58.Jia L, Sun Y. Protein asparagine deamidation prediction based on structures with machine learning methods. PLoS ONE. 2017;12(7):e0181347. doi: 10.1371/journal.pone.0181347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of proteins of Immunological Interest, 5th edn. Bethesda (MD): U.S.Department of Health and Human Services; 1991. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.